Article Text

Abstract
Introduction Intratumoral injections of novel therapeutics can activate tumor antigen-specific T cells for locoregional tumor control and may even induce durable systemic protection (against distant metastases) via recirculating T cells. Here we explored the possibility of a universal immunotherapy that promotes T-cell responses in situ and beyond, upon intratumoral injection of nanoparticles formulated with micron-sized crystals.
Methods Cucumber mosaic virus-like particles containing a tetanus toxin peptide (CuMVTT) were formulated with microcrystalline tyrosine (MCT) adjuvant and injected directly in B16F10 melanoma tumors. To further enhance immunogenicity, we loaded the nanoparticles with a TLR7/8 ligand and incorporated a universal tetanus toxin T-helper cell peptide. We assessed therapeutic efficacy and induction of local and systemic immune responses, including RNA sequencing, providing broad insight into the tumor microenvironment and correlates of protection.
Results MCT crystals were successfully decorated with CuMVTT nanoparticles. This ‘immune-enhancer’ formed immunogenic depots in injected tumors, enhanced polyfunctional CD8+ and CD4+ T cells, and inhibited B16F10 tumor growth locally and systemically. Local inflammation and immune responses were associated with upregulation of genes involved in complement activation and collagen formation.
Conclusions Our new immune-enhancer turned immunologically cold tumors into hot ones and inhibited local and distant tumor growth. This type of immunotherapy does not require the identification of (patient–individual) relevant tumor antigens. It is well tolerated, non-infectious, and affordable, and can readily be upscaled for future clinical testing and broad application in melanoma and likely other solid tumors.
- Melanoma
- Immunotherapy
Data availability statement
Data are available upon reasonable request. Data are available upon request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Basic and clinical data overwhelmingly show that immune responses against cancer are blocked by the hostile tumor microenvironment, which can be reversed by therapeutic interventions that enable T cells to overcome local immune suppression. However, current anticancer treatments are often too short-lived for sustaining T-cell responses. A new approach with immunogenic nanoparticles formulated in micron-sized crystals provides long-lasting T-cell support successfully inhibiting cancer growth. The components of this formulation have already been used in patients, accelerating the clinical translation of the novel therapy.
WHAT THIS STUDY ADDS
However, current anti-cancer treatments are often too short-lived for sustaining T cell responses.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
A new approach with immunogenic nanoparticles formulated in micron-sized crystals provides long-lasting T cell support successfully inhibiting cancer growth. The components of this formulation have already been used in patients, accelerating the clinical translation of the novel therapy.
Introduction
In situ (intratumoral) immunotherapy aims at generating local and systemic antitumor effects. This can mainly be achieved by priming an immune response in a tumor lacking pre-existing immunity or priming and boosting insufficiently immunogenic tumors via activating tumor-infiltrating lymphocytes (TILs).1 Intratumoral immunotherapy has several advantages such as increasing the local concentration of immunogenic drugs, enabling combination drug therapy, decreasing systemic adverse effects, and enhancing antigenicity and immunogenicity of tumor antigens recognized by T cells.2 Therefore, intratumoral immunotherapy can be a versatile approach for concomitant targeting T cells and modulating key components of the tumor microenvironment. In addition, it may be considered as ‘patient individualized’ treatment because tumor antigens are provided by the individual tumor itself.
Malignant melanomas usually arise from melanocytes in skin, mucosa, or retina. The majority of cutaneous or mucosal melanomas grow in visible and palpable locations. Early-stage surgical excision is the first-line treatment. Melanoma prognosis may be good in case of total tumor resection, but local, regional, or distant progression remains unfortunately relatively frequent.3 The therapeutic accessibility of melanoma lesions permits increased interest for developing in situ and local therapies.4 The concept is not new as Dr Coley has made the first attempts in 1891 by performing intratumoral injections of Coley’s toxin consisting of inactivated Streptococcus pyogenes and Serratia marcescens.5 In 1975, a case report was published of a patient in his late 70s with 64 cutaneous melanoma lesions as well as several pulmonary metastases who was treated with intratumoral injections of BCG. The patient experienced regression of all treated cutaneous lesions, in addition to 50% regression of distant metastases.6 Some recent examples of intratumoral treatment in melanoma includes CMP-001, a bacteriophage virus-like particle (VLP) loaded with A-type CpGs (NCT02680184/NCT03084640, formerly called QßG107) without any tumor antigens. This therapy is currently being examined in combination with systemic anti-programmed cell death protein-1(PD-1) in several clinical trials for patients with advanced melanoma with previous resistance to anti-PD-1 treatment. The recently collected data from the ongoing trials show manageable toxicity and potential to reverse resistance to anti-PD-1.8 Preclinically, Lizotte et al have shown that intratumoral injection of cowpea mosaic virus is partially effective in treating dermal B16F10 melanomas, forming central tumor necrosis.9 Another study has combined flexuous plant VLPs, potato virus x (PVX) with the chemotherapeutic drug doxorubicin (DOX) either packaged into the VLPs or admixed together to treat B16F10 melanoma. The results revealed that intratumoral injection of PVX VLPs could delay tumor progression, and coadministration of the VLPs with DOX enhanced antitumor immune responses and prolonged survival.10
The plant-virus derived CuMVTT VLPs (CuMVTT) are characterized by the triangulation number (T=3), which describes the symmetry of icosahedral particles built up by 180 monomers.11 CuMVTT lacks genetic material for replication and is accordingly non-infectious.11 The capsid monomer of CuMVTT has a size of 24 kDa and contains a tetanus toxin (TT)-derived peptide with a universal T-helper (TH) cell epitope to which most people have previously activated memory TH cells.12 CuMVTT also contain ssRNA that stimulates TLR7/8. It is derived from Escherichia coli and is packaged into the VLPs during the expression process. Once CuMVTT assembles into nanoparticles, it is long-term stable at 4°C as well as at 20°C.13 In several previous studies, we have demonstrated the immunogenicity of these VLPs in several animal models, including mice, cats, dogs, and horses.14–19
Microcrystalline tyrosine (MCT) is a biodegradable depot adjuvant that is being used since decades in allergy immunotherapy (AIT). Specifically, it is formulated with different native or modified allergens for subcutaneous injections in humans.20 21 Formulating MCT with different allergoids has been shown to be safe for more than 50 years in millions of people, including vulnerable populations and long-term treatment courses. MCT consists of micron-sized L-tyrosine (L-tyr) natural crystals that cannot readily enter the lymphatics and form a depot which persists at the injection site for days, causing local inflammation.22–24 Half-life at subcutaneous injection sites is 48 hours, and biodegradation is completed by roughly 1 week.21 Generally, depot-forming adjuvants such as MCT may enhance T-cell responses through several mechanisms but mostly through the ability to protect antigens from degradation and clearance and prolong antigen exposure to antigen-presenting cells (APCs), thereby enhancing T-cell activation and clonal expansion.25 In contrast to its extended use in AIT, MCT remains largely unexplored in cancer immunology. In our previous studies, we have shown that subcutaneous vaccination with CuMVTT-p33 displaying gp33 epitope derived from lymphocytic choriomeningitis virus and formulated with MCT controlled the growth of murine p33-transfected B16F10 tumors. Mechanistically, we observed strong activation of cytotoxic T lymphocytes and favorable changes in T-cell and granulocyte populations in the tumor microenvironment.26
In light of the still substantial challenges to improve immunotherapies against cancer, there is a need for effective, affordable, fast, and safe immune therapies that can be widely used for different types of solid tumors. Here, we demonstrate efficient antitumor responses induced by intratumoral injections of our novel ‘immune-enhancer’ consisting of CuMVTT nanoparticles formulated with micron-sized MCT.
Methods
Expression and production of CuMVTT VLPs
The expression and production of CuMVTT were carried out as described in Zeltins et al.12 Endotoxin level was measured and was confirmed to be <1000EU/mL.
Electron microscopy
Physical stability and integrity of CuMVTT were visualized by transmission electron microscopy using Philips CM12 EM. For imaging, sample grids were glow discharged and 5 µL of VLP solution was added for 30 s. The grids were then washed three times with ddH2O and negatively stained with 5 µL of 5% uranyl acetate for 30 s. Finally, excess uranyl acetate was removed by pipetting, and the grids were air dried for 10 min. Images were taken with ×84,000 and ×110,000 magnification.
Dynamic light scattering (DLS)
CuMVTT VLP sample solution (1 mg/mL) was used for DLS analysis on Zetasizer nano ZS instrument (Malvern Instruments, UK). Results of three measurements were analyzed by DTS software (Malvern V.6.32).27
Mass spectrometry (MS)
MS analysis was performed as described in Zeltins et al.27
Microcrystalline tyrosine
We used amino acid L-tyr that is a proprietary formulation manufactured under the registered trademark MCT. MCT was kindly provided by Allergy Therapeutics, UK/Bencard Adjuvant Systems. MCT crystals were imaged using bright field on a Zeiss AxioImager A2 microscope with Plan-NEOFLUAR ×20 objective (Zeiss).
