Article Text

Abstract
Background Although immune checkpoint inhibitors (ICIs) have been shown to yield promising therapeutic outcomes in a small subset of patients with triple negative breast cancer (TNBC), the majority of patients either do not respond or subsequently develop resistance. Recent studies have revealed the critical role of TP53 gene in cancer immunology. Loss or mutation of p53 in cancer cells has been found to promote their immune escape. Given the high mutation frequency of TP53 in TNBC cells, restoration of p53 function could be a potential strategy to overcome their resistance to anti-programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1) therapy. Herein, we have assessed the use of Pos3Aa crystal-based platform to mediate the intracellular delivery of p53 protein to restore p53 activity in p53 null tumors and consequently augment anti-PD-1 activity.
Methods The efficiency of Pos3Aa-p53 crystals in delivering p53 protein was evaluated using confocal imaging, immunofluorescence staining, flow cytometry and RNA-seq. The ability of Pos3Aa-p53 crystals to remodel tumor microenvironment was investigated by examining the markers of immunogenic cell death (ICD) and the expression of PD-L1, indoleamine 2,3-dioxygenase 1, tryptophan 2,3-dioxygenase 2 and type I interferon (IFN). Finally, both unilateral and bilateral 4T1 tumor mouse models were utilized to assess the efficacy of Pos3Aa-p53 crystal-mediated p53 restoration in enhancing the antitumor activity of ICIs. T cells in tumor tissues and spleens were analyzed, and the in vivo biosafety of the Pos3Aa-p53 crystal/anti-PD-1 antibody combination was also evaluated.
Results Delivery of p53 protein into p53-null TNBC 4T1 cells via Pos3Aa-p53 crystals restored the p53 activity, and therefore led to the induction of ICD, activation of type I IFN signaling and upregulation of PD-L1 expression. Pos3Aa-p53 crystals significantly enhanced T cell infiltration and activation in 4T1 tumors, thereby sensitizing them to anti-PD-1 therapy. The combination of Pos3Aa-p53 crystals with anti-PD-1 antibody also induced a systemic antitumor immunity resulting in the inhibition of distal tumor growth with minimal toxicity.
Conclusion This study validates that p53 restoration can be an effective approach to overcome ICI resistance and demonstrates that intracellular delivery of p53 protein can be an efficient, safe and potentially universal strategy to restore p53 activity in tumors carrying TP53 mutation.
- Immunotherapy
- Drug Therapy, Combination
- Breast Neoplasms
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The raw data required to reproduce these findings are available from the corresponding authors on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
TP53 is the most frequently mutated gene in human triple negative breast cancer (TNBC), and p53 deficiency is strongly associated with the immune evasion of cancer cells. Thus, restoration of p53 activity may be a viable strategy to remodel the immunosuppressive tumor microenvironment and convert immunogenically ‘cold’ tumor to ‘hot’. Current strategies toward restoring p53 function involve small molecules and gene therapies, which have shown promising outcomes but also have their own drawbacks, such as drug resistance, off-target toxicity and safety concerns. New strategies that can overcome these limitations are therefore needed for the reactivation of p53 pathway in cancer cells.
WHAT THIS STUDY ADDS
We demonstrate that the efficient intracellular delivery of p53 protein mediated by in vivo produced Pos3Aa-p53 fusion protein crystals is able to restore p53 activity in p53-null TNBC 4T1 cells, resulting in the immunogenic cell death of cancer cells, remodeling of tumor microenvironment and enhancement of the response of 4T1 tumors to anti-programmed cell death protein 1 (PD-1) therapy. Combining p53 protein delivery with anti-PD-1 antibody also induces systemic antitumor immunity and memory in mice. The protein-based approach employed in this study is distinctly different from the previously developed small molecule-based or vector-based approaches, thus providing an alternative effective means for activating p53 function to enhance immunosurveillance.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
P53 protein can serve as a potential therapeutic molecule when delivered with an efficient delivery platform, and protein delivery should be considered as a viable strategy for restoring p53 function in p53-inactivated cancer cells. The combination of p53 restoration and immune checkpoint inhibitors should be considered as a therapeutic intervention for patients with TNBC .
Background
Breast cancer is the leading cause of cancer-related death in women.1 Triple negative breast cancer (TNBC) which lacks the expression of estrogen receptor, progesterone receptor and human growth factor receptor 2 accounts for approximately 15%–20% of all breast carcinomas.2 Compared with other subtypes of breast cancer, TNBC typically has poorer prognosis and limited treatment options due to its inherently aggressive clinical behavior and lack of targets for therapeutic agents.2–4 Chemotherapy remains the mainstay of treatment, although positive outcomes are frequently hindered by the intrinsic and/or acquired chemoresistance.3 5
In recent years, immunotherapy has revolutionized cancer therapy, among which immune checkpoint inhibitors (ICIs), such as antibodies targeting cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1) and its ligand PD-L1, have been shown to elicit highly promising antitumor responses against a broad spectrum of cancers.6 By blocking the checkpoint proteins, these antibodies relieve T cells from the immunosuppression by cancer cells, and thereby restore their antitumor activity.7
The efficacy of ICIs is largely dependent on the immunogenic nature of tumors, and thus varies by cancer type and patient subset.8–11 Inflamed tumors (also termed immunogenically ‘hot’ tumors) with high tumor mutational burden (TMB) and T cell infiltration have a higher probability of responsiveness to ICIs.10 12 13 Breast cancer is generally considered immunogenically ‘cold’ and less likely to benefit from immunotherapy.10 An exception is TNBC which appears to be more immunogenic than other subtypes of breast cancer due to its greater genomic instability and high TMB which lead to higher levels of tumor-infiltrating lymphocytes (TILs) and PD-L1 expression.2 10 Anti-PD-1/PD-L1 antibody has demonstrated promising durable responses in a small subset of patients with TNBC, especially when combined with chemotherapy.13 In clinical trials, however, PD-1/PD-L1 checkpoint blockade exhibited significant reductions in efficacy from the first to second and/or subsequent lines of therapies.3 13–15 Researchers have consistently found that advanced TNBC is associated with a depletion of effector immune cells, an indication of tumor resistance to ICI therapy.13 Hence, the development of an effective strategy to conquer immunotherapy resistance in advanced TNBC would be of significant impact for the treatment of this disease.
TP53, which encodes the tumor suppressor p53 protein, is the most frequently mutated gene in invasive breast cancer with an overall mutation rate of 30%–35% and an extremely high mutation rate of around 80% in TNBC.16 There is increasing evidence that the status of p53 in cancer cells has a profound impact on tumor immunogenicity.17 18 P53 has been shown to directly activate immune response genes including interferon regulatory factor 9 (IRF9) and interferon-stimulated gene 15 (ISG15) to enforce antivirus response, which in turn facilitates the production of type I interferon (IFN).17 On the contrary, loss of p53 function promotes the recruitment of immunosuppressive cells and the reduction of cytokine-producing T cells, enabling tumor progression.18 Notably, several recent studies have shown that reactivation of p53 by blocking the p53–MDM2 interaction was effective in promoting antitumor immunity via the induction of a viral mimicry pathway.19 20 Considering that MDM2 inhibitors are limited to tumors expressing wild-type p53, we reasoned that direct delivery of p53 protein into cancer cells would be a more broadly applicable strategy to restore p53 function, and thereby overcome the resistance of TNBC to ICI therapy.
We have previously developed a protein delivery platform based on engineered Cry3Aa protein crystals (Pos3Aa) that are naturally formed within the bacterium Bacillus thuringiensis (Bt).21–23 Recently, we showed that Pos3Aa-p53 fusion protein crystals could be used to deliver p53 into cancer cells with high cell permeation and endosome escape efficiency, resulting in the efficacious restoration of p53 function.22 In this report, we show that delivery of p53 protein mediated by Pos3Aa-p53 crystals into p53-null 4T1 cells—a widely used preclinical model for advanced TNBC24—results in the restoration of p53 function and induction of immunogenic cell death (ICD) in vitro and in vivo. RNA-seq data verifies that the reestablishment of p53 activity by these crystals leads to autocrine activation of type I IFN signaling pathway via a viral mimicry mechanism. Notably, intratumoral delivery of Pos3Aa-p53 crystals is found to remodel the microenvironment of 4T1 tumors, as indicated by the increased expression of PD-L1 and enhanced CD8+ T lymphocyte infiltration, thereby enhancing their sensitivity to anti-PD-1 therapy. Furthermore, using a bilateral tumor model, we demonstrate that in combination with anti-PD-1 antibody, treatment of the local tumor with Pos3Aa-p53 crystals can induce systemic antitumor immunity resulting in a modest, though statistically significant, growth suppression of the distal untreated tumor. Taken together, these promising results provide strong support that intracellular p53 delivery can be an effective strategy to increase the immunogenicity of tumors carrying p53 mutation and hence overcome their intrinsic resistance to ICIs.
Materials and methods
Cell culture
The mouse 4T1 breast cancer cells were a gift from Prof Thomas Y C Leung (The Hong Kong Polytechnic University, HKSAR). 4T1 cells were maintained in RPMI-1640 medium supplemented with 10% Fetal Bovine Serun (FBS), HyClone and 1× Penicillin-Streptomycin (P/S) at 37°C in a humidified incubator containing 5% CO2.
Expression and purification of Pos3Aa-p53 fusion crystals and soluble p53 protein
Pos3Aa-p53 protein crystals and soluble p53 protein were produced as previously described.22 Briefly, the pHT315-pos3Aa-p53 plasmid was transformed into Bt 407-OA cells by electroporation for the expression of the corresponding protein crystal. In vivo formed protein crystals were then isolated from the Bt cells by sucrose gradient centrifugation, and the purity of the purified crystals was examined using phase contrast microscope and analyzed by SDS-PAGE. For soluble p53 protein expression, the pETSUMO-p53 plasmid was transformed into Escherichia coli BL21 (DE3), and the protein expression was induced by the addition of Isopropyl-β-D-1-thiogalactopyranoside (IPTG) at OD600~0.6 to a final concentration of 0.1 mM. Cells were then cultured at 25°C overnight and harvested by centrifugation at 8000 rpm for 7 min. After harvest, cell pellets were resuspended and lysed by sonication, and the His-SUMO-p53 proteins were purified in 20 mM Tris-HCl, 300 mM NaCl, pH 8.0 buffer by nickel affinity chromatography. The His-SUMO tag was removed from the purified His-SUMO-p53 protein by overnight incubation with SUMO protease Ulp1 at 4°C. Purified p53 protein was concentrated and stored at −80°C for subsequent use.
Characterization of Pos3Aa-p53 protein crystals
SEM was used to visualize the morphology of Pos3Aa-p53 crystals. Briefly, 1 µL 0.4 mg/mL Pos3Aa-p53 crystals in 40% ethanol were added to a round glass coverslip and air dried. The dehydrated samples were coated with gold by a Sputter Coater S150B (Edwards) and imaged using a Prisma E environmental scanning electron microscope (Thermo Fisher).22 For the determination of the crystal’s hydrodynamic diameter and zeta-potential, 40 µg/mL purified Pos3Aa-p53 crystals were prepared in ddH2O or 1× Phosphate-Buffered Saline (PBS), respectively, and analyzed at 25°C using a Malvern Nano ZS90 (Malvern Instruments, UK).
