Article Text

Abstract
Background and aims The role of inflammatory immune responses in colorectal cancer (CRC) development and response to therapy is a matter of intense debate. While inflammation is a known driver of CRC, inflammatory immune infiltrates are a positive prognostic factor in CRC and predispose to response to immune checkpoint blockade (ICB) therapy. Unfortunately, over 85% of CRC cases are primarily unresponsive to ICB due to the absence of an immune infiltrate, and even the cases that show an initial immune infiltration can become refractory to ICB. The identification of therapy supportive immune responses in the field has been partially hindered by the sparsity of suitable mouse models to recapitulate the human disease. In this study, we aimed to understand how the dysregulation of the complement anaphylatoxin C3a receptor (C3aR), observed in subsets of patients with CRC, affects the immune responses, the development of CRC, and response to ICB therapy.
Methods We use a comprehensive approach encompassing analysis of publicly available human CRC datasets, inflammation-driven and newly generated spontaneous mouse models of CRC, and multiplatform high-dimensional analysis of immune responses using microbiota sequencing, RNA sequencing, and mass cytometry.
Results We found that patients’ regulation of the complement C3aR is associated with epigenetic modifications. Specifically, downregulation of C3ar1 in human CRC promotes a tumor microenvironment characterized by the accumulation of innate and adaptive immune cells that support antitumor immunity. In addition, in vivo studies in our newly generated mouse model revealed that the lack of C3a in the colon activates a microbiota-mediated proinflammatory program which promotes the development of tumors with an immune signature that renders them responsive to the ICB therapy.
Conclusions Our findings reveal that C3aR may act as a previously unrecognized checkpoint to enhance antitumor immunity in CRC. C3aR can thus be exploited to overcome ICB resistance in a larger group of patients with CRC.
- Immunity, Innate
- Immunotherapy
- Gastrointestinal Neoplasms
Data availability statement
Data are available in a public, open access repository. Raw RNA-Seq data have been deposited at ArrayExpress under the accession number E-MTAB-8500.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The presence of an inflammatory immune infiltrate in colorectal cancer (CRC) is a positive prognostic marker and can confer susceptibility to immune checkpoint blockade therapy. Despite the inflammatory nature of the majority of CRC, only a minority of these cancers are currently amenable to immunotherapy and often develop resistance. This raises the question whether additional mechanisms associated with tumor development can be exploited to enhance the efficacy of immunotherapy and extend its application to a larger number of patients with CRC.
WHAT THIS STUDY ADDS
In this study, we found that the complement anaphylatoxin C3a receptor (C3aR) is downregulated in human CRC. Patients with C3aR downregulation show enhanced infiltration with innate and adaptive activated immune cells. Our in vivo studies demonstrate that the loss of C3aR in a novel spontaneous model of CRC promotes the development of a microbiota-dependent inflammatory signature in typically cold tumor and makes them responsive to immunotherapy.
As such, our study unravels a previously unrecognized role of C3aR as an immune checkpoint in CRC, which could be exploited to enhance antitumor immunity and potentially extend immunotherapy to a larger number of patients with CRC.
Introduction
Inflammation in colorectal cancer (CRC) is a double-edged sword as inflammatory immune responses can either promote tumor development and progression or be exploited for therapy. On the one hand, independently of the nature of the tumors, sporadic or genetic, inflammation is a well-known driver of CRC development1: about 20% of individuals with persistent gastrointestinal inflammation, such as patients with inflammatory bowel disease, develop CRC, and in sporadic CRC, the sequential accumulation of genetic alterations in tumor suppressors and oncogenes supports the development of an intrinsic inflammatory gene signature.2 3 On the other hand, the notion that immunosurveillance and associated immune responses can also be exploited for treatment is supported by the observation that Crohn’s-like lymphoid infiltrates are a positive prognostic factor in patients with CRC.4 However, despite the inflammatory nature of most CRCs, since the introduction of immunotherapy with immune checkpoint blockade (ICB), only a minority of CRC proved to be amenable to this treatment.5 6
To date, the Food and Drug Administration has granted approval of CRC to ICB with anti-programmed cell death-1 (PD-1) alone or in combination with anti-cytototoxic T-lymphocyte-associated protein 4 (CTLA-4) only in patients with microsatellite unstable high CRC (MSI-H CRC), which represent 15% of all CRC and 5% of metastatic CRC.7 Most importantly, independently of the PD-L1 expression, BRAF, KRAS mutations, and Lynch syndrome status, over 50% of MSI-H CRCs do not respond or develop resistance to ICB.8 Further, approximately 21% of patients with microsatellite stable (MSS) CRC show an immune signature similar to MSI-H CRC, which may render them susceptible to ICB.9 Therefore, while it is evident that there can be a favorable and an unfavorable inflammation in CRC, identifying the underlying mechanisms that determine one or the other to broaden ICB responsiveness requires a deeper understanding of the events associated with CRC development.
Alterations in innate sensing mechanisms play a crucial role in intestinal inflammation and CRC.10 The complement system is a central player because it can establish extensive networks with other innate immune and adaptive pathways.11 12 We previously reported that activation of the alternative complement pathway C3a–C3a receptor (C3aR) during spontaneous small intestinal tumorigenesis is involved in tumor-associated thrombosis via the promotion of neutrophil extracellular trap (NET) formation.13 Furthermore, in an intestinal ischemia/reperfusion injury model, the C3a–C3aR axis proved essential for tissue regeneration and protection from oxidative damage.14 These findings suggest a critical function for the C3a–C3aR axis in intestinal homeostasis, but its role in CRC remains understudied.
In the present study, by mining publicly available datasets, we found that C3ar1 gene methylation and downregulation of the C3aR expression occur in subsets of human CRC independently of their MSI or MSS status. We also found that in patients, reduced levels of C3aR expression correlate with increased accumulation in the tumor microenvironment (T(ME) of innate and adaptive immune responses, which support antitumor immunity. When generating a new in vivo mouse model, we found that APCMin/+ mice, which usually develop tumors almost exclusively in the small intestine, mainly developed tumors in the colon. The TME analysis revealed that colon tumors originating in the absence of C3aR signaling showed marked innate and adaptive immune infiltrates with prominent Th1, Th17, and cytotoxic T-cell signatures. Both immune infiltration and tumor development in the colon of APCMin/+/C3aR−/− mice could be recapitulated in APCMin/+ mice by fecal microbiota transplantation (FMT) experiments. Finally, we showed that the absence of C3aR treatment with anti-PD-1 effectively reduced tumor growth in otherwise ICB unresponsive APCMin/+ tumors, therefore demonstrating that the inflammatory infiltrate induced by loss or downregulation C3aR in CRC can be successfully exploited to enhance response to therapy.
Altogether our data indicate that loss or downregulation of C3aR could represent the Achille’s heel of CRC by rendering cold tumor hot and serve as a previously unexplored additional immune checkpoint in CRC.
Materials and methods
Extended materials and methods can be found in the online supplemental information.
Animals
C57BL/6J-ApcMin/J (referred to as APCMin/+), C57BL/6J (referred to as WT), C3aR−/− and APCMin/+/C3aR−/− mice were bred and maintained in our specific pathogen free (SPF) animal facility at the European Institute of Oncology and the Medical University of South Carolina. Animal Research Reporting of In Vivo Experiments (ARRIVE) guidelines were used.15
CRC models
For inflammation-driven CRC, mice were treated with azoxymethane (AOM, Sigma-Aldrich) and dextran sulfate sodium (DSS) as previously described.16 For the spontaneous tumor model, APCMin/+ and APCMin/+/C3aR−/− mice were euthanized at the indicated time points, tumor counted, and used for downstream applications.
Flow cytometry and preparation of single-cell suspensions
Single-cell suspensions were prepared from mLN following standard protocols. In addition, colon lamina propria (cLP) and tumor cells were isolated as previously described with minor modifications.17 Protocols for the generation of single-cell suspensions and antibodies used can be found in the online supplemental information. Samples were acquired with FACSCanto II or Fortessa LSR (BD Bioscience) and analyzed with FlowJo software (TreeStar).
Fecal microbiota transplantation
For FMT, fecal material was obtained from APCMin/+ and APCMin/+/C3aR−/− donor mice. Fecal pellets and cecal contents were harvested and frozen in Columbia broth with 20% glycerol. At the administration time, aliquots were thawed, centrifuged to eliminate the glycerol-containing medium, and gavaged. This procedure allowed us to use the same material throughout the experiment, avoiding confounding effects due to fecal material coming from different animals.
Microbiota profiling
V5–V6 hypervariable regions of bacterial 16S rRNA gene were amplified and processed with a modified version of the Nextera protocol.18 The obtained metabarcoding libraries were sequenced using the MiSeq Illumina platform. In addition, metagenomic amplicons were analyzed by applying the BioMaS19 pipeline.