CuMVTT VLPs and MCT formulation
Formulating the nanoparticles CuMVTT with MCT adjuvant was carried out by mixing both ingredients in a shaker at 500 rpm in room temperature (RT) for 1 hour. Such procedure allows the nanoparticles CuMVTT to decorate MCT crystals. For visualization of the formulation, CuMVTT VLP samples (3 mg/mL) were labeled with AF488 as per the manufacturer’s instructions, and mixed with MCT, 10 µL was added to a glass slide and covered with a cover slip. The samples were imaged using fluorescent light illumination with a Zeiss AxioImager A2 microscope with Plan-NEOFLUAR ×20 and ×40 objectives (Zeiss).
Mice
Wild-type C57BL/6 female mice (8–12 weeks, Harlan) were used in all experiments. RAG−/− C57BL/6 mice were kindly provided by Professor A Ochsenbein and were bred in our pathogen-free animal facility at University of Bern. All animal experiments were performed in accordance with the Swiss Animal Act (455.109.1 September 5, 2008) under license no. BE10/18 and BE43/21.
Treatment regimen and doses
Wild-type C57BL/6 mice were treated intratumorally on day 3 post B16F10 tumor implantation (2 mm3 fragments). Intratumoral injections were performed three times over 15 days. Treatment doses were 100 µL phosphate buffered saline (PBS) (mock), 50 µg of CuMVTT alone diluted in a final volume of 100 µL PBS/dose,or 50 µg CuMVTT formulated with 4% MCT in a final volume of 100 µL/dose. Intratumoral injections were performed three or four times as indicated in the figure legends. Mice were monitored every 2 days to assess tumor volume and general health score. Tumors in the control groups reached the ethically maximal tolerated size of 1 cm3. Tumors were collected as indicated in the Results section, and mice were monitored every 2 days.
Tumor experiments
One million B16F10 melanoma cells were injected into the flank of RAG−/− C57BL/6 mice. Tumors were allowed to grow into palpable tumors for 15 days until they reached 1 cm3. Tumors were dissected and processed into ~2 mm2 fragments and kept in complete medium on ice. A tumor’s fragment was then implanted into a flank of wild-type C57BL/6 mice (8–12 weeks old, Harlan) under full anesthesia. The implanted tumor was allowed to grow for 3 days before treatment start. Implanted tumors were treated intratumorally three times over 13 or 15 days as indicated in the Results section. Tumor growth was monitored every 2 days and measured using calipers. Tumors were collected and weight was recorded. Tumors were kept in Dulbecco's Modified Eagle Medium (DMEM) medium containing 10% Fetal Bovine Serum (FBS) and 1% penicillin/streptomycin on ice. TILs were collected as follows: tumors were dissected into pieces, digested with collagenase at 37°C for 30 min and smashed using a 70 µM cell strainer. Cells were washed during the process using DMEM medium containing 10% FBS and 1% penicillin/streptomycin in falcon 50 mL tubes. Collected cells were added to 15 mL tubes containing 2 mL of 35% Percoll slowly. The tubes were centrifuged at 1800 rpm for 25 min at RT to isolate TILs. TILs were then resuspended in 200 µL PBS, 0.1% BSA, and 100 µL was transferred to a 96-well plate v-bottom and centrifuged at 1200 rpm for 5 min. The supernatant was discarded, and red blood cells (RBCs) were lysed using 500 µL Ammonium-Chloride-Potassium (ACK) buffer (Sigma-Aldrich) on ice for 2–3 min. TILs were stained with anti-mouse CD16/CD32 (mouse BD Fc block) mAb clone 2.4G2 (BD Bioscience) for 10 min in the dark, centrifuged as described previously, and stained with live/dead 7-AAD, PE anti-mouse CD8α mAb clone 53–6.7 (BD Bioscience), APC anti-mouse Ly6G mAb clone 1A8 (BioLegend), FITC anti-mouse Ly6C clone HK1.4 (BioLegend) and APC/Cyanine7 anti-mouse CD11b clone M/170 (BioLegend). Plates were centrifuged at 1200 rpm for 5 min; the supernatant was discarded; TILs were resuspended in PBS and 0.1% BSA and were added to 5 mL round-bottom tubes with a cell strainer to remove excess tumor debris. Samples were read by FACSCaliber and analysis was done using GraphPad Prism V.8.4.2 (464). Cell density was measured by dividing the total number of cells in each tumor by its weight.
Costimulation experiments
To perform costimulation experiments, we adapted a similar method as published in van Vloten et al.28 Briefly, 1×105 cells/well of B16F10 and MC57 (fibrosarcoma) cell lines were platted in U-bottom plates with 10 µg/mL of rIFN-γ for 48 hours. Tumor cells were pelleted by centrifugation for 5 min at 1200 rpm in complete DMEM medium. The supernatant was discarded. TILs were collected from a mock group and another group treated with CuMVTT+MCT as described in the Tumor experiments section. TILs were cocultured with tumor cells (B16F10 or MC57) for 3 days. On day 4, intracellular cytokine (ICS) was performed as explained next for detection of interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) production by CD8+ or CD4+ T cells.
Depletion experiments
Anti-CD8α mAb (clone 53–6.7, 10 µg) or anti-CD4 mAb (clone GK1.5) was administered intravenously 48 hours prior to tumor implantation and subsequently every 2 days using a low dose of 10 µg. Depletion of CD8+ or CD4+ T cells was confirmed 48 hours post first administration and on day 12. Blood (150 µL) was collected from the tail vein in 500 µL 1× PBS containing heparin and kept on ice. Cells were centrifuged at 1200 rpm for 5 min. RBCs were lysed using 500 µL ACK buffer (Sigma-Aldrich) on ice for 2–3 min. Cells were collected by centrifugation for 5 min at 1200 rpm. The supernatant was aspirated and cells were resuspended with 1× PBS containing 0.1% BSA and centrifuged again. Pelleted cells were stained with anti-mouse CD16/CD32 (mouse BD Fc block) mAb clone 2.4G2 (BD Bioscience) for 10 min in the dark, centrifuged as described earlier, and stained with PE anti-mouse CD8α mAb clone 53–6.7 (BD Bioscience) or FITC anti-mouse CD4 mAb clone (RM4-5). Samples were read by a FACSCaliber and analysis was done using GraphPad Prism V.8.4.2 (464).
ICS staining for IFN-γ and TNF-α
TILs were isolated as described in the Tumor experiments section and transferred to sterile 96-well flat-bottom plates. TILs were incubated with mouse interleukin (IL)-2 (mIL2-Ref: 11271164001-MERCK) 100 U/mL in DMEM medium containing 10% FBS and 1% penicillin/streptomycin at 37°C for 2 days. TILs were washed three times with DMEM medium containing 10% FBS and 1% penicillin/streptomycin, and a stimulation cocktail was added 1 µg/mL of ionomycin/PMA / Brefeldin and Monensin (1:1000) at 37°C for 6 hours. TILs were washed three times with DMEM medium and then transferred to 96-well v-bottom plates for staining. TILs were stained with anti-mouse CD16/CD32 (mouse BD Fc block) mAb clone 2.4G2 (BD Bioscience) for 10 min in the dark, centrifuged as described previously and stained with PE anti-mouse CD8α mAb clone 53–6.7 (BD Bioscience). The plate was centrifuged at 1200 rpm for 5 min; the supernatant was discarded; and TILs were fixed using 100 µL of the fixation buffer (BD Cytofix) at 4°C for 15 min. The plate was centrifuged at 1200 rpm for 5 min; the supernatant was discarded; and TILs were washed with 100 µL of 1× diluted permeabilization wash buffer (BioLegend) and centrifuged immediately at 1200 rpm for 5 min; the supernatant was discarded. TILs were then stained with APC anti-mouse IFN-γ mAb clone XMG1.2 (MERCK) and PerCP-Cyanine5.5 anti-mouse TNF-α mAb clone MP6-XT22 (BioLegend). Plates were centrifuged at 1200 rpm for 5 min; the supernatant was discarded; TILs were resuspended in PBS and 0.1% BSA and added to 5 mL round-bottom tubes with cell strainer to remove excess tumor debris. Samples were read by FACSCaliber and analysis was done using GraphPad Prism V.8.4.2 (464).
Antibody measurements (ELISA)
The induced antibody response after intratumoral injections was assessed by ELISA. Mice were kept warm for up to 15 min; 100 µL of whole blood was collected from mice tail in BD Microtainer tubes. Serum was separated by centrifugation at 8000 rpm for 1 min and stored at −20°C. To determine total IgG antibody titres against CuMVTT proteins; ELISA plates were coated overnight with 2 µg/mL CuMVTT. Plates were washed with PBS 0.01% Tween and blocked using 100 µL PBS–casein 0.15% for 2 hours. Sera from treated mice were diluted 1/20 initially and then serially 1/3. Plates were incubated for 1 hour in RT. After washing with PBS 0.01% Tween, goat anti-mouse IgG conjugated to horseradish peroxidase was added 1/1000 and incubated for 1 hour at RT. Plates were developed and OD450 reading was performed.