Cellular uptake of Pos3Aa-p53 protein crystals
Purified Pos3Aa-p53 crystals were labeled with Alexa Fluor 568 C5 Maleimide (Alexa568-Pos3Aa-p53). 4T1 cells were seeded at 35,000 cells/well in confocal dishes (MatTek) and incubated overnight at 37°C in a 5% CO2 incubator for cell attachment. On the next day, the cells were washed with 1× PBS, and then treated with 200 nM Pos3Aa-p53 crystals for 24, 48 or 72 hours. At the end of each incubation period, cells were washed three times with 20 U/mL heparin in PBS and then stained with 0.2 µg/mL Hoechst 33 342 (Life Technologies) and 50 nM LysoTracker Green DND-26 (Thermo Scientific) at 37°C for 10 min prior to imaging on a Leica SP8 confocal microscope.
Cell viability assay
4T1 cells were seeded into a 96-well flat bottom microtiter plate at 1500 cells/well in 100 µL culture medium (with 10% FBS and 1× P/S). A graded dose (50–800 nM) of p53 protein, Pos3Aa-mCherry crystals or Pos3Aa-p53 crystals was added into each well and incubated for 48 hours. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent was used to determine the cell viability.
Caspase-3/7 activity
4T1 cells were seeded in 12-well plates at 20,000 cells/well and incubated overnight at 37°C. After the removal of non-adherent cells, the adherent 4T1 cells were treated with 800 nM p53 protein, Pos3Aa-mCherry crystals or Pos3Aa-p53 crystals for 48 hours. At the end of the incubation period, cells were washed with PBS, collected by trypsin digestion and stained with 500 nM CellEvent Caspase-3/7 Green Reagent (Thermo Fisher, Cat No: C10427) for flow cytometric analysis as previously described.23
Calreticulin assay
In 12-well plates, 35,000 cells/well of 4T1 cells were plated and incubated for overnight cell attachment. On the next day, the adherent cells were treated with 800 nM p53 protein, Pos3Aa-mCherry crystals or Pos3Aa-p53 crystals. After 24 hours of incubation, the free protein/crystals were washed away using 1× PBS, and then the cells were incubated in fresh culture medium for another 24 hours. Following this final incubation period, the cells were harvested, washed twice with 1× PBS and fixed in 0.25% paraformaldehyde (PFA) in 1× PBS for 5 min at room temperature (RT). After washing twice with 1× cold PBS, cells were incubated with Alexa Fluor 488-conjugated calreticulin (CRT) antibody (1:100 dilution, Santa Cruz, Cat No: sc-166837) on ice for 1 hour, followed by washing with 1× PBS before subject to flow cytometric analysis on a BD FACSVerse flow cytometer.
ATP release assay
4T1 cells were seeded at 7500 cells/well in a 24-well plate for overnight attachment. The next day, the cells were treated with 500 nM p53 protein, Pos3Aa-mCherry crystals or Pos3Aa-p53 crystals in 500 µL culture medium. After 48 hours of incubation, the culture media was collected and centrifuged at 12,000 rpm for 10 min at 4°C. Fifty microliters supernatant of each sample was mixed with 50 µL ATP detection solution (10 µg/mL luciferase, 1 mM luciferin and 30 mM MgCl2 in 50 mM HEPES buffer, pH 7.5) in the well of a black 96-well plate with flat glass bottom (Nunc), and the luminescence was quantitated using an Infinite M1000 pro (TECAN) spectrophotometer.
Immunofluorescence staining
In confocal dishes (MatTek), 35,000 cells/well of 4T1 cells were seeded and incubated overnight at 37°C in a 5% CO2 incubator for cell attachment. After the removal of non-adherent cells, adherent cells in 1 mL fresh RPMI-1640 medium containing 10% FBS and 1× P/S were treated with 800 nM Pos3Aa-p53 crystals. At the end of a 48-hour incubation period, cells with or without Pos3Aa-p53 treatment were washed thrice with 1× PBS and fixed in 4% PFA for 15 min, followed by washing with 1× PBS and then wash buffer (1% Triton X-100 in 1× PBS) for 15 min each. Cells were then blocked using blocking buffer (5% NGS, 0.3% Triton X-100 in 1× PBS) at RT for 2 hours and incubated with high-mobility group box-1 (HMGB1) antibody (1:50 dilution, Santa Cruz, Cat No: sc-56698) overnight at 4°C. The next day, cells were washed twice with 1× ice cold PBS and incubated with Alexa Fluor 488-conjugated secondary antibody (Invitrogen, Cat No: A28175) at RT for 2 hours, then imaged under a Leica SP8 confocal microscope.
Frozen sections of tumor tissues were fixed in pre-chilled methanol for 20 min and blocked using blocking solution (5% NGS/3% BSA/0.1% Triton X-100/PBS) at RT for 1 hour, followed by overnight incubation at 4°C with different primary antibodies (P53, Cleaved Caspase-3, BAX, CRT, HMGB1, indoleamine 2,3-dioxygenase 1 (IDO1), tryptophan 2,3-dioxygenase 2 (TDO2), PD-L1, CD3, CD4, CD8) at optimized concentrations (online supplemental table S1). After the removal of primary antibodies, slides were washed with 1× PBST and incubated overnight at 4°C with the corresponding fluorescent secondary antibodies (online supplemental table S1). Confocal imaging was performed on a Leica SP8 confocal microscope.
Supplemental material
Determination of HMGB1 levels in tumor tissues
Tumors from mice treated with PBS or Pos3Aa-p53 crystals were homogenized in 100 µL/100 mg RIPA buffer. After centrifugation at 12,000 rpm for 10 min at 4°C, the supernatants were collected and diluted for the detection of HMGB1 by ELISA. Briefly, 100 µL/well diluted supernatant or purified HMGB1 protein (for standard curve generation) was coated on a Nunc Maxisorp 96-well plate and blocked with 5% non-fat milk in 1× PBST at RT for 1 hour. The plate was then washed with 1× PBST for three times and incubated with HMGB1 antibody (1:100 dilution, Santa Cruz, Cat No: sc-56698) at 37°C for 2 hours. After washing the plate to remove unbound primary antibodies, HRP-conjugated secondary antibody (1:1000 dilution) was added to the plate and incubated at 37°C for 2 hours. After the removal of secondary antibodies and washing three times with 1× PBST, 0.4 mg/mL o-phenylenediamine (Thermo Fisher, Cat No: 34005) in 1× peroxide substrate buffer (Thermo Fisher, Cat No: 34062) was applied to develop the color. The reaction was terminated using concentrated sulfuric acid, and the absorbance at 492 nm was measured using an Infinite M1000 pro (TECAN).
Quantitative real-time PCR
4T1 cells seeded at 35,000 cells/well in a 6-well plate were treated with 800 nM p53 protein, Pos3Aa-mCherry or Pos3Aa-p53 crystal for 48 hours. The total RNA was extracted using TRIzol Reagent (Invitrogen). The quality and quantity of total RNA were determined by measuring the absorbance at 260 nm/280 nm and 260 nm/230 nm using an Eppendorf BioSpectrometer. Fifteen micrograms RNA was reverse transcribed to cDNA using the iScript cDNA Synthesis Kit (Bio-rad) according to the manufacturer’s instructions. RT-qPCR analysis was performed with the SYBR Green Master Mix (Bio-rad) in a Bio-rad CFX96 Real-Time PCR System. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as internal control. Samples were run in triplicate technical replicates. The RT-qPCR primers are listed in online supplemental table S2.
RNA-seq
4T1 cells were treated with 500 nM Pos3Aa-p53 crystals for 48 hours and then collected for bulk RNA sequencing. RNA extraction and DNBSEQ eukaryotic strand-specific transcriptome sequencing using a 100 bp paired-end mode were performed at the Beijing Genomics Institute (BGI Genomics, Shenzhen, China). The RNA-seq data were processed and analyzed using Galaxy (https://usegalaxy.org/). Briefly, raw reads were quality-controlled using FastQC, trimmed using Trim Galore! and mapped to the mouse mm39 reference genome using HISAT2. Gene counts were quantified using htseq-count, and the differential expression analysis was performed using DESeq2. Genes with a Log2FC value larger than 0.7 (p<0.05) or smaller than −0.7 (p<0.05) were identified as significantly upregulated or downregulated, respectively. Pathway enrichment of differentially expressed genes was performed using the Gene Set Enrichment Analysis (GSEA) website (http://www.gsea-msigdb.org/gsea/index.jsp). Heatmap was plotted using Morpheus software (https://software.broadinstitute.org/morpheus/).
Determination of TNF-α levels
Collected serum samples were assayed using a tumor necrosis factor α (TNF-α) ELISA kit (Invitrogen, Cat No: 88-7324) according to the manufacturer’s protocol. Tumor tissues were homogenized in RIPA buffer (100 µL/100 mg tissue) and centrifuged at 10,000 g for 10 min. The supernatant after centrifugation was collected and diluted for the determination of TNF-α level using an ELISA kit (Invitrogen, Cat No: 88-7324).
Flow cytometric analysis of PD-L1 expression
4T1 cells were seeded into a 12-well plate at 35,000 cells/well and treated with 500 nM p53 protein, Pos3Aa-p53 crystals or Pos3Aa control crystals for 48 hours. At the end of incubation period, cells were harvested and stained with 0.25 µg/sample anti-PD-L1 antibody (eBioscience) for 45 min on ice. After removing the primary antibody and washing with 1× PBS, cells were incubated with 1:400 diluted Dylight549-anti-Rat secondary antibody (Bio-rad) for 30 min on ice, followed by washing with 1× PBS before subject to flow cytometric analysis.
To determine the role of type I IFN in Pos3Aa-p53-induced PD-L1 upregulation, 4T1 cells were treated with either 500 nM Pos3Aa-p53 crystals or 500 nM Pos3Aa-p53 crystals plus IFNAR1 blocking antibody (5 or 10 µg/mL) for 24 hours, and the resultant PD-L1 expression levels were determined by flow cytometry as described above.
In vivo xenograft models
BALB/c mice were housed in a temperature-controlled room on a 10:14 hour light/dark cycle in the animal house maintained by the Laboratory Animal Service Centre, The Chinese University of Hong Kong, Shatin, Hong Kong SAR. The unilateral 4T1 breast cancer mouse model was established as previously described.22 Treatments were initiated 9 days after inoculation (tumor volume reached around 100 mm3). Mice were administered with 20 mg/kg Pos3Aa-p53 crystals (intratumorally), 200 µg/injection/mouse anti-PD-1 antibodies (BioXcell, Clone: RMP1-14, Cat No: BE0146) (intraperitoneally) or a combination of both according to the treatment schedule depicted in figure 5A. Tumor size was measured throughout the experimental period, and the tumor volume was calculated based on the formula: tumor volume = (length × width2)/2. Two days after the last treatment, mice were sacrificed, and the tumor tissues were collected, weighted and dissected into two pieces for preparing frozen sections and analyzing tumor-infiltrating immune cells, respectively. Blood samples and major organs were also collected and processed for subsequent toxicity studies as previously described.25
A bilateral 4T1 tumor mouse model was established by subcutaneously injecting 1×106 4T1 cells into the left flank and 3×105 4T1 cells into the right flank of female BALB/c mice. When the volume of the right tumor reached around 60 mm3, treatments were initiated by injecting 20 mg/kg Pos3Aa-p53 crystals intratumorally into the left tumor (local tumor), followed by intraperitoneal injection of 200 µg/injection/mouse of anti-PD-1 antibodies (BioXcell, Clone: RMP1-14, Cat No: BE0146) the day after (figure 6B). The length and width of the right tumor (distal tumor) was measured every 2 days and the tumor volume was calculated based on the formula: tumor volume = (length × width2)/2. At the end of the experiment, mice were sacrificed, and tumors and spleens were collected for subsequent analyses.