RNA sequencing (RNA-Seq) and RNA-Seq data analysis using the edgeR framework
According to manufacturer instructions, the proximal and distal colon and the single tumors were immediately preserved in RNAlater stabilization solution (Qiagen) and stored at −20°C. For RNA extraction, tumors and colons were homogenized in Trizol. The supernatant was loaded on Qiagen Mini Kit columns and treated as indicated in the manufacturer instructions. RNA was quantified at the Nanodrop and used to generate libraries using the TruSeq RNA kit (Illumina). Quality and integrity of the obtained libraries were evaluated using a bioanalyzer (Agilent Technologies). Samples were sequenced on an Illumina MiSeq instrument at a depth of 35×106 reads per sample.
All analyses were performed using the R environment for statistical computing (R V.3.3.1) using Bioconductor Release V.3.3 as well as other packages.20 Alignment to the mouse reference genome (GRCm38.82) was performed using STAR V.2.5.0a followed by gene-level counting using the featureCounts function of the Rsubread package V.1.22.3.21 Raw counts were normalized using edgeR V.3.14.0, and differential expression was determined using generalized linear models and likelihood ratio tests from the edgeR package.22 23 Significance was determined using the edgeR function decideTestsDGE with Benjamini-Hochberg correction for false discovery rate (FDR); a default FDR threshold of 0.05 and a log2 fold change threshold of 0.6 were applied. Gene set enrichment analyses were conducted using the camera function in the limma package V.3.28.21.24
Analysis of C3aR expression, methylation and correlation with immune cells in patients with rectal and colon cancers
Data on C3aR expression were obtained from the R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl) or TCGA database. Data on C3aR methylation were obtained from the human pan-cancer methylation database (http://methhc.mbc.nctu.edu.tw/php/index.php).25 The correlations of C3aR expression and immune cells on a retrospective cohort of 231 patients with rectal cancer in the S:CORT WS3 Grampian Set and 97 patients with colon cancer in the FOxTROT cohort were visualized and downloaded through the private S:CORT cBioPortal. The data deconvolution was performed using CIBERSORT, xCell, and MCP.
Mass cytometry
Mass cytometry antibodies were either labeled in-house using antibody-labeling kits and protocols or purchased from Fluidigm according to the manufacturer’s instructions. For live-cell barcoding, we followed the protocol by Mei et al26 using six metal isotopes and adapted it to live-cell barcoding of samples with CD45.2 following the protocol published by Lai et al.27 Briefly, individual samples were incubated with unique choose three out of six CD45–platinum or CD45–indium barcodes (table 1) in cell staining buffer for 15 min at 4°C, washed twice, and combined into one composite sample. To identify dead cells, 2.5 µM live/dead marker Pt198 in phosphate-buffered saline (PBS) was added for 2 min at RT. After washing, the composite sample was stained with the cocktail of primary antibodies as described in the online supplemental information. Data from different days and across acquisition time were normalized by adding five-element beads to the sample immediately before acquisition and using the MATLAB-based normalization software, as described previously.28
Cytometry by time of flight (CyTOF) barcodes and antibodies
Statistical analysis
Data were analyzed for normal distribution before performing statistical analyses. Values are presented as means±SEM of multiple individual experiments, each carried out at least in duplicate or as means±SEM of replicates in a representative experiment. Student’s t-test determined a comparison between two groups. Comparison of multiple groups was carried out by one-way or two-way analysis of variance followed by Bonferroni post-test correction using GraphPad Prism software V.8 as indicated in the figure legends. All statistical tests were two-sided, and p<0.05 was considered statistically significant unless otherwise specified.
Results
c3ar1 downregulation occurs in patients with CRC
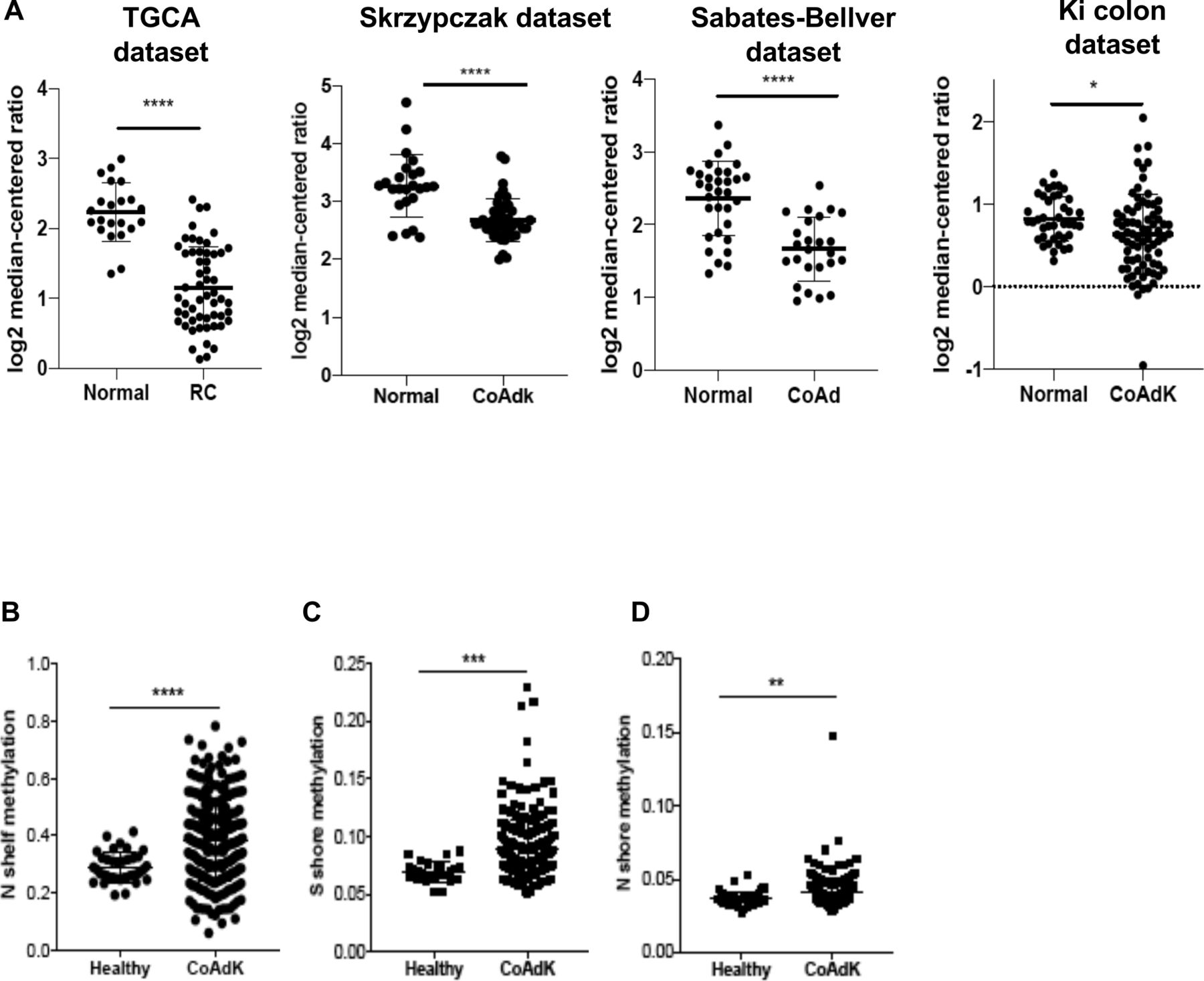
The analysis of the gene expression libraries provided through the Skrzypczack Colorectal cohort, Sabates-Bellver Colon cohort, Ki Colon cohort, and the Cancer Genome Atlas (TCGA) cohort (https://www.oncomine.org) revealed that downregulation of C3aR occurs in patients with CRC compared with controls (figure 1A). We found that C3aR downregulation occurred in approximately 33% of patients with MSI-H CRC and 34% of patients with MSS CRC. Additionally, although patients with stages II and III showed significantly lower expression than stage I patients, overall, the reduction in C3aR expression was not specific for a defined tumor stage (online supplemental figure 1). Since mutations of single genes in the complement pathway are rare, we reasoned that downregulation of the c3ar1 gene might be mediated by epigenetic modifications.29 Methylation occurs at a high frequency in CRC.30 We mined the MethHC database of DNA methylation and gene expression in human cancer and found that the c3ar1 gene is highly methylated in CRC tissues compared with normal mucosa.25 Remarkably, we found significant hypermethylation in the N and S shores (regions up to 2 kb away from CpG islands) and in the shelves (figure 1B–D), which, as shown by Irizarry and collaborators, inversely correlates with gene expression levels.31 These results suggest that the downregulation of C3aR in CRC may be driven by CpG island methylation events, which are prominent for CRC development.32 These novel findings suggest that C3aR plays an important role in the human intestinal tract. In support of this hypothesis, loss or reduction of C3aR caused by DNA methylation events, as we reported in patients, may favor the establishment of CRC.
