Article Text

Abstract
Background Cancer immunotherapy has revolutionized cancer treatment. However, considering the limited success of immunotherapy to only some cancer types and patient cohorts, there is an unmet need for developing new treatments that will result in higher response rates in patients with cancer. Immunoglobulin-like transcript 2 (ILT2), a LILRB family member, is an inhibitory receptor expressed on a variety of immune cells including T cells, natural killer (NK) cells and different myeloid cells. In the tumor microenvironment, binding of class I MHC (in particular HLA-G) to ILT2 on immune cells mediates a strong inhibitory effect, which manifests in inhibition of antitumor cytotoxicity of T and NK cells, and prevention of phagocytosis of the tumor cells by macrophages.
Methods We describe here the development and characteristics of BND-22, a novel, humanized monoclonal antibody that selectively binds to ILT2 and blocks its interaction with classical MHC I and HLA-G. BND-22 was evaluated for its binding and blocking characteristics as well as its ability to increase the antitumor activity of macrophages, T cells and NK cells in various in vitro, ex vivo and in vivo systems.
Results Collectively, our data suggest that BND-22 enhances activity of both innate and adaptive immune cells, thus generating robust and comprehensive antitumor immunity. In humanized mice models, blocking ILT2 with BND-22 decreased the growth of human tumors, hindered metastatic spread to the lungs, and prolonged survival of the tumor-bearing mice. In addition, BND-22 improved the antitumor immune response of approved therapies such as anti-PD-1 or anti-EGFR antibodies.
Conclusions BND-22 is a first-in-human ILT2 blocking antibody which has demonstrated efficient antitumor activity in various preclinical models as well as a favorable safety profile. Clinical evaluation of BND-22 as a monotherapy or in combination with other therapeutics is under way in patients with cancer.
Trial registration number NCT04717375.
- Immunotherapy
- Tumor Escape
- Macrophages
- Lymphocyte Activation
- Translational Medical Research
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
While cancer immunotherapy has revolutionized cancer treatment, there is a constant need for developing new treatments that will result in higher response rates in patients with cancer.
WHAT THIS STUDY ADDS
The study described herein presents evidence for the role of immunoglobulin-like transcript 2 (ILT2) as a ‘multi-immune cell checkpoint’ which serves as an important immune evasion mechanism for tumors. In addition, the design and pharmacological activity of a novel, first-in-class, humanized ILT2 blocking monoclonal antibody, BND-22, is described.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
The results show that the inhibition of the ILT2 axis holds a great promise as monotherapy and in combination with established cancer therapies.
Background
Cancer immunotherapies, which target tumor immune evasion mechanisms, have transformed clinical cancer care.1 Immune checkpoint inhibitors, such as antibodies against programmed cell death protein-1 (PD-1), programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated protein-4 are now approved for the treatment of multiple cancer types. Despite this, only a small percentage of patients with cancer benefit from checkpoint-inhibitor therapies. In addition, late relapses are emerging with longer follow-up, suggesting the emergence of acquired resistance.2 Such resistance develops due to either the inability of T cells to infiltrate into tumors or to tumor-derived suppressive mechanisms overcoming antitumor functions of T cells. Most current immunotherapy regimens target T cells, yet myeloid cells, which constitute a larger cellular milieu of the tumor microenvironment, also play an important role in controlling overall tumor immunity.3 Therefore, immunotherapies that can promote functions of both T cells (adaptive immunity) and macrophages (innate immunity) may generate a more robust antitumor response, thus overcoming the limitations of current checkpoint inhibitors.4
Ig-like transcripts (ILTs) are a family of immune-modulating receptors expressed on various immune cells.5 Immunoglobulin-like transcript 2 (ILT2) (leukocyte Ig-like receptor 1/CD85j/LILRB1), a member of the ILT family, is an inhibitory receptor expressed on both adaptive and innate immune cells including T cells, natural killer (NK) cells and various myeloid cells.6–10 In T cells, ILT2 is highly expressed in a distinct CD8 T-cell population called effector memory T cells re-expressing CD45RA (TEMRA cells).11 12 ILT2 binds to the classical (HLA-A and -B) and non-classical (HLA-G, HLA-E, and HLA-F) major histocompatibility complex (MHC) class I molecules; however, its binding affinity to HLA-G is threefold to fourfold higher than to classical MHC I molecules.10 Besides its physiological role in maternal–fetal tolerance, HLA-G is commonly expressed by solid tumors and has been associated with worse prognosis in multiple cancers including colorectal, esophageal, gastric, liver, lung, and cervical cancers.13–16
The interaction of ILT2 with its ligands in the context of an immune reaction can lead to impairment of immune cell proliferation, differentiation, cytotoxicity, cytokine secretion, and chemotaxis; and induction of regulatory T cells and myeloid-derived suppressor cells (MDSC) or M2-type macrophages,16–20 thus allowing tumor cell evasion. ILT2 has been shown to inhibit multiple T-cell functions critical for antitumor immunity including cytokine production, cytotoxicity, and proliferation.11 21–25 ILT2 inhibits NK-cell cytotoxicity and their ability to secret proinflammatory cytokines,7 21 26–28 which can be restored by blocking ILT2.7 In addition, it has been recently demonstrated that interaction of ILT2-expressing macrophages with class I MHC expressing tumor cells can convey a ‘do not eat me’ signal, which prevents the phagocytosis of tumor cells by macrophages.18 These collective observations identify the ILT2-mediated signaling axis as a potential novel target for anticancer immunotherapy.
Here we present evidence for the role of ILT2 as a novel immune checkpoint, which serves as an important immune evasion mechanism for tumors. Furthermore, we describe the design, pharmacological activity, and non-clinical safety assessment of a novel, first-in-class, humanized ILT2 blocking monoclonal antibody. Collectively, the results provide the foundation for clinical testing of BND-22 in patients with solid tumors as monotherapy or in combination with PD-1 or tumor targeting therapeutics.
Materials and methods
Cells
Cell media and supplements were obtained from Biological Industries. Cell lines used in this study include A375 (ATCC, CRL-1619), A253 (ATCC, HTB-41), BW 5147.3 (BW; ATCC, TIB-47), 721.221 (ATCC, CRL-1855), 786-O (ATCC, CRL-1932), Jurkat (ATCC, TIB-152), COLO-320 (ATCC, CCL-220.1), and MEL526.
A253 and A375 were transfected to stably express human HLA-G1 using jetPEI (Polyplus). BW cells were transfected to express the extracellular part of human ILT2 or ILT4 and the intracellular part of mouse zeta chain using the Amaxa Nucleofector (Lonza) and Ingenio Kit (Mirus). 721.221 cells were transfected to express human HLA-G1, BW cells to express cynomolgus ILT2, COLO-320 to express human HLA-G1 and Jurkat cells to express human ILT2 using the Amaxa Nucleofector and V Kit (Lonza). G418 (Sigman-Aldrich) was used for selection and generation of stable clones. A375 WT cells or A375-HLA-G cells were also transfected to express OKT3scFv-CD1429 followed by selection with hygromycin (Sigman-Aldrich). Primary cells used in this study include tumor-infiltrating lymphocytes (TILs) expanded from a melanoma patient (purchased from Sheba Medical Center).
Generation of single-cell suspensions from tumor samples
Tumor samples were obtained from biobanks of Rambam Medical Center and the Galilee Medical Center. Samples were digested into single-cell suspensions using DNase 1 type IV (30 U/mL), hyaluronidase type V (100 µg/mL) and collagenase (1 mg/mL). The samples were digested for 2 hours at 37°C followed by filtration through a cell strainer (70 µm) and washing in PBS to generate a single-cell suspension.