Ex vivo analysis of immune cells in tumor tissues from unilateral tumor-bearing mice
Tumor tissues were fragmented and digested in 1× TrypLE (Gibco, Cat No: 12604021) supplemented with DNase (Roche, Cat No: 10104159001) at 37°C for 30 min. Single cell suspension was prepared by passing the dissociated cell suspension through a 40 µm cell strainer (SPL, Cat No: 93040) placed atop a 50 mL falcon tube. Cells were then stained with different antibodies (CD3, Biolegend, Cat No: 100312; CD4, Biolegend, Cat No: 100406; CD8, Biolegend, Cat No: 100723; Granzyme B, Biolegend, Cat No: 372208) according to the manufacturer’s protocol for subsequent flow cytometric analyses on a BD FACSVerse flow cytometer.
Ex vivo analysis of splenocytes from bilateral tumor-bearing mice
Spleens were collected and minced using the flat end of the plunger from a 10 cc syringe in a 100 mm petri dish containing 5 mL dissociation solution (1 mM EDTA in PBS). Single cells were prepared by passing the cell suspension through a 40 µm cell strainer (SPL, Cat No: 93040). Red blood cells (RBCs) were lysed using the RBC Lysis Buffer (eBioscience, Cat No: 00-4333-57). Splenocytes were then stained with different antibodies (CD45, CD3, CD4, CD8 and CD44) at optimized concentrations as indicated in online supplemental table S1 for flow cytometric analysis on a BD FACSVerse flow cytometer.
Statistical analysis
Data are presented as mean±SE of the mean. Statistical significance and the number of samples are noted in figures or legends where appropriate. Student’s t-test was performed when only two value sets were compared. Ordinary one-way analysis of variance was performed for the comparisons between multiple groups using GraphPad Prism 8 software (V.8.4.3). The tumor growth curves were analyzed using the TumGrowth tool.26 A value of p<0.05 was considered significant (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Results
Pos3Aa-p53 crystals promote efficient cytosolic delivery of p53 protein into 4T1 cells
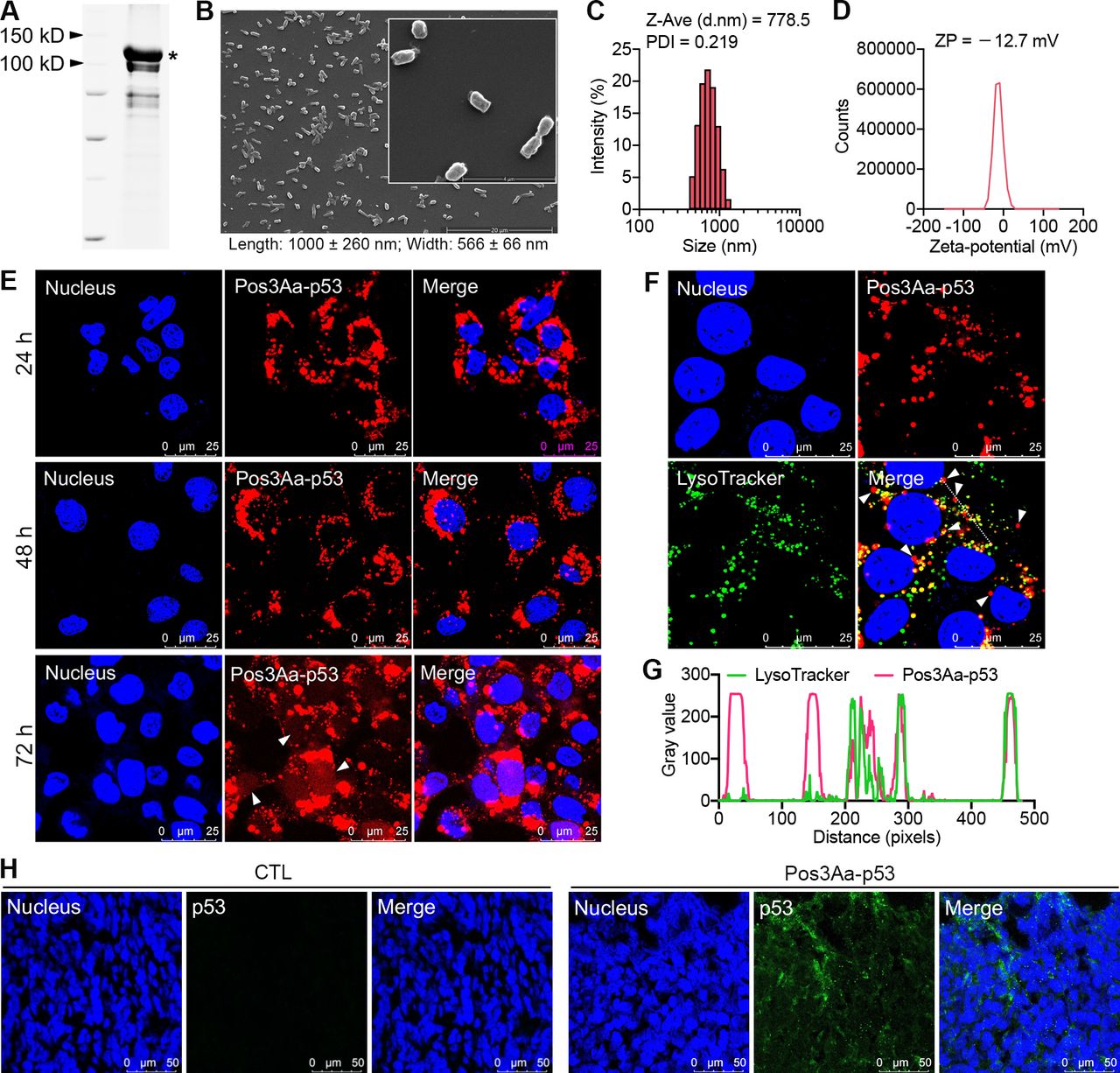
Pos3Aa-p53 fusion protein crystals were isolated from Bt cells and characterized by scanning electron microscopy (SEM) and dynamic light scattering (figure 1A–D). These uniform submicrometer-sized rod-shaped crystals were similar to those reported in our earlier study,22 confirming their reproducible production in Bt cells. Given that inefficient cytoplasmic delivery of biotherapeutics is a major hurdle to their efficacy, the cellular uptake of Pos3Aa-p53 crystals by 4T1 cells and their endosomal escape efficiency were evaluated by confocal fluorescence microscopy. Alexa 568-labeled crystals were readily internalized by 4T1 cells, as indicated by the intense fluorescence signal in nearly all cells (figure 1E). Most notably, significant diffuse fluorescence signal was clearly displayed in the cytosol, and more important, in the nucleus where the p53 protein acts as a transcription factor (figure 1E and online supplemental figure S1), suggesting that Pos3Aa-p53 fusion protein crystals could efficiently escape from endo/lysosomal entrapment. This observation was further supported by the fact that a considerable amount of the Pos3Aa-p53 crystals were not co-localized with the LysoTracker-stained intracellular vesicles (figure 1F,G).

Characterization of Pos3Aa-p53 crystals and their intracellular delivery. (A) SDS-PAGE gel of purified Pos3Aa-p53 crystals solubilized in SDS loading buffer. The asterisk indicates the full length band of Pos3Aa-p53. (B) Representative SEM images (scale bar, 20 µm; inset scale bar, 4 µm), (C) size distribution and (D) zeta potential (ZP) distribution of purified Pos3Aa-p53 crystals. (E) Confocal images showing the efficient uptake of Alexa 568-Pos3Aa-p53 crystals by 4T1 cells after 24, 48 and 72 hours of incubation. White arrows indicate the diffuse signal in cytoplasm and nucleus. (F) Confocal images showing the subcellular localization of internalized Pos3Aa-p53 crystals. White arrows indicate Pos3Aa-p53 crystals in the cytoplasm. (G) Corresponding fluorescence intensity profiles of the dashed white line in (F). (H) Representative confocal images of 4T1 tumor sections stained with anti-p53 antibodies. Tumors injected with Pos3Aa-p53 crystals showed a positive staining of p53 protein. CTL, control.
The in vivo delivery of Pos3Aa-p53 crystals to 4T1 tumors was then evaluated. While tumor tissues from control mice showed no positive signal after staining with anti-p53 antibodies, obvious intracellular fluorescence was observed from the tumors that had received Pos3Aa-p53 injections, indicating the successful in vivo delivery of p53 protein into 4T1 cells (figure 1H).
Pos3Aa-p53 crystals restore p53 activity and induce cancer cell death and apoptosis
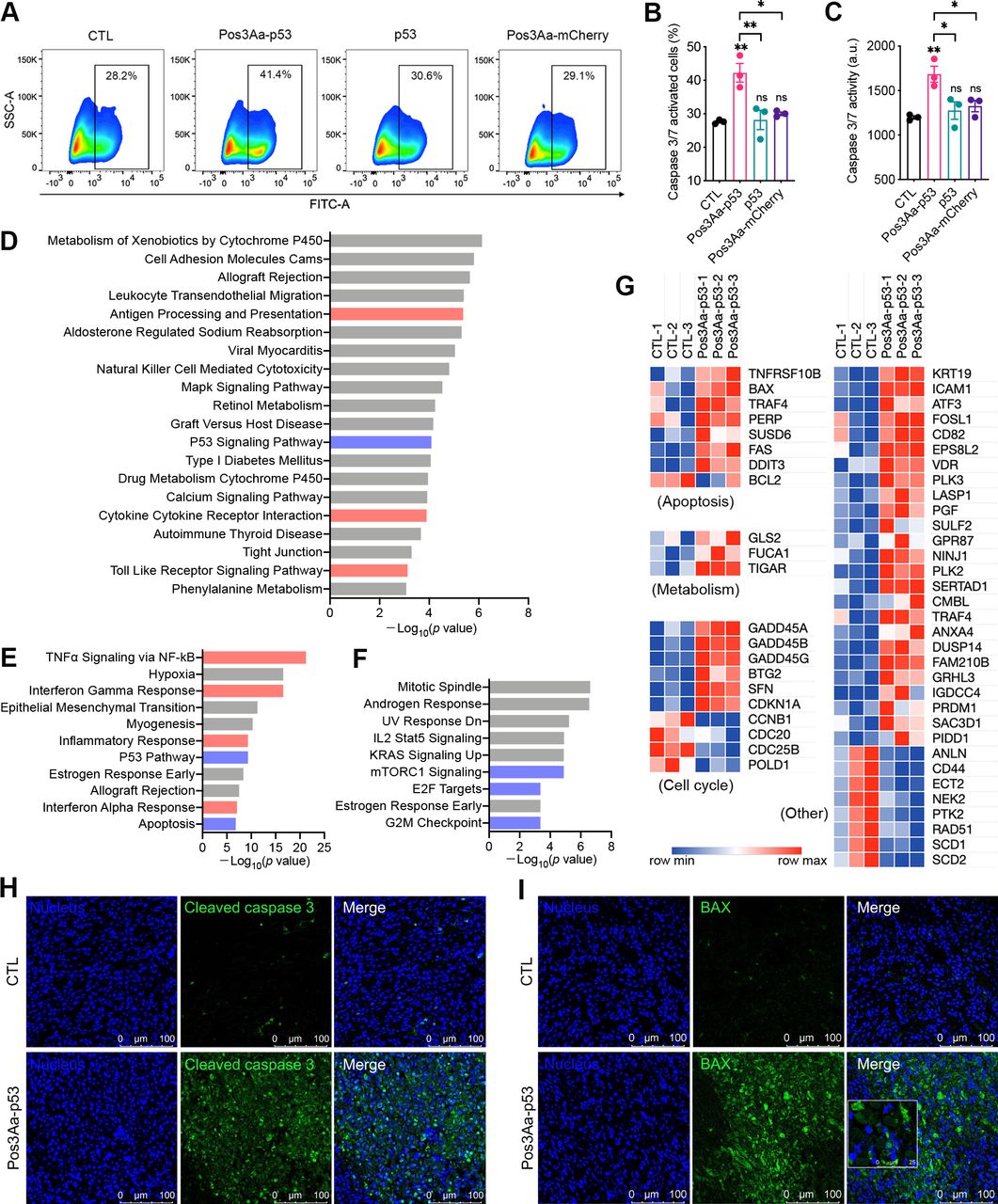
Restoration of p53 function has long been touted as a promising therapy for cancer treatment, given that p53 is found to be inactivated in more than half of all human cancers. As a tumor suppressor, functional wild-type p53 promotes cell cycle arrest, apoptosis and cell death to prevent cancer formation. The ability of the delivered p53 protein by Pos3Aa-p53 crystals to promote cancer cell death was therefore evaluated by incubating 4T1 cells with a graded dose of Pos3Aa-p53 crystals, free p53 protein or Pos3Aa-mCherry crystals for 48 hours. Pos3Aa-p53 crystals were found to significantly inhibit the cell growth in a dose-dependent manner, while neither free p53 protein nor Pos3Aa-mCherry control crystals were effective against the cancer cells (online supplemental figure S2). This restoration of p53 function was further supported by the increased caspase-3/7 activity—a marker of apoptosis—in the 4T1 cells treated with Pos3Aa-p53 crystals, as well as by the enhanced cleaved caspase-3 positive staining of the tumor tissues from the Pos3Aa-p53-treated mice (figure 2A–C and H).