Supplemental material
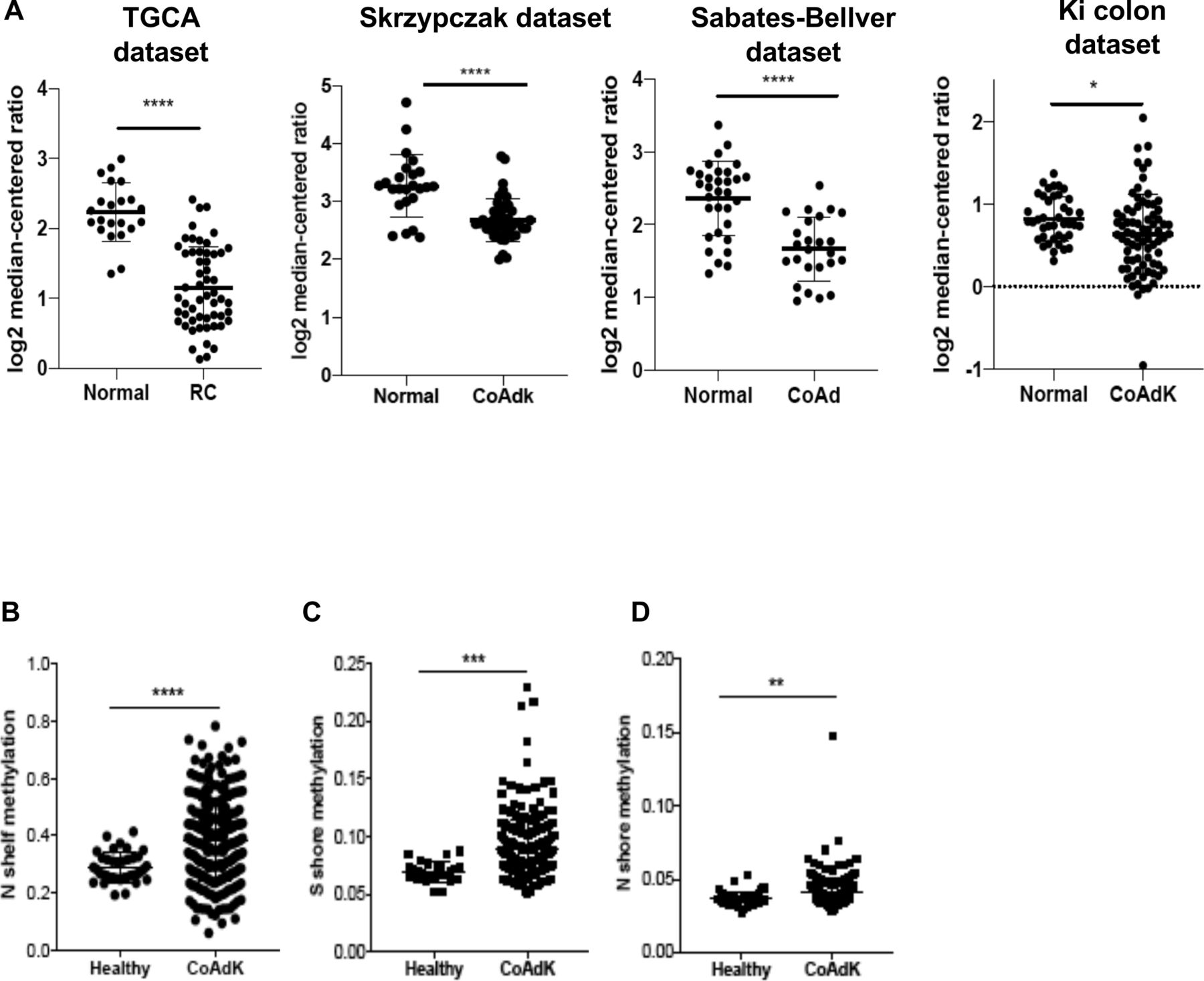
C3aR methylation and downregulation in patients with CRC. (A) C3aR expression in patients with RC and colon cancer from four independent datasets (TGCA: 22 healthy, 55 RC; Skrzypczak cohort: 24 healthy, 45 CoAdK; Sebates-Bellver cohort: 32 healthy, 25 CoAd; Ki cohort: 41 healthy, 76 CoAdK). (B) N shelf, (C) S shore and (D) N shore methylation of c3ar1 in patients with CRC. Significance was calculated using t-test. *=P>0.05, ****=P>0.0001. C3aR, C3a receptor; CoAd, colon adenoma; CoAdK, colon adenocarcinoma; CRC, colorectal cancer; RC, rectal cancer.
C3a–C3aR axis plays a previously unappreciated role in CRC development
To test the role of the C3a–C3aR axis in CRC development, we reverse-translated our findings from the database and investigated CRC development in vivo in mice lacking C3aR. First, age-matched and sex-matched C3aR−/− and WT mice were administered the carcinogen AOM followed by three cycles of the inflammatory agent DSS. Relative to WT, C3aR−/− mice showed increased weight loss (online supplemental figure 2A), suggesting that C3aR protects from excessive intestinal inflammation. This result agrees with previous literature showing that lack of complement activation in C3−/− mice exacerbates chronic intestinal inflammation.33 Notably, as shown in online supplemental figure 2B-D, C3aR−/− mice developed significantly higher tumor numbers and higher tumor load in the colon than the WT mice, with a predominance of larger tumors (>2 mm). As innate and adaptive immune responses are involved in colon inflammation and tumorigenesis, we set up three interlinked flow cytometry panels to characterize the immune infiltrates in the colon adenocarcinoma generated following AOM/DSS treatment and the mesenteric lymph nodes (mLNs). As shown in online supplemental figure 2E–G, we found higher numbers of CD11c+ macrophages in the mLN of C3aR−/− mice than WT, with no significant differences in the other cell populations. In the tumors, in addition to higher numbers of CD11c+ macrophages (online supplemental figure 2H), in the C3aR−/− mice, we detected significantly enhanced levels of activated CD4+ T cells (CD4+CD25+ FoxP3) and interferon gamma (IFN-γ)+ CD4 (Th1) (online supplemental figure 2I,J) compared with WT mice.
Although more than 20% of individuals with IBD develop CRC, intestinal inflammation accounts for only about 2% of total CRC.34 Therefore, we next investigated the impact of C3aR loss in the APCMin/+ spontaneous model, which, like 80% of human CRC, carries a mutation of the apc gene.35 We generated APCMin/+ mice lacking the c3ar1 gene (APCMin/+/C3aR−/−) and compared tumor development in the colon from 5 weeks to 28 weeks of age APCMin/+ and APCMin/+/C3aR−/− mice. We and others have shown that in APCMin/+ mice, CRC development is seldom, with the highest tumor burden in the small intestine.13 36 In line with the human data and the results in the model of inflammation-driven CRC, we found that in the absence of C3aR, starting at 10 weeks of age, APCMin/+ mice showed a shift of tumorigenesis from the small intestine to the colon with the highest localization in distal colon and rectum (figure 2A,B). Similar to the approach used in the AOM/DSS model, we performed flow cytometry to quantify the immune infiltrate associated with tumor development. Due to the insufficient number of colon tumors in APCMin/+ mice, we analyzed the mLN and the cLP. In agreement with our findings in the AOM/DSS model, loss of C3aR resulted in significantly higher numbers of Th17, Th1, Th1/Th17, and CD8+ T cells in the cLP of APCMin/+/C3aR−/− mice compared with the APCMin/+ counterpart (figure 2C–H). In the mLN, we found higher numbers of CD4+ and CD8+ T cells in the APCMin/+/C3aR−/− mice compared with APCMin/+ mice, but functional differences were only limited to higher numbers of IFN-γ-producing CD8+ T cells in APCMin/+/C3aR−/− mice (online supplemental figure 3A–F). Altogether, these results demonstrate that loss of C3aR unleashes a colon-specific inflammatory program.

Loss of C3aR switches tumorigenesis from small intestine to colon in APCMin/+ mice and promotes increased immune cell infiltration. (A) Tumor numbers in the colon of APCMin/+ and APCMin/+/C3aR−/− from weeks 5 to 28 weeks of age. (B) Representative picture of tumor number and distribution in the colon of APCMin/+ and APCMin/+/C3aR−/− mice. Single-cell suspensions from cLP of APCMin/+ and APCMin/+/C3aR−/− mice were analyzed by flow cytometry. Shown are the total number of (C) CD4+ T cells, (D) Th17 cells, (E) Th1 cells, (F) Th1/Th17 cells, (G) CD8+ T cells, and (H) Tc cells (CD3+CD8+IFN-γ+). A significance was calculated using two-way analysis of variance with Bonferroni post-test and a minimum of 10 mice/group. In panels, (C–H) Results are pooled from two independent experiments with a minimum of nine mice/group. Significance was calculated using unpaired t-test. *P<0.05, **P<0.01, ****P<0.0001. C3aR, C3a receptor; cLP, colon lamina propria; IFN-γ, interferon gamma; IL, interleukin; ns, not significant.