Depot assessment
CuMVTT were labeled with AF488 (Fisher Scientific) for intratumoral injection with or without formulation with MCT in RAG−/− mice. Twelve days later, intratumoral injections with AF488-CuMVTT or AF488-CuMVTT+MCT were performed. Injected tumors were collected 1 and 5 days post injection, and analysis was performed to detect labeled VLPs.
Histology
Histology staining and assessment were performed at the COMPATH Institute at University of Bern. H&E staining was performed on the median-sized tumors preserved in 4% paraformaldehyde for 1 day before sending them to the COMPATH Institute for histological analysis. One full section of each collected tumor was examined after melanin bleach followed by H&E staining. The melanomas were digitally quantified for the extent of microscopic tumor necrosis (software QuPath V.0.1.2). The following parameters were measured digitally and calculated: total area of the tumor cross section, total area of tumor necrosis and percentage of tumor necrosis. Formalin-fixed slides were also assessed by immunohistochemistry staining for CD4+ T cells using CD4 (clone 4SM95, rat, Thermo Fisher Scientific, 14-9766). One full cross section for each tumor was examined by quantitative evaluation for peritumoral and intratumoral CD4+ T cells using the software QuPath V.0.2.3.
RNA isolation, sequencing and bioinformatics
B16F10 in vivo tumors were collected and stored in RNAlater solution (Sigma-Aldrich) according to the manufacturer’s recommendations. Samples were sent to Microsynth AG for RNA isolation and sequencing. Illumina’s TruSeq stranded RNA library preparation kit including polyA enrichment was used to construct libraries from total RNA. Subsequently, the Illumina NextSeq 500 and 550 platforms and a high-output V.2.5 kit (75 cycles) were used to sequence the libraries. The produced single-end reads which passed Illumina’s chastity filter were subject to demultiplexing with zero mismatches allowed and trimming of Illumina adaptor residuals using Illumina’s bcl2fastq software V.2.20.0.422 (no further refinement or selection). Quality of the reads in fastq format was checked with the software FastQC V.0.11.8. The reads were filtered by removing PhiX controls while keeping reads with an average minimum Phred quality score of 20 and 0 ambiguous bases (Ns). The splice aware RNA mapping software STAR V.2.7.7 a was used to map the reads to the reference genome mm10 (UCSC genome browser assembly ID, based on GRCm38.p6) as downloaded from iGenomes. To count the uniquely mapped reads to annotated genes, the software htseq-count (HTSeq V.0.13.5) was used. Normalization of the raw counts and differential gene expression analysis were carried out with the help of the R software package DESeq2 V.1.26.0. Based on the differential gene expression results, KEGG pathway and Gene Ontology (GO) geneset analyses are carried out with the help of the R software package GAGE V.2.36.0, and visualization of the results was achieved by R software package ggplot2 V.3.3.5. Libraries, sequencing and data analysis described in this section were performed by Microsynth AG (Balgach, Switzerland).
Statistics
Data are presented as mean±SEM. Comparisons between more than two groups were performed by one-way analysis of variance; comparisons between two groups were performed by Student’s non-parametric t-test. P values were ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05.
Results
CuMVTT nanoparticles decorate the surface of MCT micron-sized crystals
CuMVTT consists of icosahedron T=3 nanoparticles and has a size of ~30 nm. Indeed, our CuMVTT comprises pentamers (subunit A) and hexamers (subunits B and C) assembled together into an icosahedron (figure 1A). We have immunologically improved these nanoparticles by incorporating a universal tetanus toxin (TT) TH cell epitope (Gln Tyr Ile Lys Ala Asn Ser Lys Phe Ile Gly Ile Thr Glu) to the interior surface of the VLPs. This addition may enhance the immune response due to activation of pre-existing TT specific TH cells present in most humans, particularly useful in elderly and immunocompromised patients.11 12 Additionally, CuMVTT nanoparticles package ssRNA during the expression process in E. coli, which serves as TLR7/8 ligands facilitating APCs activation. CuMVTT nanoparticles were produced as detailed previously.12 Successful production and confirmation of integrity of the nanoparticles were assessed by SDS-PAGE (figure 1B), electron microscopy (figure 1C) and MS (figure 1D), all demonstrating a homogeneity of the product. Incorporation of ssRNA was visualized by agarose gel (figure 1E). DLS of the nanoparticles confirmed successful assembly of CuMVTT (figure 1F). The average hydrodynamic diameter (Z(av)) of CuMVTT was ~38 nm.

CuMVTT nanoparticles decorate the surface of MCT micron-sized crystals. (A) Geometry of the icosahedral CuMVTT VLPs illustrating the capsid protein in T=3 configuration. VLPs are formed of 60 of each subunits A, B and C (illustrated here in shades for clarity). (B) SDS-PAGE analysis of CuMVTT VLPs, lane 1: protein size marker (Thermo Scientific), lane 2: 6 µg VLPs. A monomer of CuMV-protein has a size of ~(25–30 kDa). (C) Electron microscopy image of purified VLPs, VLP sample (1.5 mg/mL) examined by JEM-100C electron microscopy, VLP’s size between 26 and 28 nm. (D) Mass spectrometry analysis confirmed a major peak of ~24,477.586. (E) Agarose gel analysis of the VLPs, lane 1: DNA marker (Thermo Fisher), lane 2: 10 µg purified VLPs, VLP sample (1.5 mg/mL). (F) Dynamic light scattering analysis of the VLP nanoparticles, VLP sample (1.5 mg/mL). Three measurements were performed and analyzed by DTS software. (G) MCT crystals visualized with light microscopy, exhibiting a micron size of ca=4.5 µm in length. (H) CuMVTT VLPs labeled with AF488 and formulated with MCT micron-sized adjuvant, referred to as immune-enhancer, ×40 objective scale bar 20 µm. (I) CuMVTT VLPs labeled with AF488 and formulated with MCT micron-sized adjuvant, ×40 objective scale bar 20 µm. (J) A cartoon illustrating CuMVTT VLPs; packaging ssRNA (in red) and decorating MCT crystals. CuMVTT, cucumber mosaic virus-like particles incorporating a tetanus toxin peptide; MCT, microcrystalline tyrosine; VLP, virus-like particle.
MCT is an L-tyr-based TH1-polarizing adjuvant15 consisting of micron-sized crystals (figure 1G). MCT is considered a classical, biodegradable depot-forming adjuvant that is used since decades in combination with modified or native allergens for subcutaneous immunotherapy in humans.20 21 Due to their micron size of ~5 μM, MCT cannot readily enter the lymphatics and remain at the injection site and forms a depot causing local inflammation.22 29
Together, we successfully formulated CuMVTT nanoparticles and MCT micro-sized adjuvant, resulting in CuMVTT coated microparticles, our new immune-enhancer (figure 1H–J).
Administering the novel immune-enhancer intratumorally hinders B16F10 progression
We tested the effects of this novel immune-enhancer (CuMVTT+MCT) in an aggressive melanoma murine model which we have previously established in our laboratory.26 30 This model facilitates intratumoral injections as it is based on tumor fragment implantation rather than injection of cell suspensions. Intratumoral injections with the immune-enhancer started few days post tumor implantation (figure 2A), allowing physiological stroma and tumor vascularization to be established before initiating the treatment. Appropriate control treatments were included such as mock (PBS), CuMVTT (alone) and MCT (alone).

Administering the novel immune-enhancer intratumorally hinders B16F10 progression. (A) A cartoon illustrating the treatment regimen and the groups of mice treated with intratumoral injections. (B) Tumor weight (mg) measured on day 15 post B16F10 (2 mm3) implantation; each dot represents an individual tumor. Combined (C) and individual (D) tumor growth curves of B16F10 melanoma in the different mouse groups measured by caliper, as tumor volume mm3. (E) Density of CD8+ T cells in tumors as determined by flow cytometry. The densities were determined by dividing the total number of CD8+ T cells in each tumor by the tumor weight (in mg), pregated on TILs. Using FS/SS (F) FACS plots showing the total numbers of CD8+ T cells in a tumor, pregated on TILs. (G) Correlation between density of CD8+ T cells and tumor weight (mg). (H) Density of IFN-γ (I) and IFN-γ/TNF-α producing CD8+ T cells in tumors as determined by flow cytometry upon stimulation with 1 µg /mL PMA/ionomycin. The densities were determined by dividing the total number of CD8+ T cells in each tumor by the tumor weight (in mg). (J) Representative FACS plots showing the total numbers of IFN-γ/TNF-α producing CD8+ T cells in tumors. Statistical analysis (B,C,E,G–I) by ordinary one-way analysis of variance (multiple comparison) (mean±SEM). Statistical analysis in F by simple linear regression. n=12 (B–G) and n=5 (H,I). One representative of two similar experiments is shown. ****p<0.0001; ***p<0.001;, **p<0.01;, and *p<0.05. APC, antigen-presenting cell; IFN-γ, interferon gamma; MCT, microcrystalline tyrosine; ns, not significant; TIL, tumor-infiltrating lymphocyte; TNF-α, tumor necrosis factor alpha.