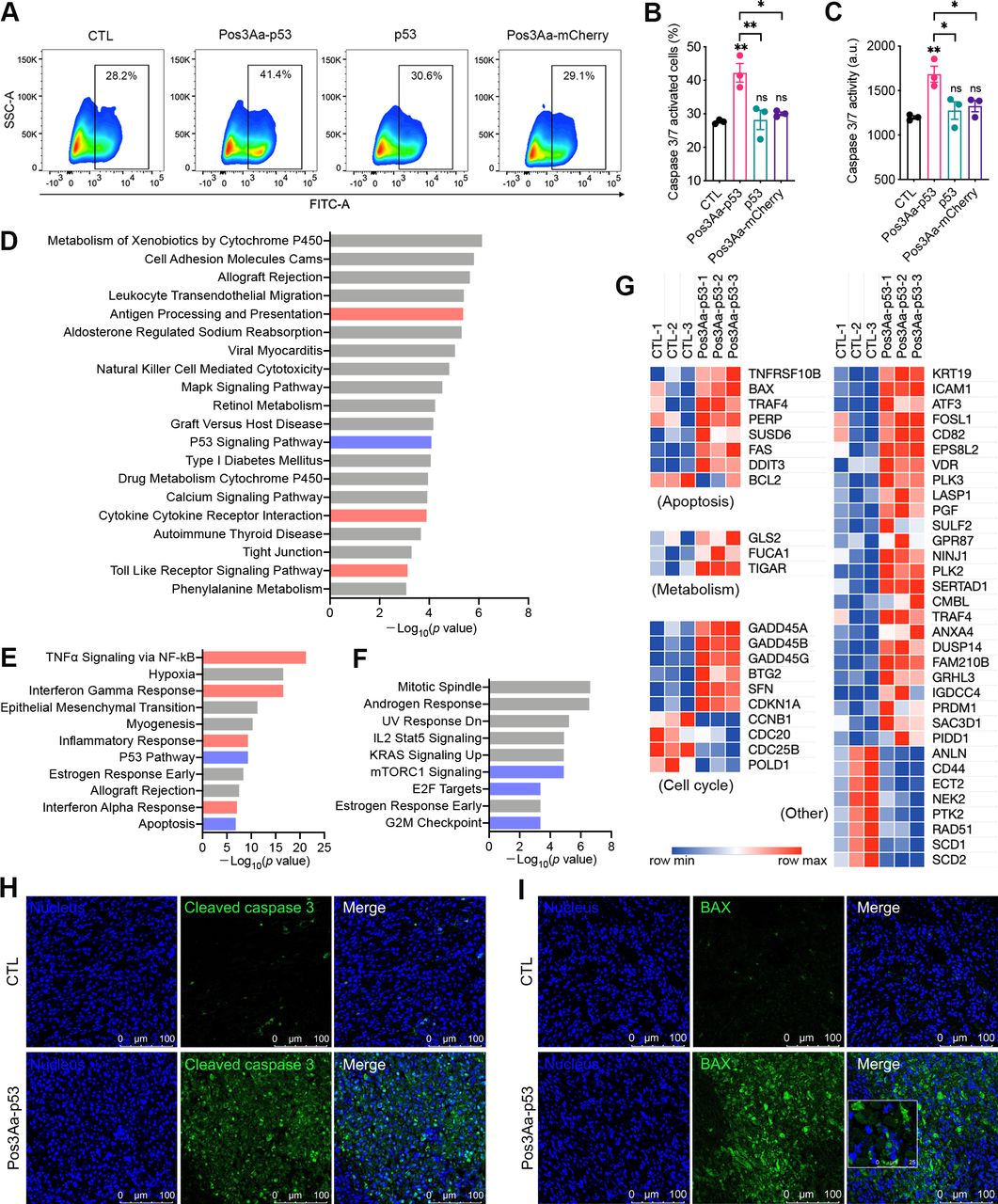
Pos3Aa-p53 crystals restore p53 function in 4T1 cells. (A) Representative flow cytometry plots of 4T1 cells with different treatments. Cells were stained with the Caspase-3/7 Green Reagent which generates a green fluorescence on the activation of caspase-3/7. (B) Corresponding percentage of caspase-3/7-activated cells and (C) relative caspase-3/7 activity. (D) KEGG pathway enrichment analysis of significantly upregulated genes. Hallmark gene set analysis of (E) significantly upregulated genes and (F) significantly downregulated genes. (G) Heat map showing the normalized expression of differentially expressed p53 target genes. Representative confocal images of 4T1 tumor sections stained with (H) anti-cleaved caspase-3 antibodies and (I) anti-BAX antibodies. Tumors injected with Pos3Aa-p53 crystals exhibited higher level of cleaved caspase-3 and BAX. Data are presented as mean±SE of the mean. ns, no significant difference; *p<0.05; **p<0.01. BAX, bcl-2-associated X protein; CTL, control; FITC, fluorescein isothiocyanate; KEGG, kyoto encyclopedia of genes and genomes; KRAS, kirsten rat sarcoma viral oncogene homologue; mTORC, mammalian target of rapamycin complex; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells. SSC-A, side scatter-area; TNF-α, tumor necrosis factor α.
To gain additional insight into the impact of Pos3Aa-p53-mediated restoration of p53 function, bulk RNA sequencing was performed on the treated and untreated 4T1 cells. Differentially expressed genes with a p value <0.05 and a log2FC value >0.7 or <−0.7 were identified as significantly upregulated or downregulated, respectively. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of the significantly upregulated genes identified the p53 signaling pathway among the top 20 most activated pathways (figure 2D). GSEA against ‘hallmark’ gene sets also highlighted the enrichment of upregulated genes in gene sets associated with p53 pathway and apoptosis, and the enrichment of downregulated genes in those associated with mTORC1 signaling, E2F targets and G2M checkpoint—known regulatory targets of p53 for tumor suppression—in the Pos3Aa-p53-treated samples (figure 2E and F).27 28 A list containing 54 known p53 direct targets29 was compiled and their mRNA expression levels were analyzed. Consistent with the results from the enrichment analyses, the mRNAs of these well-characterized p53 targets including BAX, TIGAR, GADD45, CDKN1A (p21) and CCNB1 were significantly altered on the treatment of Pos3Aa-p53 crystals (figure 2G), indicative of p53 activation. Moreover, the increased expression of one of the aforementioned p53 targets, BAX, was validated in 4T1 tumors injected with Pos3Aa-p53 crystals (figure 2I). Taken together, these results confirmed the successful restoration of p53 function by Pos3Aa-p53 crystals in cancer cells.
Pos3Aa-p53 crystals induce ICD in vitro and in vivo
An intriguing finding from the RNA sequencing analysis was the highly enriched pathways related to antigen processing and presentation, TNF-α signaling and IFN response identified for the significantly upregulated genes in the Pos3Aa-p53-treated cells (figure 2D–E), suggestive of the potential involvement of Pos3Aa-p53 crystals in the stimulation of antitumor immunity. And since Pos3Aa-p53 crystals have shown to be effective in inducing apoptosis and cell death of 4T1 cells (online supplemental figure S2), we speculate whether this cell death is immunogenic in nature.