Loss of C3aR results in altered gut microbiota that supports tumor growth in the colon of APCMin/+ mice
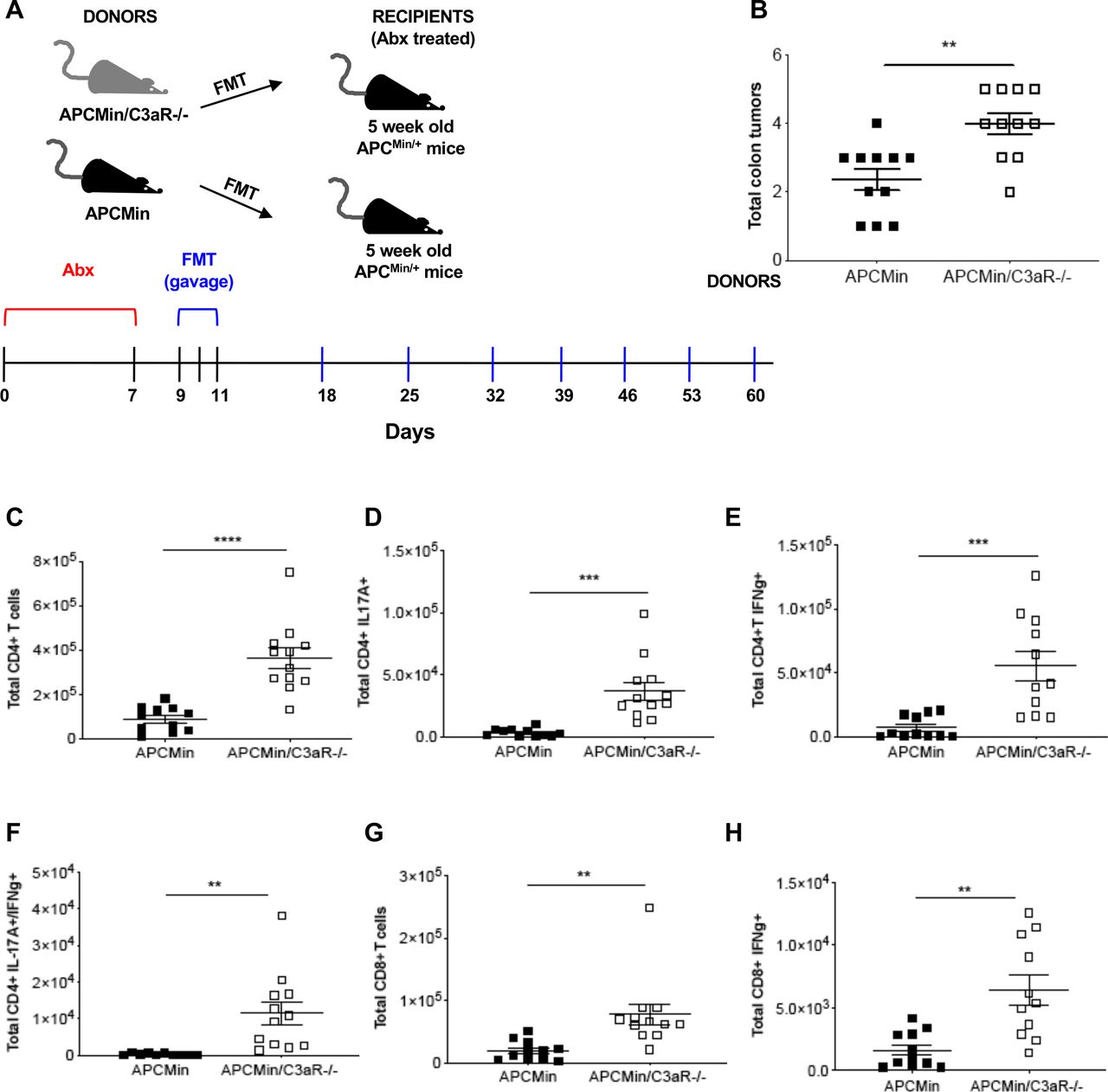
Several evidences suggest that the microbial flora strictly influences CRC development and immune signature.37–39 Therefore, to assess the functional role of the microbiota in tumor development in our model, we first set up to understand whether the transfer of APCMin/+/C3aR−/− microbiota could induce tumor development in the colon of APCMin/+ mice. To this aim, tumor-free, 5-week-old APCMin/+ mice were treated with broad-spectrum antibiotics, then administered via gavage with fecal and tumor-associated microbiota from 12-week-old APCMin/+/C3aR−/− mice or APCMin/+ mice and sacrificed at the age of 12 weeks (figure 3A). As shown in figure 3B, while APCMin/+ mice receiving the APCMin/+ microbiota developed very few tumors in the colon, the transfer of the APCMin/+/C3aR−/− microbiota resulted in increased tumor burden in the colon, with no changes in the total number of small intestinal tumors (online supplemental figure 4A,B). To test whether the microbiota could also affect the phenotype and functions of the immune cells in the colon of microbiota-transplanted mice as observed in the donors, we characterized the immune cell infiltrates in APCMin/+ mice receiving APCMin/+ or APCMin/+/C3aR−/− microbiota. As shown in figure 3C–H and online supplemental figure 4C–H, APCMin/+ mice receiving APCMin/+/C3aR−/− microbiota showed higher tumor numbers in the colon and increased Th1, Th17, and Th1/Th17 cells as well as IFN-γ-producing CD8+ T cells.
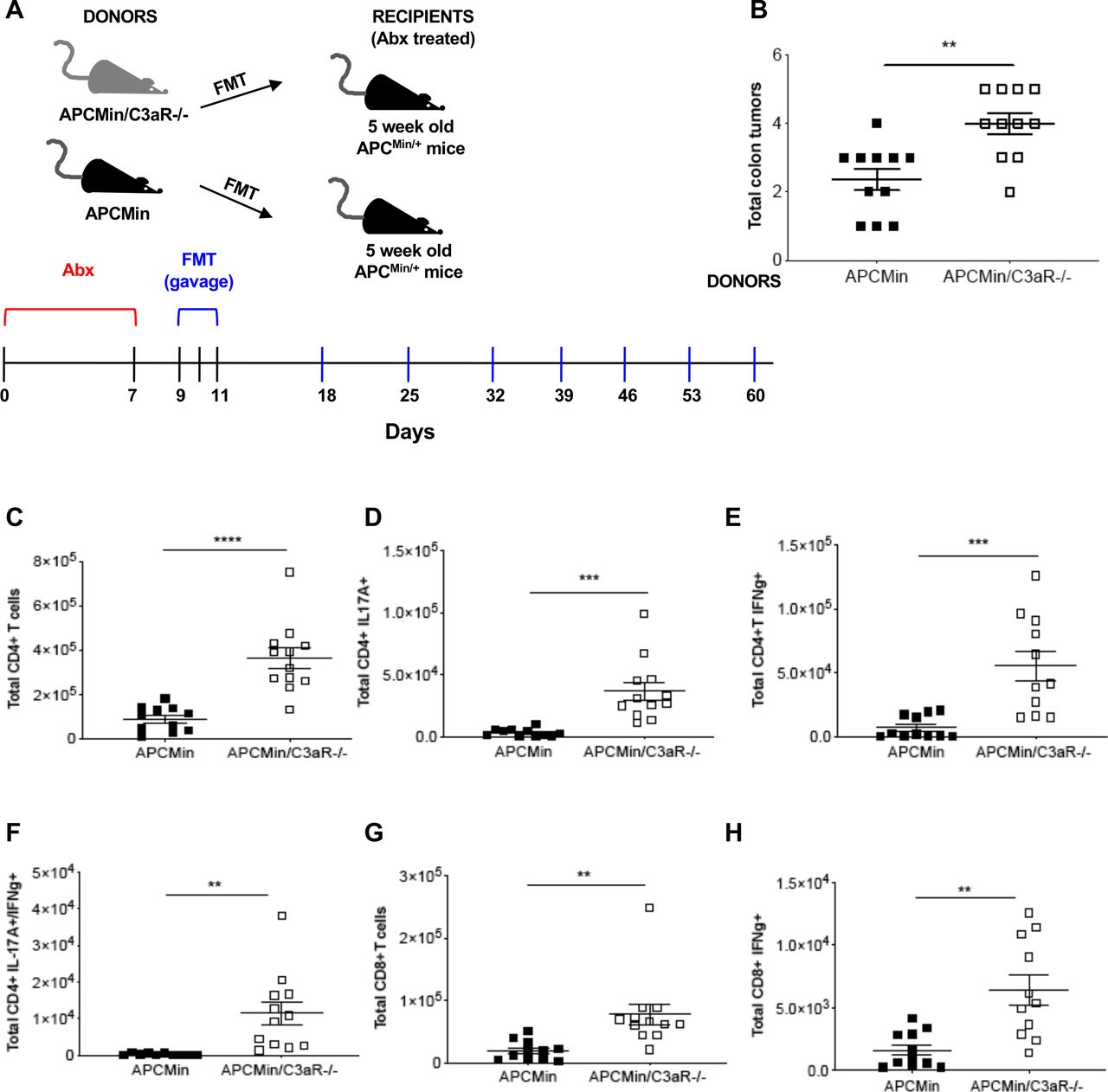
Transplantation of the APCMin/+/C3aR−/− microbiota transfers inflammation and colon tumorigenesis to APCMin/+ mice. (A) Treatment scheme for 5-week-old APCMin/+ mice (recipients) transplanted with the microbiota of 12-week-old APCMin/+ or APCMin/+/C3aR−/− mice (donors). (B) Tumor counts in the colon of recipient mice. (C–H) Flow cytometry analysis of cLP infiltrating lymphocytes showing total cell number of (C) CD4+ T cells, (D) Th17 cells, (E) Th1 cells, (F) Th1/17 cells, (G) CD8+ T cells, and (H) cytotoxic T (Tc) cells. Shown are the results of two independent experiments with 11 mice/group. Significance was calculated using unpaired t-test. **P<0.01, ***P<0.001, ****P<0.0001. C3aR, C3a receptor; cLP, colon lamina propria; FMT, fecal microbiota transplantation; IFN-γ, interferon gamma; IL, interleukin
Next, to understand whether the results obtained with the microbiota transplantation were the consequence of a tumor-induced microbiota or a change in microbiota composition driven by C3aR deficiency, we characterized the microbiota before and after tumor development. For this purpose, we collected the fecal pellets from tumor-free (8-week-old) and tumor-bearing (12-week-old) APCMin/+ and APCMin/+/C3aR−/− mice. Feces from age-matched and sex-matched WT and C3aR−/− were used as controls. The V5–V6 hypervariable regions of bacterial 16S rRNA genes in feces were amplified, and the obtained libraries were sequenced on the MiSeq Illumina platform. Compared with controls, we observed higher alpha diversity in 8-week-old APCMin/+/C3aR−/− and C3aR−/− mice measured by observed ASV, Faith, and Shannon indices (figure 4A). The alpha diversity was normalized in 12-week-old mice, although there was still a clear separation among the two mouse strains in terms of beta diversity (figure 4B). These results support the concept that the fecal microbiota that precedes tumor growth is highly diversified within the same group.