The tumor weight on day 15 showed that the treatments with MCT or CuMVTT alone significantly hindered B16F10 tumor progression (p=0.0018 and 0.0012, respectively). However, the best response was achieved with the immune-enhancer (p≤0.0001) (figure 2B–D). Since CD8+ T-cell infiltration often correlates with favorable therapeutic outcome and prognosis,31–33 we determined the density and total number of tumor-infiltrating CD8+ T cells. No significant increase in CD8+ T-cell density was found in the groups treated with MCT or CuMVTT alone. Interestingly, T-cell infiltration was increased in mice treated with our immune-enhancer (figure 2E–F), significantly correlating with reduced tumor weight (p=0.0025) (figure 2G). IFN-γ producing CD8+ T cells were also significantly increased after treatment with the immune-enhancer (p=0.0046) (figure 2H). The density of TNF-α+ CD8+ T cells producing cells in tumors showed no significant increase in CuMVTT+MCT; however, the polyfunctional CD8+ T cells producing both IFN-γ and TNF-α were significantly increased (p=0.0054) in this group (figure 2I,J).
To evaluate specific CD8+ T-cell reactivity against the B16F10 melanoma cell line, we performed a coculture experiment. Briefly, TILs were isolated from mock-treated mice as well as from mice treated with the immune-enhancer. The isolated TILs were cocultured with the B16F10 cell line or with the irrelevant MHC-matched MC57 fibrosarcoma cell line for 3 days. To induce MHC-I expression on tumor cells, B16F10 and MC57 cells were incubated with rIFN-γ for 48 hours prior coculturing. Online supplemental figure 1A shows tumor weight on day 15 from both groups. TILs isolated from the mock group did not show reactivity against B16F10 nor MC57. TILs from the group treated with CuMVTT+MCT could enhance IFN-ɣ production by CD8+ and CD4+ T cells when cocultured with B16F10 but not with MC57 cells (online supplemental figure 1B,C). These data confirm the specific reactivity of CD8+ T cells following intratumoral treatment.
Supplemental material
In a separate experiment, we measured the density of CD4+ T cells. Results showed that intratumoral treatment with the immune-enhancer increased CD4+ T-cell density in comparison to the control group (online supplemental figure 2A–C). We performed H&E staining to assess intratumoral and peritumoral CD4+ T cells. Histological sections showed a significant increase (p=0.0248 and 0.0046, respectively) in the treated group (online supplemental figure 2D–F).
Supplemental material
CD8+ T cells play an essential role in the antitumor impact of the immune-enhancer
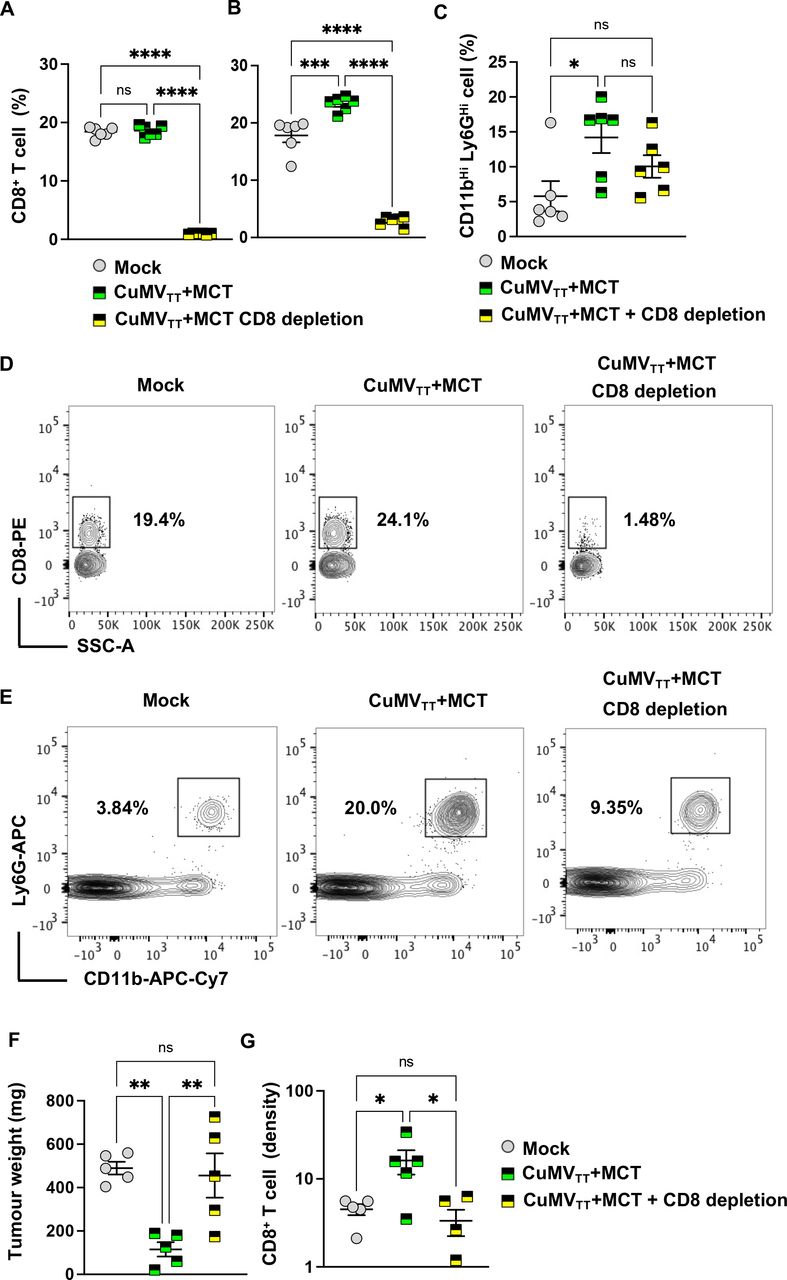
To better understand the role of CD8+ T cells in the antitumor impact of the immune-enhancer (CuMVTT+MCT), we depleted CD8+ T cells. Briefly, anti-CD8α mAb was administered intravenously 48 hours prior to tumor implantation and subsequently every 2 days. Effective depletion of CD8+ T cells in the blood was confirmed before tumor implantation and on day 12 post tumor implantation. Tumor-bearing mice were treated intratumorally with mock (PBS), CuMVTT+MCT alone or in combination with anti-CD8α mAb.
Treatment with anti-CD8α mAb depleted ~95% of CD8+ T cells (figure 3A). Interestingly, intratumoral injection of CuMVTT+MCT resulted in a significant increase in the systemic (blood) percentage of CD8+ T cells, which was abolished after CD8+ T-cell depletion (figure 3B,D), as well as granulocytic cells characterized by CD11bHi Ly6GHi 12 days post tumor implantation (after two injections) (figure 3C,E). Tumors were collected on day 15 post tumor implantation. Results were consistent with the aforementioned findings, and tumors treated with the immune-enhancer showed significant tumor weight reduction (p=0.0037) in comparison to the control group or the group with CD8+ T-cell depletion (p=0.0072) (figure 3F). The CD8+ T-cell density was increased only in the group treated with CuMVTT+MCT (figure 3G). In the next step, we depleted CD4+ T cells to determine their possible contribution to the induced antitumor effect (online supplemental figure 3A,E). Tumors collected on day 15 showed a small trend of weight increase which was, however, not statistically significant (p=0.2857) (online supplemental figure 3B). The density of CD8+ T cells was higher in the groups treated with the immune-enhancer with or without CD4+ T cell depletion than the mock (online supplemental figure 3C) but not in the groups treated with CuMVTT+MCT. When assessing the density of CD4+ T cells in the tumor microenvironment, the results showed a significant increase in the group treated with the immune-enhancer in comparison to the mock (online supplemental figure 3D). As expected, there were no intratumoral CD4+ T cells upon depletion (online supplemental figure 3D). Collectively, these data confirm that CD8+ but not CD4+ T cells are key for the induced antitumor effects.
Supplemental material

CD8+ T cells play an essential role in the antitumor impact of the immune-enhancer. (A,B) Percentage (%) of CD8+ T cells in peripheral blood 48 hours post anti-CD8+ mAb administration and on day 12 post tumor implantation; each dot represents an individual tumor. Depletion efficacy was ~95%. (C) Percentage (%) of CD11bHi Ly6GHi cells in peripheral blood; each dot represents an individual tumor. (D) Representative FACS plots showing the percentage of CD8+ T cells in peripheral blood. (E) Representative FACS plots showing the percentage of CD11b+ Ly6G+ cells in peripheral blood. (F) Tumor weight (in mg) measured on day 15 post B16F10 (2 mm3) implantation; each dot represents an individual tumor. (G) Density of CD8+ T cells in tumors as determined by flow cytometry. The densities were determined by dividing the total number of CD8+ T cells in each tumor by the tumor weight (in mg), pregated on TILs. Statistical analysis by ordinary one-way analysis of variance (multiple comparison) (mean±SEM) n=5 or 6, one representative of two similar experiments is shown.****p<0.0001; ***p<0.001; **p<0.01; and *p<0.05. MCT, microcrystalline tyrosine; ns, not significant. TIL, tumor-infiltrating lymphocyte.