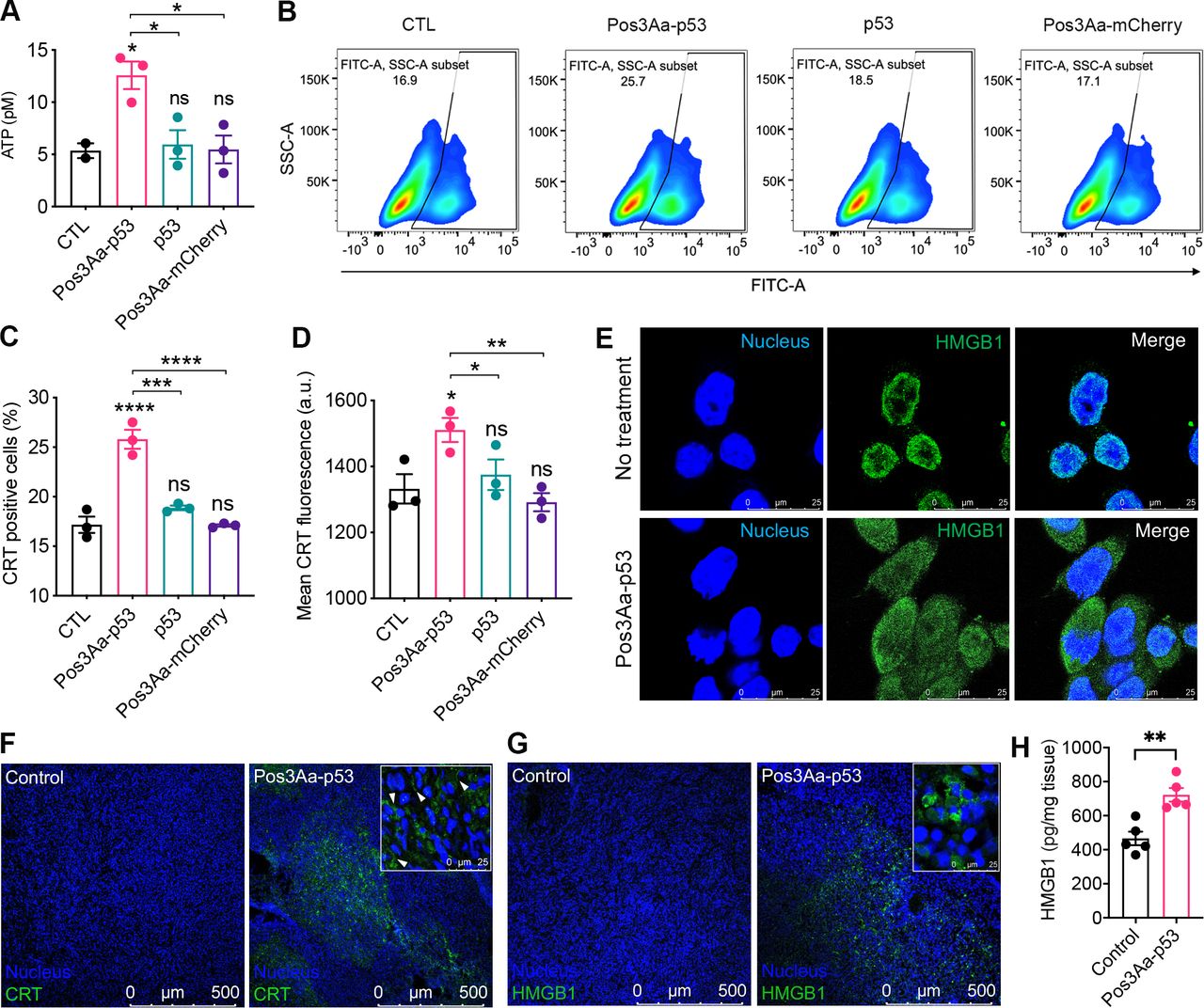
Cancer cells undergoing ICD surface-expose or release damage-associated molecular patterns (DAMPs), such as ATP, CRT and HMGB1, which can stimulate an antitumor immune response featuring tumor infiltration by lymphocytes.30 31 To ascertain whether the cell death caused by Pos3Aa-p53 crystals was immunogenic, 4T1 cells were incubated with Pos3Aa-p53 crystals and the resultant levels of different DAMPs were evaluated. 4T1 cells treated with Pos3Aa-p53 crystals were found to release more ATP extracellularly than cells in other groups (figure 3A). Likewise, flow cytometric analysis showed that Pos3Aa-p53 crystals elicited increased CRT exposure, while p53 protein or Pos3Aa-mCherry control crystals showed no effect (figure 3B–D). Nuclear HMGB1 release, another hallmark of ICD, was also observed on the immunofluorescence (IF) staining of Pos3Aa-p53 crystals-treated 4T1 cells (figure 3E).

Pos3Aa-p53 crystals induce immunogenic cell death of 4T1 cells. (A) Levels of ATP released into the culture medium of 4T1 cells on the treatment of Pos3Aa-p53 crystals. (B) Representative flow cytometry plots of 4T1 cells stained with Alexa 488-CRT antibody, (C) corresponding percentage of CRT-positive cells and (D) their mean CRT fluorescence intensities showing increased CRT exposure in cells treated with Pos3Aa-p53 crystals. (E) IF staining of cultured 4T1 cells showing the release of nuclear HMGB1 after Pos3Aa-p53 treatment. (F,G) IF staining of frozen sections of tumors treated with PBS (control) or Pos3Aa-p53 crystals. Significant (F) CRT and (G) HMGB1 signals were observed after intratumoral administration of Pos3Aa-p53 crystals. White arrows indicate surface-exposed CRT. (H) HMGB1 protein levels in the microenvironment of 4T1 tumors determined by ELISA. Data are presented as mean±SE of the mean. ns, no significant difference; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. ATP, adenosine triphosphate; CTL, control; CRT, calreticulin; FITC-A: fluorescein isothiocyanate-area; HMGB1, high-mobility group box-1; IF, immunofluorescence; PBS, phosphate-buffered saline; SSC-A, side scatter-area.
We then examined the in vivo ability of Pos3Aa-p53 crystals in inducing ICD in 4T1 tumors. Consistent with our in vitro observations, tumor tissues excised from mice treated intratumorally with Pos3Aa-p53 crystals exhibited significantly increased CRT exposure on the cell membrane and HMGB1 release from nucleus (figure 3F,G). ELISA quantification of HMGB1 levels in the microenvironment of 4T1 tumors further confirmed that Pos3Aa-p53 crystals treatment led to the release of higher levels of HMGB1 compared with the PBS control (figure 3H). These findings indicated that Pos3Aa-p53 crystals could restore p53 function in 4T1 cells and thus lead to the induction of ICD in vitro and in vivo.
Pos3Aa-p53 crystals induce a type I IFN response
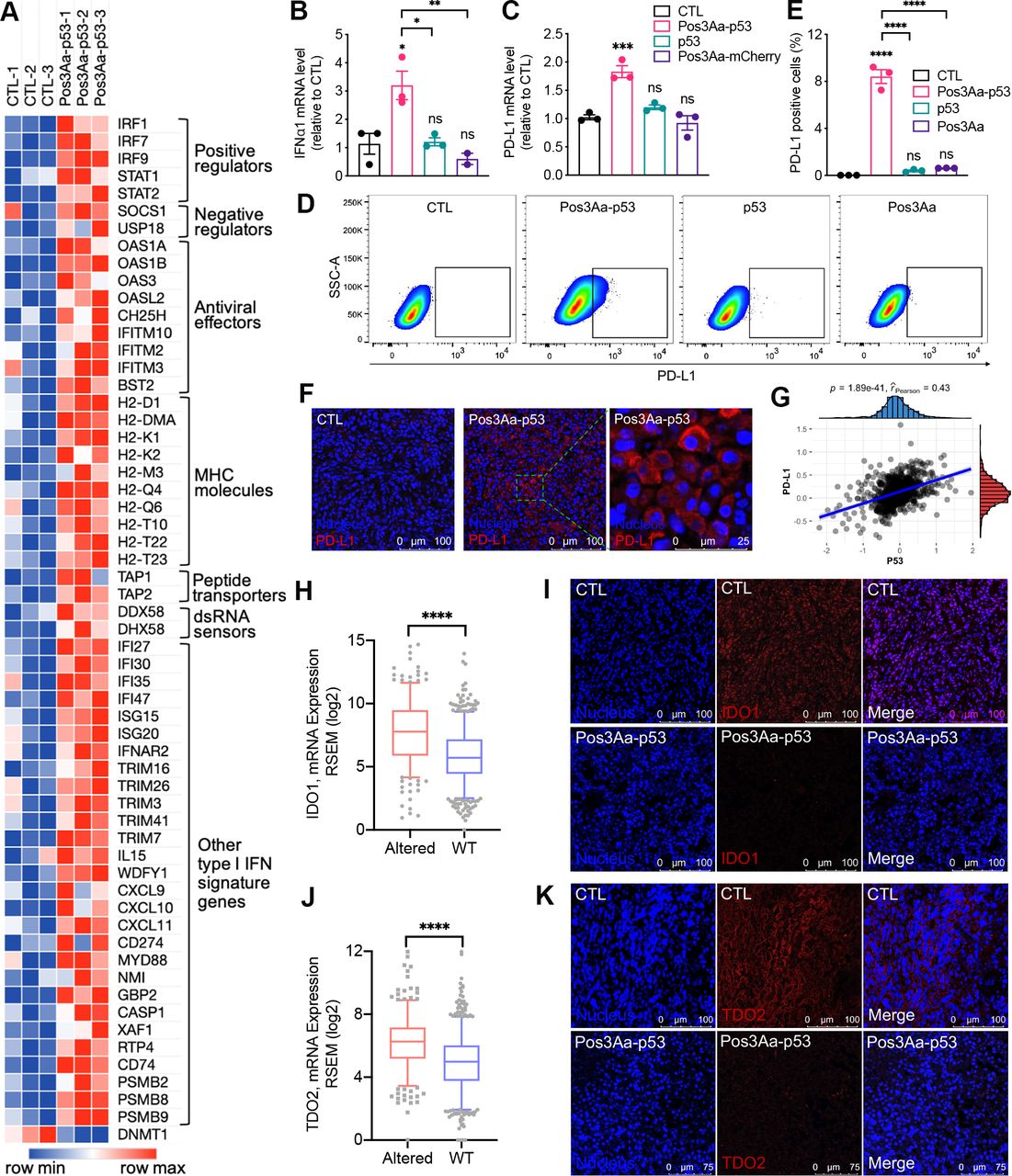
ICD is generally accompanied by a type I IFN response.32 33 The findings that Pos3Aa-p53 crystals could induce ICD of 4T1 cells together with our RNA-seq analysis showing the enrichment of upregulated genes in IFN-related pathways in Pos3Aa-p53 crystals-treated cells (figure 2E) suggested that Pos3Aa-p53 crystals might have a role in type I IFN induction. Therefore, a deeper analysis of the RNA-seq data was performed to better characterize the effect of Pos3Aa-p53 crystals on the expression levels of the type I IFN signature genes. This analysis revealed that cells treated with Pos3Aa-p53 crystals exhibited increased mRNA levels of interferon alpha and beta receptor subunit 2 (IFNAR2)—a key component that binds extracellular IFNs and triggers intracellular signaling cascades, and typical IFN-stimulated genes (ISGs) including positive regulators (eg, IRFs, STAT1 and STAT2), negative regulators (eg, SOCS1 and USP18) and antiviral effectors (eg, OAS and BST2) (figure 4A).34 We subsequently confirmed by q-PCR that the expression of IFNα1 and IFN-inducible cytokine IL15 was indeed upregulated in 4T1 cells treated with Pos3Aa-p53 crystals (figure 4B and online supplemental figure S3). Concurrent with the upregulation of the IFN-signaling pathways, enhanced mRNA expression of classical antigen processing and presenting molecules such as the major histocompatibility complex (MHC) proteins H2-D1, H2-K1, H2-Q4 and H2-T23, and peptide transporter associated with antigen processing 1 (TAP1) and TAP2 whose upregulation is known to improve the presentation of tumor antigens in enhancing antitumor immunity were also observed (figure 4A and online supplemental figure S4).35 Furthermore, mRNAs of other IFN-inducible genes such as CXCL9 and CXCL10—two important chemokines that favor tumor infiltration by T cells36 37—were also upregulated in Pos3Aa-p53 crystal-treated cells (figure 4A). Taken together, these data indicated that delivery of p53 protein into p53-null cancer cells by Pos3Aa-p53 crystals induced a type I IFN response.