Isolation of peripheral blood mononuclear cells (PBMC)
Buffy coat samples were obtained from blood bank of healthy donors and patients with cancer following informed consent. Samples were diluted 1:2 in PBS and PBMCs were isolated using LSM gradient (MP Biomedicals) by centrifuging (1000×g, for 15 min) at room temperature without brakes. PBMCs were harvested, treated with red blood cell lysis buffer (Biological Industries), washed twice with PBS and used fresh or kept frozen in liquid nitrogen. The characteristics of the patients with cancer included in the study are detailed in the following table. The healthy donor samples acquired from the blood bank (n=6) were anonymous.
Immunohistochemistry (IHC) of HLA-G
Tissue microarrays from different cancer types (Biomax) were subjected to antigen retrieval in citric buffer followed by staining with HLA-G antibodies (Sigma Aldrich, SAB2702386) and detection with ZytoChem Plus HRP-Polymer Anti-Rabbit (Zytomed, ZUC032-100) and DAB (3,3′-Diaminobenzidine) staining. Sections were then lightly counterstained with hematoxylin, dehydrated through ascending ethanols and mounted in DPX. IHC score was calculated by staining intensity multiplied by percentage of positive cells (Smart Assays). The following arrays were used in the study: ES809 (n=80, median age 59±8, male:female ratio (M:F) 75%:25%); HN802b (n=67, median age 55±12.2, M:F 87%:13%); ST1001a (n=57, median age 58.0±10.0, M:F 82%:18%) and CO20813a (n=143, median age 60±12.6, M:F 55%:45%). Healthy tissues constituted of the normal samples included in ES809 and HN802b (n=14, median age 34±13.1, M:F 57%:43%).
Soluble HLA-G ELISA
Levels of soluble HLA-G were detected in plasma samples from patients with cancer and healthy controls (DX biosamples) using a commercial ELISA according to the manufacturer’s instructions (AVIVA, HKEH00979) (n=20 for each cancer type; characteristics are stated as follows).
Epitope mapping of BND-22
Initial in silico epitope mapping was performed by MAbSilico. The structure of ILT2 used was modeled using the structures 6AEE (four Ig-like domains, some loops missing), 1VDG (unpublished, domains 1 and 2), 1G0X (domains 1 and 2), and 4LL9 (domains 3 and 4).30 31 A three-dimensional model of the antibody was built using Modeler. Empirical mapping was performed by Neoproteomics using hydroxyl radical footprinting (HRF) and mass spectrometry techniques.
Xenograft mice tumor models
For the melanoma lung lesions model, 5–12 SCID-NOD mice/group were engrafted IV with MEL526-HLA-G cells (4×106). PBMCs (15×106) from different healthy donors were injected intravenously into the mice on days 2, 10 and 18 or on days 15, 25, 35 and 51. Interleukin (IL)-2 (75×103 U/ mouse; Proleukin Novartis Pharma) was administered intraperitoneally every other day. A blocking ILT2 antibody (maILT2, hybridoma derived 19E3), BND-22, anti-PD1 (nivolumab, BMS), or a control IgG (BioXcell, BE0019-1 or Sino, HG4K) was administered at 10 mg/kg two times a week according to the treatment groups. Mice were monitored for weight and well-being by a person blinded to the experimental procedure. At the endpoint, full-body evaluation of metastasis was conducted; the lungs were photographed, harvested and weighed in order to record tumor burden.
For the subcutaneous (SC) colon cancer macrophage model, 1×106 COLO-320-HLA-G were injected subcutaneously to the right flank of 9 SCID NOD mice/group together with human macrophages which were generated from monocytes isolated from the blood of healthy donors (2×106). The mice were administered BND-22 or a control IgG4 (BioLegend, 403702), 20 mg/kg each. The antibodies were given SC on tumor inoculation day and four additional injections intravenously at days 3, 7, 10 and 14. Mice were monitored three times a week for weight, survival and for tumor volume using caliper measurement by a person blinded to the experimental procedure. Tumor volume was calculated as (small diameter2×large diameter)/2.
For the humanized head and neck (H&N) cancer model, humanized NSG mice (Jackson, #705557/NSG; 17–19 weeks) were engrafted with CD34+ immune cells from three different human donors (12 mice/group). After 14 weeks, the mice were examined for the presence of immune cells by flow cytometry (FACS) analysis. Only mice with at least 25% human CD45+ cells in their peripheral blood were included in the study. The mice received subcutaneous injections of 1×106 A253-HLA-G mixed with an equal volume of Matrigel. Once the tumors were established (average of 80 mm3), the mice were randomly divided into two groups that were administered intravenously with BND-22 or control hIgG4 (Sino, HG4K) at 10 mg/kg two times a week. Mice were monitored two times a week for signs of morbidity and tumor volumes were measured.
Tumor growth inhibition (TGI) was calculated by the following formula: 100−(group average tumor weight/control average tumor weight)×100.
Statistical analysis
Data are presented as means±SEM as stated in the figure legends. Statistically significant differences were tested using specific tests as indicated in the figure legends. A p value of <0.05 was considered statistically significant. Quantitative and statistical analyses of the data were preformed using Graphpad Prism V.8.1 software. EC50 values were determined using non-linear regression curve, fit variable slope (four parameters). In in vivo experiments, outliers were removed from different evaluations according to Grubb’s test.
Results
ILT2 and HLA-G are highly expressed in the tumor microenviorment (TME) and peripheral blood of patients with cancer
The expression of ILT2 was first evaluated in peripheral blood samples from patients with cancer. Interestingly, it was found that the percentages of CD4 T cells, CD8 T cells, and NK cells that express ILT2 are significantly higher in the periphery of patients with cancer in comparison to healthy controls (figure 1A). These results suggest that ILT2 upregulation can serve as a mechanism for immune evasion in several tumor indications. To understand which immune cell populations express ILT2 in the tumor microenvironment, fresh tumor samples from patients with cancer were evaluated by flow cytometry. Overall, ILT2 was found to be expressed in about 20% of the total immune cell population (CD45+) present in the TME of various patients with cancer. A high percent of macrophages and NK cells expressed ILT2 in TME (85%±3.2% and 53%±6.5%, respectively) (figure 1B). Lower frequencies of ILT2-expressing cells were observed in CD4 and CD8 T cells. However, within T cells, highly differentiated CD8 TEMRA cells (CD45RA+CCR7−) selectively exhibited ILT2 expression. Interestingly, a low frequency of PD-1 expression was detected in this CD8 T-cell population, whereas PD-1 expression was mostly confined to ‘non-TEMRA’ CD8 T-cell population (figure 1C).

ILT2 and HLA-G are highly expressed in the TME and the periphery of patients with cancer. (A) PBMCs were isolated from the blood of the indicated patients with cancer or from healthy donors. The percentage of ILT2-expressing NK cells, CD8 and CD4 T cells was evaluated by flow cytometry. *P<0.05; Unpaired Student’s t-test. (B) Single-cell suspensions were prepared by enzymatic digestion of fresh tumors isolated surgically from patients with various cancer indications. The percentage of ILT2 expression in total intratumoral immune cells, TAMs, CD8, CD4 T cells and NK cells was evaluated by flow cytometry as mentioned in the online supplemental material. The median percentage of ILT2-positive cells for each immune cell population is indicated. (C) Dot plots representative from evaluation of the levels of CD8 TEMRA cells (left) and ILT2 and PD-1 expression in intratumoral CD8 TEMRA cells (right) from an esophageal cancer patient. (D) Tissue microarrays from the indicated cancer types were stained with anti-HLA-G antibody for IHC. The percentages in the graph indicate patients with cancer samples that had a higher staining score (above 100). (E) Soluble HLA-G protein was evaluated in plasma samples collected from patients with cancer (n=20, each group) by ELISA. *P<0.05. H&N, head and neck; IHC, immunohistochemistry; ILT2, immunoglobulin-like transcript 2; MSS, microsatellite stable; NK, natural killer; NSCLC, non–small cell lung cancer; PD-1, programmed cell death protein-1; RCC, renal cell carcinoma; TAM, tumor-associated macrophage. TEMRA cells, effector memory T cells re-expressing CD45RA.