Intratumoral administration of CuMVTT+MCT forms a depot, enhances inflammation, and reduces tumor necrosis
The observed strong T-cell responses may be enabled by the capability of MCT to form depots, prolonging immune stimulation and antigen exposure to APCs, thereby enhancing T-cell activation and clonal expansion. To explore this possibility in vivo, we formulated fluorescently labeled AF488-CuMVTT with and without MCT and performed intratumoral injections in RAG−/− mice harboring B16F10 melanoma (online supplemental figure 4A). Tumors were collected 1 and 5 days post treatment, and flow cytometry was performed to assess the percentage of CD11b+ cells positive for AF488-CuMVTT. The labeled VLPs could be detected 1 day after intratumoral injection in both groups at similar levels. However, 5 days later, the percentages of positive cells in the group injected with CuMVTT+MCT were significantly higher compared with the group injected with CuMVTT alone (p=0.0047) (online supplemental figure 4B). These data confirm that MCT promotes depot persistence in situ.
Supplemental material
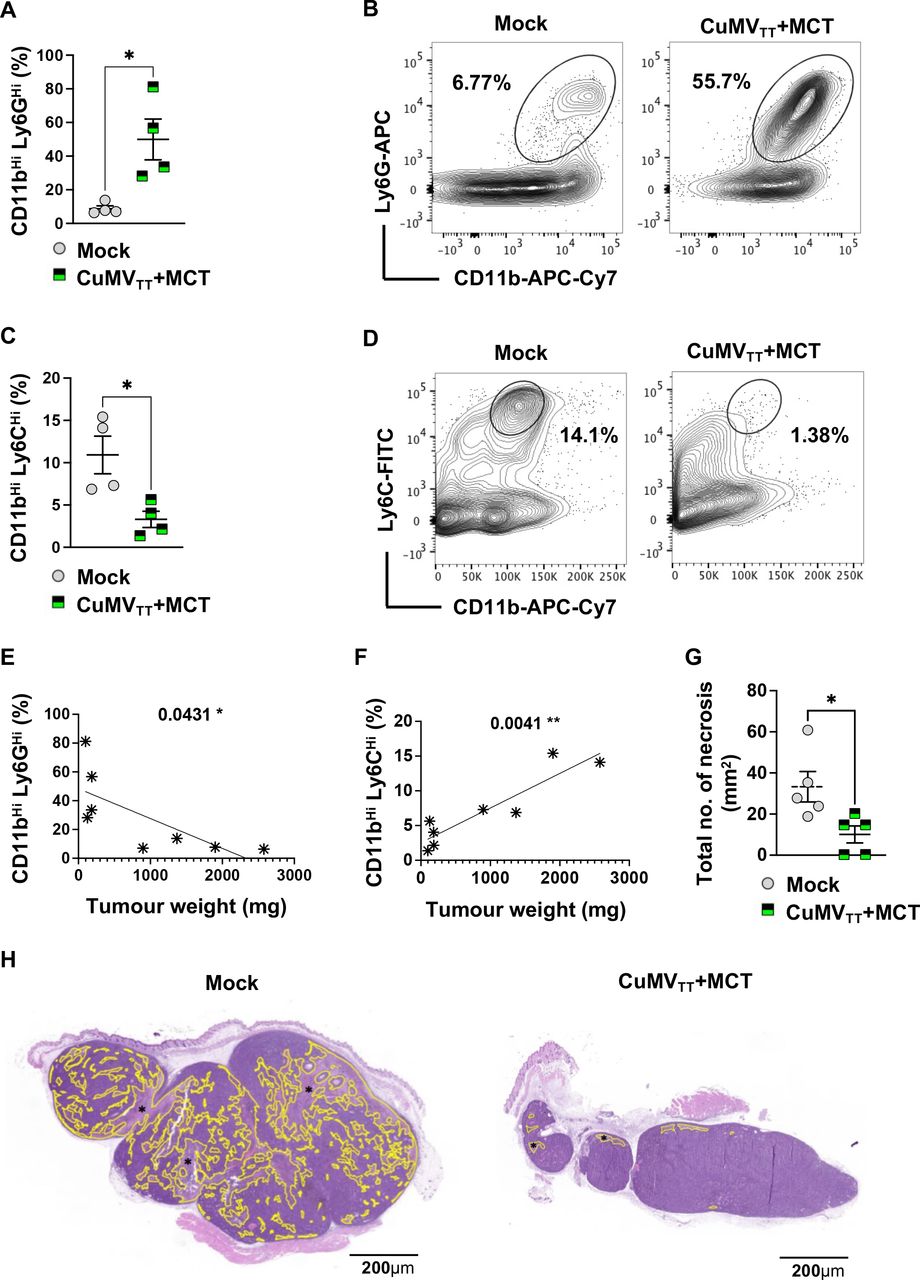
To assess whether intratumoral administration of the immune-enhancer (CuMVTT+MCT) impacts on immunosuppressive cells, we measured the percentages of granulocytic and monocytic myeloid-derived cells in the tumor by focusing on the CD11bHi Ly6GHi and CD11bHi Ly6CHi populations. Due to the short life span of neutrophils, we collected the treated tumors 1 day after the third intratumoral injection (day 13). Interestingly, we found a significant increase in the granulocytic population (p=0.0286) (figure 4A,B), while the monocytic population showed a significant reduction (p=0.0286) (figure 4C,D) compared with the mock (PBS) group. Both the increase of granulocytic cells and the decrease of monocytic cells correlated significantly with the tumor weight (p=0.04131 and 0.0041, respectively) (figure 4E,F). We were also interested in examining the histological changes of the treated tumors on day 15 after third intratumoral injections. We quantified necrotic tumor areas by digital histological assessment. Several studies have shown that tumor necrosis reflects the presence of hypoxia in the tumor, which can indicate an aggressive phenotype of rapid cellular proliferation.34 We have found that the intratumoral administration of the immune-enhancer could significantly reduce (p=0.0159) the total number of necrosis in the tumor in comparison to the mock group (figure 4G,H), which is likely related to the reduced size of the tumors.

Intratumoral administration of CuMVTT+MCT forms a depot, enhances inflammation and reduces tumor necrosis. (A) Percentage (%) of CD11bHi Ly6GHi cells in tumors; each dot represents an individual tumor. (B) Representative FACS plots showing the percentage (%) of CD11bHi Ly6GHi cells in a tumor. (C) Percentage (%) of CD11bHi Ly6CHi cells in tumors; each dot represents an individual tumor. (D) Representative FACS plots showing the percentage (%) of CD11bHi Ly6CHi cells in a tumor. (E) Correlation between percentage (%) of CD11bHi Ly6GHi cells and tumor weight (in mg). (F) Correlation between percentage (%) of CD11bHi Ly6CHi cells and tumor weight (in mg). (G) Total number of necrosis (mm2); each dot represents an individual tumor. (H) Example of a mock tumor (left): necrosis comprises 30% of the tumor section, example of a treated tumor with the immune-enhancer (right): necrosis comprises 2% of the tumor section. Necrosis is indicated in yellow (*). The entire tumor cross section was assessed for necrosis, H&E staining. Statistical analysis (A,C,G) by Student’s t-test (mean±SEM). Statistical analysis (E,F) by linear regression. n=4 (A–C) and n=5 (E,F,G), one representative of two similar experiments is shown. MCT, microcrystalline tyrosine.
Systemic therapeutic effects induced by the treatment with the immune-enhancer
We have shown previously that, when injected subcutaneously in the footpad of mice, the VLPs drain readily into the lymphatic system, as expected for such particles with a size of ~30 nm.35 Furthermore, CuMVTT-p33 displaying p33 T-cell epitope formulated with MCT crystals formed depots at subcutaneous injection sites. These depots facilitated slow release and were detectable for at least 9 days.26 Accordingly, to confirm that in situ injections of CuMVTT+MCT can induce a systemic response, we first measured anti-CuMVTT VLP antibodies in the sera of treated mice after two intratumoral injections. As expected, the treatment with CuMVTT or CuMVTT+MCT readily induced anti-CuMVTT antibodies (figure 5A). No antibodies were detected in the groups after mock or MCT-alone treatments. Together with the increased T-cell frequencies in blood (figure 3B), these results indicate that intratumoral treatment with VLPs can effectively induce a systemic immune response.