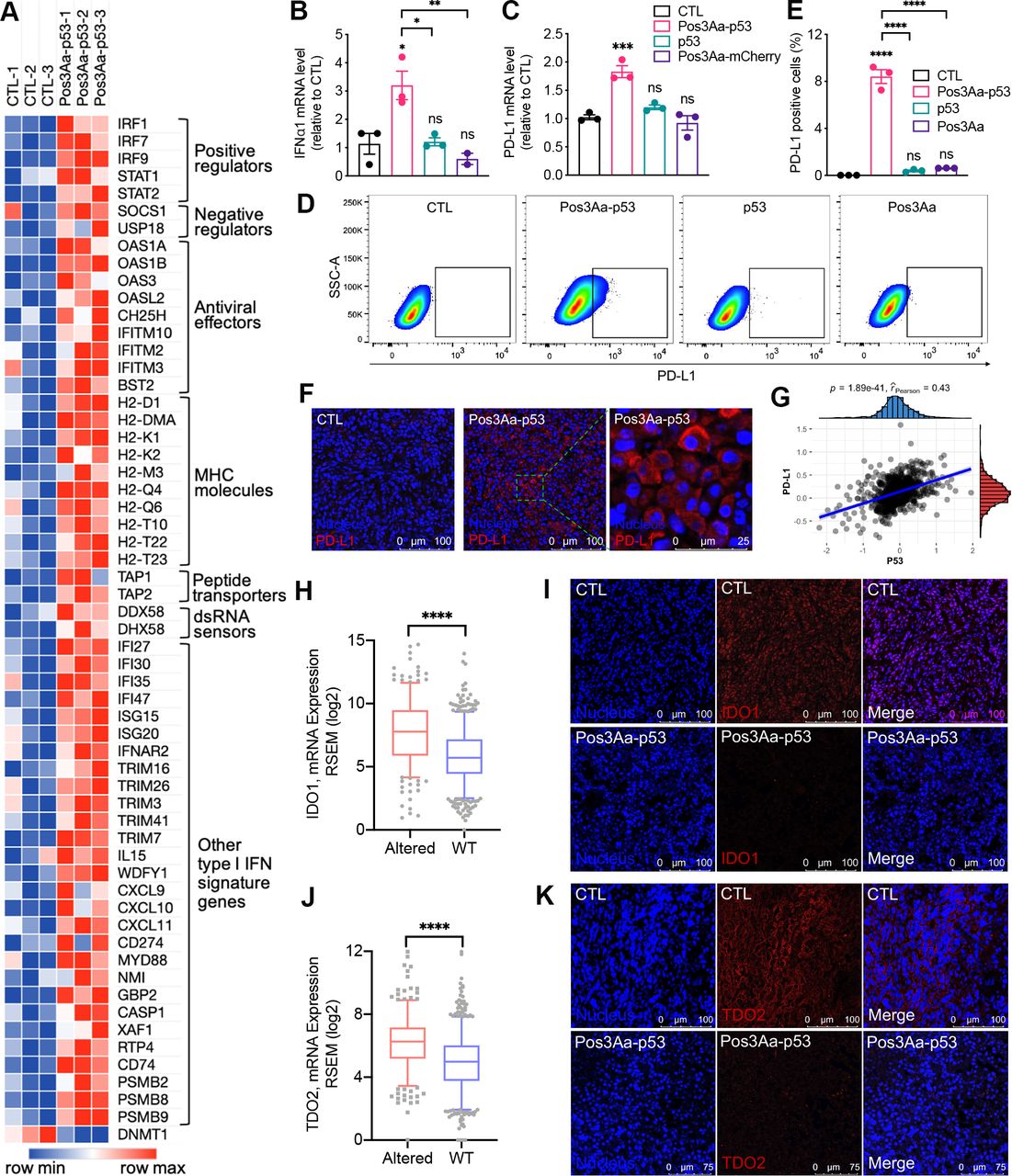
Pos3Aa-p53 crystals increase the immunogenicity of 4T1 tumors. (A) Heatmap showing the normalized expression level of type I IFN signature genes. The mRNA levels of (B) IFNα1 and (C) PD-L1 determined by q-PCR in 4T1 cells with different treatments. (D) Representative flow cytometry plots of 4T1 cells stained with PD-L1 antibody and (E) percentage of PD-L1-positive cells showing increased PD-L1 expression in cells treated with Pos3Aa-p53 crystals. (F) IF staining of frozen sections of tumors treated with PBS (CTL) or Pos3A-p53 crystals showing Pos3Aa-p53 crystals elevated PD-L1 expression in vivo. (G) Protein levels of p53 and PD-L1 are positively correlated in human BRCA. Analysis of TCGA RNA-seq datasets indicated that the mRNA expression of (H) IDO1 and (J) TDO2 were downregulated in human BRCA carrying wild-type (WT) p53. Correspondingly, IF staining indicated that Pos3Aa-p53 crystals treatment led to diminished expression of (I) IDO1 and (K) TDO2 in 4T1 tumors. Data are presented as mean±SE of the mean. ns, no significant difference; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. CTL, control; IDO1, indoleamine 2,3-dioxygenase 1; IF, immunofluorescence; MHC, major histocompatibility complex; PBS, phosphate-buffered saline; PD-L1, programmed cell death ligand 1; SSC-A, side scatter-area; TCGA, The Cancer Genome Atlas; TDO1, tryptophan 2,3-dioxygenase 2.
Pos3Aa-p53 crystals elevate PD-L1 expression
Increasing evidence from preclinical and clinical studies has shown that elevated PD-L1 expression is correlated with improved efficacy of anti-PD-1 therapy for many cancers including metastatic TNBC.14 38–44 Consistent with the fact that IFNs can induce the expression of PD-L1 (CD274),45–47 RNA-seq analysis showed the upregulation of CD274 in the Pos3Aa-p53 crystal-treated 4T1 cells (figure 4A). We subsequently confirmed this upregulated expression of PD-L1 at both mRNA and protein levels using q-PCR and flow cytometry (figure 4C–E). While free p53 protein and control Pos3Aa crystals had no effect, Pos3Aa-p53 crystals significantly increased the mRNA level and cell surface PD-L1 staining of 4T1 cells (figure 4C–E). Moreover, the in vivo augmented expression of PD-L1 on the treatment of Pos3Aa-p53 crystals was evidenced by the IF staining of 4T1 tumor sections (figure 4F).
The relevance of our findings to human cancer was substantiated by analyzing the reverse phase protein array data of breast invasive cancers (BRCA) from The Cancer Proteome Atlas (TCPA),48 which showed a significant positive correlation between PD-L1 and p53 protein expression in patients with breast cancer (figure 4G). Furthermore, analysis on the RNA-seq data of BRCA from The Cancer Genome Atlas (TCGA) database indicated that the mRNA level of PD-L1 is highly correlated with those of STAT1, STAT2 and IRF9—components of interferon-stimulated gene factor 3 (ISGF3) in the canonical type I interferon signaling pathway, as well as their downstream transcription factor IRF1 (online supplemental figure S5). Based on this analysis, it appears that Pos3Aa-p53 crystals might indirectly upregulate PD-L1 expression via the IFN signaling pathway, given that PD-L1 is not a direct transcriptional target of p53. To validate this hypothesis, 4T1 cells were treated with Pos3Aa-p53 crystals in the presence of interferon-α/β receptor (IFNAR) blocking antibodies. Flow cytometric analysis showed that anti-IFNAR1 antibodies attenuated the PD-L1 upregulation mediated by Pos3Aa-p53 crystals in a dose-dependent manner (online supplemental figure S6), thus confirming that Pos3Aa-p53 crystal-stimulated IFNα secretion promoted the PD-L1 expression on 4T1 cells via an autocrine and/or paracrine manner.
Pos3Aa-p53 crystals suppress IDO1 and TDO2 overexpression
The overexpression of IDO1 and TDO2 in cancer cells has been found to promote the formation of Tregs and myeloid-derived suppressor cells (MDSCs) while concurrently suppress the proliferation and function of effector T cells, thereby resulting in tumor evasion from immunosurveillance.49 50 Combinations of anti-PD-1/PD-L1 antibodies with IDO1 inhibitors have been shown to elicit an improved efficacy of anti-PD-1/PD-L1 therapy.49 51 Interestingly, studies on lung cancers have demonstrated that p53 could attenuate IDO1 signaling,52 suggesting that restoration of p53 function could be a potential strategy to downregulate IDO1 and overcome immunotherapy resistance. We therefore first examined if there was any correlation between p53 status and IDO1/TDO2 expression in human breast cancer. We found that BRCA tissues harboring wild-type p53 exhibited a significantly lower mRNA level of IDO1 and TDO2 than tumors with p53 mutations (figure 4H,J). This observation led to the hypothesis that Pos3Aa-p53 crystals might be able to decrease IDO1 and TDO2 expression in p53-null 4T1 tumors. As expected, IF staining of tumor sections indicated that the expression of IDO1 and TDO2 was suppressed in tumors received Pos3Aa-p53 treatment (figure 4I,K). Collectively, our results revealed that intracellular delivery of p53 protein by Pos3Aa-p53 crystals could increase the immunogenicity of 4T1 tumors and remodel the immunosuppressive tumor microenvironment via multiple mechanisms.
Pos3Aa-p53 crystals enhance the efficacy of anti-PD-1 therapy
Numerous preclinical and clinical studies have revealed that tumors with higher CD8+ T cells frequency would have better response to anti-PD-1/PD-L1 therapy.12 41 53 As a typical model for late-stage human breast cancer, 4T1 tumors are deficient in CD8+ tumor-infiltrating T cells and therefore resistant to PD-1/PD-L1 blockade.54 PD-L1 expression has been shown to be geographically associated with infiltrating immune cells in multiple types of cancer.55 Given the demonstrated ability of Pos3Aa-p53 crystals to restore p53 function in the p53-null 4T1 cells (figure 2) and the resultant enhancement in tumor immunogenicity and PD-L1 expression (figure 4), we hypothesized that tumor delivery of these crystals might be able to enhance T cell infiltration and thus overcome the resistance of 4T1 tumors to ICIs. To test this conjecture, mice bearing subcutaneously established 4T1 tumors were treated with anti-PD-1 antibody alone or in combination with Pos3Aa-p53 crystals (figure 5A). While the anti-PD-1 monotherapy had no effect on tumor growth, its combination with Pos3Aa-p53 crystals led to a significant growth inhibition as evidenced by the reduction of tumor size and weight, indicating the efficacy of Pos3Aa-p53 crystals in sensitizing 4T1 tumors to anti-PD-1 antibody (figure 5B,C and online supplemental figure S7). Moreover, the levels of TNF-α in the tumor tissues and serum of mice injected with Pos3Aa-p53 crystals, both individually and in combination, were significantly elevated (figure 5D), indicating a strong immune response stimulated by Pos3Aa-p53 crystals.