Supplemental material
Similarly, when performing a gene set enrichment analysis bioinformatic analysis of the The Cancer Genome Atlas (TCGA) database using The Cancer Immunome Atlas (www.tcia.at), a correlation was observed between higher ILT2 expression and enrichment of MDSC and ‘M2-type’ macrophages in patients with cancer (online supplementary figure S1A,B). In addition, when examining enrichment of CD8 TEMRA cells across various tumor indications using the TCGA database, 22.8%–47.3% of patients had high levels of TEMRA cells (online supplementary figure S1C). Furthermore, 57%–94% of the patients with high TEMRA cells also showed high levels of ILT2 expression, indicating a strong association between ILT2 and TEMRA cells (online supplementary figure S1D). Collectively, these results demonstrate that in the TME, ILT2 expression is increased and mainly expressed by myeloid cells, NK cells and CD8 TEMRA cells.
Expression of HLA-G, a high-affinity ligand of ILT2, was also evaluated in patients with cancer by IHC. An IHC score was calculated based on staining intensity and number of stained cells, and a score of >100 was considered as representative of high expression of HLA-G. Using this criterion, high expression levels of HLA-G were observed in 55% of patients with microsatellite stable colon cancer, 45% of squamous cell carcinoma of the patients with H&N cancer, 37% of patients with esophageal cancer and 53% of patients with gastric cancer (figure 1D). Furthermore, higher levels of soluble HLA-G were detected in the plasma of various patients with cancer in comparison to healthy controls (figure 1E). Taken together, these results demonstrate that upregulation of ILT2 and HLA-G is a common mechanism of immune suppression within the TME and periphery of patients among many types of cancers.
BND-22 binds ILT2 with high affinity and selectivity
BND-22, a novel humanized immunoglobulin G4 (IgG4) monoclonal antibody with antagonist characteristics against human ILT2, was generated using hybridoma technology. Evaluation of the binding kinetics of BND-22 to human ILT2 using SPR BIAcore technology showed that BND-22 has strong and stable binding to ILT2 with an equilibrium dissociation constant (kd/ka) ranging from 1.26 nM to 4.48 nM (figure 2A). A similar dose-dependent binding to ILT2 expressed by a transfected cell line (BW-ILT2) was observed by flow cytometry (EC50=0.05 µg/mL, figure 2B). BND-22 is selective to ILT2 and did not bind to other ILT family members that are highly homologous to ILT2 (eg, ILT4, ILT6, and LILRA1) (online supplementary figure S2).

BND-22 selectively binds to ILT2 and blocks its interaction with HLA-G and HLA-A2 by blocking ILT2 binding to the B2M-binding region. (A) The binding of BND-22 to human ILT2 as determined by multicycle analysis using the Biacore T200. Sensograms and fitted curves (1:1 binding model) are displayed as well as the summary of the kinetic parameters. Recombinant human ILT2 in six points in the range of 25–0.78 nM in twofold dilutions, represented by the different colors in the sensogram, was added to the system. (B) The binding of BND-22 to BW cells transfected with ILT2 as evaluated by flow cytometry. (C) A375-HLA-G cells were incubated with biotinylated ILT2-FC in the presence of various concentrations of BND-22 followed by staining with streptavidin–PE and FACS analysis. A graphical representation of the percentage of blocking compared with cells without antibody is shown. BW WT cells or BW cells transfected with ILT2-zeta (BW-ILT2) were incubated with 721.221 wildtype cells (221 W.T), or 721.221 transfected to express (221-HLA-G) or HLA-A2 (221-HLA-A2) (D) or with A375-HLA-G cells (E) in in the presence of various concentrations of BND-22, control IgG or PBS for 48 hours. The secretion of a reporter mouse cytokine was measured by ELISA. *P<0.05, unpaired Student’s t-test. Three-dimensional ribbon or surface diagrams of ILT2 showing the epitope of BND-22 (pink) and its interaction on ILT244 with B2M (lilac) in complex with (F) HLA-A or (G) HLA-G.45 B2M, beta-2-microglobulin; BW ILT2, BW cells transfected with ILT2-zeta; BW WT, BW wild type; FAB, fold above (isotype control) background; ILT2, immunoglobulin-like transcript 2; Ka, association rate constant (1/Ms); Kd, dissociation rate constant (1/s); KD, equilibrium dissociation constant (kd/ka).
BND-22 blocks binding of ILT2 to class I MHC molecules by inhibiting interactions in the B2M-binding region
When examining the binding of recombinant ILT2 to HLA-G-expressing A375 cells in the presence of varying doses of BND-22, BND-22 prevented the binding of ILT2 to HLA-G in a dose-dependent manner (IC50=0.314 µg/mL, figure 2C). In addition, the ability of BND-22 to functionally block the interaction between either HLA-G or HLA-A2, and ILT2 was further examined in a BW ILT2/mouse Z-chain chimera cell-based reporter assay. Using this assay, it was demonstrated that BND-22 can efficiently block the interaction of ILT2 with both classical (HLA-A2) (figure 2D) and non-classical (HLA-G) MHC class I molecules (IC50=0.179 µg/mL, figure 2E).
In silico epitope mapping revealed that BND-22 interacts with ILT2 at four epitope regions of ILT2 that are all found in the hinge section between domain 1 (D1) and domain 2 (D2). Interestingly, these epitopes have been identified as the interaction domains of ILT2 that specifically bind to beta-2-microglobulin (B2M) on HLA-A or HLA-G32 (figure 2F,G). In addition, empirical epitope mapping using HRF and mass spectrometry techniques confirmed these results and identified binding residues within the D1 D2 domains of ILT2 that are known to be involved in the interaction of ILT2 with B2M (online supplementary table S1,S2).
BND-22 enhances the phagocytosis of cancer cells by macrophages
The ability of BND-22 to enhance the phagocytosis of tumor cells by macrophages was examined in vitro and ex vivo using two different experimental systems. Using a real-time analysis system, a dose-dependent enhancement of phagocytosis of HLA-G-positive cancer cells (A375) was observed over time (figure 3A). This is apparent when looking through the course of 18.0 hours (figure 3A) and at 12.5 hours (figure 3B). It is also evident when looking at the accumulation of the red signal that represents phagocytosis, which is higher in the samples treated with BND-22 versus control IgG (figure 3C). It was found that BND-22 can enhance phagocytosis of tumor cells expressing HLA-G or classical MHC-I (figure 3C,D).