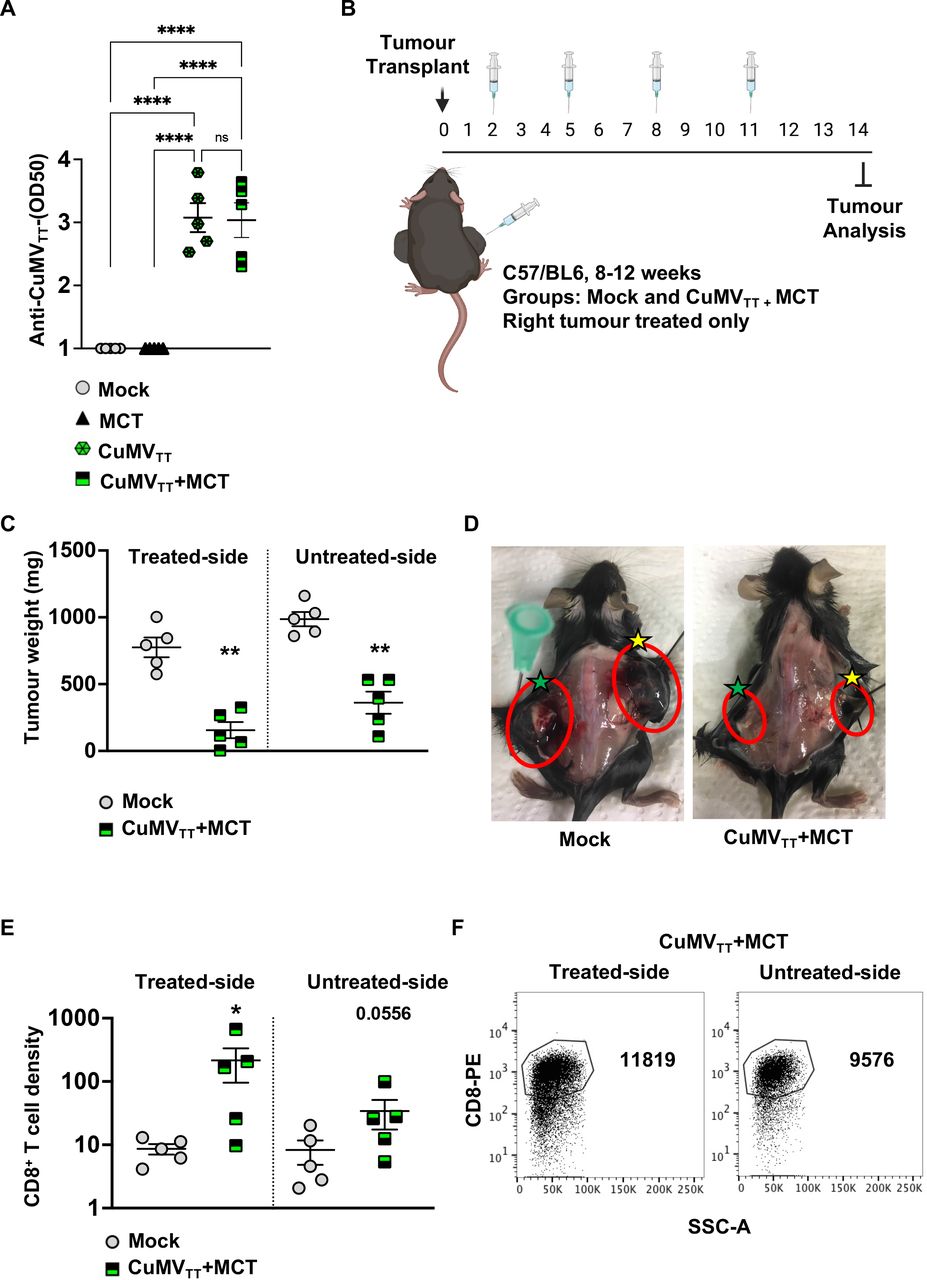
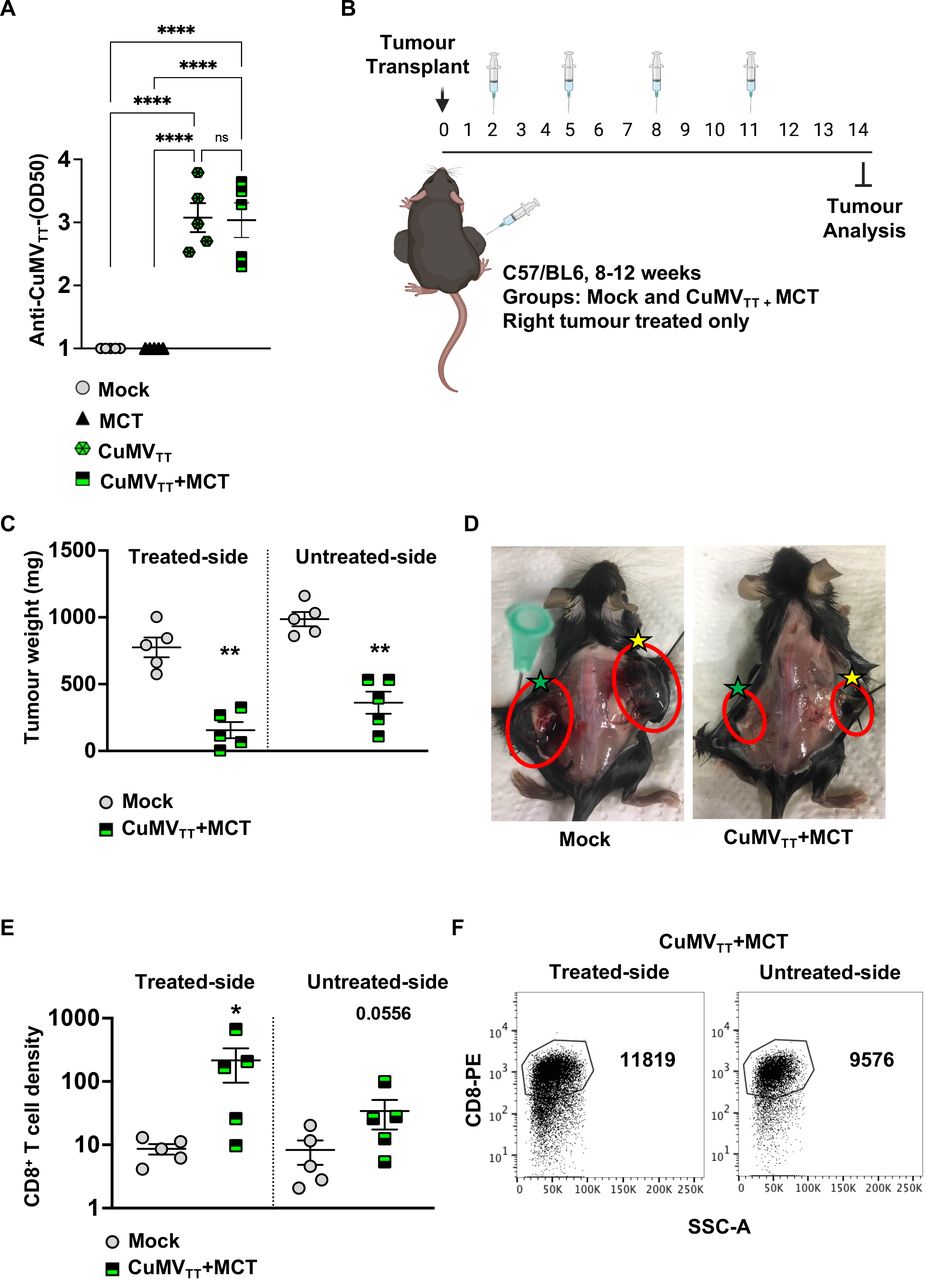
Systemic therapeutic effects induced by the treatment with CuMVTT+MCT. (A) Log OD50 of CuMVTT specific IgG antibodies in serum of treated mice. (B) A cartoon illustrating the treatment regimen and mouse groups using intratumoral injections. (C) Tumor weight (mg) measured on day 14 post B16F10 (2 mm3) implantation; each dot represents an individual tumor. (D) Representative images of B16F10 tumors on both flanks in a mock and a treated mouse; right treated tumors are marked with yellow stars and left untreated tumors with green stars. (E) Density of CD8+ T cells in treated and untreated tumors in each group, as determined by flow cytometry. The densities were determined by dividing the total number of CD8+ T cells in each tumor by the tumor weight (in mg), pregated on TILs. (F) Representative FACS plots showing the total number of CD8+ T cells in treated and untreated tumors in each group, pregated on TILs. Statistical analysis (A) by one-way analysis of variance (multiple comparison) (mean±SEM). Statistical analysis (C,E) by Student’s t-test (mean±SEM) n=5. One representative of two similar experiments is shown. MCT, microcrystalline tyrosine; TIL, tumor-infiltrating lymphocyte.
To test for a potential abscopal effect, two tumors were implanted in each mouse flank as illustrated in figure 5B. Only the right tumor was treated twice intratumorally, while the left one was left untreated. As the total tumor burden was higher in this experiment, we increased the treatment regimen to two injections weekly.
The obtained results revealed that intratumoral treatment with the immune-enhancer in one implanted tumor can induce an antitumor response in the untreated side of the tumor as well. This was shown by the decreased tumor weight in both right and left implanted tumors in the treated group in comparison to the mock group (figure 5C,D). The density of CD8+ T cells in the right (treated) tumor was significantly higher than that in the right tumor in the control group (p=0.0317). The density of CD8+ T cells in the left untreated tumor of the treated mice showed a trend but not a statistically significant increase (p=0.0556) likely related to relatively large heterogeneity (figure 5E,F).
