Article Text

Abstract
Background Circulating tumor cells (CTCs) can survive in the circulation and return to primary tumors through a self-seeding process. However, the mechanisms underlying CTCs escape from natural killer (NK) cell-mediated immune surveillance remain unclear.
Method Self-seeded tumor cells were isolated and characterized using a modified contralateral seeding model. A comparison of transcriptional profiles was performed between the parental cells and self-seeded cells. The molecular mechanism of self-seeded tumor cells escaping from NK cell was demonstrated through in vitro experiments and verified in a CTC-mimicking in vivo model. Then, the expression level of key protein mediating CTCs immune escape was detected in 24 paired primary and recurrent tumor samples of patients with oral cancer by the immunohistochemical method.
Result Self-seeded cells displayed resistance to NK cell-mediated lysis and a higher tumor seeding ability than their parental cells. Elevated expression levels of the CDH2 gene and its protein product, N-cadherin were found in self-seeded cells. NK cells secreted cytokines, and fluid shear stress facilitated N-cadherin release by promoting A disintegrin and metalloprotease 10 (ADAM10) translation or converting the precursor ADAM10 to the mature form. Soluble N-cadherin triggered NK cell functional exhaustion by interacting with the killer cell lectin-like receptor subfamily G member 1 (KLRG1) receptor and therefore protected tumor cells from NK cell killing in the circulation. In vivo experimental results showed that overexpression of N-cadherin promoted tumor self-seeding and facilitated the survival of CTCs. Compared with primary tumors, N-cadherin expression was significantly increased in matched recurrent tumor tissues.
Conclusion Together, our findings illustrate an unknown mechanism by which CTCs evaded NK cell-mediated immune surveillance, and indicate that targeting N-cadherin is an effective strategy to prevent CTCs from homing to primary tumor.
- immune evation
- head and neck neoplasms
- biomarkers, tumor
- gene expression profiling
- killer cells, natural
Data availability statement
Data are available on reasonable request. The RNA-seq data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE186082.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immune evation
- head and neck neoplasms
- biomarkers, tumor
- gene expression profiling
- killer cells, natural
What is already known on this topic
Thousands of circulating tumor cells (CTCs) are released daily into the bloodstream, however, only a small fraction of them can evade immunocyte-mediated killing and form secondary lesions. Our understanding of this immune escape mechanism remains poor.
What this study adds
The cross-talk between tumor cells and natural killer (NK) cells in the circulation can trigger NK cell functional exhaustion via N-cadherin/KLRG1 axis, thus enabling CTCs to avoid immune attack.
How this study might affect research, practice or policy
Our study provided a basis for development of therapeutic strategies targeting N-cadherin/KLRG1 axis to promote NK cell antitumor activity, especially against CTCs.
Background
Circulating tumor cells (CTCs) are believed to play critical roles in cancer metastasis.1–3 Even in the early stage of tumor formation, a large number of tumor cells may enter the circulation.4 Most CTCs are eliminated by immune cells and various stresses in the circulation; thus, only a minority of CTCs are capable of surviving and eventually contribute to the spread of tumor.5 Understanding of the immune evasion mechanisms of ‘surviving CTCs’ will provide new insights into tumor cell biology and cancer therapy. However, because CTCs are extremely rare in the circulation, it is difficult to obtain surviving CTC populations with conventional in vitro methods, which impedes such research.
Kim et al revealed a distinctive pathological outcome of CTCs in which these cells can return to and grow in the primary tumors through a process termed ‘self-seeding’.6 This finding showed that the flow of CTCs is more complicated than a one-way path. In addition to causing distant metastasis, CTCs can also affect host tumor growth.7 Most importantly, self-seeded tumor cells originate from CTCs capable of surviving in the circulation, and thus offer an unique opportunity to trace and obtain the surviving CTC subpopulations for investigating their immune evasion mechanism.
Natural killer (NK) cells exert crucial roles in innate immune responses. Unlike cytotoxic T lymphocytes, NK cells mediate cytotoxic functions independent of major histocompatibility complex-mediated antigen presentation.8 Therefore, NK cells exhibit a rapid response against infected and malignant target cells, which are critical for immune surveillance, particularly in the control of metastasis and hematological cancers.9 10 NK cells can directly kill tumor cells or secrete various cytokines, such as interferon (IFN)-γ and tumor necrosis factor (TNF)-α, to eliminate tumor cells.9 NK cell function is regulated by a balance between activating and inhibitory signals. Positive signals for cytolysis are provided by the ligation of several activating receptors, such as NKG2D, NKp46, NTB-A, and natural cytotoxicity receptors NKp30, NKp80. Conversely, NK cell inhibitory signaling ‘exhausts’ NK cells by suppressing the cytokine secretion and cytotoxicity of NK cells. Inhibitory receptors include NKG2A, killer immunoglobulin receptors, KLRG1, and CD85j.11–13 Tumor cells can effectively suppress NK cell function by upregulating inhibitory molecules, which is believed to be one of the major mechanisms by which tumor cells escape immune surveillance.14 15 Functional impairment of circulating NK cells has been reported in multiple types of tumors.16–19 However, whether CTCs have immunosuppressive effect on NK cells is still unknown.
In this study, we sought to demonstrate the immune escape mechanisms mediated by the cross-talk between tumor cells and NK cells in the circulation. We provide evidence that self-seeded tumor cells can release soluble N-cadherin, which contributes to KLRG1 signaling-induced NK cell functional exhaustion and facilitates tumor cells escape from NK cells-mediated immune surveillance. We also extended our findings from the self-seeded tumor cells to CTCs in an animal model. Furthermore, targeting N-cadherin has been proved to be an effective strategy to clear CTCs and prevent tumor cells from homing to primary tumor.
Methods
Patient samples
Primary NK cells were isolated from 10 healthy donors and 10 patients with oral cancer. Human CTCs sample were also isolated from these 10 patients with oral cance. Primary and matched recurrent tumor tissue samples were collected retrospectively from 24 patients with oral cancer (stage T3–T4) who underwent surgical resection from 2015 to 2020 in the Department of Oral and Maxillofacial Surgery. Recurrent was defined as any local or regional recurrence. Tissue samples were obtained by biopsy or surgical resection.
Self-seeding animal model
Rag1-/- mice were purchased from Shanghai Model Organisms. Contralateral seeding experiments were performed on Rag1-/- mice (n=6) to establish the ‘self-seeding’ model as described previously.6 Briefly, 2×106 paired GFP-labeled cells (designated recipient tumor cells) or RFP-labeled cells (designated donor tumor cells) were subcutaneously implanted in the contralateral flank regions. All mice were sacrificed after 30 or 60 days, with dissections of bilateral tumors. The seeding of tumor cells was observed under a fluorescence microscope. In the functional analysis groups, the donor tumor cells were changed into specific cells. The animal study was reviewed and approved by the Animal Ethics Committee of Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (2017-314).
Additional methods are detailed in online supplemental material, including cell lines and cell culture, fluorescence activated cell sorting (FACS), flow cytometry analysis, RNA sequencing (RNA-seq), blocking antibody preparation, Western Blot, immunofluorescence, NK cells isolation, NK cell cytotoxicity assay, RNA interference and lentiviral expression, ELISA, capturing and staining of CTCs, in vivo flow cytometry (IVFC), microdissection, capillary Western assay, etc and statistical analysis.
Supplemental material
Result
NK cell-dependent self-seeding model
Previously, tumor self-seeding was confirmed in several tumor types, such as breast cancer, colon adenocarcinomas and melanomas, in nude mouse models.6 To determine the self-seeding capacity of oral cancer, a type of cancer that frequently metastasizes via the lymphatic rather than the hematogenous pathway,20 contralateral seeding experiments were performed. We modified the xenograft model by using Rag1-/- mice, which have normal NK cells but are deficient in functional T cells and B cells, to focus on the role of NK cells in the control of tumor self-seeding. To specifically trace the self-seeded tumor cells, RFP-labeled SCC9 cells were defined as donor cells, and GFP-labeled SCC9 cells were defined as recipient cells. Sixty days after inoculation, obvious seeding of donor SCC9 cells could be detected in the recipient tumor mass (figure 1A, left, online supplemental figure 1A). To assess the potential preventive effect of NK cells on tumor self-seeding, Rag1-/- mice were treated with anti-asialo GM1 antibodies to deplete NK cells (online supplemental figure 1B). The depletion of NK cells significantly promoted tumor self-seeding. Increased self-seeded cells (4.28-fold higher) were observed in NK cell-depleted Rag1-/- mice on day 60 postinoculation (figure 1A, middle and right). In addition, NK cell depletion accelerated tumor self-seeding. Thirty days after inoculation, rare tumor cell seeding formation was observed in the control group (figure 1B, left). However, the presence of numerous seeding cells was found at the same time point in NK cell-depleted Rag1-/- mice (figure 1B, middle and right). These results indicate that tumor self-seeding is a common event in tumors, regardless of the pathways they metastasize, and NK cells play critical roles during this process.