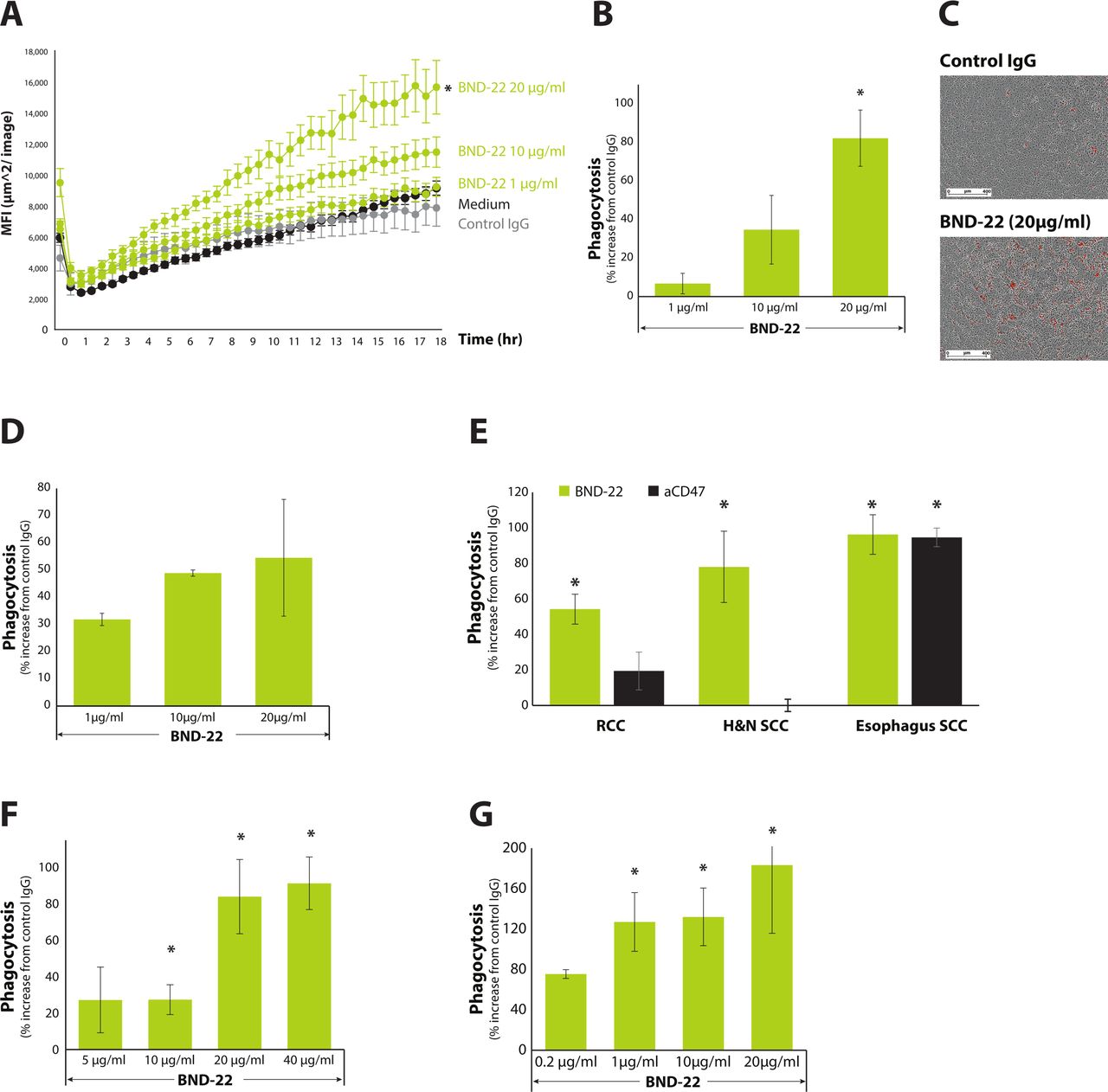
BND-22 enhances the phagocytosis of cancer cells by macrophages. A375-HLA-G cells were stained with pHrodo red cell labeling dye and added to monocyte-derived macrophages generated from healthy donors, along with the various indicated treatments in triplicates. (A) A representative kinetic quantification of phagocytosis is presented in a time-course of 18 hours as analyzed using Incucyte S3.(B) A single representative time point of the experiment at 12.5 hours is presented. Percent increase in macrophage-mediated phagocytosis with respect to control IgG (20 µg/mL) is presented. (C) Images of cells incubated in the presence of control IgG or BND-22 are presented. The red signals captured represent phagocytosis events. (D) Phagocytosis of A375 wild-type cells by macrophages. Single-cell suspensions were generated by enzymatic digestion of fresh tumors surgically isolated from patients with cancer (indicated in E,F), patients with RCC, and (G) patients with H&N cancer. The cells were stained with pHrodo red cell labeling dye and mixed with monocyte-derived macrophages generated from healthy donors or (E) from autologous patients (F,G), along with the various indicated treatment doses in triplicates. The percent increase in macrophage-mediated phagocytosis of tumor cells compared with control IgG is presented. MFI - mean fluorescence intensity. *P<0.05; unpaired Student’s t-test compared with cells with control IgG. H&N, head and neck; RCC, renal cell carcinoma.
BND-22 was also found to enhance the phagocytosis of patient-derived tumor cells that were obtained from patients with renal cell carcinoma (RCC), H&N cancer, and esophagus cancer. The magnitude of increase in phagocytosis differed between patients and were up to 1.2-fold compared with the isotype antibody (online supplementary figure S3). Interestingly, the ability of BND-22 to increase phagocytosis was comparable to CD47 blockade, which is a predominant mechanism to augment tumor cell phagocytosis (figure 3E). Additionally, to a higher relevance to the clinical settings, BND-22 also enhanced the phagocytosis of tumor cells from RCC and H&N patients by respective autologous macrophages in a dose-dependent manner (figure 3F,G).
BND-22 enhances the activity of antitumor T cells
The ability of BND-22 to alleviate ILT2-mediated inhibition on T cells was examined in different cellular systems. As depicted in figure 4, coculturing ILT2-expressing Jurkat T cells with A375 engineered to express OKT3 (anti-CD3) and HLA-G (figure 4A) or endogenous classical MHC-I (figure 4B) led to increased production of human IL-2 by BND-22 in a dose-dependent manner. Interestingly, on blocking HLA-G, with an HLA-G-specific antibody, the effect was much less significant than that of BND-22, suggesting that the ILT2 interaction with both classical and non-classical MHC-I plays a major role in suppressing T-cell function in this system (figure 4C). Using this system, it was also demonstrated that the increase in T-cell function by BND-22 is specific to its ability to block ILT2, as in ILT2-deficient wild-type Jurkat cells, BND-22 had no effect on IL-2 production. Furthermore, the basal IL-2 secretion was notably higher when comparing wild-type Jurkat cells to that of ILT2-transfected Jurkat cells, further demonstrating the inhibitory effect of ILT2 (figure 4C,D). The ability of BND-22 to increase T-cell function was also demonstrated in a primary human T cell-based mixed lymphocyte reaction (MLR) assay. The addition of BND-22 to cultures of allogenic T cells and dendritic cells (DCs) from various donors significantly increased interferon gamma (IFN-γ) secretion from the T cells (figure 4E). Thus, in addition to the results observed in Jurkat cells, it was demonstrated using primary cells that ILT2 mediates inhibitory activity in T cells, which could be reversed by BND-22.

BND-22 enhances T-cell activity against cancer cells. Jurkat cells transfected with ILT2 (Jurkat-ILT2, A–C) or wild-type Jurkat cells (Jurkat-WT, D) were incubated with mitomycin-treated OKT3-expressing A375 cells that were either MHC class I positive (A375-WT-OKT3, B) or HLA-G positive (A,C,D) in the presence of BND-22 control IgG or a HLA-G-specific blocking antibody. The amount of secreted human IL-2 was evaluated by ELISA kits following 48 hours. *P<0.001, one-way analysis of variance46 followed by Dunnett’s multiple comparisons test compared with control IgG. (E) DCs generated from monocytes and CD8 T cells (T cells) were isolated from healthy human donors. The cells were combined in an E:T of 5:1 together with the indicated treatments followed by detection of IFN-γ secretion on day 5 Representative results are shown. The mean±SE of values from three repeats per treatment is displayed. TILs were coincubated with A375-HLA-G-OKT3 cells for 5 min and immediately placed in 2% Paraformaldehyde (PFA) on ice followed by detection of phosphorylated ZAP70/Syk by flow cytometry. (F) Representative FACS analysis of the levels of phosphorylated ZAP70 in the indicated conditions. The cells were gated on ILT2+CD8 T cells. (G) A graphical representation of the results shown in F. (H) A summary of three experiments performed in similar conditions. DC, dendritic cell; E:T, effector-to-target ratio; IFN-γ, interferon gamma; IL, interleukin; TIL, tumor-infiltrating lymphocyte.