Treatment with the novel immune-enhancer inhibits tumor recurrence
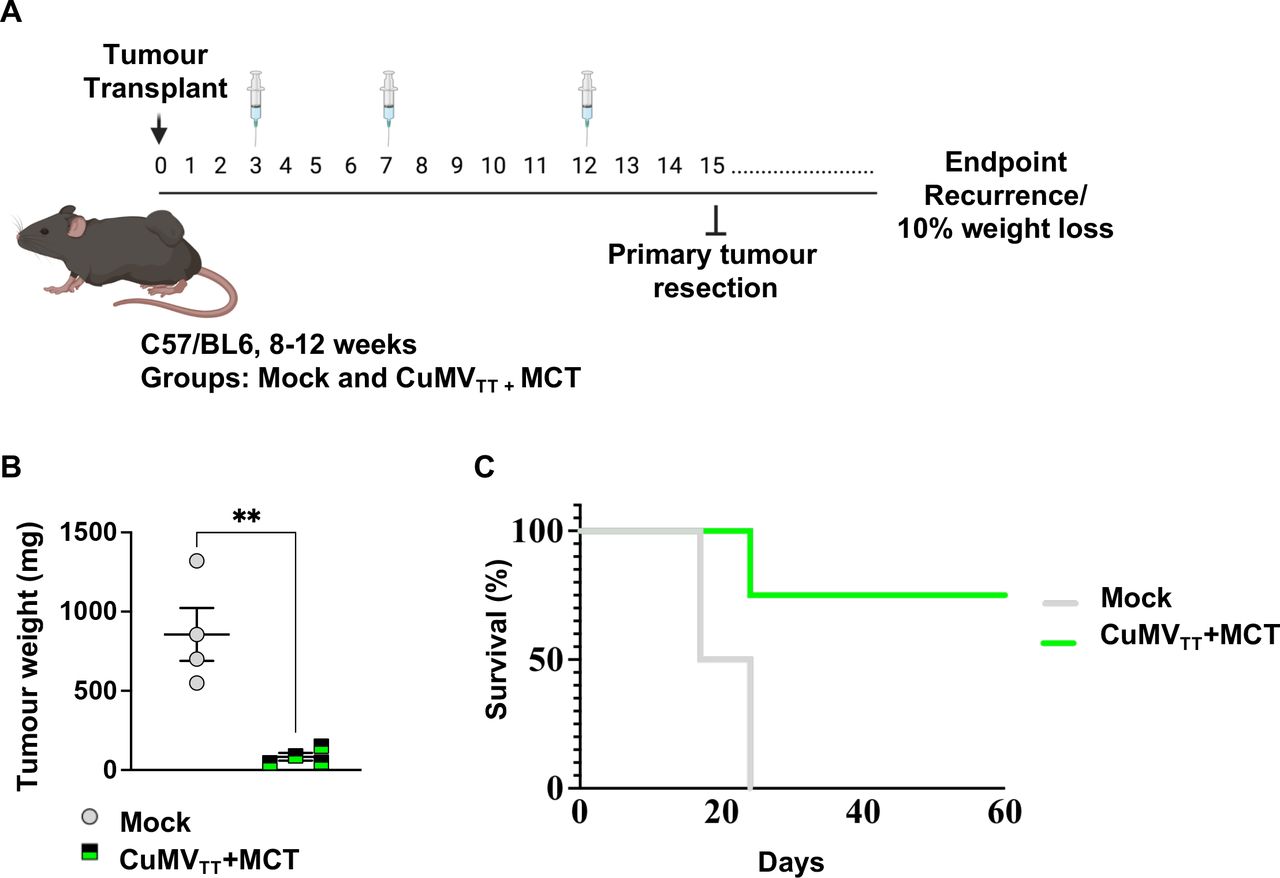
The observed antitumor immune response in vivo of the immune-enhancer promoted us to determine whether it could prevent recurrence after primary melanoma resection, an endpoint with clinical relevance. To this end, a similar experimental setting as described in the section (Administering the novel immune-enhancer intratumorally hinders B16F10 progression) was used, and primary tumors were resected on day 15 as illustrated in figure 6A. The tumor weight in the group treated with the immune-enhancer was significantly lower (p=0.0038) (figure 6B). The mice were subsequently monitored for tumor local and metastatic recurrence. We found that 75% of mice treated with the immune-enhancer had disease-free survival, while only 25% had local melanoma recurrence (figure 6C). In contrast, all of the non-treated animals succumbed to lethal tumor recurrence. Thus, intratumoral treatment with the immune-enhancer was capable of inducing a systemic effect that protected against tumor recurrence.

Treatment with the novel immune-enhancer inhibits tumor recurrence. (A) Mice were implanted with B16F10 tumor fragment on day 0 and treated three times intratumorally over 15 days. The primary tumor was surgically resected on day 15 under isoflurane anesthesia. (B) Tumor weight (in mg) measured on day 15 post B16F10 (2 mm3) implantation; each dot represents an individual tumor. Statistical analysis by Student’s t-test (mean±SE). (C) Mouse survival; mice were euthanized when the volume of recurrent tumors reached 1 cm3, statistical analysis by log-rank test. n=4, one representative of two similar experiments is shown. MCT, microcrystalline tyrosine.
Transcriptional changes induced by the intratumoral treatment
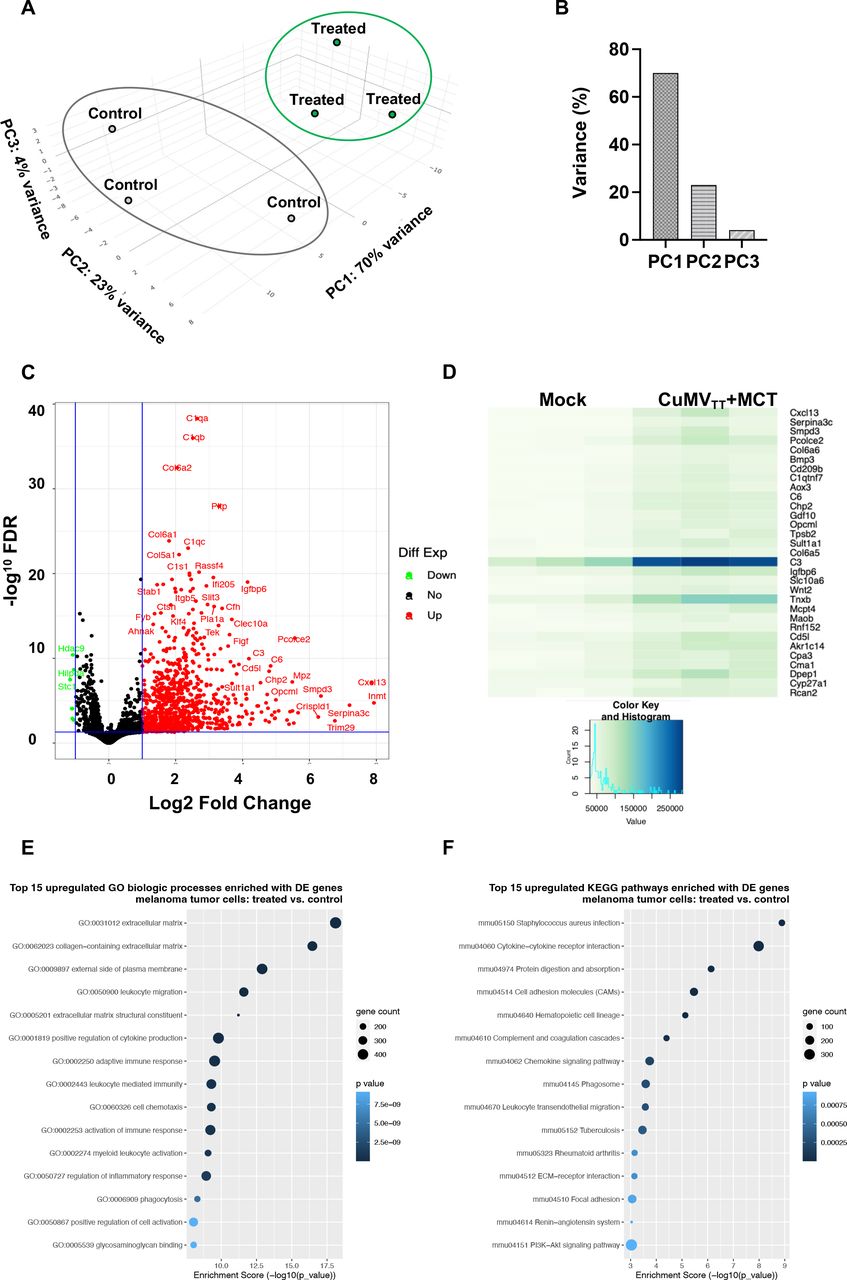
To identify the molecular changes in the tumor microenvironment following intratumoral administration of the immune-enhancer and to obtain broad insight in the transcriptional landscape, we performed RNA sequencing (RNA-Seq) profiling. We compared B16F10 tumors treated intratumorally with the immune-enhancer (CuMVTT+MCT) versus mock (PBS). Differential gene expression was analyzed using the first principal component (PC), revealing a 70% distinct gene expression on intratumoral treatment compared with mock. In situ treatment with the immune-enhancer also formed gene groups that were most distant from mock-treated tumors for PC2 (23% variance) and PC3 (4% variance) (figure 7A,B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Transcriptional changes induced by the intratumoral treatment. (A,B)Principal component analysis (PC1, PC2 and PC3—together representing 97% of the variance in the data) of genes differently expressed between the two biological groups (mice with B16F10 tumors treated with immune-enhancer CuMVTT+MCT vs mock group). (C)Volcano plot of differentially expressed genes (treated vs mock). The plot illustrates the log2 fold change threshold and the threshold for p values adjusted for multiple testing (Benjamini-Hochberg procedure). Upregulated genes are presented in red and downregulated in green. (D)Heat map of differential gene expression of mice treated with immune-enhancer versus mock group. RNA-Seq analysis was performed on RNA isolated from triplicate samples for each biological group. (E,F)Analyses of GO and KEGG pathway enrichment were performed to identify biological processes and pathways significantly enriched with upregulated and downregulated genes in tumors for each biological group. Enrichment scores show gene count and statistical significance determined with Fisher’s exact test and presented for the top 15 biological process-related GO terms and the top 15 KEGG pathways. FDR, false discovery rate; GO, gene ontology; MCT, microcrystalline tyrosine; RNA-Seq, RNA sequencing.
