Article Text

Abstract
Background Tumor-draining lymph nodes (TdLNs) are critical in the regulation of local and systemic antitumor T cell immunity and are implicated in coordinating responses to immunomodulatory therapies.
Methods Biomaterial nanoparticles that deliver chemotherapeutic drug paclitaxel to TdLNs were leveraged to explore its effects in combination and immune checkpoint blockade (ICB) antibody immunotherapy to determine the benefit of TdLN-directed chemoimmunotherapy on tumor control.
Results Accumulation of immunotherapeutic drugs in combination within TdLNs synergistically enhanced systemic T cell responses that led to improved control of local and disseminated disease and enhanced survival in multiple murine breast tumor models.
Conclusions These findings suggest a previously underappreciated role of secondary lymphoid tissues in mediating effects of chemoimmunotherapy and demonstrate the potential for nanotechnology to unleashing drug synergies via lymph node targeted delivery to elicit improved response of breast and other cancers.
- Drug Therapy, Combination
- Immunotherapy
- Immunomodulation
- Breast Neoplasms
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
While chemoimmunotherapy is known to mediate direct effects within the tumor microenvironment, its effects within secondary lymphoid tissues have not been fully elaborated. This study sought to elucidate how chemoimmunotherapy influences lymph nodes and how, due to the role the lymph node plays in the ongoing antitumor immune response, these effects influence tumor control.
WHAT THIS STUDY ADDS
Directing chemoimmunotherapy to tumor-draining lymph nodes using lymph node-targeting nanotechnology resulted in mobilization of T lymphocytes into the circulation that was associated with improved control of both local and disseminated tumors as well as animal survival in multiple murine models of triple negative breast cancer. Tumor-draining lymph nodes thus appear to play previously underappreciated roles in mediating the effects of chemoimmunotherapy.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
These findings can inform future clinical investigations into combination chemoimmunotherapies for triple negative breast cancer and how delivery strategies and/or technologies that target drug effects to within lymph nodes may improve treatment efficacy and patient outcomes.
Introduction
Breast malignancies continue to be the leading cause of cancer-related deaths in women worldwide, leading to nearly half a million deaths per year.1 Triple negative breast cancer (TNBC) is a particularly aggressive subtype due to its rapid growth rate and propensity to metastasize, and generally results in poor prognoses for patients with advanced disease. Additionally, by definition TNBC lacks expression of estrogen (ER), progesterone (PR), and human epidermal growth factor-2 (HER2) receptors used as therapeutic targets for other breast cancer (BC) subtypes, limiting the treatment modalities available to patients.2 Until recently, chemotherapy was used to treat local and disseminated TNBC alongside surgery or radiation therapy as available depending on disease stage, but as of late, immune checkpoint blockade (ICB) monoclonal antibodies (mAbs) have emerged as a potent treatment modality.3–5 Response rates for advanced TNBC remain low (~10–20%),5 however, which has motivated the development of combined regimens of chemotherapy and immunotherapy in order to take advantage of the benefits of both treatment modalities. While combination chemoimmunotherapy has demonstrated modestly higher survival rates over monotherapies in the clinic (~20–30%),6 there is still a clear need for improving these therapies to further improve patient outcomes.