Supplemental material
Supplemental material

Isolation and characterization of self-seeded cells. (A, B) Seeded tumors were visualized by fluorescence microscopy at 60 days (A) or 30 days (B) after inoculation using contralateral seeding model (left) or NK cell-depleted contralateral seeding model (middle), and fluorescence-based quantification was measured (Right) (n=6 per group). RFP-labeled SCC9 cells were defined as donor cells and GFP-labeled SCC9 cells were defined as recipient cells. Bars: 500 µm. (C) Isolation of SCC9-seeded cells by FACS. (D) Bright field and fluorescence images of isolated SCC9-seeded cells. Bars: 50 µm. (E–G) Comparison of wound healing (E), colony formation (F) and proliferation ability (G) between SCC9-parental and SCC9-seeded cells. (H) The expression of EMT markers in SCC9-parental and SCC9-seeded cells. (I) Cleaved-PARP expression in monolayer cultured or suspension cultured SCC9-seeded and SCC9-parental cells. (J) Lysis of SCC9-seeded and SCC9-parental cells by fresh NK cells at E/T ratios of 5:1, 10:1, and 20:1. (K, L) Seeded tumors were visualized by fluorescence microscopy at 60 days (K) or 30 days (L) after inoculation using contralateral seeding model (Left, middle), and fluorescence-based quantification was measured (Right) (n=6 per group). SCC9-seeded cells were inoculated as donor cells. Bar: 500 µm. Data represent three independent experiments done in triplicate (in vitro model) or with 6 mice per group (in vivo model) (mean±SD). *p<0.05, **p<0.01, ***p<0.001; ns, no significance; unpaired Student’s t-test (A, B, E, F, K and L), two-way ANOVA analysis (G, J). ANOVA, analysis of variance; E/T, effector/target; FACS, fluorescence activated cell sorting; GFP, green fluorescent protein; NK, natural killer; PARP, poly ADP-ribose polymerase; RFP, red fluorescent protein; shN-cad, short hairpin N-cadherin.
Isolation and characterization of self-seeded cells
These self-seeded tumor cells represent distinct subpopulations that can evade NK cell-mediated immune surveillance in the circulation and finally home to primary tumor. We, therefore, isolated and cultured these seeded cells (SCC9-seeded) from a single cell suspension of digested recipient tumors by FACS (figure 1C,D). No significant differences in the migration ability, colony formation or proliferation were observed between the SCC9-seeded cells and SSC9-parental cells (figure 1E–G). SCC9-seeded cells exhibited molecular changes consistent with epithelial to mesenchymal transition (EMT). Decreased expression of E-cadherin and increased expression of Vimentin, Snail, ZEB1 and ZEB2 were observed in SCC9-seeded cells (figure 1H). Analysis of cleaved PARP, a marker for apoptosis, was tested in both monolayer-cultured and suspension-cultured cancer cells. A decreased cleaved PARP level was observed in suspension-cultured SCC9-seeded cells compared with suspension-cultured SCC9-parental cells, although these two cell lines had similar cleaved PARP levels in monolayer cultures (figure 1I). This result indicates that SCC9-seeded cells are more resistant to anoikis, a form of apoptosis resulting from the loss of cell-cell interactions. In the cytotoxicity assay, SCC9-seeded cells and SCC9-parental cells were cocultured with freshly isolated NK cells (online supplemental figure 1C). Lower lysis of SCC9-seeded cells relative to SCC9-parental cells was observed (figure 1J). The tumor seeding ability of SCC9-seeded cells was also examined. SCC9-seeded cells displayed an approximately 3.97-fold higher seeding ability than SCC9-parental cells (figure 1K). In addition, SCC9-seeded cells developed self-seeding much sooner (30 days after inoculation) than did SCC9-parental cells (no tumor cell seeding formation at the same time point) (figure 1L). These data indicate that the isolated SCC9-seeded cells are subpopulations with the ability to avoid NK cell killing and anoikis in the circulation.
High expression of N-cadherin in self-seeded cells
To reveal the underlying mechanism that triggers the escape of SCC9-seeded cells from NK cell mediated immune surveillance, transcriptional profiling and comparative analysis were performed (figure 2A). Differential expression of a set of genes (62 genes upregulated and 74 genes downregulated in SCC9-seeded cells compared with SCC9-parental cells) was observed (fold change more than 20 times) (set 1). Then, we reviewed 11 published studies and determined 67 membrane molecules related to NK cell function (set 2) (online supplemental table 1). A Venn diagram analysis indicated three overlapping genes (CDH2, FN1 and HLA-E) between set 1 and set 2 (figure 2B). Using qRT-PCR, we verified the upregulatiom of CDH2, but not FN1 and HLA-E, in SCC9-seeded cells (figure 2C, online supplemental figure 1D). Therefore, CDH2 was considered to be potentially involved in tumor cells escaping immune surveillance of NK cells. Two other classical members of the CDH family, E-cadherin (CDH1) and R-cadherin (CDH4), are also known as ligands for KLRG1,21 and the homology between the human and murine N-cadherin proteins was the highest (96.55%) (figure 2D). Elevated CDH2 mRNA, but not CDH1 and CDH4 mRNA, and increased N-cadherin were confirmed in SCC9-seeded cells (figure 2E–G). To determine whether self-seeded cells express N-cadherin during the circulation process, CTCs were harvested from 10 patients with oral cancer as well as the contralateral seeding model and stained with N-cadherin antibody. More than 60% of CTCs were found to express N-cadherin in both patients and animal model (figure 2H, online supplemental figure 1E and table 2). In addition to N-cadherin, the expression of its receptor, KLRG1, was also measured in NK cells isolated from patients with oral cancer, healthy individuals and Rag1-/- mice. Positive KLRG1 expression was observed on numerous human and mice NK cells (figure 2I and online supplemental figure 1F). Moreover, an increased proportion of KLRG1 (+) NK cells was observed in patients with oral cancer (64.22%±13.11%) compared with healthy individuals (39.89%±14.53%), and these increased KLRG1 (+) NK cells were mainly CD56dim subsets, which are known to have higher cytotoxicity than CD56bright subsets (figure 2I).22

High expression of N-cadherin in self-seeded cells. (A) A comparison of transcriptional profiles was performed between SCC9-seeded and SCC9-parental cells. (B) Venn diagram analysis indicated three overlapping genes (CDH2, FN1 and HLA-E) between set 1 and set 2. Set 1 includes differentially expressed genes between SCC9-seeded and SCC9-parental cells; and set 2 includes membrane molecules related to NK cell function based on published literature. (C) The mRNA expression of CDH2, FN1 and HLA-E in SCC9-seeded and SCC9-parental cells was assessed using qRT-PCR analysis. (D) Homology of CDH1, CDH2 and CDH4 between humans and mice. (E) The mRNA expression of CDH1, CDH2 and CDH4 in SCC9-seeded and SCC9-parental cells was assessed using qRT-PCR analysis. (F) Protein levels of N-cadherin in SCC9-seeded and SCC9-parental cells. (G) Immunofluorescence staining of N-cadherin in SCC9-seeded and SCC9-parental cells. Bars: 25 µm. (H) Detection of N-cadherin in CTCs harvested from patients with oral cancer. From left to right: CK staining; CD45 staining; DAPI staining; N-cadherin staining; merged image. Bars: 50 µm. (I) Comparison of KLRG1 expression status on NK cells in healthy individuals and patients with oral cancer. Data represent three independent experiments done in triplicate (in vitro model) or with 10 patients with oral cancer (H) or 20 individuals (I, 10 patients with oral cancer and 10 healthy control) (mean±SD). **p<0.01, ****p<0.0001; ns, no significance; unpaired Student’s t-test (C, E and I). CK, cytokeratin; CTCs, circulating tumor cells; DAPI, 4’,6-diamidino-2-phenylindole; mRNA, message RNA; NK, natural killer; qRT-PCR, quantitative real time-PCR.
N-cadherin protects self-seeded cells from NK cytolysis through KLRG1 receptor
High expression of N-cadherin was observed in self-seeded cells, and because N-cadherin binding to its receptor KLRG1 reportedly impaired the NK-cell functional and immune responses.23 We assume that the interaction between N-cadherin and the KLRG1 receptor may contribute to the inhibition of NK cell antitumor activity and immune evasion of self-seeded tumor cells. To verify the influence of tumor cell N-cadherin expression on NK cell cytotoxicity, N-cadherin was silenced in SCC9-seeded cells (SCC9-seeded shN-cad) (online supplemental figure 2A, B). SCC9-seeded cells, SCC9-parental cells and SCC9-seeded shN-cad cells were cocultured with NK cells. Silencing of N-cadherin resulted in a much higher lysis rate in SCC9-seeded shN-cad cells than in SCC9-seeded cells and SCC9-parental cells (figure 3A, left). With the N-cadherin blocking antibody treatment, lysis of SCC9-seeded cells by NK cells was significantly increased. This result suggested that the N-cadherin expression levels in tumor cells determined their susceptibility to NK cell killing. To clarify that binding to its receptor KLRG1 is required for N-cadherin-induced impaired antitumor efficacy of NK cells, KLRG1 (−)/(+) NK subsets were freshly isolated (online supplemental figure 2C), and cocultured with SCC9-seeded cells, SCC9-parental cells and SCC9-seeded shN-cad cells. Notably, KLRG1 (+) NK subsets produced lysis patterns resembling those of total NK populations (figure 3A, middle). However, no difference in lysis rates was found in the KLRG1 (−) NK subset group (figure 3A, right). These results indicate that the expression of N-cadherin can protect tumor cells from NK cytolysis, which is mediated by interacting with the KLRG1 receptor.