Loss of C3aR affects the composition of the fecal microbiota in APCMin/+mice. Bacterial DNA was extracted from the feces of 8-and 12-week-old APCMin/+, APCMin/+/C3aR−/−, C3aR−/− and WT mice, and bacterial profiling was performed by sequencing the V5-V6 hypervariable of 16S rDNA using Illumina MiSeq platform. (A) Plots showing alpha diversity evaluated by observed ASV, Faith, and Shannon Index in 8-and 12-week-old mice. (B) Principal Coordinates Analysis (PCoA) showing the beta diversity assessed by the three inferred Beta Diversity metrics (weighted UniFrac, unweighted UniFrac, and Bray-Curtis) in mice 8 and 12 weeks old. (C) Doughnut charts showing phylum abundance at 8 and 12 weeks. Four to eight mice/group were used. Significance was calculated using (A) the Kruskal-Wallis test, followed by a pairwise Wilcoxon as post-test; (B) PERmutational Multivariate ANOVA (PERMANOVA) test; and (C) DESeq R-package. C3aR, C3a receptor.
In contrast, the selection of defined bacterial species reduced intragroup and enhanced intergroup diversification on tumor development. When examining the most abundant phyla differences, we observed that already at 8 weeks, when the mice in our colony are devoid of tumors in both small intestine and colon, in APCMin/+/C3aR−/− mice, there was an increase in Bacteroidetes and Proteobacteria. Further, there was an evident reduction of Firmicutes compared with APCMin/+ mice. These differences became even more striking at 12 weeks when we could see the first differences in colon tumorigenesis (figure 4C). Notably, Bacteroidetes are significantly upregulated in the stool-associated microbiota of patients with CRC and have been correlated with elevated Th17 in their tumor-adjacent mucosa.40 41 To determine whether the differences found in the fecal microbiota were mirrored at the tumor site, we additionally sequenced the mucus-associated and tumor-associated bacteria in 12-week-old APCMin/+ and APCMin/+/C3aR−/− mice. As shown in online supplemental figure 5A,B, despite the bacterial composition in tumor and mucus of the two mouse strains being more similar than that in the stool and was dominated by Firmicutes, the tumor-associated microbiota in APCMin/+/C3aR−/− mice still showed a higher abundance of Bacteroidetes. These results closely mirror the bacterial composition in CRC, where Firmicutes is the predominant phylum in the cancerous tissues, followed by Bacteroidetes.42
Altogether, these results suggest that lack of C3aR promotes the establishment of a fecal microbiota that is among the drivers of tumor development and proinflammatory immune responses.
Loss of C3aR converts cold tumors into hot tumors
To more thoroughly determine how the lack of C3aR shapes the TME and the adjacent mucosa in our model of spontaneous colon tumorigenesis, we performed RNA-Seq analysis. First, we compared the transcriptomic profile of colon tumors developed by 12-week-old APCMin/+ and APCMin/+/C3aR−/− mice. We found 72 downregulated genes and 369 upregulated genes in colon tumors from APCMin/+/C3aR−/− mice, clearly indicating that loss of C3aR affects the transcriptomic profile of the polyps (figure 5A,B).

Tumors in APCMin/+/C3aR−/− show enrichment in innate and adaptive inflammatory pathways. (A) Heatmap from log counts per million values using data from colon polyps from APCMin/+ and APCMin/+/C3aR−/− mice. (B) Summary plot showing the number of significant differentially expressed genes in polyps of APCMin/+ and APCMin/+/C3aR−/− mice. The plot displays log fold change against log counts per million for each gene. The red points represent significant differentially expressed genes, and the horizontal blue lines indicate a twofold increase or decrease in expression. (C) Visualization of the gene-associated GO biological processes in the polyps. (D) Volcano plot showing the fold change magnitude for all genes differentially expressed in the polyps of APCMin/+C3aR−/− versus APCMin/+ mice (n=4). Significantly upregulated and downregulated genes belonging to the GO biological processes (B) are highlighted in yellow. Vertical and horizontal red dotted lines indicate the threshold. Significance was calculated using the edgeR function decideTestsDGE with Benjamini-Hochberg correction for FDR; a default FDR threshold of 0.05 and a log2 fold change threshold of 0.6 were applied. C3aR, C3a receptor; FDR, false discovery rate; GO, Gene Ontology.
Notably, iPathway analysis revealed that the top 25 Gene Ontology (GO) biological processes associated with differentially expressed genes in the polyps were related to innate and adaptive immune responses (figure 5C). Accordingly, using the edgeR-GLM framework (see Materials and methods), we found significant upregulation of genes associated with defense response. These gens were cd8, ccr6, il21, cd19, and il22ra2; IFN-γ-mediated signaling pathway-related genes, such as if and guanylate-binding protein family genes tap-1, ciita, b2-microglobulin, and stat1; genes associated with development and maturation of innate immune cells and response to type I interferon such as irf4 and irf8 and cd83; negative regulators of the immune response, such as btla, cd274 (pd-l1), cd40lg, and ido1; genes associated with leukocyte migration such as cxcl9, cxcl10, cxcl11, and cxcr5; and NK cell activation genes such as il-12b and ncr1. Among the significantly downregulated genes, we found genes for transporters including slc47 and slc44a5 and Wnt signaling pathway inhibitors such as dkk2 and dkk3 (figure 5D). The latter mediates the failure of anti-PD-1 treatment in patients with MSS-high CRC by dampening NK and CD8-mediated antitumor immune responses.43
Similar to the approach used for the TME, we performed RNA-Seq analysis on the distal colon of the same APCMin/+ mice and APCMin/+/C3aR−/− using WT and C3aR−/− as respective controls. The comparison revealed that APCMin/+ mice had a transcriptomic profile closer to WT mice than to APCMin/+/C3aR−/− mice, and similarly, healthy colons of C3aR−/−mice exhibited a transcriptomic profile more similar to the colons of APCMin/+/C3aR−/− than to the colons of WT mice (online supplemental figures 6A–F and 7A). Notably, iPathway analysis revealed that the top 25 GO biological processes associated with differentially expressed genes were related to innate and adaptive immune responses (online supplemental figure 7B,C).
Since tumor development in APCMin/+ mice is driven by Wnt signaling, which results in immune exclusion and MSS-like phenotype, our findings indicate that the loss of C3aR is sufficient to promote a vigorous immune signature in the colon and the tumors of APCMin/+ mice, therefore converting cold tumors into hot tumors.