Differential gene expression showed an overexpression of several genes involved in complement activation as well as collagen formation. The overexpressed genes of the complement pathway were C1qa, C1ab, C1qc, C1s, C3, and C6, while the overexpressed genes associated with collagen formation included Col6a2, Col6a1, and Col5a1 (figure 7C,D).
GO and KEGG pathway analyses were performed to identify the biological processes significantly enriched with differentially upregulated and downregulated genes in both treated tumors. We report here the enrichment of the top 15 GO biological processes and KEGG pathways in the tumors treated with CuMVTT+MCT (figure 7E,F, and online supplemental figure 5A,B). Upregulated GO biological processes were collagen-containing extracellular matrix, leukocyte migration, cytokine production, cell chemotaxis, myeloid leukocyte activation and phagocytosis. KEGG pathway analysis showed significant upregulation in genes involved in cytokine receptor interaction, cell adhesion molecules, complement and coagulation pathways, as well as chemokine signaling pathways and leukocyte migration.
Supplemental material
Discussion
In this study, we developed an immune-enhancer for intratumoral administration with the aim to promote immunity and reverse immune suppression in the tumor microenvironment. Our immune-enhancer consists of the modified plant-derived CuMVTT and packaged with ssRNA (TLR7/8 ligand), formulated together with a micron-sized MCT adjuvant. The novel immune-enhancer is applicable to individuals with solid tumors without the need to know tumor-specific antigens. Our data show efficient local and systemic tumor control of aggressive implanted B16F10 tumors through activation of CD8+ T cells and reduction of immunosuppressive myeloid cells.
Intratumoral injection of immunostimulatory adjuvants such as CpG-oligonucleotides, a TLR-9 ligand, has shown promising therapeutic effects through activation of both innate and adaptive immune system components.30 36 In murine models, TLR-9 is expressed by almost all dendritic cells (DCs); however, in humans its expression is much more limited, essentially to plasmacytoid DCs and B cells. In contrast, targeting of TLR7/8 also leads to the highly desired activation of myeloid DCs in both mice and humans. We took advantage of the fact that ssRNA (a TLR7/8 ligand) is naturally packaged during expression of VLP proteins such as our CuMVTT in E. coli. Our newly optimized nanoparticles also incorporate a TT peptide in its interior surface which enhances TH-cell activation, which is particularly powerful in TT-immune elderly people and individuals with otherwise weak immune responses.13 37 B cells are also activated by our immune-enhancer, likely further enhancing tumor inflammation by local immune complex formation. Several recent studies have pointed out the potential importance of B cells and tertiary lymphoid structures (TLSs) for the activation of tumor-specific CD8+ T cells in patients with melanoma, correlating with patient survival.38 We are currently investigating whether our novel immune-enhancer is capable of inducing TLS formation.
In the tumor microenvironment, neutrophils represent a heterogenous population with different phenotypes and opposing functions. Mouse models were used to classify tumor-associated neutrophils in antitumor neutrophils (N1) and tumor-promoting neutrophils (N2).39 The antitumorgenic function of N1 cells is exerted by direct cytotoxicity, antibody-dependent cytotoxicity, and/or activation of different innate and adaptive immune cells including DCs, natural killer cells, and T and B cells.40 41 Lizotte et al have demonstrated that in situ immunotherapy with cowpea mosaic virus by inhalation resulted in activation of Ly6G+ CD11b+ neutrophils 24 hours later.9 Previous studies have shown the ability of MCT to induce the secretion of caspase-dependent IL-1β from human monocytes in vitro, which was inhibited by zVAD, indicating that MCT can cause an acute transient inflammatory response that recruits granulocytes.29 This is supported by the finding that intraperitoneal injections of MCT in murine models promoted an inflammatory response of Ly6G+ CD11b+ cells which was inflammasome independent and lasted for 24 hours.29 Neutrophils can be attracted to tumors by several mechanisms including chemokines produced by tumor cells.42 This is consistent with RNA-Seq data, suggesting that intratumoral administration of the immune-enhancer supports neutrophil infiltration through upregulation of several neutrophil attracting chemokines such as CXCL1 (KC), CXCL2 (MIP-2),42 and CXCL16.43
Several studies have shown that the prognostic impact of tumor necrosis may represent a paradoxical relationship whereby evidence of increased tumor cell death indicates a more aggressive cancer and decreased necrosis may predict a more benign situation.44 45 This relationship can be explained by rapid tumor growth that has outgrown its own blood supply, creating a hypoxic microenvironment and subsequently causing tumor cell death. The presence of necrosis was significantly associated with advanced stage, poor differentiation, vascular invasion, and large tumor size.46 We found reduced tumor necrosis in the treated tumors, likely reflecting reduced growth and, in particular, size of these tumors.
We found that our immune-enhancer induced upregulation of several collagen genes such as COL6A1, COL6A2, as well as COL5a1. Interestingly, a recent study has shown that type VI-α collagens such as COL6A1 and COL6A2 may act as classical collagens in bladder carcinoma and form a physical barrier to inhibit tumor growth and invasion.47 COL6A1 and COL6A2 could effectively inhibit human bladder carcinoma BCaEJ cells proliferation in vitro, induced cell cycle arrest, and inhibited wound healing and invasion by suppressing matrix metalloproteinase MMP-2 and MMP-9.47 It remains to be determined whether similar mechanisms apply in our model.
The complement system is considered a phylogenetically conserved branch of the innate humoral immune system, which rapidly responds to microbial intruders and triggers the release of several inflammatory mediators and consequently cell lysis. Therapeutic targeting of CD20 by rituximab or ofatumumab is known to elicit complement-mediated cytotoxicity. VLPs activate complement via their pathogen-associated molecular and structural patterns.48 Our results show a fold increase of 1000–100,000 of RNA expression of several complement components including C1qa, C1qb, C1qc, C1s, C3, and C6, indicating activation of both classical and alternative complement pathways. Powerful complement activation may lead to tumor cell opsonization and mobilization of potent-inflammatory mediators which activate immune cells with phagocytic properties such as neutrophils that may contribute to tumor control.49 50
The observed systemic therapeutic effect, beyond the injected tumor, was intended and to some degree expected, as intratumoral injection approaches have been shown to bear such potential.4 6 8 Given the strong immunogenicity of our novel therapy and the consequent systemic T-cell activation, it was perhaps not surprising that distant tumors were also inhibited. Even after surgical removal of the primary tumor, we found an efficient systemic effect preventing tumor recurrence, a finding of direct potential clinical relevance.
Together, our new immune-enhancer triggers multiple immune pathways likely explaining its considerable antitumor effects. As good manufacturing practice GMP-grade MCT is available and production of GMP-grade CuMVTT is planned for the near future, this novel approach will be readily translated for clinical application for the treatment of the increasing numbers of tumor types that are accessible for intratumoral injection.
Supplemental material
Data availability statement
Data are available upon reasonable request. Data are available upon request.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
We thank Marc Gasser from Microsynth for RNA sequencing data and pathway enrichment analyses, and Dr Simone Danielle De Brot at COMPATH, University of Bern, for histology.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Design of experiments, acquisition of data, interpretation and analysis of data: MOM. Writing, revision and editing of manuscript: MOM, DES, MH, and MFB. Technical, material, and tool support: MOM, MH, TCV, AB, and MFK. Study supervision: MOM and MFB. All authors read and approved the final manuscript. MOM is the guarantor.
Funding This work was supported by Allergy Therapeutics PLC and Swiss Cancer Research (KFS 5246-02-2021R).
Competing interests MOM, DES, and MFB are shareholders of DeepVax GmbH, involved in the development of cancer immunotherapy.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.