Supplemental material
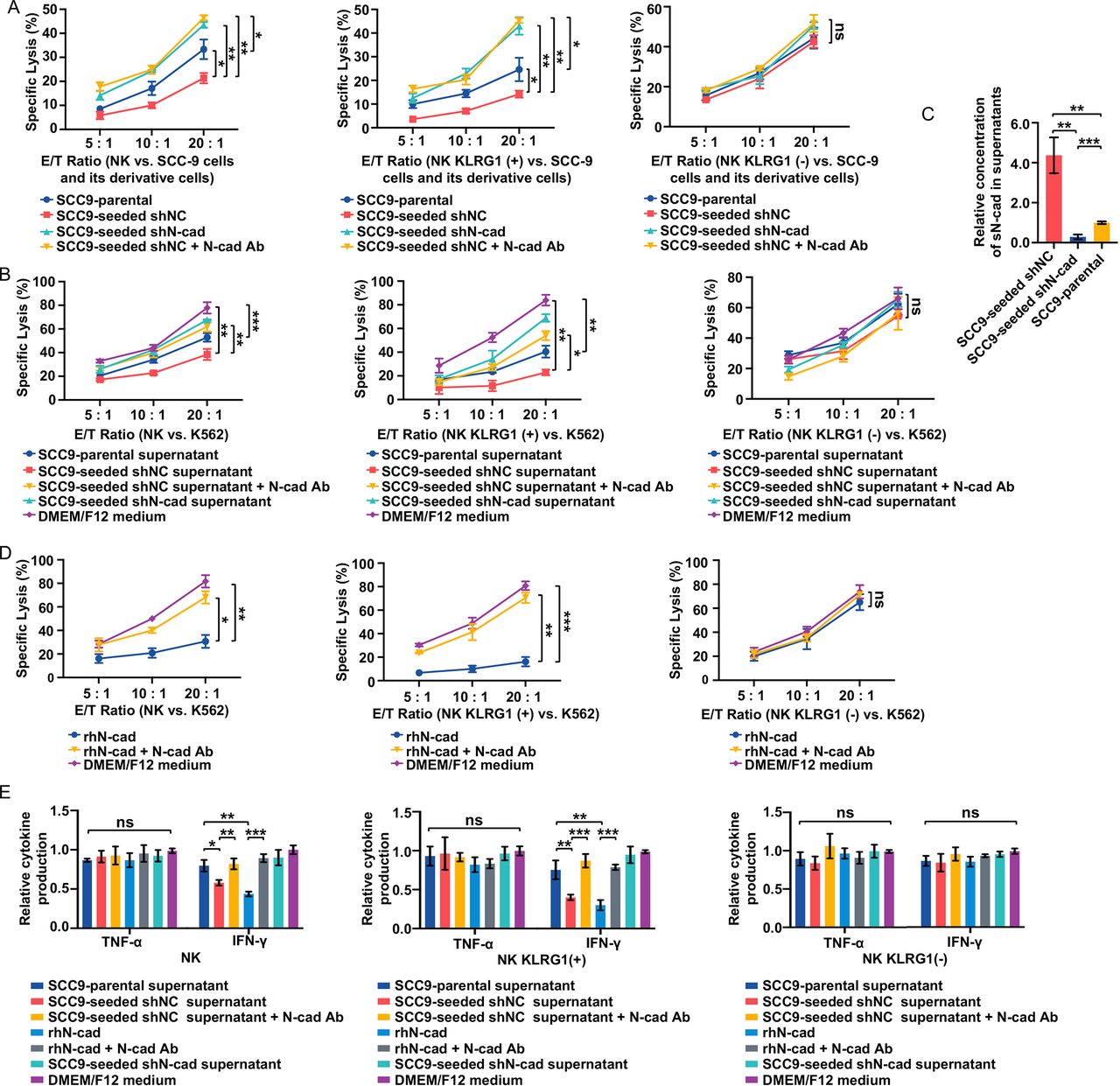
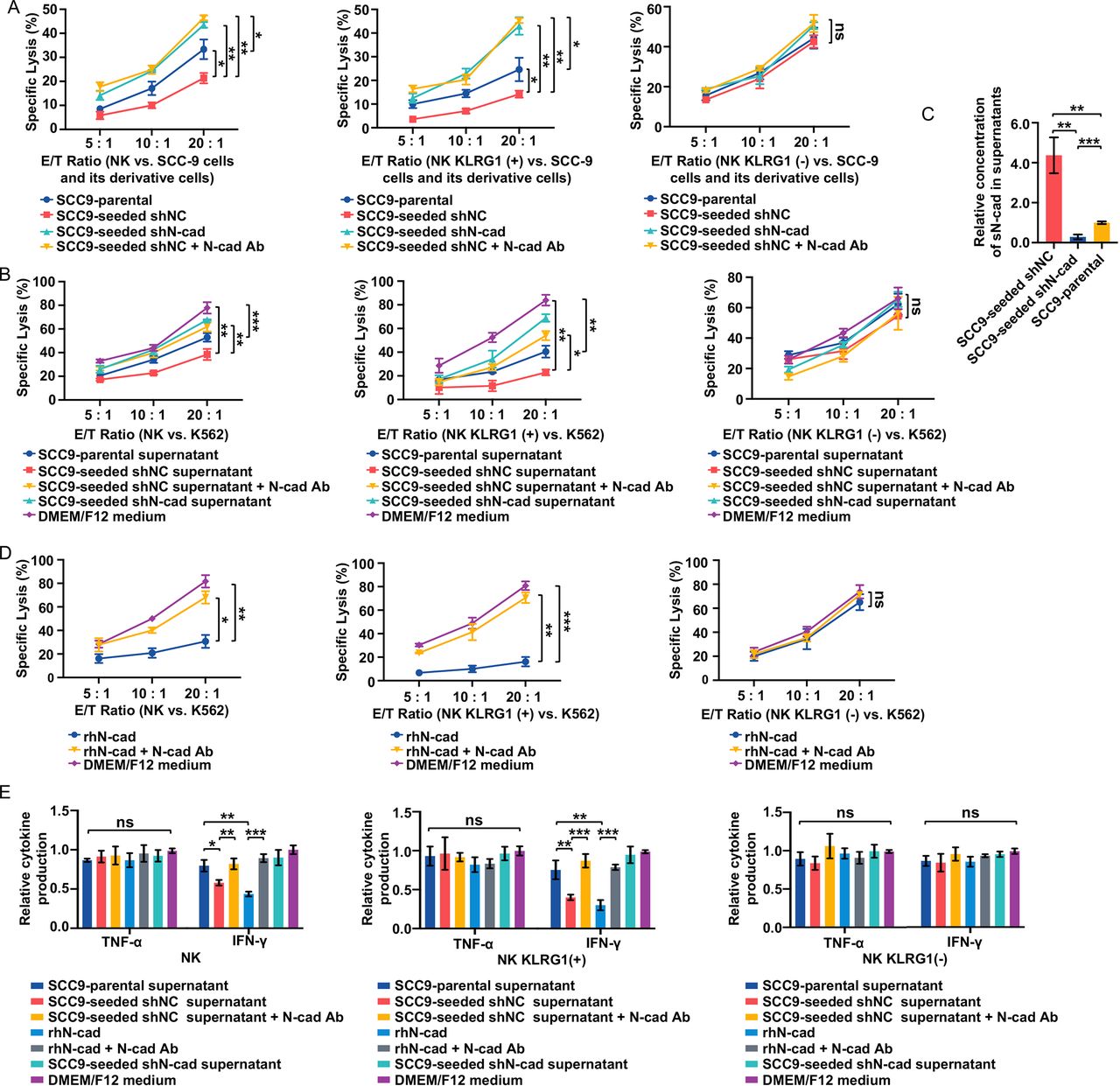
Self-seeded cell-derived soluble N-cadherin impaired cytotoxicity by interacting with the KLRG1 receptor. (A) Lysis of the indicated tumor cell lines by total NK populations (left), KLRG1 (+) subsets (Middle) and KLRG1 (−) subsets (Right) at E/T ratios of 5:1, 10:1 and 20:1. (B) Comparison of total NK populations (Left), KLRG1 (+) subsets (Middle) and KLRG1 (−) subsets (right) cytotoxicity against K562 cells after exposure to various tumor cell culture supernatants at E/T ratios of 5:1, 10:1 and 20:1. (C) Soluble N-cadherin levels in supernatants of the indicated cells. sN-cad: soluble N-cadherin. (D) Comparison of total NK populations (left), KLRG1 (+) subsets (Middle) and KLRG1 (−) subsets (Right) cytotoxicity against K562 cells with the indicated treatments at E/T ratios of 5:1, 10:1 and 20:1. (E) Cytokine production of total NK populations (left), KLRG1 (+) subsets (Middle) and KLRG1 (−) subsets (right) after the indicated treatments. Data represent three independent experiments done in triplicate (mean±SD). *p<0.05, **p<0.01, ***p<0.001; ns, no significance; two-way ANOVA analysis (A, B, D), one-way ANOVA and Tukey-Kramer multiple comparison tests (C, E) ANOVA, analysis of variance; E/T, effector/ target; IFN, interferon; NK, natural killer; N-cad Ab, N-cadherin blocking antibody; rhN-cad, recombinant human soluble N-cadherin; shNC, short hairpin negative control; shN-cad, short hairpin N-cadherin; TNF, tumor necrosis factor.