C3aR expression in patients with cancer affects the infiltration of innate and adaptive immune cells
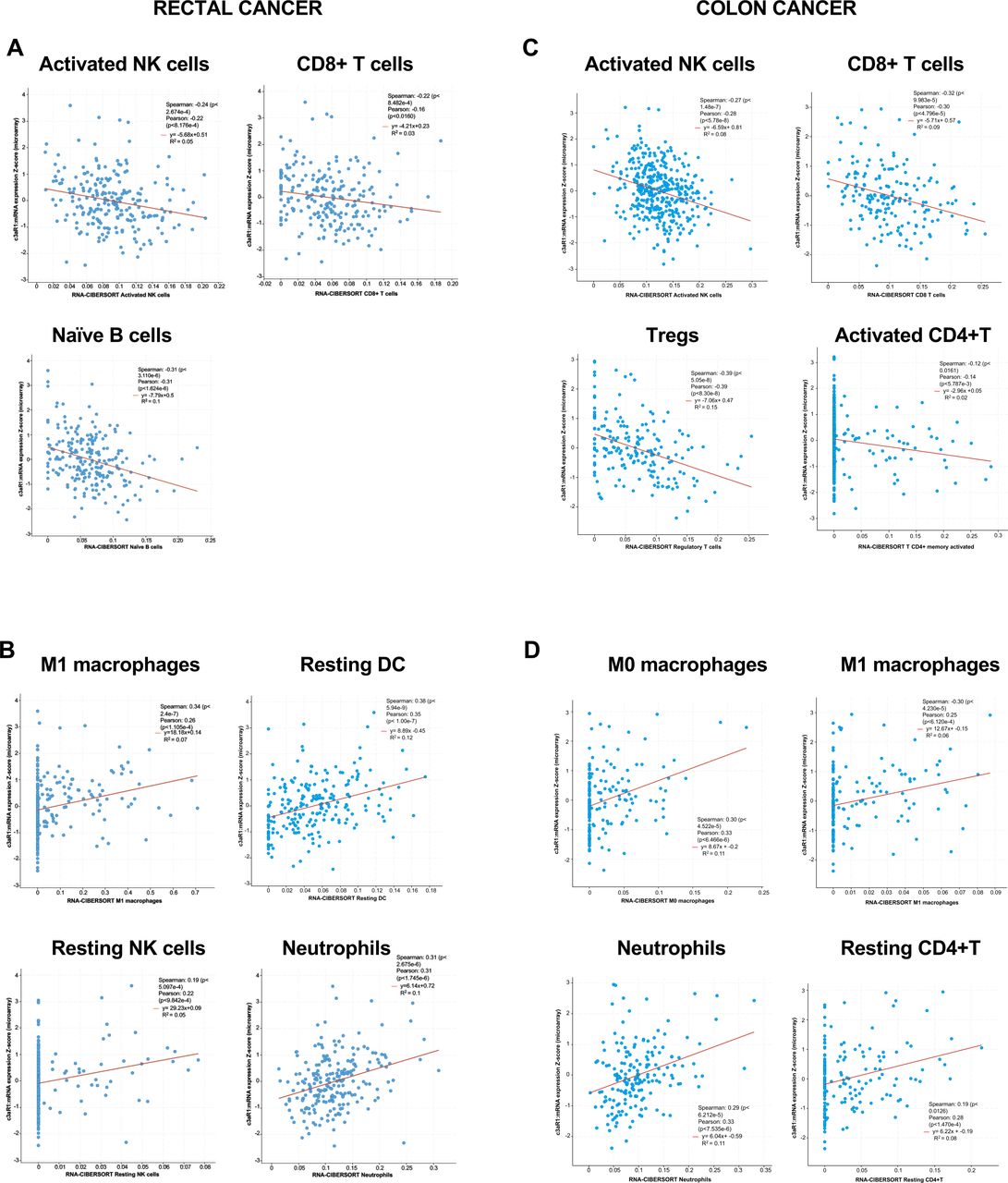
Our C3aR expression data in the patients with colon and rectal cancers and our mouse model prompted us to search the CBIOportal database for how C3aR expression affects the immune contexture in the human TME. We obtained the RNA expression for c3ar1 and immune cell signatures from 231 cases of rectal cancer and 97 cases of colon cancer in the FOxTROT cohort (https://ascopubs.org/doi/abs/10.1200/JCO.2019.37.15_suppl.3504) using CIBERSORT, xCell and MCP (online supplemental table 1).44–47 Cell populations showing significant positive or negative correlation with C3aR with two out of three methods in both patients with rectal and colon cancers are reported in figure 6. Pearson and Spearman correlation analyses revealed that in patients with rectal and colon cancers reduced expression of C3ar1 resulted in an increased level of innate and adaptive immune cells, which have been associated with better response to anti-PD1 immunotherapy in several solid malignancies, including CRC, such as activated NK, CD8+ T, B cells, memory CD4+ T cells and Tregs5 45 46 (figure 6A,C). Conversely, our analysis revealed that higher expression of C3ar1 positively correlated with several immune cells that can contribute to the generation of an immunosuppressive TME or can directly inhibit cytotoxic immune responses by inducing tolerance such as resting NK cells, macrophages, neutrophils, and resting dendritic cells (DCs) (figure 6B,D).48 49 Therefore, confirming our earlier results in our novel mouse model, results showed that the C3aR status may have substantial implications for treating patients with colon and rectal cancers with immunotherapy using ICB.

C3aR expression in patients with cancer affects the infiltration of innate and adaptive immune cells. Plots showing negative (A–C) and positive (B–D) correlation between C3ar1 expression and innate and adaptive immune cell populations assessed by CIBERSORT in a retrospective cohort of 231 patients with rectal cancer in the S:CORT WS3 Grampian Set and 97 patients with colon cancer in the FOxTROT cohort. Data were visualized and downloaded through the private S:CORT cBioPortal and analyzed using the Spearman and Pearson correlation tests. C3aR, C3a receptor; DC, dendritic cell; NK, natural killer.
Lack of C3aR confers susceptibility to treatment with anti-PD-1
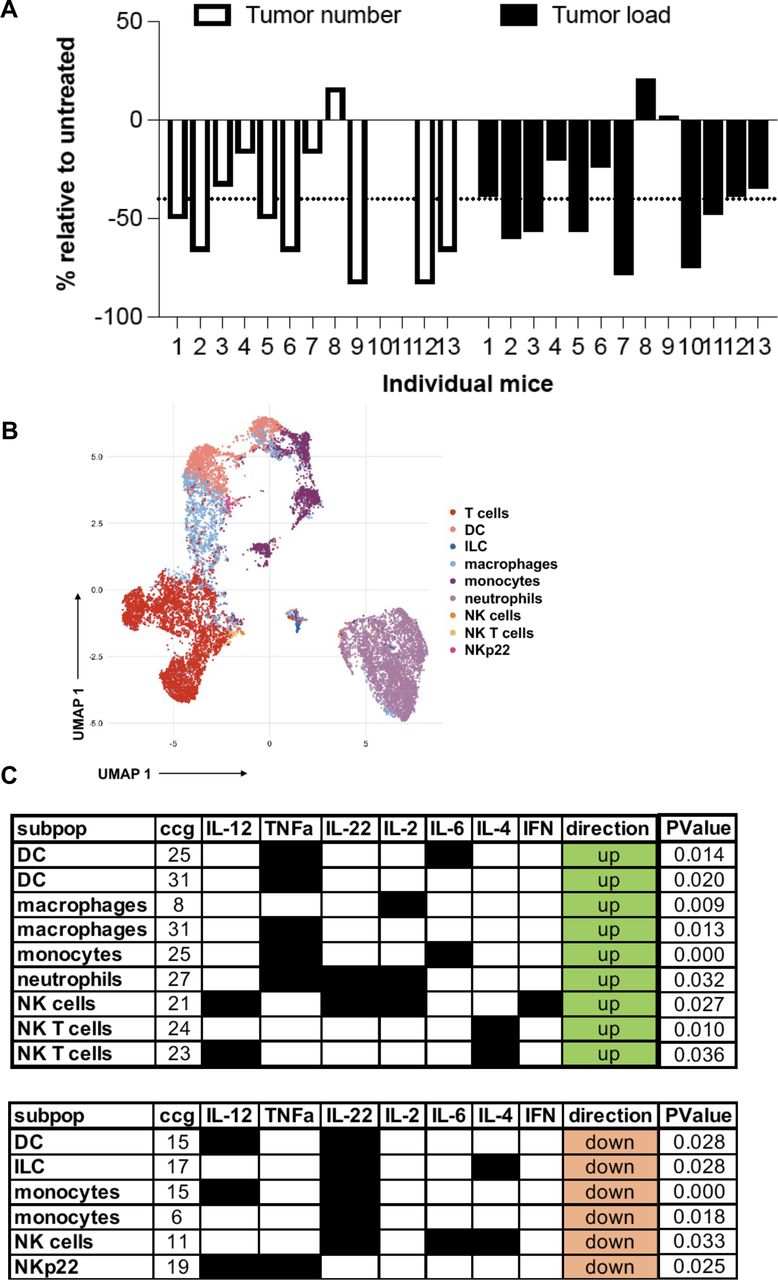
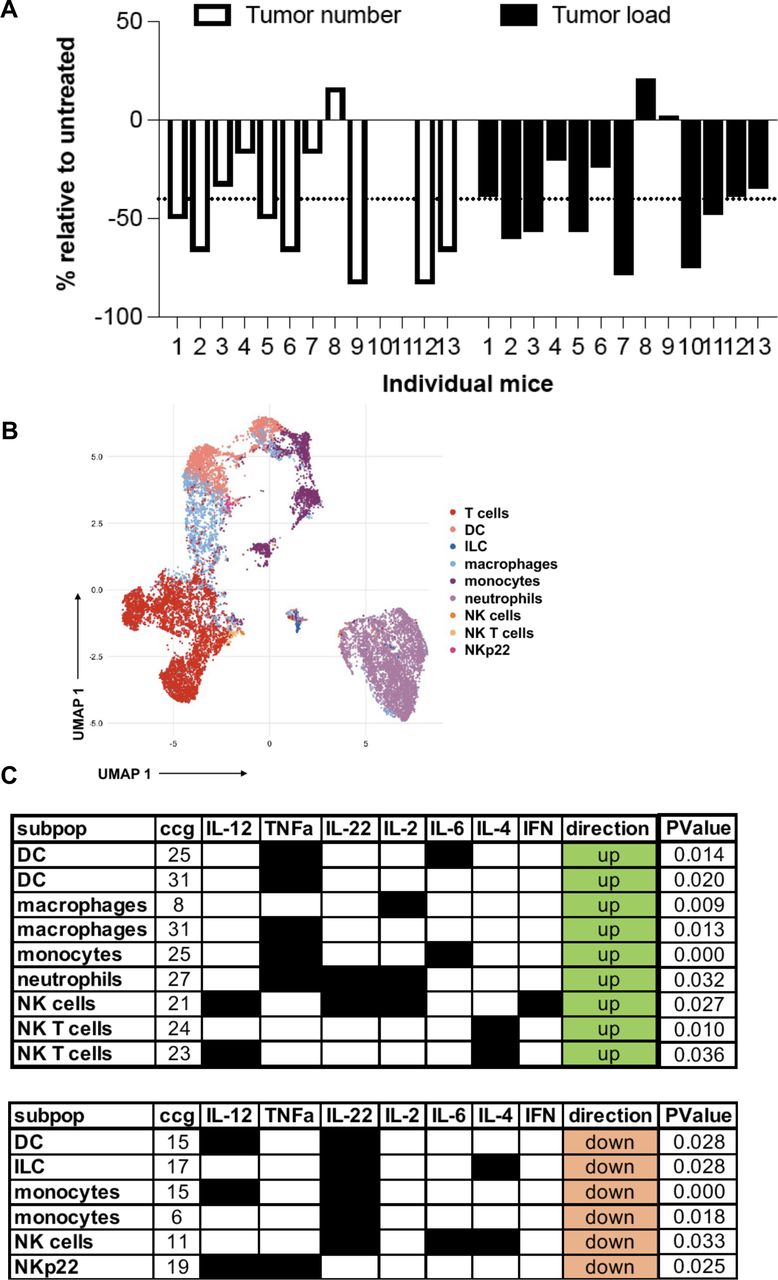
Based on the results on the human dataset and the findings in our mouse model showing an increased innate and adaptive immune response and the expression of checkpoint blockade molecules such as PD-L1 and an IFN signature, we next investigated whether this signature confers response to ICB therapy in APCMin/+/C3aR−/− mice. To perform a treatment regimen that would more closely resemble the clinical setting, APCMin/+/C3aR−/− mice were treated with a-PD-1 or with the control antibody twice a week for 5 weeks starting at the age of 12 weeks when colon tumors are already established in our colony. As shown in figure 7A, at sacrifice, we found that relative to their untreated controls, the vast majority of APCMin/+/C3aR−/− mice treated with a-PD-1 showed a significant reduction in tumor number and load, which demonstrates that a-PD-1 is not only effective at reducing the development of new tumors but also can significantly reduce the size of already established tumors. After ICB treatment, mass cytometry was used to determine tumor-infiltrating immune cells’ overall composition and function. To this aim, we developed and validated a mass cytometry panel encompassing markers to define cell types and a set of cytokines to define immune cell functions, which were analyzed following our previously published bimatrix analysis (table 1 and online supplemental figure 8).50
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lack of C3aR confers susceptibility to treatment with a-PD-1 treatment. (A) Tumor number and load in individual APCMin/+/C3aR−/− mice treated for 5 weeks with a-PD1 expressed as % reduction over the mice treated with the control antibody. (B) Visualization of 70 000 pooled events from a-PD1 and control mice using the uMAP algorithm. (C) The functional profile of cell clusters that were significantly upregulated or downregulated in anti-PD1-treated mice compared with control was visualized by using the bimatrix approach. The results of two independent experiments with a minimum of 11 mice/group are shown. DC, dendritic cell; IFN, interferon; IL, interleukin; ILC, innate lymphoid cells; NK, natural killer.