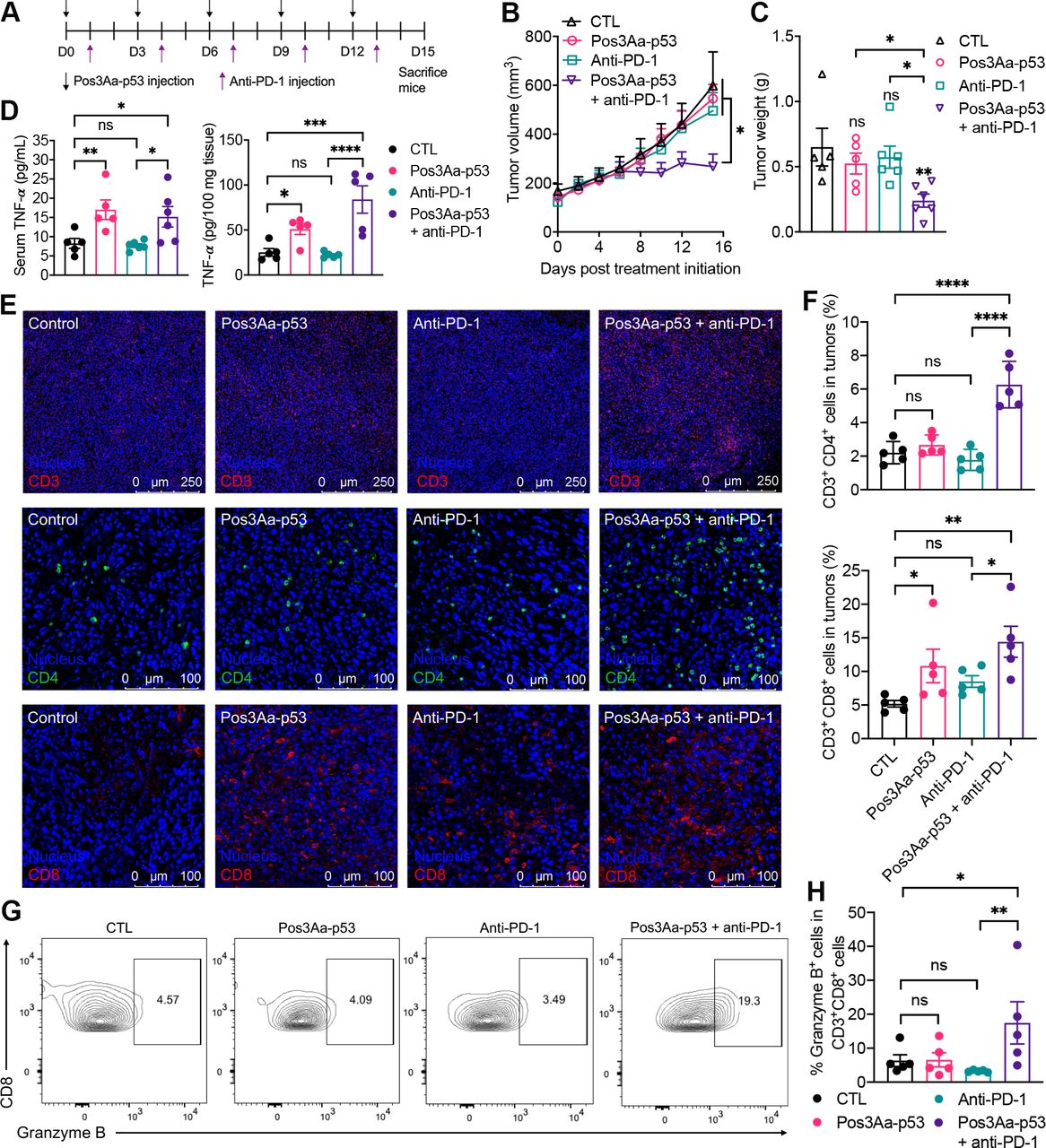
Pos3Aa-p53 crystals sensitize 4T1 tumors to anti-PD-1 therapy. (A) Treatment schedule. (B) Tumor growth curve of 4T1 tumors treated with PBS (n=5), 20 mg/kg Pos3Aa-p53 crystals (n=5), 200 µg/injection/mouse anti-PD-1 (n=6) and Pos3Aa-p53 crystals plus anti-PD-1 (n=6). (C) Corresponding 4T1 tumor weight on day 15. (D) TNF-α levels in serum and tumor tissues determined by ELISA. (E) Representative confocal images of IF staining of CD3+, CD4+ and CD8+ cells in 4T1 tumors. (F) Flow cytometric quantification of tumor-infiltrating helper T cells (CD3+CD4+) and cytotoxic T cells (CD3+CD8+). (G) Representative flow cytometry plots of granzyme B-positive cytotoxic T cells (CD3+CD8+ GrB+). Numbers in the plots indicate the percentage of gated cells. (H) Corresponding percentage of GrB+ cells in CD8+ T cells. Data are presented as mean±SE of the mean. ns: no significant difference; *p<0.05; **p<0.01. ****p<0.0001. CTL, control; PBS, phosphate-buffered saline; PD-1, programmed cell death protein 1; TNF-α, tumor necrosis factor α.
To validate that Pos3Aa-p53 crystals can enhance the tumor infiltration of T cells, tumor tissues were collected for IF staining and flow cytometric analysis. Tumors treated with Pos3Aa-p53 crystals alone showed higher numbers of positively stained CD3+and CD8+ cells but not CD4+ cells while its combination with anti-PD-1 significantly boosted the numbers of all three (figure 5E). Flow cytometric quantification further confirmed that intratumoral administration of Pos3Aa-p53 crystals led to markedly increased tumor-infiltrating CD8+ cytotoxic T cells but did not affect the numbers of CD4+ helper T cells (figure 5F). Meanwhile, tumors from the combination group showed significantly higher levels of both CD4+ helper and CD8+ cytotoxic T cells, whereas tumors from mice treated with anti-PD-1 antibody alone only exhibited a slight but non-statistically significant increase in the number of CD8+ T cells (figure 5F).
While Pos3Aa-p53 crystals were able to promote CD8+ T cell infiltration, treatment with these crystals alone was insufficient for tumor suppression (figure 5B–F). This phenomenon is consistent with the results from previous studies which showed no difference in tumor growth between tumors carrying wild-type TP53 gene and those with p53 loss.56 57 Based on our findings, one possible reason could be that Pos3Aa-p53 crystals failed to facilitate the infiltration of CD4+ helper T cells (figure 5E,F), given the important role of CD4+ T cells in driving and sustaining antitumor immune responses.58 59 Another potential explanation could be that the cytotoxicity of the CD8+ T cells were suppressed by the upregulated PD-L1 protein in the absence of anti-PD-1 antibody.60 This latter hypothesis is partially supported by the fact that only tumor tissues from the combination group showed an increased proportion of granzyme B-positive CD8+ T cells (CD3+CD8+GrB+) (figure 5G,H). Similar results were also found for the splenic tissues in which only the combination group exhibited elevated levels of CD8+GrB+ T cells (online supplemental figure S8A). Of note is the fact that the percentage of CD8+GrB+ T cells was negatively correlated to the tumor size, implying that the induced T cell responses might be tumor-specific (online supplemental figure S8).
We then performed toxicity studies to assess the safety of the Pos3Aa-p53/anti-PD-1 combination therapy. No obvious weight change was observed in the treatment groups compared with the PBS control group during the treatment period (online supplemental figure S9A). Major organs including liver, spleen, kidney, heart and lung were collected and sliced for histopathological examination (online supplemental figure S9B). All tumor-bearing mice developed splenomegaly in comparison with normal mice. This was due to the extramedullary hematopoiesis (EH) during the progression of 4T1 tumors, as evidenced by the prominent presence of megakaryocytes in spleen (online supplemental figure S9B). Perivascular accumulations of granulocytes were concurrently observed in liver tissues (online supplemental figure S9B). Hematoxylin and eosin (H&E)-stained sections of collected organs indicated that no apparent tissue damage was caused by the combination treatment (online supplemental figure S9B). We also examined the levels of serum aspartate aminotransferase (AST), alanine aminotransferase (ALT) and creatinine, which are closely related to liver and kidney function. None of the treatment groups exhibited any significant difference in AST, ALT and creatinine levels when compared with the control group (online supplemental figure S9C,D). All these results suggested that Pos3Aa-p53 crystals had minimal toxicity on mice even when combined with anti-PD-1 antibody.
Pos3Aa-p53 crystals combined with anti-PD-1 antibody induce a systemic antitumor immunity
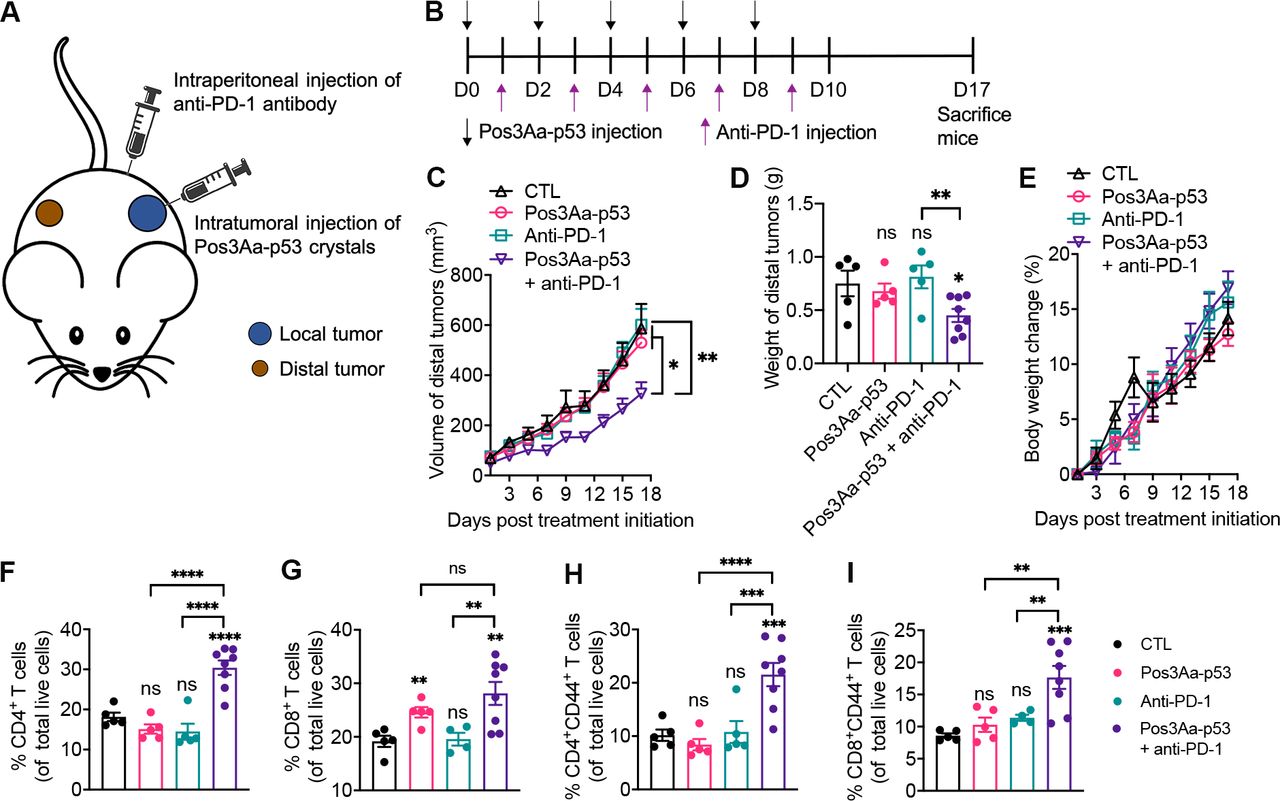
A durable cancer remission will require not only elimination of treated tumors but also clearance of distant metastases, the latter of which necessitates effective systemic antitumor immunity. Furthermore, peripheral immunity is essential to the efficacy of anti-PD-1/PD-L1 therapy since intratumoral T cells have been found to acquire terminally exhausted status over time, and thus need to be replenished with fresh, non-exhausted cells outside the tumor to maintain antitumor activity.61 62 Given the significantly increased TNF-α level in the serum of mice treated with Pos3Aa-p53 crystals (figure 5D), we hypothesized that localized administration of these crystals might be able to induce a systemic immunity. To test this conjecture, mice bearing bilateral 4T1 tumors were injected with either Pos3Aa-p53 crystals or PBS (control) at the primary (local) tumors followed by intraperitoneal administration of anti-PD-1 antibody or PBS (control) the next day (figure 6A) per the treatment schedule depicted in figure 6B. In mice that received the combination therapy, localized injection of Pos3Aa-p53 crystals to the primary tumors sensitized the untreated distal tumors to anti-PD-1 therapy (figure 6C,D and online supplemental figure S10), indicating the successful induction of systemic antitumor immunity. No difference in body weight change was observed in any of the treatment groups compared with the control group (figure 6E), again confirming the minimal toxicity of the combination therapy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pos3Aa-p53 crystals combined with anti-PD-1 antibody induce a systemic antitumor immunity. (A) Schematic illustration of the establishment of bilateral 4T1 tumor model where only the local tumor received intratumoral injection of Pos3Aa-p53 crystals. (B) Treatment schedule. (C) Tumor growth curve of distal 4T1 tumors from mice treated with PBS (n=5), 20 mg/kg Pos3Aa-p53 crystals (n=5), 200 µg/injection/mouse anti-PD-1 (n=5) and Pos3Aa-p53 crystals plus anti-PD-1 (n=8). (D) Corresponding tumor weight on day 17. (E) Body weight change of mice during the experiment. Flow cytometric quantification of (F) helper T cells (CD3+CD4+) and (G) cytotoxic T cells (CD3+CD8+) in the spleen. Flow cytometric quantification of (H) CD4+CD44+ and (I) CD8+CD44+ memory T cells in the spleen. Data are presented as mean±SE of the mean. ns, no significant difference; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. CTL, control; PD-1, programmed cell death protein 1.