Finally, when adding A375-HLA-G-OKT3 cells to TILs expanded from a patient with melanoma, BND-22 blocked the inhibitory activity of ILT2, leading to an increase in the phosphorylation levels of ZAP70 (figure 4F–H). These results demonstrate that BND-22 can directly disrupt ILT2-mediated signaling events that occur on ligand binding, thus leading to increased phosphorylation of key signaling elements which are involved in downstream activation of T cells.
BND-22 enhances NK-cell activity against cancer cells
The ability of BND-22 to enhance the cytotoxic capacity of NK cells was tested in both an NK cell line and primary human NK cells. A dose-dependent increase in NK cell-mediated target cell killing was observed against HLA-G and MHC class I-expressing cancer cells in the presence of BND-22 (figure 5A–D). Additionally, a corresponding increase in IFN-γ and granzyme B levels was also observed in the presence of BND-22 (online supplementary figure S4). The ability of BND-22 to increase the activity of primary NK cells against cancer cells demonstrated that BND-22 increased both the levels of intracellular IFN-γ as well as membranal CD107a (figure 5E,F). These results indicate an enhancement in the activity and cytotoxicity of primary NK cells in response to BND-22 in the presence of target HLA-G+ cancer cells. Furthermore, a correlation between the percentage of ILT2-expressing NK cells (CD56+ILT2+) and the ability to increase both IFN-γ and CD107a levels was observed (figure 5G). In a separate set of experiments, on adding A253-HLA-G cells to NK cells, BND-22 increased the phosphorylation of Syk in comparison to control IgG (figure 5H–J), suggesting blockade of the NK inhibitory activity of ILT2. Collectively, these data show that ILT2-mediated signaling inhibits NK-cell function and can be reversed by ILT2 blockade by BND-22.
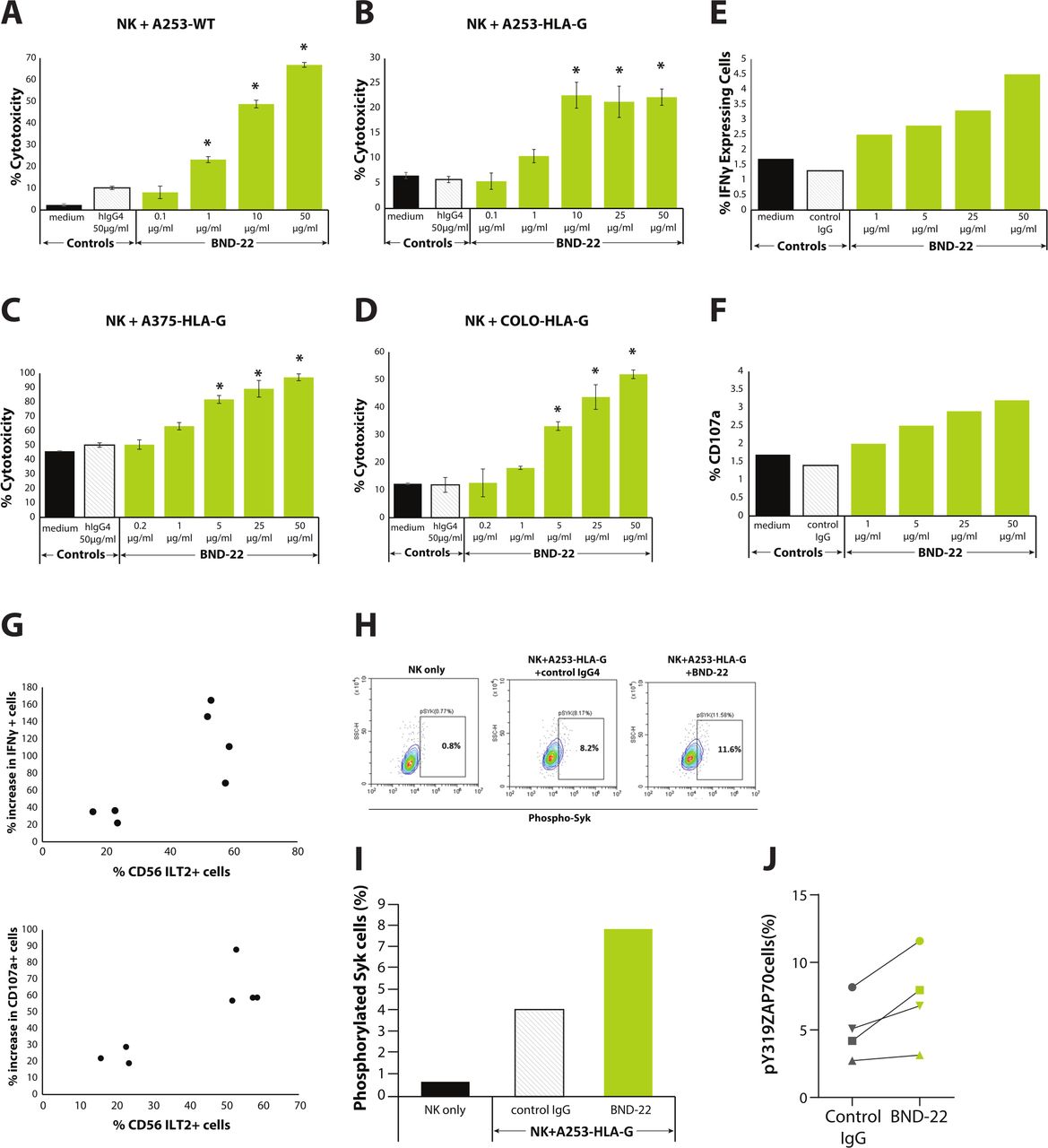
BND-22 enhances NK-cell activity against cancer cells. NK cells were incubated with the indicated concentrations of BND-22 or a control IgG; Target cells including A253-WT (A), A253-HLA-G (B), A375-HLA-G (C) and COLO 320-HLA-G (D) were added for an additional 5 hours at an E:T of 7.5:1. Representative results are shown. Results represent an average of % cytotoxicity±SE determined by LDH release assay in triplicates. WT cells express MHC-I; HLA-G transfected cells express both MHC-I and HLA-G. *p<0.02, One-way analysis of variance followed by Dunnett’s multiple comparisons test compared with NK+target cells+control IgG. NK cells isolated from healthy donors were incubated with the indicated concentrations of BND-22 or a control IgG. A375-HLA-G cells were added for additional 5 hours at E:T of 8:1 followed by staining for intracellular IFN-γ, membranal CD107a, ILT2 and CD56 and analysis by flow cytometry. (E) The percentage of IFN-γ-positive NK cells in the CD56+ILT2+ population. (F) The percentage of CD107a-positive NK cells in the CD56+ILT2+ population. (G) Correlation between percentage of CD56+ILT2+ cells in the isolated NK cells and percent increase in IFN-γ (top) or CD107a (bottom) in the presence of A375-HLA-G and BND-22 (50 µg/mL) is presented for seven different donors tested. NK cell lines were cocultured with A253-HLA-G cells for a minute followed by immediately fixing in 4% PFA on ice and ZAP70/Syk phosphorylation was examined by flow cytometry. (H) Representative FACS plots showing phosphorylated SyK in the indicated conditions. (I) A graphical representation of the results shown in H. (J) A summary of four experiments performed in similar conditions. IFN-γ, interferon gamma; ILT2, NK, natural killer; WT, wild type.