In addition to direct killing, self-seeded cell-induced dysfunction in NK cells was also evaluated in an indirect culture system using the NK-sensitive cell line K562. We compared the NK cell cytotoxicity after exposure to various tumor cell culture supernatants. Incubation of the SCC9-parental cell supernatant slightly inhibited the cytotoxic activity of NK cells against K562 target cells when compared with normal culture medium. Treatment of SCC9-seeded cell supernatant, but not SCC9-seeded shN-cad cells supernatant, caused a more profound inhibitory effect, which could be reversed by the application of an N-cadherin blocking antibody (figure 3B, left). Similar results were observed in KLRG1 (+) NK subsets exposed to tumor cell supernatants (figure 3B, middle). However, none of the tumor supernatants had any effect on the KLRG1 (−) NK subsets (figure 3B, right). These data suggest that soluble molecules released by SCC9-seeded cells are involved in the suppression of NK cell antitumor function in a KLRG1 receptor-dependent manner. Given that blocking N-cadherin could restore NK cell function, we reasoned that this effect factor might be soluble N-cadherin. The levels of soluble N-cadherin in these tumor cell supernatants were determined by ELISA. Soluble N-cadherin levels were elevated in SCC9-seeded cell supernatants compared with SCC9-parental cells and SCC9-seeded shN-cad cells (figure 3C). We next sought to determine whether the higher expression of soluble N-cadherin could induce the impaired NK cell cytotoxicity function. Total NK populations and KLRG1 (−)/ (+) NK subsets were administered recombinant human soluble N-cadherin (rhN-cadherin) (500 ng/mL) with or without an N-cadherin blocking antibody. RhN-cadherin treatment resulted in an obvious cytotoxicity inhibition in the total NK cell and KLRG1 (+) NK cell subset groups but not in the KLRG1 (−) NK cell subset group, and the application of an N-cadherin blocking antibody reversed this effect (figure 3D). This result shows that self-seeded cell-derived soluble N-cadherin could suppress NK cell cytotoxicity by interacting with the KLRG1 receptor.
Soluble N-cadherin-induced and KLRG1 interaction-induced functional exhaustion of NK cells
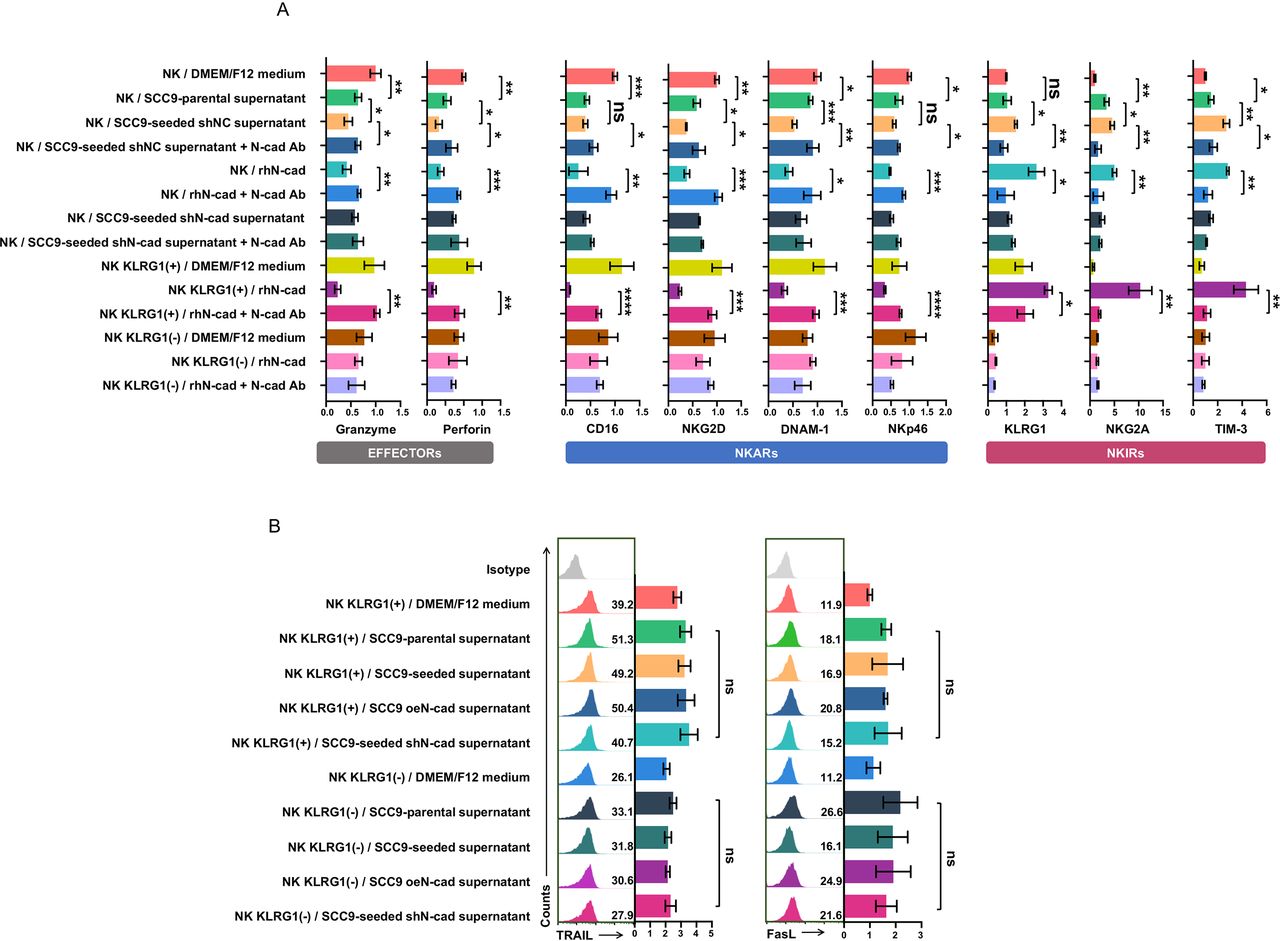
Inhibitory signals may facilitate the exhausted status of NK cells. Typically, exhausted NK cells are characterized by (1) diminished cytolytic activity; (2) decreased production of effector cytokines; and (3) an exhausted phenotype characterized by downregulated expression of activating receptors and upregulation of inhibitory receptors.24 To verify that self-seeded cell-derived soluble N-cadherin could trigger NK cells functional exhaustion, the expression of cytolytic molecules (granzymes and perforin), cytokine production (IFN-γ and TNF-α) and the exhausted phenotype of NK cells were measured. NK cells incubated with SCC9-seeded cell supernatant or treated with rhN-cadherin displayed an obvious exhausted phenotype, as evidenced by decreased expression of granzymes and perforin and lower IFN-γ (but not TNF-α) production, along with the downregulation of four main activating receptors (CD16, NKG2D, DNAM-1 and NKp46) but upregulation of three inhibitory receptors (NKG2A, TIM-3 and KLRG1 itself) when compared with the SCC9-parental cell supernatant-treated NK cells (figure 3E, left, figure 4A and online supplemental figure 3). In rescue experiments, the SCC9-seeded cell supernatant-cadherin-induced or rhN-cadherin-induced NK cell exhaustion was abrogated with the use of N-cadherin blocking antibody. These experiments were also tested in KLRG1 (−)/(+) NK subsets. Due to the limitation of our specimens, we only assessed the influence of rhN-cadherin on the phenotype of these two subsets. Similar but more pronounced alterations were observed in KLRG1 (+) NK subsets. In contrast, the rhN-cadherin treatments had no effect on the KLRG1 (−) NK subsets (figure 3E, middle and right, and figure 4A and online supplemental figure 3). These findings demonstrate that self-seeded cell-derived soluble N-cadherin facilitates the functional exhaustion of NK cells, which is dependent on the presence of the KLRG1 receptor.