Briefly, after annotating immune cell types using only the type markers (figure 7B), we considered an entire cytokine set profile for each cell type. The cells were then clustered using the self-organizing map (SOM) method in 10 times 10 grid to generate a profile of potential 100 cytokine-producing clusters per cell type. Following a-PD-1 treatment, we found increased frequencies of intratumoral polyfunctional NK cells expressing interleukin (IL)-2, IL-12, and IFN-γ and downregulation of an NK cluster expressing IL-6, IL-4, and IL-22. In addition, although our mass cytometry panel was not designed to study ILC specifically, we also found a parallel reduction of an anti-inflammatory ILC-like cell cluster producing IL-4 and IL-22 (figure 7C). These results agree with a recent study in human CRC, which described in the tumor tissues of MSI-H CRC an innate immune signature characterized by increased levels of cytotoxic innate lymphoid cells and NK cells.51 Furthermore, we found increased frequencies of DC, macrophage, and monocyte clusters expressing IL-6 and tumor necrosis factor alpha (TNF-α). Notably, six out of seven significantly downregulated lymphoid and myeloid cell clusters expressed IL-22. Further, TNF-α and IL-12 producing NKp22, a cell type that has been shown to support CRC development, were downregulated following a-PD-1 therapy.
Overall, our results show that the inflammatory signature in APCMin/+/C3aR−/− tumors confer susceptibility to ICB therapy.
Discussion
Inflammation in CRC plays a paradoxical role. For instance, inflammatory immune responses fuel tumor development and progression, and the use of anti-inflammatory agents lowers sporadic CRC-related mortality.52 Conversely, the presence of an inflammatory immune infiltrate is essential to promote antitumor immunity and correlates with increased survival in patients with CRC.8 This evidence suggests that there may be options to exploit the underlying mechanisms of cancer-promoting inflammation to treat the disease more efficiently.
We describe the downregulation of the receptor for the complement anaphylatoxin C3a (C3ar1) in subsets of patients with CRC. Furthermore, we found that reduced C3aR expression in patients with cancer correlates with increased numbers of innate and adaptive antitumor immune cells in the TME. Reverse-translation in vivo uncovered that loss of C3aR enhances tumor development in a model of inflammation-driven CRC. Additionally, loss of C3aR was sufficient to induce colon tumorigenesis in the spontaneous APCMin/+ model, which usually shows a prevalence of tumors in the small intestine and seldom colon tumors. As such, our novel mouse of colon carcinogenesis represents an important contribution to the field where progress toward chemoprevention and treatment has been largely hindered by the sparsity of reliable models to closely mirror the human disease. Specifically, the generation of the APCMin/+ mouse following mutagenesis screening historically enabled the critical discovery of the central role of apc inactivation for the development of CRC.53 54 However, the development of multiple tumors in the small intestine rather than in the colon of APCMin/+ mice does not only poorly recapitulate the anatomical location of the human disease but also carries several additional shortcomings. Relevant publications in the field of CRC have tried to recapitulate the human disease by developing APC-based models of colorectal tumorigenesis such as the CPC-APC mice obtained by crossing the APCflox/wt with the CDX2-cre, which targets APC inactivation to the colon, therefore resulting in the development of adenomas in the distal colon.55 More recent data have also shown that the treatment with the carcinogen AOM can induce the development of tumors also in absence of the cre recombinase, but the underlying mechanism is poorly understood.56 Although the use of the cre-lox system deserves cautions and can have many pitfalls,57 it would be highly relevant to assess how the loss of C3aR in the CPC-APC system affects the number and the immune infiltration of colon tumors. In the herein described model by knocking out the receptor for the complement anaphylatoxin C3a (C3aR) in the APCMin/+ mice, we were not only able to achieve the development of several polyps in the colon but were also able to achieve a drastic reduction of tumors in the small intestine13 (online supplemental figure 9). As result, APCMin/+/C3aR−/− mice are devoid of several shortcomings that we and others have described in the APCMin/+ model such as ulcerations of the gastrointestinal tract that cause progressive increase in blood loss, development of hypercoagulation, anemia and splenomegaly and have a longer lifespan compared with APCMin/+ mice.13
Furthermore, similar to the findings in patients, the tumors developing in the absence of C3aR showed enhanced infiltration by innate and adaptive immune cells. This signature depended on the fecal and tumor-associated microbiota, and we provided evidence that this tumor-associated immune cell signature can be exploited for ICB therapy.
Several mechanisms could potentially mediate C3aR downregulation or inactivation in human CRC. In this context, it is essential to note that, while genetic mutations in the complement system as a group occur at a significantly higher rate than in any other gene for several cancers, most individual complement genes mutate at a lower rate than many canonical oncogenes and oncosuppressors.29 Accordingly, we found that in patients with CRC compared with healthy controls, there is a significantly higher frequency of C3aR methylation occurring in those regions likely to result in mRNA expression changes.