To further understand the effect of Pos3Aa-p53 crystals on stimulating a systemic immune response, we analyzed the T cell populations in the mouse spleens using flow cytometry (figure 6F–I). Consistent with the results of tumor-infiltrating T cells from the unilateral tumor model (figure 5F), Pos3Aa-p53 crystals markedly increased the amount of CD8+ T cells but not CD4+ T cells in spleens, whereas its combination with anti-PD-1 antibody was able to significantly enhance both T cells (figure 6F,G). To determine whether this combination strategy would generate a long-lasting immunological memory, we examined the frequencies of CD4+CD44+ and CD8+CD44+ memory T cells in the spleens. While mice treated with only Pos3Aa-p53 crystals or anti-PD-1 antibody exhibited no difference in memory T cells compared with the control mice, combination therapy significantly increased the population of these memory T cells (figure 6H,I). Altogether, these results demonstrated that Pos3Aa-p53 crystals could stimulate a systemic immune response and induce an immunological antitumor memory when combined with anti-PD-1 therapy.
Discussion
Despite the positive clinical outcomes reported for responders of ICI therapies, many patients and multiple cancer types do not or subsequently cease to respond to ICIs. Although patient-specific factors, such as age, HLA genotype, tumor-associated stroma or even gut microbiota, may affect patients’ responses to ICIs, tumor-intrinsic factors referring to the genetic, transcriptional or functional profile of the cancer cells are considered to be the critical determinants of response and resistance.8 For instance, cancer cells can impair neoantigen presentation by downregulating the expression of surface MHC, leading to diminished formation of tumor-reactive T cells and consequently their escape from T cell-mediated cytotoxicity.8 As one of the most frequently mutated genes in human cancer, TP53 has a profound impact on antitumor immunity. Activation of p53 has been found to enhance the expression of TAP1, an essential protein for generating MHC-I-bound peptides, resulting in increased levels of MHC-I-peptide complex on the surface of cancer cells.18 On the contrary, loss or mutation of p53 in cancer cells modulates their cytokine production, leading to an immunosuppressive tumor microenvironment promoting cancer progression.18
Given the crucial role of p53 in immune regulation, therapeutic strategies that can directly and effectively restore p53 function are highly desirable but challenging to achieve. Most reported studies have been focused on the development of adenovirus/liposome-based TP53 gene therapies and small molecules that inhibit the MDM2-p53 interaction or promote the refolding of mutant p53.19 20 22 56 63 Intracellular delivery of wild-type p53 protein could be a more direct and safe approach since it can completely avoid the potential insertional mutagenesis associated with gene therapy. P53 protein delivery, however, has been rarely reported so far, due in large part to the protein’s intrinsic instability and poor cell permeability.22 The Pos3Aa crystal-based delivery platform we have developed allows for the simple in vivo production of uniform p53-encapsulated microcrystals with exceptional stability and cellular uptake efficiency.22 As demonstrated in this proof-of-concept study, the Pos3Aa platform can effectively deliver bioactive p53 protein into p53-null TNBC tumor and successfully reestablish wild-type p53 function in tumor cells, and thereby conquer tumor resistance to ICIs. The next step would be to further optimize the Pos3Aa-p53 system to improve the release of p53 protein and thus achieve comparable therapeutic effect at lower doses of Pos3Aa-p53 crystals. Recently, we have advanced our crystal-based delivery platform by designing a mutant of the Pos3Aa protein, Pos3AaTM, that retains its in vivo crystallizability and whose fusion protein crystals exhibited enhanced cargo release.21 The work to test this rationally engineered Pos3AaTM platform on delivering p53 is already underway.
One limitation of the current study is that the effect of Pos3Aa-p53 crystals and anti-PD-1 combination on the distal untreated tumor was modest. In light of the fact that most patients with metastatic TNBC have multiple sites of disease,64 some of which might be challenging for direct intratumoral injection, a more potent systemic antitumor immunity induced by this combination therapy would be of significant importance for its potential clinical applications. We have previously demonstrated that Pos3Aa-p53 crystals can improve the efficacy of chemotherapy on TNBC.22 Given that some chemotherapeutics can also serve as ICD inducers, a triple-drug combination of Pos3Aa-p53 crystals, chemo drugs and anti-PD-1 antibodies can probably boost a stronger systemic immune response against metastatic tumors.
Another limitation to the clinical application of Pos3Aa-p53 crystals is the relatively frequent dosing (figures 5A and 6B), which needs to be optimized in future investigations. In our previous studies, we have shown that as much as 20% p53 protein remained within Pos3Aa-p53 crystals after 3 days incubation in PBS at 37℃.22 We have also found that Pos3Aa crystals could last for more than 4 days after being taken up by mammalian cells based on western blot analysis with anti-Pos3Aa antibodies (unpublished data). Based on these results, it appears that Pos3Aa-p53 crystals might still be effective 3–4 days after intratumoral injection, thus making it possible to lengthen the dosing interval. Another potential strategy is to use specially designed bacteria such as those demonstrated by the work of Gurbatri et al.65 In their study, the researchers designed and bioengineered a quorum sensing-based lysis circuit into E. coli, which enabled the programmed E.coli cells to keep colonizing tumors and undergoing intratumoral quorum lysis, resulting in the continuous in situ production and release of ICIs with just a single injection.65 Pos3Aa-p53 crystal-producing Bt cells can be similarly engineered with such a synthetic population control circuit and directly delivered into tumors to allow for long-term production and retention of Pos3Aa-p53 crystals within tumors, thereby minimizing administration frequency.
Type I IFNs are known for their critical role in the antiviral immune response. Recently, increasing evidence has indicated that type I IFNs exert direct anticancer effects by activating IFNAR signaling and stimulate antitumor immune response by promoting the cross-priming of dendritic cells and reactivation of CD8+ T cells.66–68 Induction of type I IFNs within tumors has been reported to overcome innate immune resistance and activate antitumor immunity.66 67 In this study, our RNA-seq and qPCR results on the Pos3Aa-p53-treated cells have shown the upregulation of type I IFN and ISGs including PD-L1 with the concomitant restoration of the p53 pathway (figure 4). We have also demonstrated that Pos3Aa-p53 crystals significantly boost the infiltration of CD8+ T cells in 4T1 tumors, which are considered as immunologically ‘cold’ and sensitize them to ICIs (figure 5). We speculate that such enhancement can be attributed in part by the induction of type I IFN pathways.
Further insights into how Pos3Aa-p53 crystals induce the expression of type I IFNs come from the recent study by the Selivanova group at the Karolinska Institutet 20which demonstrated that p53 effected IFN responses by turning on the expression of the normally repressed endogenous retroviruses (ERVs) via increased promoter occupancy, thus giving rise to double-stranded RNA (dsRNA) stress which in turn triggered the IFN response and related cellular pathways.20 The study also found that the p53-mediated inhibition of two major repressors of ERVs, histone demethylase LSD1 and DNA methyltransferase DNMT1 was critical to the derepression of ERVs.20 We thus mapped our RNA-seq reads to a mouse reference genome with a unique annotation file containing known transposable elements (TEs)69 and discovered that there was indeed an increased expression of ERVs including ERV1, ERVL, ERVL-MaLR and long interspersed nuclear element (LINE) subfamilies in the Pos3Aa-p53 crystal-treated cells (online supplemental figure S11), which was accompanied by the downregulation of the ERV repressor—DNMT1 whose expression is reported to be negatively regulated by wild-type p53 in previous studies (figure 4A).70 The genes of the key sensors of cytosolic dsRNA, retinoic acid-inducible gene I (RIG-I)-like receptors, which includes RIG-I (encoded by DDX58) and LGP2 (encoded by DHX58), were also significantly upregulated (figure 4A). Based on this analysis, we speculate that Pos3Aa-p53 crystals induce type I IFNs in 4T1 cells most likely via the viral mimicry mechanism. It should be noted that induction of IFNs is not the only mechanism by which Pos3Aa-p53 crystals remodel the immunosuppressive tumor microenvironment since ICD and the downregulation of IDO1 and TDO2 were also observed within 4T1 tumors on the treatment of these crystals (figures 3 and 4H–K).
In summary, we have demonstrated the intracellular delivery of wild-type p53 and its resultant restoration of p53 activity in cancer cells as an effective strategy to sensitize immunologically ‘cold’ tumors to ICIs. P53 protein was successfully delivered into p53-null TNBC 4T1 cells via Pos3Aa-p53 fusion protein crystals, resulting in ICD, activation of IFN signaling pathway and induction of PD-L1 expression. In mouse 4T1 xenograft models, Pos3Aa-p53 crystals increased tumor immunogenicity, facilitated the tumor infiltration by immune cells and altered the immunosuppressive tumor microenvironment, thereby augmenting anti-PD-1 therapy. Our results provide a widely applicable strategy to restore p53 function in cancers involving p53 deficiency and reinforce the potential of p53 restoration and ICIs combination as a therapeutic intervention for patients with cancer.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The raw data required to reproduce these findings are available from the corresponding authors on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
ZY would like to thank Dr Yinshan Fang and Dr Junjie Zhu for their kind help and suggestions. The authors also thank Prof Didier Lereclus (Institut Pasteur in Paris) and Dr Daniel Ziegler (Bacillus Genetic Stock Center, The Ohio State University) for kindly providing them with the Bt strains. RNA-seq analysis of patient samples is based on data generated by the TCGA Research Network: https://www.cancer.gov/tcga
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors ZY, MML and MKC conceived and planned the experiments. ZY carried out the experiments. JK-LS helped with RNA-seq data analysis. ZY, MML and MKC wrote the manuscript. MML and MKC supervised the project, and act as guarantors of this manuscript.
Funding We acknowledge the financial support from Hong Kong Research Grants Council AoE grant AoE/P-705-16, and the Center of Novel Biomaterials, The Chinese University of Hong Kong.
Competing interests MKC, MML and ZY are named inventors on patent application PCT/CN2020/084939.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.