Supplemental material

Soluble N-cadherin induces NK cell functional exhaustion in a KLRG1 receptor-dependent manner. Freshly isolated total NK populations were exposed to the indicated tumor cell culture supernatants or the recombinant human soluble N-cadherin with or without an N-cadherin blocking antibody. KLRG1 (+) subsets and KLRG1 (−) subsets were administered recombinant human soluble N-cadherin with or without an N-cadherin blocking antibody. The NK populations were then phenotyped. (A) Statistics for FACS data of cytolytic effector molecules expression (left), NKARs expression middle) and NKIRs expression (right) in NK-cell populations following indicated treatments. (B) FACS analysis of surface FasL and TRAIL expression levels in KLRG1 (−) and KLRG1 (+) NK subsets after exposure to various tumor cell culture supernatants. Data represent three independent experiments done in triplicate (mean±SD). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; ns, no significance; one-way ANOVA and Tukey-Kramer multiple comparison tests. ANOVA, analysis of variance; DMEM, dulbecco‘s modified eagle medium; FACS, fluorescence activated cell sorting; NKARs, NK cell activation receptors; N-cad Ab, N-cadherin blocking antibody; NK, natural killer; NKIRs, NK cell inhibitory receptor; rhN-cad, recombinant human soluble N-cadherin; shNC, short hairpin negative control; shN-cad, short hairpin N-cadherin.
In addition to activating and inhibitory receptors, the expression of death ligands also affects the antitumor function of NK cells. Therefore, we examined the expression of FasL and TRAIL in KLRG1 (−) /(+) NK subsets after exposure to various tumor cell culture supernatants. No significant difference in surface expression of FasL and TRAIL was observed (figure 4B), suggesting that death ligands may not be involved in N-cadherin/KLRG1-mediated inhibition of NK cell cytotoxicity.
NK cell-secreted cytokines and fluid shear stress facilitate N-cadherin release from tumor cells by promoting A disintegrin and metalloprotease 10 translation or converting the pro-ADAM10 to the m-ADAM10
A disintegrin and metalloprotease 10 (ADAM10), a metalloproteinase, has previously been reported to cleave and induce ectodomain shedding of cadherin family members.25 Similar levels of ADAM10 protein and mRNA transcript corresponding to full-length ADAM10 with proteolytic activity were observed in SCC9-parental and SCC9-seeded cells (figure 5A and online supplemental figure 4A). A 90 KD band corresponds to the precursor protein (pro-ADAM10), and a 70 KD band corresponds to the mature and catalytically active form of ADAM10 (m-ADAM10). To determine whether ADAM10 mediated soluble N-cadherin release from tumor cells, we silenced ADAM10 in SCC9-seeded cells (SCC9-seeded shADAM10) (figure 5A and online supplemental figure 4B). Knockdown of ADAM10 significantly suppressed the soluble N-cadherin levels in SCC9-seeded cell supernatants (figure 5B) and abolished the inhibitory effect of tumor supernatants on NK cell cytolytic activity (figure 5C), thus supporting that ADAM10-mediated production of soluble N-cadherin was responsible for the self-seeded cell-induced NK cell dysfunction.
Supplemental material
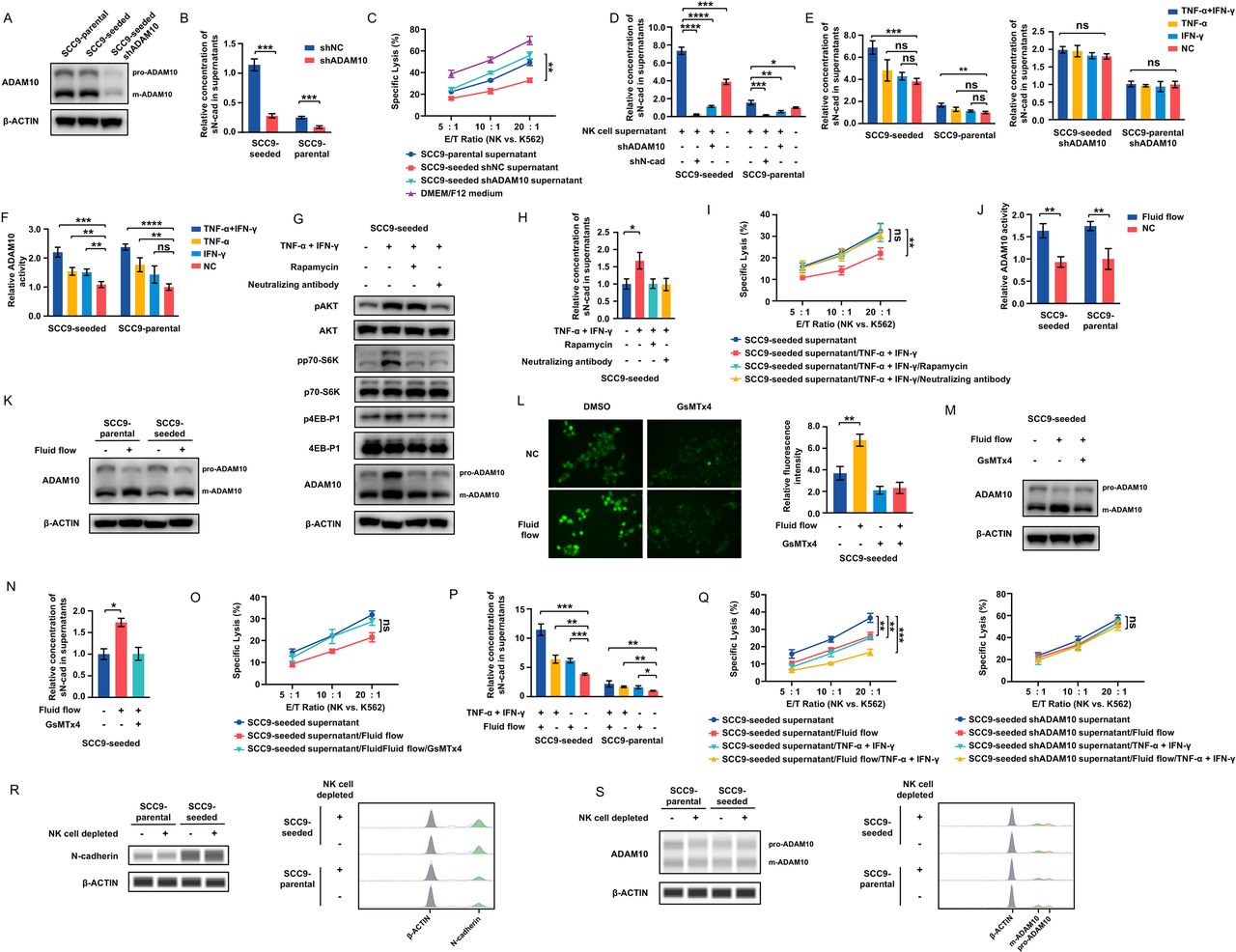
NK cell-secreted cytokines and fluid shear stress facilitate N-cadherin release from tumor cells by enhancing the activity of ADAM10. (A) ADAM10 expression in SCC9-parental, SCC9-seeded and SCC9-seeded shADAM10 cells. (B) sN-cad levels in the supernatants of SCC9-seeded, ADAM10-silenced SCC9-seeded, SCC9-parental and ADAM10-silenced SCC9-parental cells. (C) NK cell cytotoxicity against K562 cells with the indicated treatments at E/T ratios of 5:1, 10:1 and 20:1. (D) sN-cad levels in the supernatants of SCC9-seeded, ADAM10-silenced SCC9-seeded, N-cadherin-silenced SCC9-seeded, SCC9-parental, ADAM10-silenced SCC9-parental and N-cadherin-silenced SCC9-parental cells after exposure to NK cell supernatants. (E) sN-cad levels in the supernatants of SCC9-seeded, ADAM10-silenced SCC9-seeded, SCC9-parental and ADAM10-silenced SCC9-parental cells with the indicated treatments. (F) ADAM10 activity in SCC9-seeded and SCC9-parental cells with the indicated treatments. (G) Western blots of AKT, phospho-AKT, p70-S6K, phospho-p70-S6K, 4EB-P1, phospho-4EB-P1 and ADAM10 levels in SCC9-seeded cells with the indicated treatments. (H) sN-cad levels in the supernatants of SCC9-seeded cells with the indicated treatments. (I) NK cell cytotoxicity against K562 cells with the indicated treatments at E/T ratios of 5:1, 10:1 and 20:1. (J, K) ADAM10 activity (J) and expression (K) in tumor cells cultured in a shaker incubator for 24 hours. (L) Calcium influx in SCC9-seeded cells cultured in a shaker incubator for 24 hours. GsMTx4, a specific inhibitor of the Piezo1, abrogated the shear stress-induced calcium influx. (M–O) ADAM10 expression (M), sN-cad levels (N) and the effect of cell supernatants on NK cell cytotoxicity (O) in SCC9-seeded cells cultured in a shaker incubator with or without GsMTx4 treatment. (P) sN-cad levels in the supernatants of tumor cells with the indicated treatments. (Q) NK cell cytotoxicity against K562 cells with the indicated treatments at E/T ratios of 5:1, 10:1 and 20:1. (R, S) Gel-like image representation (left) and electropherograms representation (right) of N-cadherin (R) and ADAM10 (S) expression in SCC9-parental and SCC9-seeded cell enriched samples obtained from both Rag1-/- and NK cell-depleted Rag1-/- self-seeding models by Capillary Western analysis. Data represent three independent experiments done in triplicate (mean±SD). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; ns, no significance; unpaired Student’s t-test (B, J); two-way ANOVA analysis (C, I, O and Q); one-way ANOVA and Tukey-Kramer multiple comparison tests (D–F, H, L, N and P). ANOVA, analysis of variance; E/T, effector/target; NK, natural killer; shADAM10, short hairpin ADAM10; shNC, short hairpin negative control; shN-cad, short hairpin N-cadherin; sN-cad, Soluble N-cadherin; TNF, tumor necrosis factor.