One of the most striking findings in our study is that lack of C3aR resulted in significant upregulation of immune signatures in the tumors and adjacent mucosa. Specifically, we found increased Tregs, Th17 and Th1/Th17, and IFN-γ-producing CD8 T cells. The role of IL-17 in CRC is highly debated. Data in patients and mouse models of CRC showed that IL-17 production by different cell sources promotes the inflammatory process and fosters cancer cell proliferation.58 59 On the other hand, the production of IL-17A is essential to preserve the integrity of the epithelial barrier, and published literature showed that Th17 in human CRC could recruit CD8+ T cells.60 The higher abundance of Th17 cells could be directly linked to the increased levels of il-21 in APCMin/+/C3aR−/− compared with APCMin/+ mice as shown by our RNA-Seq data. There is abundant evidence in the literature showing that IL-21 plays a crucial role in the differentiation of Th17 cells.61 62 Interestingly, the data on the effect of IL-21 in antitumor immunity and CRC development are somewhat conflicting. Specifically, in several preclinical models, IL-21 has been shown to potentiate antitumor immunity by supporting the cytotoxic activity of CD8+ T and NK cells, the proliferation of Th1 and Th17 cells and the antigen presentation by DCs.63 As such, it has undergone phase I and II clinical trials for several cancers with overall promising results.64–66 However, in the context of CRC, while one study in APCMin/+ mice confirmed the antitumorigenic role of IL-21,67 other studies have shown that both in sporadic and inflammation-driven CRC, IL-21 supports CRC development.68–70These findings were confirmed in patients with CRC by De Simone et al, who additionally showed that IL-21 upregulation in the tumors of patients with CRC is mostly driven by the infiltrating CD8+ T cells.71
In addition, lack of C3aR also resulted in upregulation of innate immune signatures suggestive of enhanced antigen presentation. Overall, these findings are surprising because apc mutations result in Wnt-driven tumors that have an MSS phenotype, with the exclusion of immune cells from the TME and consequent resistance to ICB therapy.72
The finding that immune responses can both promote the tumor development and be exploited to increase the efficacy of current anticancer treatments was also emphasized in a recently described dual oncogene mouse model of CRC. In this model, DeStefano Shields and collaborators showed that the administration of the enterotoxigenic Bacteroides fragilis promoted the development of colon tumors in mice carrying both the APCMin/+ (MinApcΔ716/+) and the human BRAFV600E mutations.73 Interestingly, in these mice, the increased tumorigenesis was accompanied by a Th1 signature in the TME with increased CD8+ T cells and expression of checkpoint inhibition molecules, which rendered the tumor responsive to anti-PD-L1 treatment.
The mechanistic reasons underlying these opposing roles of the immune system in cancer are the object of active investigations in the field. Two recent reports suggested that cancer cells can exploit even transient inflammatory conditions to undergo epigenetic reprogramming, and once they established the tumor, they exploit the selective pressure of the immune system to develop genetic alterations that enable their immune evasion.74 75
Our finding that MSS colon tumors lacking C3aR are responsive to anti-PD-1 treatment indicates that the identified immune signature has a positive prognostic value and that C3aR may serve as an additional checkpoint in ICB therapy of CRC. Previous studies have shown that inhibition of the complement anaphylatoxin C5aR enhances ICB response in immunologically hot tumors, such as lung cancer and melanoma.76 77 Based on our findings, we propose that dysregulation of the C3a–C3aR axis in CRC renders ‘cold’ tumors ‘hot’ rather than simply deblocking infiltrating immune cells. Notably, while we found that almost all animals responded to a-PD-1 therapy, with half of them achieving over 50% reduction in tumor number and tumor load, we also observed a lack of response in a few cases. These results are compatible with the ICB treatment in human CRC, where a-PD1 alone confers an overall response rate of 31% in patients with MSI-H CRC, which can be increased to 55% by the combination with a-CTLA-4.8
While CD4 and CD8 T cells are the main targets of anti-PD-1 therapy, checkpoint therapies’ efficacy depends on innate immunity.78 Specifically, increased frequencies of DC, monocytes, and macrophages producing TNF-α and IL-6 on treatment with anti-PD-1 indicate a direct effect of ICB on these myeloid cells. This is supported by published data showing that PD-1-expressing macrophages have a protumorigenic phenotype and that a-PD-1 therapy reverses this trend, increasing the expression of TNF-α, iNOS, and IL-6, which potentiate antitumor immunity.79 Additionally, we found that among the most downregulated cytokines in innate lymphoid and myeloid cell clusters, there was IL-22, which can support CRC growth by directly acting on epithelial cells.80 This finding is particularly interesting in light of our RNA-Seq data showing significantly higher expression of il-22ra2 in the polyps of APCMin/+/C3aR−/− mice compared with APCMin/+ mice. IL-22RA2 is also known as IL-22 binding protein (IL-22BP) and limits IL-22 bioavailability by preventing its binding to the IL-22-ra1. Seminal work in the field has shown that IL-22RA2−/− mice develop more colon tumors as a result of an enhanced IL-22-driven epithelial cell proliferation.81 DCs have been initially described as the main cells expressing IL-22ra2. However, Pelczar and collaborators unraveled a pathogenic role for the IL-22RA2 produced by CD4+ T cells, therefore suggesting that the cellular source of IL-22RA2 may be relevant for its function.82 Accordingly, based on our finding that the tumors from APCMin/+/C3aR−/− mice are highly infiltrated by T cells, it is likely that the IL-22RA2-producing CD4+ T cells fuel the pre-existing inflammation associated with the protumorigenic microbiota ultimately supporting tumor growth. It remains to be determined whether, similar to the anti-TNF-α treatment, a-PD1 therapy can selectively affect the expression of IL-22RA2 in CD4+ T cells.
A caveat of our finding on il-22ra2 is that the increased RNA levels may not necessarily correlate with the protein levels.
Our study also unraveled that the fecal microbiota of APCMin/+/C3aR−/− and C3aR−/− mice carried a higher abundance of Gram-negative bacteria such as Bacteroidetes and Proteobacteria. Based on the results of our FMT experiments, the fecal microbiota established in the absence of C3aR is likely the main driver of tumor development and the immune infiltrate in the colon of APCMin/+ mice. Conversely, the similarity between APCMin/+ and APCMin/+/C3aR−/− mice may suggest that the physiological and metabolic alterations occurring in the TME represent the main constraint that dictates the specific composition of the microbial community. The similarity in the overall composition does not exclude the blooming in the TME of APCMin/+/C3aR−/− of more pathogenic bacterial strains that can further support tumor growth and therefore play a more pronounced role at the later stages in agreement with the driver–passenger theory.83 However, considering the significant differences described in the fecal microbiota of patients with CRC and healthy controls, it is tempting to speculate that in the clinical setting, the composition of the indigenous rather than the tumor-associated microbiota may be a more relevant indicator for the risk of developing CRC.
Altogether, we reported here for the first time that the complement C3a–C3aR axis represents the Achilles’ heel of CRC. The complement system in the gastrointestinal tract is essential to avoid overt inflammation in health conditions. However, this regulatory mechanism may restrain the activation of immune responses during tumor development. Thus, the C3a–C3aR axis can be a previously unrecognized checkpoint to enhance antitumor immunity in CRC.
Data availability statement
Data are available in a public, open access repository. Raw RNA-Seq data have been deposited at ArrayExpress under the accession number E-MTAB-8500.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human subjects, and all samples were obtained following individual informed consent and ethical approval by the National Research Ethics Service in the UK (ref 15/EE/0241). The subjects gave informed consent to participate in the study before taking part. All animal experiments were performed following the guidelines established in the Principles of Laboratory Animal Care (directive 86/609/EEC) and approved by the Italian Ministry of Health and under approved protocols by the Institutional Animal Care and Use Committee at MUSC (IACUC-2017–00165; IACUC-2020–01022).
Acknowledgments
The authors thank Graeme Murray, Leslie Samuel, Dion G Morton and Nicholas P West for provision of the S:CORT cohorts of Grampian and Foxtrot and to all the patients and their families who gave consent to further research on their samples. S:CORT was funded by the Medical Research Council and Cancer Research UK. The authors also thank Riyue 'Sunny' Bao for discussion.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @brunofosso, @olcina_mm
Contributors SG conceived the study and the experimental setup, performed and analyzed the experiments, wrote the manuscript with input from all authors and is responsible for the overall content of this manuscript; CK performed the experiments and the data analysis and wrote the manuscript; LMW, MDR, and GH performed the analysis of RNA-Seq and mass cytometry data; BF and MM performed the microbiota sequencing and analysis; MMO and ED performed the analysis on human datasets; KM assisted with the mouse experiments; and SEA gave critical input for microbiota experiments and interpretation.
Funding This work was supported by 'Lenino Fontana and Maria Lionello' AIRC fellowship, Umberto Veronesi fellowship, ACS-IRG, and NIH P20GM130457 and NIH R01CA258882-01A1 to SG. The S:CORT consortium is a Medical Research Council (MRC)-stratified medicine consortium jointly funded by the MRC and CRUK (UKRI grant number MR/M016587/1). MMO is funded by the MRC (MC_UU_00001/10). ED is supported by the S:CORT Consortium funded by a grant from the MRC and Cancer Research UK.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.