BND-22 enhances the antitumor activity of anti-PD-1 and tumor targeting antibodies
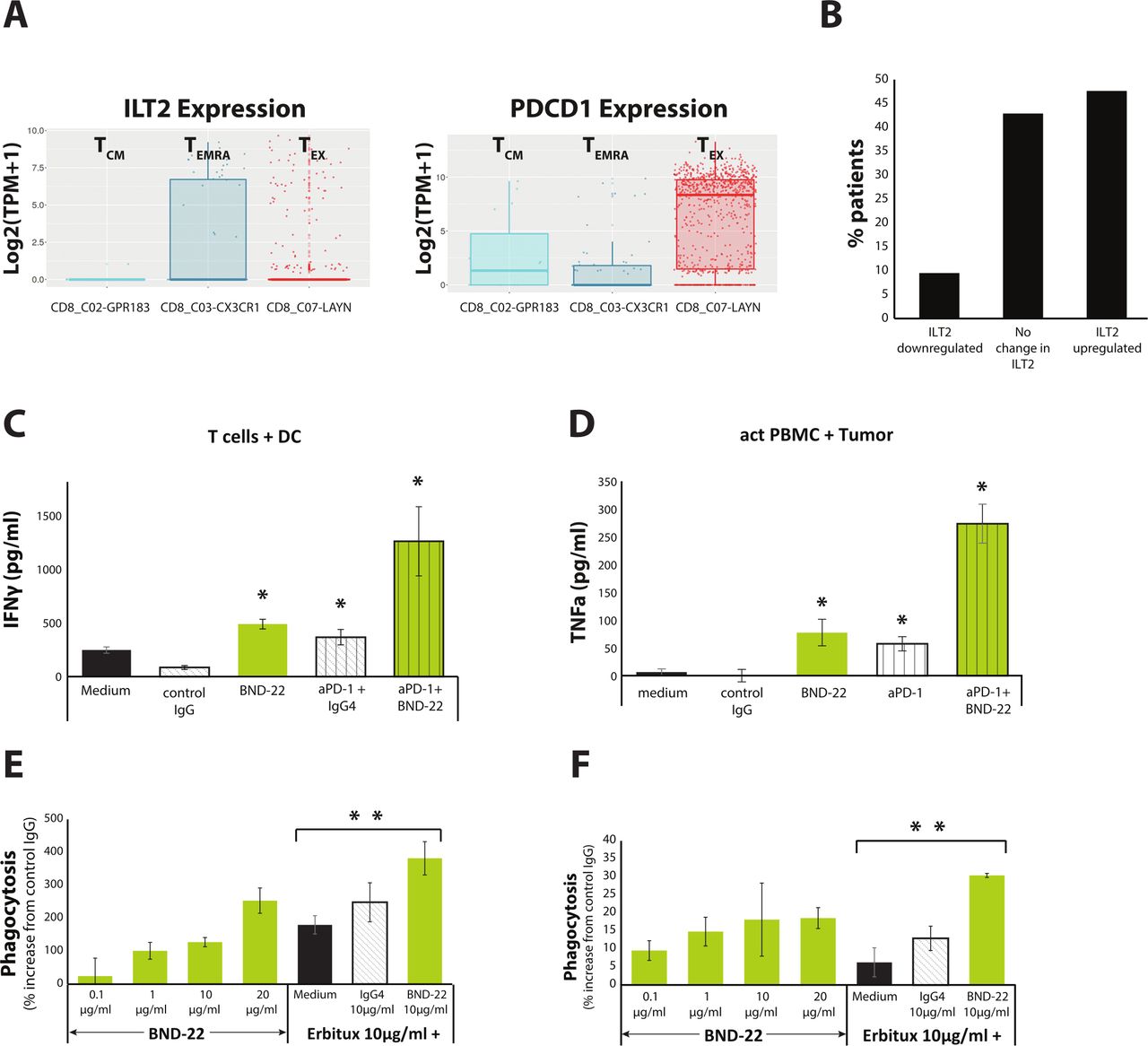
In order to evaluate the rationale to combine BND-22 with anti PD-1, the expression of PD-1 and ILT2 was examined on immune cells from patients with cancer. An analysis of an available database for T cells isolated from patients with colorectal cancer (CRC) (http://crctcell.cancer-pku.cn) revealed that ILT2 and PD-1 are expressed on different CD8 T-cell populations in tumors. ILT2 is expressed in the TEMRA cell population, whereas PD-1 expression was confined to central memory T cells and exhausted T cells (TEX) (figure 6A).33 Similar results were observed in intratumoral T cells from a patient with esophageal cancer (figure 1C). The intratumoral TEMRA cells in this patient were found to express ILT2 but not PD-1. Additionally, bioinformatic analysis of published data revealed that ILT2 expression was increased in 48% of melanoma patients after treatment with anti-PD-1 (Nivolumab) (figure 6B), which suggested that ILT2 upregulation is a potential mechanism for developing anti-PD1 resistance.

BND-22 enhances anti-tumor functions of anti-PD-1 or tumor targeting antibodies. (A) Analysis of ILT2 and PD-1 expression in intratumoral CD8 T cells from patients with CRC documented in http://crctcell.cancer-pku.cn/. (B) A bioinformatic analysis of ILT2 expression in melanoma patients treated with Nivolumab from.47 The RNAseq data was used to calculate expression changes in patients with cancer post versus pre-Nivolumab treatment. Log2 ILT2 fold-change differences above 0.5 or below 0.5 were considered as increase and decrease in expression, respectively. (C) DCs were generated from monocytes and CD8 T cells were isolated from PBMCs of healthy human donors. The cells were combined in E:T of 5:1 ratio with the indicated treatments and IFN-γ was measured in culture supernatants at day 5. (D) Single-cell suspensions were prepared by enzymatic digestion of a fresh tumor isolated from a patient with colon cancer. PBMCs were isolated from the same patient and cocultured with the indicated treatments in presence of IL-2. TNFα secretion was detected by ELISA. Representative results are shown. The mean±SE of values from three repeats per treatment are displayed. Unpaired Student’s t-test compared with control IgG for BND-22 or anti-PD-1, compared with anti-PD-1+IgG for anti-PD-1+BND-22.*P<0.05; (E) 786-O cells, or (F) A253-HLA-G cells were stained with pHrodo red cell labeling dye, washed and added to macrophages generated from monocytes isolated from healthy donors, along with the various treatments in triplicates. A single representative time point of the experiment at 9.5 hours is presented. The percentage of increase in phagocytosis compared with control IgG is presented.Unpaired Student’s t-test compared with erbitux+medium. **P<0.01. CRC, colorectal cancer; DC, dendritic cell; IFN-γ, interferon gamma; ILT2, immunoglobulin-like transcript 2; PD-1, programmed cell death protein-1; TCM, central memory T cells; TEMRA, T cells re-expressing CD45RA; TEX, exhausted T cells.
The effect of combining BND-22 with anti-PD-1 was first evaluated in an MLR assay. On testing for single agent activity, both BND-22 and anti PD-1 antibody enhanced IFN-γ secretion from T cells in the presence of allogeneic dendritic cells. Interestingly, IFN-γ production by T cells was further enhanced when a combination of both antibodies was tested (figure 6C). Similarly, co-culture of tumor cells with autologous PBMCs from a patient with colon cancer demonstrated that BND-22 and anti-PD-1 antibody can increase tumor necrosis factor alpha (TNFα) secretion. The combination of these two antibodies resulted in a significant synergistic effect (figure 6D).
Antibody Dependent Cellular Phagocytosis (ADCP) is one of the main mechanisms of anti-tumor activity for tumor-targeting antibodies (eg, cetuximab, rituximab).34 Since ILT2 is highly expressed by macrophages and can suppress their phagocytic activity,18 there is a rationale for combining BND-22 with ADCP-inducing antibodies. The ability of BND-22 to further enhance anti-EGFR antibody (cetuximab) mediated ADCP was evaluated against an EGFR expressing cell line. Using a real-time live-cell analysis system, it was observed that when tested as a single agent, both BND-22 and Cetuximab increased phagocytosis of the EGFR-expressing cancer cell lines. Interestingly, the combination of the antibodies enhanced the phagocytosis to a higher level than single agent activity (figure 6E,F).