Most interestingly, when tumor cells (but not N-cadherin-silenced or ADAM10-silenced tumor cells) were pre-exposed to NK cell supernatants, a significant increase in soluble N-cadherin levels was observed (figure 5D). This result suggested that some NK cell-sourced signaling molecules can promote ADAM10-induced N-cadherin shedding from tumor cells. Therefore, the influence of IFN-γ and TNF-α, two major cytokines secreted by NK cells,26 on ADAM10 proteolytic activity and soluble N-cadherin production in tumor cells was tested. As shown in figure 5E,F, combined treatment with IFN-γ and TNF-α was sufficient to increase ADAM10 activity and soluble N-cadherin levels. In contrast, no soluble N-cadherin expression alteration was observed in ADAM10-silenced tumor cells that received treatment with IFN-γ and TNF-α.
To clarify the activation mechanism of ADAM10 by IFN-γ/TNF-α, the full-length mRNA and protein expression levels of ADAM10 were measured in tumor cells following IFN-γ/TNF-α treatment. We found that IFN-γ/TNF-α treatment did not significantly change ADAM10 mRNA levels, but increased the protein levels of both active and precursor forms of ADAM10 (figure 5G, online supplemental figure 4C,D). In addition, the translation inhibitor cycloheximide (CHX) but not the transcription inhibitor actinomycin D (ActD) abrogated the IFN-γ/TNF-α-induced increase in ADAM10 expression (online supplemental figure 4E). These results indicate that IFN-γ/TNF-α affects ADAM10 expression at the translational level. IFN-γ and TNF-α were reported to activate PI3K/AKT/mTOR signaling,27 28 which has been well characterized to control protein translation via its two downstream effectors, p70 ribosomal protein S6 kinase (p70-S6K) and eukaryotic translation initiation factor 4E (eIF4E) binding protein 1 (4E-BP1).29 As shown in figure 5G, IFN-γ/TNF-α treatment resulted in enhanced phosphorylation of AKT, p70-S6K and 4E-BP1, indicating the activation of PI3K/AKT/mTOR signaling in response to IFN-γ/TNF-α. Treatment of the mTOR inhibitor, rapamycin, significantly blocked the IFN-γ/TNF-α-induced phosphorylation of p70-S6K and 4E-BP1 but had no effect on the phosphorylation status of the upstream protein Akt. In addition, pretreatment of SCC9-seeded cells with rapamycin and IFN-γ/TNF-α neutralizing antibodies completely abrogated the IFN-γ/TNF-α-induced enhancement of ADAM10 protein and soluble N-cadherin levels (figure 5G,H), and reversed the NK cell supernatants-induced and IFN-γ/TNF-α-induced cytotoxicity inhibition in NK cells (figure 5I, online supplemental figure 4F). These results demonstrate that IFN-γ/TNF-α promotes ADAM10 protein translation by activating PI3K/AKT/mTOR signaling, thus promoting N-cadherin shedding and causing NK cell exhaustion.
Previous studies reported that shear stress could activate ADAM10 via the Piezo1 force sensor.30 We then tested the effect of fluid flow on ADAM10 activity and expression. ADAM10 activity and m-ADAM10 expression levels, but not the mRNA levels, were elevated in tumor cells cultured in a shaker incubator compared with those cultured under normal conditions (figure 5J,K and online supplemental figure 4G). Piezo1 is a calcium-permeable non-selective cationic channel. Activation of piezo1 leads to calcium influx. Our results showed that shear stress did not alter the expression of Piezo1 (supplemental figure 4H), but caused elevation of the cytosolic calcium concentration, which could be blocked by GsMTx4, a specific inhibitor of the Piezo1 channel (figure 5L). This result suggests that shear stress can trigger Piezo1 activation. Accompanied by the activation of Piezo1, the levels of m-ADAM10 increased, while the levels of pro-ADAM10 decreased (figure 5K,M), which could be reversed by GsMTx4, suggesting that shear stress converted the pro-ADAM10 to the m-ADAM10 via activating Piezo1. GsMTx4 treatment also abrogate the shear stress-induced enhancement of soluble N-cadherin and inhibitory effect on NK cell cytotoxicity (figure 5N,O).
A time-course analysis showed that m-ADAM10 expression returned to its original value within 72 hours or 36 hours after removing IFN-γ/TNF-α or shear stress treatment, indicating that IFN-γ/TNF-α and shear stress might only exert a short-lasting effect on ADAM10 expression (supplemental figure 4I).
Combined treatment with cytokines and fluid flow led to more soluble N-cadherin release and enhanced inhibitory effect on NK cell cytolysis, but had no effect on tumor cells lacking ADAM10 (figure 5P,Q and online supplemental figure 4J).
NK cell challenging is not sufficient to cause a SCC9-seeded cell like phenotype in SCC9-parental cells
To demonstrate whether exposure to NK cells may induce a SCC9-seeded-like phenotype in SCC9-parental cells, we cocultured SCC9-parental cells with freshly isolated NK cells in the indirect culture system. The expression of N-cadherin and ADAM10 was detected in the tumor cells. The N-cadherin expression level remained at a low level, while ADAM10 expression increased in these cells (online supplemental figure 5A and figure 4C). In spite of the increase of ADAM10 expression, we observed only small increases in the levels of soluble N-cadherin (figure 5D), which may be attributed to the low expression of N-cadherin in these cells. These cells displayed a similar susceptibility to NK-mediated lysis as the untreated SCC9-parental cells, and their supernatants only had a weak inhibitory effect on the cytotoxic activity of NK cells when compared with SCC9-seeded cells (online supplemental figure 5B). These results show that NK challenging is not sufficient to cause a SCC9-seeded cell like phenotype in SCC9-parental cells.
Supplemental material
SCC9-parental and SCC9-seeded cell enriched samples were obtained from both Rag1-/- and NK cell-depleted Rag1-/- self-seeding models by laser capture microdissection (online supplemental figure 5C), and the expression of N-cadherin and ADAM10 were assessed by capillary Western assays. There was no significant difference in the expression of N-cadherin and ADAM10 between two groups (figure 5R,S), further indicating that the sustained expression of N-cadherin and ADAM10 in tumor cells is independent of the presence of NK cells.
N-cadherin and ADAM10 expression affects the tumor self-seeding process
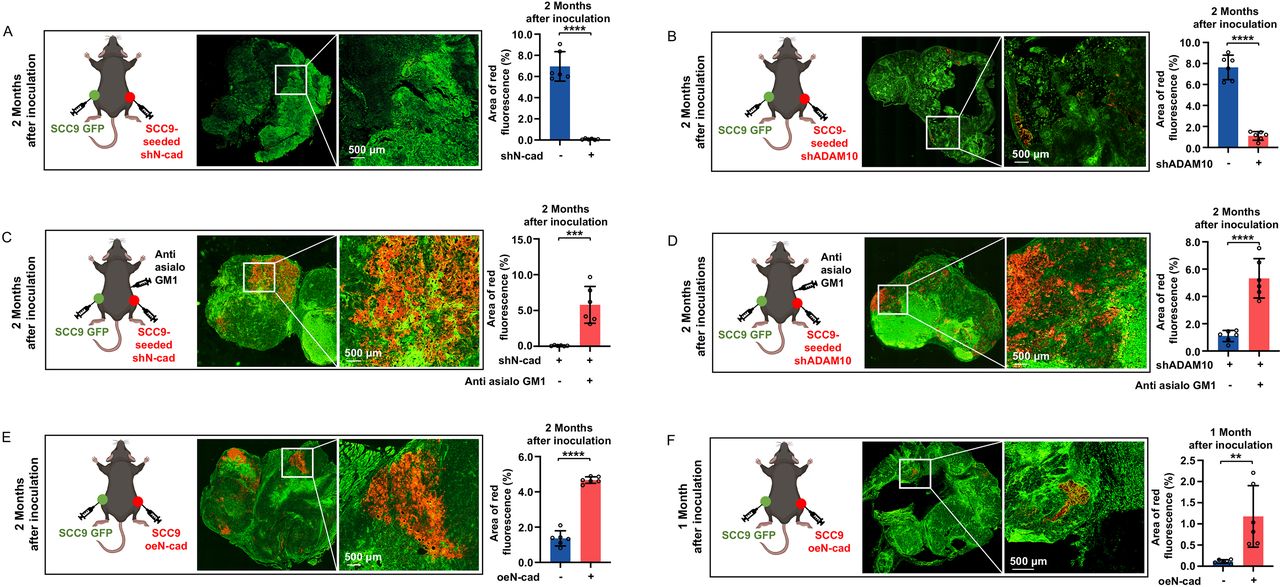
Next, we assessed whether the expression levels of N-cadherin and ADAM10 on tumor cells could affect tumor self-seeding in vivo. Contralateral seeding experiments were performed in Rag1-/- mice with or without anti-asialo GM1 antibody treatment. We forced SCC9 cells to stably express N-cadherin (SCC9 oeN-cad) (online supplemental figure 2B). SCC9-parental, SCC9-seeded, SCC9-seeded shN-cad, SCC9-seeded shADAM10 and SCC9 oeN-cad cells were inoculated as donor cells, and GFP-labeled SCC9-parental cells were inoculated as recipient cells. Silencing of N-cadherin dramatically inhibited the seeding ability of SCC9-seeded cells (tumor cell seeding formation was hardly observed 60 days after inoculation) (figure 6A and online supplemental figure 6A). ADAM10 silencing also greatly suppressed tumor cell seeding ability (6.93-fold lower than that of the SCC9-seeded cells, 60 days after inoculation) (figure 6B and online supplemental figure 6B). Depletion of NK cells could restore the seeding ability of these tumor cells (figure 6C,D, online supplemental figure 6C,D). SCC9 oeN-cad cells showed a stronger (3.43-fold higher than that of the SCC9-parental cells, 60 days after inoculation) and faster (seeding formation was observed as early as 30 days after inoculation) tumor seeding ability than SCC9-parental cells (figure 6E,F, online supplemental figure 6E,F). These results indicate that downregulation of N-cadherin and ADAM10 could inhibit tumor self-seeding, which is dependent on NK cells.