Taken together, these results demonstrate the rational to combine BND-22 with either PD-1 blockers or tumor targeting therapies such as Cetuximab to further enahnce antitumor immune activity.
BND-22 increases the antitumor activity of lymphocytes and macrophages in vivo
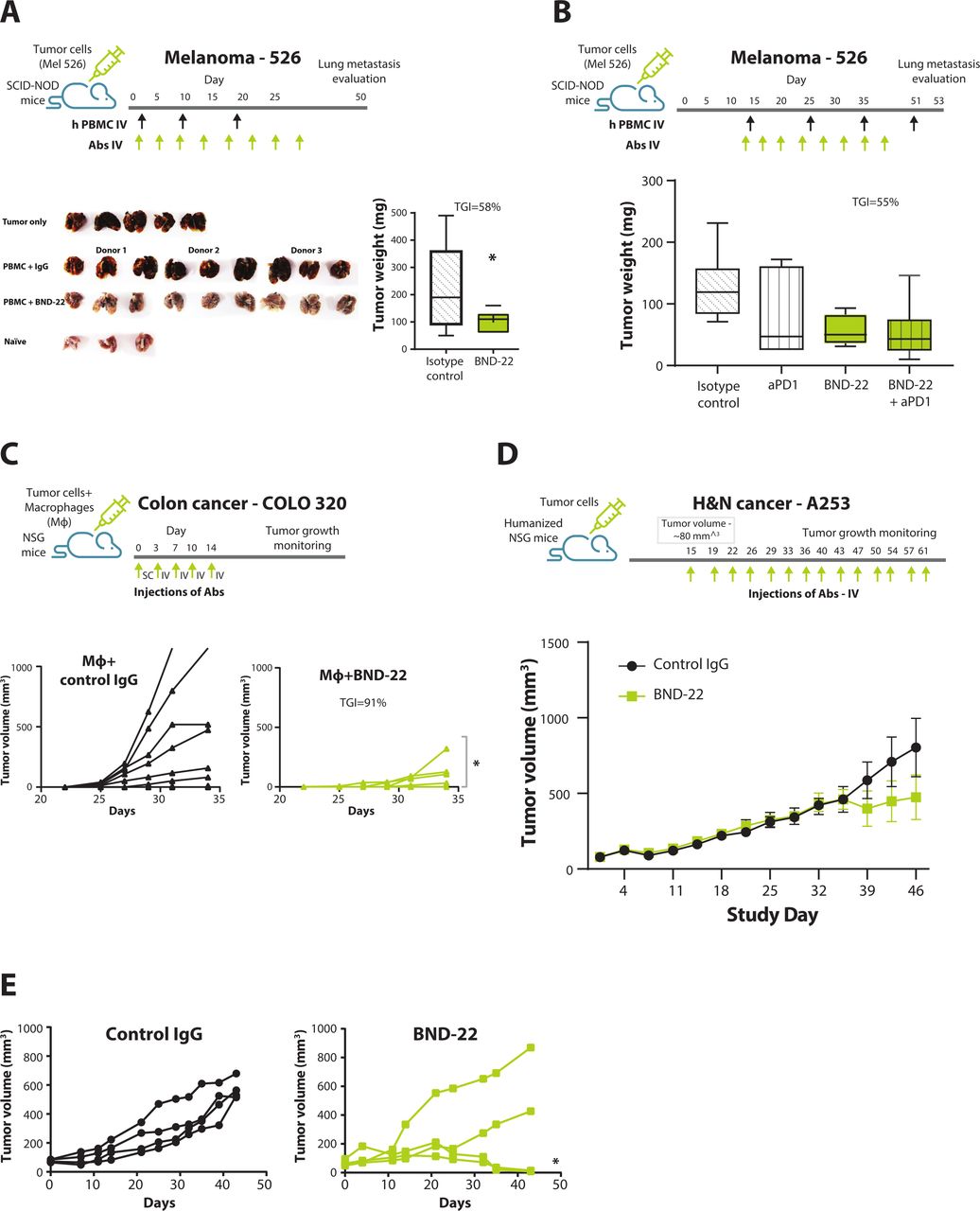
ILT2 shares low homology with the mouse homolog of ITL2, PirB,5 and BND-22 cannot bind PirB (data not shown). Therefore, several different xenograft models were used to study the mode of action of BND-22. The ability of BND-22 to elicit anti-tumor activity of lymphocytes and consequently prevent metastasis was evaluated in a human xenograft lung lesion model in NOD SCID mice with human PBMC engraftment. In this experiment, tumor burden and numerous lung lesions were developed in the lungs of untreated mice or mice that received human PBMCs with control IgG . In contrast, blockade of ILT2 resulted in significant TGI (figure 7A, TGI of 58%). ILT2 blockade in this model led to prevention of metastasis so that lungs weight and morphology appeared closer to healthy non-tumor bearing lungs (figure 7A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
BND-22 inhibits tumor growth in vivo. (A,B) SCID-NOD mice were engrafted with MEL526-HLA-G cells intravenously. Human PBMCs isolated from healthy donors were transferred to the relevant groups together with IL-2 followed by antibody treatments (10 mg/kg each) starting from day 1 (A) or day 14 (B) according to the treatment schedules as indicated in the Materials And Methods section. At the endpoint, the mice were sacrificed; the lungs were weighted; and images were captured. Images showing the lung lesions developed by melanoma cells in the mice lungs from groups are presented (B). Tumor weight was calculated by subtracting naïve mouse lung weight from the lung weight of the experimental mouse. The median value from each group is presented in the graph as a vertical line (A,B). (C) NSG mice were engrafted SC with COLO-320 cells and human macrophages differentiated from monocytes isolated from healthy donors. The animals were treated with BND-22 or with control IgG (20 mg/kg) according to the treatment schedule outlined in the illustration. The data are presented as tumor growth curves from individual mice. Unpaired t-test was used to calculate statistical significance of TGI between the groups (A), One-way ANOVA followed by Dunnett’s multiple comparisons was used (B). A two-way repeated measures ANOVA was used (C,E). Human CD34+cell-engrafted mice were inoculated with A253-HLA-G cells. The mice were treated at indicated treatment schedule with BND-22 or a control IgG (10 mg/kg) when tumors reached 80 mm3. (D) Mean of tumor growth in mice that received CD34+ cells from all three donors and were treated with either BND-22 or control IgG4. (E) Tumor growth in mice inoculated with CD34+ cells from one of the donors treated with either BND-22 or control IgG4. *P<0.05. ANOVA, analysis of variance; IL, interleukin; TGI, tumor growth inhibition.
A similar model was used to demonstrate therapeutic efficacy of BND-22 in pre-established tumors. Metastatic melanoma was established in the lungs of NOD SCID mice with human PBMC engraftment. TGI was observed in the groups treated with BND-22 alone and in combination with anti-PD-1 (figure 7B; TGI of 55% and 57% respectively compared with the IgG group). A statistical trend was observed for the group treated with BND-22 +anti-PD-1 in comparison to the isotype control group (one-way ANOVA followed by Dunnett’s multiple comparisons; p=0.0586). In addition, BND-22 alone or in combination with PD-1 blockade upregulated various activation markers such as CD69 and CD107a on T/NK cells in the tumors (online supplementary figure S5A,B). Interestingly, mice that received PBMCs from donors with a higher expression/frequency of ILT2 on CD8 T and NK cells had a more substantial increase in CD69 and CD107a post BND-22 treatment. This correlation suggests that treatment with BND-22 played a direct role in intratumoral immune cell activation (online supplementary figure S5C,D). Taken together, these results support the mode of action of BND-22 as an enhancer of the cytotoxic activity of T and NK cells against cancer cells in vivo and shows that anti-ILT2 treatment can reduce tumor burden and eliminate metastasis.