Supplemental material

N-cadherin and ADAM10 expression affects the tumor self-seeding process. (A, B) Seeded tumors were visualized by fluorescence microscopy at 60 days after inoculation using contralateral seeding model, and fluorescence-based quantification was measured (n=6 per group). SCC9-seeded shN-Cad cells (A) and SCC9-seeded shADAM10 cells (B) were inoculated as donor cells. Bars: 500 µm. (C, D) Seeded tumors were visualized by fluorescence microscopy at 60 days after inoculation using NK cell-depleted contralateral seeding model, and fluorescence-based quantification was measured (n=6 per group). SCC9-seeded shN-Cad cells (C) and SCC9-seeded shADAM10 cells (D) were inoculated as donor cells. Bars: 500 µm. (E, F) Seeded tumors were visualized by fluorescence microscopy at 60 days (E) or 30 days (F) after inoculation using contralateral seeding model, and fluorescence-based quantification was measured (n=6 per group). SCC9 oeN-Cad cells were inoculated as donor cells. Bars: 500 µm. Data represent two independent experiments with 6 mice per group (mean±SD). **p<0.01, ***p<0.001, ****p<0.0001; unpaired Student’s t-test. RFP, red fluorescent protein; GFP, green fluorescent protein; shN-cad, short hairpin N-cadherin; shADAM10, short hairpin ADAM10; oeN-cad, overexpress N-cadherin.
N-cadherin and ADAM10 expression facilitates survival of CTCs
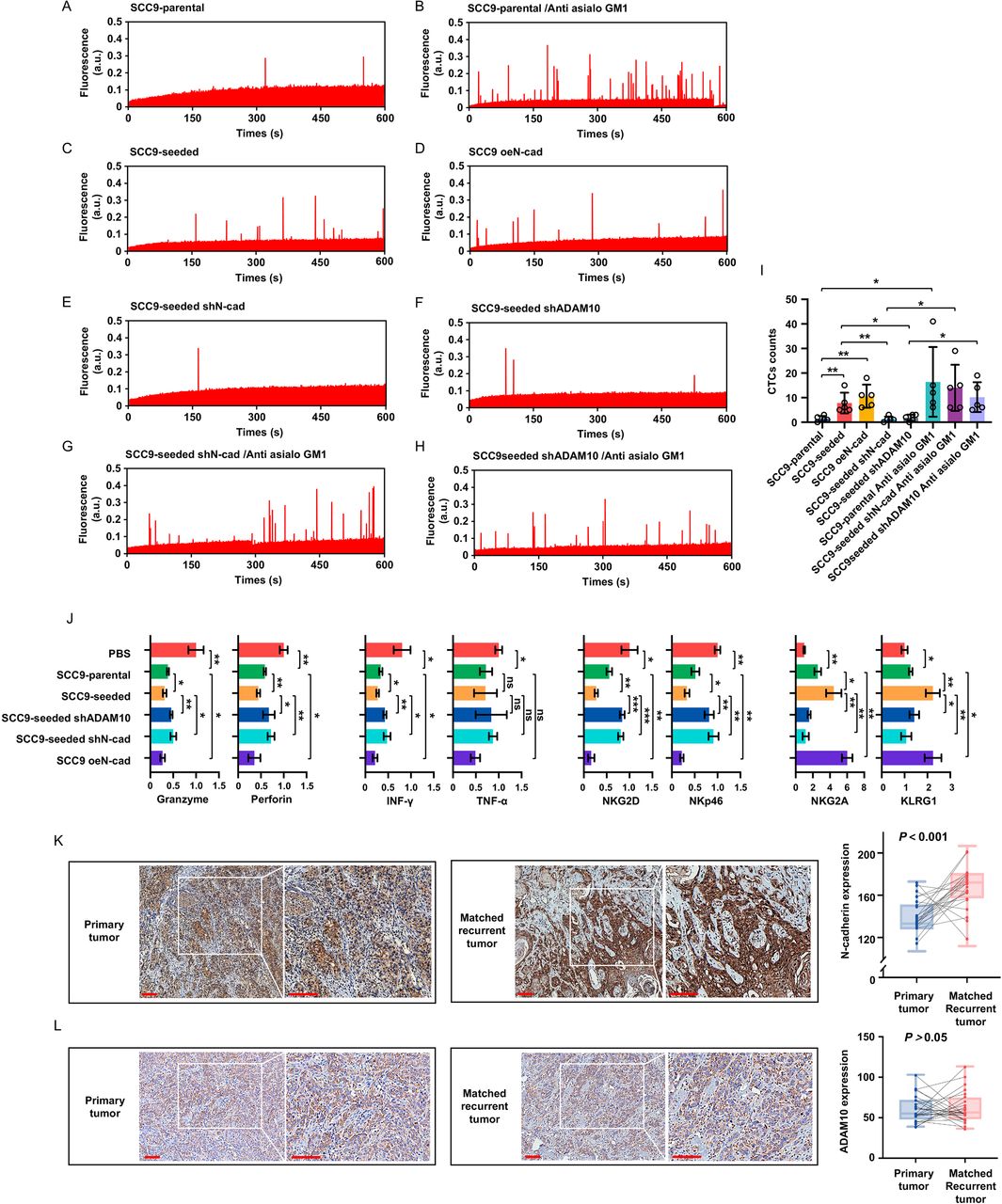
To test whether N-cadherin and ADAM10 indeed favor CTCs survival, IVFC was used to quantitatively assess the CTCs in mice at 5 hours after tail vein injections of indicated cell lines (online supplemental figure 7A). Typical data traces were presented in figure 7A–H, each fluorescence peak corresponded to a single surviving CTC. CTCs were barely detected in mice injected with SCC9-parental cells (figure 7A,I), indicating a rapid clearance of tumor cells in the circulation. The depletion of NK cells significantly enhanced the number of detected CTCs (figure 7B,I). Compared with parental cells, CTCs number was significantly increased in mice injected with SCC9-seeded and SCC9 oeN-cad cells (figure 7C,D and I). In addition, both silencing of N-cadherin and ADAM10 in SCC9-seeded cells led to a reduction of CTCs (figure 7E,F and I), which could be reversed by the treatment of anti-NK antibody (figure 7G–I). These results provide direct in vivo evidence that the expression of N-cadherin and ADAM10 in tumor cells facilitates survival of CTCs.
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
N-cadherin and ADAM10 expression facilitates survival of CTCs and elevated N-cadherin expression in recurrent oral cancer samples. (A–H) CTCs were detected by IVFC in indicated groups of mouse models, each fluorescence peak corresponded to a single surviving CTC. (n=5 per group) (I) Quantification of surviving CTCs in each group. (J) Freshly NK cells were ioslated from each group and FACS analysis of cytolytic effector molecules (granzymes and perforin) expression, cytokines (IFN-γ and TNF-α) expression, NK cell activation receptors (NKG2D and NKp46) expression and NK cell inhibitory receptors (NKG2A and KLRG1) expression in in NK cells of indicated groups. (K, L) Representative images of N-cadherin-stained (K) and ADAM10-stained (L) sections and quantification of immunohistochemical staining in both primary and matched recurrent tumor tissues (n=24 paired samples). Bars: 100 µm. Data represent three independent experiments done in triplicate with 5 mice per group (A–J) or with 24 paired samples (K, L) (mean±SD). *p<0.05, **p<0.01, ***p<0.001, ns, no significance; one-way ANOVA and Tukey-Kramer multiple comparison tests (I, J), paired Student’s t-test (K, L). shN-cad, short hairpin N-cadherin; shADAM10, short hairpin ADAM10; oeN-cad, overexpress N-cadherin; PBS, phosphate buffered saline.
The phenotype of NK cells isolated from the spleen of mice in each experimental group was also examined. NK cells from mice injected with SCC9-seeded or SCC9 oeN-cad cells exhibited an obvious exhausted phenotype, as evidenced by decreased expression of granzymes and perforin, lower cytokines production, and downregulation of two main activating receptors on mouse NK cells (NKG2D and NKp46) but upregulation of two inhibitory receptors on mouse NK cells (NKG2A and KLRG1 itself) when compared with the NK cells isolated from mice injections of SCC9-parental cells. Knockdown of N-cadherin and ADAM10 in tumor cells reversed the exhausted phenotype of NK cells (figure 7J, online supplemental figure 7B). These results show that CTCs with high expression of N-cadherin exhaust NK cells in vivo.
Elevated N-cadherin expression in recurrent oral cancer samples
We measured and compared the expression levels of N-cadherin and ADAM10 in both primary and matched recurrent tumor tissues from 24 patients. N-cadherin and ADAM10 expression was semiquantitatively assessed by immunohistochemistry. Overall, N-cadherin expression in recurrent tissues was significantly higher (172.41±14.27 score value) than that in primary disease tissues (136.87±11.14 score value) (figure 7K). N-cadherin staining in primary tumors was localized mainly on the membranes, while a wider expression of N-cadherin from the membrane to the cytoplasm was observed in the recurrent counterparts. In contrast, no significant difference in the expression of ADAM10 was found between primary and recurrent tumor tissues, which is consistent with the ADAM10 expression patterns observed in SCC9-seeded and SCC9-parental cells (figure 7L). These data indicate that tumor cells with high expression of N-cadherin were enriched during tumor recurrence, probably through tumor self-seeding.
Discussion
In our study, we successfully isolated self-seeded tumor cells and confirmed that NK cell antitumor immunity plays a critical role in eliminating tumor cells in the circulation.31 ADAM10-mediated tumor cell shedding of N-cadherin can induce NK cell functional exhaustion via the KLRG1 receptor. We documented that CTCs adopt this mechanism to protect themselves from the clearance of circulating NK cells.
The fact that NK cells depletion could accelerate and increase tumor self-seeding confirms that NK cells exert a key role in immune surveillance against tumor cells in circulation. The successfully obtained self-seeding cells exhibited molecular changes consistent with EMT and increased anoikis resistance when compared with their parental cells. The tumor seeding ability of the harvested self-seeded cells was much higher than that of their parental cells, suggesting that in addition to anoikis resistance, these subpopulations may also be able to escape NK cell-mediated immune surveillance. This notion was supported by the NK cell cytotoxicity assay results. Self-seeded cells exhibited lower susceptibility to lysis by NK cells than parental cells.
Remarkable molecular heterogeneity has been reported within CTCs or between CTCs and their primary tumors, which is related to tumor progression and responses to immunotherapy.32 However, these studies did not distinguish surviving CTCs from total CTC populations and, therefore, may not accurately reflect the molecular characteristics of ‘functional CTCs’. In this study, high expression of CDH2 gene and its protein product, N-cadherin, was observed in self-seeded cell lines, which represent ‘surviving CTCs’. N-cadherin is considered a signature of EMT through which tumor cells can shed from primary tumors into the peripheral blood.33 Given that SCC9-seeded cells display an EMT phenotype, we speculate that the EMT process may contribute to the high expression of N-cadherin in SCC9 seeded cells, via cadherin switching.34 N-cadherin has been identified as a natural ligand of the inhibitory receptor KLRG1 that is shared between NK cells and CD8 (+) T cells.21 After binding to its receptor KLRG1, N-cadherin can transduce inhibitory signals to NK cells. We, therefore, hypothesize that N-cadherin may protect tumor cells from immune surveillance through KLRG1 signaling. We found that the expression of N-cadherin in tumor cells determined their susceptibility to lysis by the total NK populations and the KLRG1 (+) NK subsets, but not by the KLRG1 (−) NK subsets. KLRG1 is expressed by 50% of NK cells.35 Our data indicated a remarkably increased proportion of KLRG1 (+) subsets in tumor patients. Recently, Sunwoo and colleagues found that NK cells in patients with head and neck squamous cell carcinoma can differentiate into distinct subsets and divergent terminal states,36 which depends on contact with tumor cells. Since KLRG1 is known to be a terminal differentiation marker of NK cells,37 we think tumor cell-induced NK cell differentiation leads to the upregulation of KLRG1 on NK cells in tumor patients. Notably, these increased KLRG1 (+) cells were mainly cytotoxic CD56dim cells. KLRG1 signaling-induced CD56dim NK cell dysfunction may partly explain the poor antitumor response of NK cells in tumor patients.
Another interesting finding in our study is that we revealed an unrecognized role for soluble N-cadherin in NK cell antitumor immunity. N-cadherin is highly expressed as an adhesion molecule in the epithelial tissue, and its soluble form is stably expressed in serum.38 Shedding of soluble inhibitory ligands from tumor cells represents a novel strategy by which tumor cells evade immune surveillance. For example, elevated soluble Galectin-3 or soluble ULBP expression suppresses NK cell activity in gastric cancer or cervical adenocarcinoma.39 40 N-cadherin and other cadherin family members have been described as ligands for KLRG1; however, it remains undetermined whether soluble N-cadherin could regulate NK cell function. Our data demonstrated that in addition to membrane-bound N-cadherin-induced inhibition of NK cell cytotoxicity, soluble N-cadherin released by CTCs can also cause NK cell functional exhaustion through the KLRG1 receptor in a cell-cell non-contact manner.
ADAM10 has been implicated as a predominant metalloprotease responsible for the ectodomain shedding of cadherin family proteins.25 A positive correlation between ADAM10 expression and soluble N-cadherin levels in culture supernatants were observed in the tumor cell lines. Of note, we found that NK cell-produced IFN-γ and TNF-α could enhance ADAM10 activity and subsequently release more soluble N-cadherin into the extracellular environment by activating PI3K/AKT/mTOR signaling. These interesting findings indicate that tumor cells can sense the signaling molecules secreted by NK cells in the surrounding environment and promote the self-release of soluble N-cadherin to avoid immune attack. In addition, fluid shear stress was found to convert the pro-ADAM10 to the m-ADAM10, suggesting that although shear force in the circulation can kill tumor cells, tumor cells could also exploit fluid stress to protect themselves from immune surveillance.
Our in vivo study showed that ectopic expression of N-cadherin triggered rapid and higher tumor self-seeding in the contralateral seeding model. Conversely, silencing of ADAM10 and N-cadherin in self-seeded cells partially or completely suppressed the tumor cell self-seeding ability, respectively. The difference in the inhibition efficiency between ADAM10 silencing and N-cadherin silencing treatment may be because ADAM10 silencing merely inhibited the production of soluble N-cadherin while N-cadherin silencing abolished both soluble and membrane-bound N-cadherin. By using IVFC and an animal experimental model of CTCs, we also provided in vivo evidence that the expression of N-cadherin facilitates survival of CTCs by inducing NK cell exhaustion.
Higher expression of N-cadherin was observed in recurrent oral cancer tissues than that in matched primary tumors, suggesting CTCs that evade immune killing by NK cells may return to primary tumors and favor local recurrence, which needs further experimental confirmation.
Our study provides new insight into the diagnostic and therapeutic potential of N-cadherin/KLRG1 axis in cancer. N-cadherin expression analysis on CTCs can develop a new strategy for liquid biopsy to stratify patients with high recurrent risk who should receive more aggressive treatment. In addition, our data supported that KLRG1 is a novel checkpoint inhibitor target to promote NK cell antitumor activity, especially against CTCs.
Collectively, our data showed that a distinct subpopulation of CTC characterized by high expression of N-cadherin can sense NK cell-secreted signaling molecules and release soluble N-cadherin by enhancing ADAM10 activity. Membrane-bound and soluble N-cadherin triggers NK cell functional exhaustion and protects CTCs from NK cell-mediated killing by interacting with the KLRG1 receptor (online supplemental figure 8). Targeting N-cadherin is an effective strategy to prevent CTCs from homing to primary tumor.
Supplemental material
Supplemental material
Data availability statement
Data are available on reasonable request. The RNA-seq data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE186082.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by Ethics Committee of Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine No. 2017-139. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank the Med-X Research Institute and School of Biomedical Engineering, Shanghai Jiao Tong University for their expert technical assistance with the IVFC.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
CL and KW contributed equally.
Contributors XQ conceived the study. LC and WK conducted experiments. DZ collected clinical samples. SJ analyzed the data. XQ and LC drafted the manuscript. XQ is responsible for the overall content as the guarantor. All the authors reviewed, edited, and approved the manuscript.
Funding This work was supported by grants from National Natural Science Foundation of China (grant No. 81972526) and the Innovative Research Team of High-level Local Universities in Shanghai (SHSMU-ZLCX20212301).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.