The ability of BND-22 to enhance the anti-tumor activity of macrophages in vivo was evaluated in a subcutaneous humanized tumor mouse model. In this model, human macrophages, and human colon cancer cells (COLO-320-HLA-G) were transferred into SCID NOD mice. Administration of BND-22 resulted in 91% TGI in comparison to mice that received control IgG (p<0. 001 two-way repeated measures ANOVA) (figure 7C). Furthermore, BND-22 administration led to a prolonged/sustained TGI (online supplementary figure S6E). These results support the notion that in the absence of T/NK cells, BND-22 can enhance phagocytic activity of macrophages against tumor cells in vivo.
BND-22 generates a potent antitumor immune response and inhibits tumor growth in humanized tumor mouse models
In order to evaluate the ability of BND-22 to mediate anti-tumor activity in an in vivo model in which the entire human immune system is present, the efficacy of BND-22 was tested in a H&N cancer model (A253-HLA-G) in fully humanized NSG mice engrafted with human CD34+cord blood cells. Notably, these mice contain cells of both the lymphoid and myeloid compartments as evaluated in the periphery and tumors of the mice (online supplementary figure S5F and data not shown). In this model, administration of BND-22 caused 41% overall TGI (figure 7D). Interestingly, in mice receiving immune cells from one of the human donors, 50% of the mice showed complete tumor regression with BND-22 treatment (p<0.001 two-way repeated measures ANOVA, responding mice versus IgG-treated mice) (figure 7E). Furthermore, intratumoral increase in CD107a on CD8 T cells, M1/M2 ratio and CD80 expression on DCs confirmed that a potent anti-tumor immune response was elicited in the mice that responded to BND-22 treatment (online supplementary figure S5F) In addition, a correlation between ILT2 expression levels in the peripheral CD8 T cells of the mice and response to BND-22 was established (online supplementary figure S5G). These results demonstrate the ability of BND-22 to generate a robust, broad anti-tumor response by orchestrating an effect on both innate and adaptive immune cells thus leading to a shift in the environment of the tumor from suppressive to inflammatory.
Discussion
LILRB family comprises of a number of regulatory, inhibitory receptors that are expressed on various immune cells. Recently, interesting data was generated in the clinic by inhibiting one of the LILRB members, LILRB2 (ILT4), in patients with cancer.35 Therefore, inhibition of LILRB family members holds promise as monotherapy and in combination with established cancer therapies. Unlike ILT4, LILRB1 (ILT2) is widely expressed by immune cells T, NK, and myeloid cells, and negatively regulates both the innate and the adaptive immune system. Our clinical analysis suggests that ILT2 and HLA-G are further upregulated in the periphery and intratumorally in patients with cancer across many indications. Therefore, in context of tumor immunotherapy, ILT2 could be considered a ‘multi-immune cell checkpoint’ and its potential role in immune suppression in the TME provides a strong rationale for its targeting.36
Our in vitro, ex vivo and in vivo results demonstrate that ILT2 promotes both tumor growth and lung metastasis by compromising activation and functions of anti-tumor T cells, NK cells and macrophages. BND-22 is a first-in-class fully humanized ILT2 blocking antibody, which reverses immune suppressive functions of ILT2 and reduced tumor growth in mice by augmenting tumor specific immune responses. In our experiments, BND-22 enhanced the phagocytic activity of macrophages and the cytotoxic capability of both NK and T cells against tumor cells. Interestingly, BND-22 increased ZAP70/Syk signaling in both T and NK cells. Overall data presented in this report suggests that blocking ILT2 with BND-22 shifts the TME towards pro-inflammation.
BND-22 demonstrated clear anti-tumor activity in different in vivo models as evident by reduction in primary tumor growth and lung metastasis and increased survival in the BND-22 treatment groups. BND-22 was shown to mediate anti-tumor activity through a number of mechanisms including increasing the effector functions of T and NK cells. Additionally, BND-22 treatment caused an increase in the M1/M2 ratio within tumors and upregulation of costimulatory marker CD80 on DCs suggesting that blocking ILT2 increased the ratio of inflammatory compared with suppressive myeloid cells intratumorally. Although these tumor models can be valuable in demonstrating potency, the small number of mice in the humanized mice model and possibility of developing allogenic responses in the PBMC-engrafted lung lesion model need to be taken into consideration. Of note, the mice did not display any physiological symptoms that could point to graft-versus-host disease (GVHD) responses.
Blockade of PD-1/PDL-1 pathway is considered as the mainstay of cancer immunotherapy however, many patients receiving this therapy do not respond at all or experience relapses.37 Therefore, there is an unmet need for additional complementary approaches, which can help improve overall response and survival in these patients. In this context, resistance to anti-PD-1/PD-L1 therapy could be mediated by additional immune checkpoint pathways. Recent findings have suggested that the ILT2/HLA-G pathway is upregulated in patients with cancer previously treated with anti-PD-1 drugs suggesting ILT2-mediated suppression as a potential resistance mechanism. It was demonstrated by us and others that PD-1 and ILT2 are expressed on different CD8 T cell sub-populations.11 12 These studies demonstrated that PD-1 is primarily expressed by effector memory T cells and TEX, whereas ILT2 expression is confined to TEMRA cells in the TME. These observations suggest that due to lack of PD-1 expression, anti-PD-1 treatment is not effective in activating the TEMRA cell population that could be present in large numbers in the TME. In addition, ILT2 expression and the number of TEMRA cells that express ILT2 were shown to be increased following anti-PD-1 treatment.38 39 Using computational biology, we observed that ILT2 expression was increased in 48% of melanoma patients that were previously treated with anti-PD-1. Considering that increased levels of soluble HLA-G are also associated with reduced response to anti-PD-1 treatment,40 we postulate that blocking of ILT2/HLA-G interactions with BND-22 will be effective to maximize the benefit of anti-PD1. Data presented in this manuscript also support this hypothesis. In addition, there is also a rationale to combine BND-22 with anti-EGFR antibody as demonstrated here. Patients expressing high levels of ILT2 were found to have an impaired ability to kill cancer cells in the presence of cetuximab and functional blockade with an ILT2 antagonist antibody restored Antibody-dependent cellular cytotoxicity (ADCC).41 42 In addition, antibody-mediated ADCP (such as mediated by cetuximab) function can also be further enhanced by BND-22.34,43 In our studies, BND-22 enhanced tumor cell phagocytosis in combination with cetuximab. Similar approaches were clinically examined, for example, the combination of CD47/SIRP1a blockers with tumor targeting antibodies such as rituximab and cetuximab. Overall, data presented here point to a potential benefit of combining BND-22 with both anti-PD1/PDL1 and additional therapies.
The data presented in the current report support clinical testing of BND-22 as a potential cancer therapy of solid tumors as monotherapy and in combination with other therapies. A phase I clinical trial of BND-22 in patients with cancer with advanced solid tumors is currently ongoing.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
This study involves samples obtained from human participants and was approved by Helsinki committee (approval numbers 0626-17-RMB and 0037-18-NHR). The participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank everyone that has contributed to the generation and characterization of BND-22. We thank Dr Ohad Ronen and the Galilee Medical Center and Dr Ronit Almog, Dr Sivan Cohen Matsliah and the Rambam Medical Center Biobank, which provided primary tumors and blood samples for this study. We would particularly like to thank the patients that consented to provide the samples. We thank Dr Donald Shaffer, Sanofi for critically reviewing the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors DHZ, IG, TP, DA, AFD, MH, RG and HQ designed, preformed, and analyzed in vitro, ex vivo and in vivo mode of action studies. IM, YS, TBM, SS, LP, DW and FON supervised the studies and discussed and interpreted results, SH supervised tissue cross reactivity and toxicology studies. IM and SS wrote the manuscript. YS and IF reviewed the manuscript. TBM is the guarantor.
Funding This study was funded and supported by Biond Biologics and Sanofi.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.