Current understandings of the mechanisms of chemoimmunotherapy efficacy focus on effects within the tumor microenvironment (TME).7 Chemotherapeutics such as paclitaxel (PXL), a first-line treatment for TNBC, are traditionally cytotoxic small molecules that directly kill tumor cells, but their use can also result in induction of immunogenic cell death whereby tumor cells release danger signals and antigen in a manner that activates antigen presenting cells (APCs) and the adaptive immune pathway. In addition, PXL has demonstrated direct enhancement of maturation and costimulatory markers in human dendritic cells,8 and treatment with PXL or other taxanes has been shown to enhance levels of proinflammatory cytokines, circulating functional T cells, and tumor infiltrating lymphocytes in human clinical studies.9–12 Given these immunomodulatory effects, ICB mAbs have recently been paired with PXL-containing chemotherapy regimens in clinical TNBC investigations in an effort to bolster antitumor therapeutic effects.6 ICB mAb immunotherapies work by blocking inhibitory pathways resulting in dampened T cell responses, such as the programmed death-1 (PD-1) receptor on T cells engaging its ligand, programmed death-ligand 1 (PD-L1), on tumor cells.13 The therapeutic efficacy of ICB mAbs is mediated by a population of Tcf1+PD-1+ stem-like CD8+ T cells which proliferate and differentiate to give rise to effector CD8+ T cells that can mediate tumor control.14–16 Immune checkpoints are active not only in the TME but also in secondary lymphoid tissues, including spleen and lymph nodes (LNs) which are responsible for housing and mediating adaptive antitumor immunity.17 18 Additionally, tumor-draining (Td) LNs contain a reservoir of Tcf1+PD-1+ stem-like CD8+ T cells which have been shown to differentiate and mobilize following stimulation of the conventional dendritic cell compartment.19 20 While the effects of combination chemoimmunotherapy on the tumor immune landscape have been studied extensively,7 21 their effects on secondary lymphoid tissues, particularly the TdLN, are less well understood. Given the widespread clinical use of combination chemoimmunotherapies and ICB immunotherapies, elucidating the immunomodulatory effects of these therapies on APCs and tumor-reactive T cells housed within TdLNs is critical to improving treatment of TNBC.
Despite an emerging appreciation for their role in the regulation of antitumor immunity22 23 and immunotherapeutic response,17 18 24 challenges exist to studying the effects of small molecule accumulation within LNs. The short half-life and rapid clearance rate of small molecules from an intravenous administration route, as well as the propensity for absorption into the bloodstream from other locoregional administration routes commonly used to allow for lymphatic uptake such as intradermal (i.d.), subcutaneous (s.c.), and intramuscular injection, limits the accumulation and retention of small molecule chemotherapies in LNs.25 26 In order to circumvent these challenges, we employed poly(propylene sulfide) nanoparticles (PPS-NPs) optimally sized at 30 nm in diameter for uptake via lymphatic capillaries and transport to/accumulation within downstream LNs from a locoregional i.d. injection scheme.27 PPS-NPs are formed via emulsion polymerization and feature a Pluronic-F127 PEG-bl-PPG-bl-PEG block copolymer corona and a hydrophobic, cross-linked PPS core.28 29 PPS-NPs are amenable to encapsulation of hydrophobic small molecules into the PPS core, which can be released in a passive manner via first order diffusion independent of external stimuli to allow multistage drug release to immune cells in the LN parenchyma, such as dendritic cells (DCs) and T cells, responsible for antitumor immunity.30 31 While the traditional focus of the use of NPs for chemoimmunotherapy has been to study the enhanced permeability and retention of therapeutics in the TME with the goal of increasing NP exposure to the tumor, and subsequently the treatment-induced changes in the tumor-immune landscape alone, in this work we use PPS-NPs and different routes of administration to leverage PXL accumulation in LNs and other relevant tissues in order to probe the effect of PXL/anti-PD-1 (aPD-1) ICB mAb chemoimmunotherapy co-delivery to LNs on the resulting local and systemic antitumor immune response. Using PPS-NPs to encapsulate and deliver PXL, we demonstrate improved therapeutic effects of PXL monotherapy and combination therapy with aPD-1 compared with free, non-NP formulated delivery from both systemic and locoregional i.d. delivery strategies. This enhanced therapeutic efficacy coincided with immune stimulation in the TdLN with combination PXL-NP/aPD-1 as well as direct accumulation of NPs within the TdLN via both administration routes. We additionally found that co-delivery of PXL and aPD-1, enabled by NP delivery of PXL via locoregional administration, has a stimulatory effect on expansion of Tcf1+PD-1+ stem-like CD8+ T cells, a population known to be critical in mediating the success of antitumor immunotherapies including ICB mAbs,14–16 within LNs and mobilization into circulation. Lastly, we demonstrated a surprizing ability of locoregional (i.d.) PXL-NP to mediate an abscopal effect that protected against secondary tumor challenge in a manner equivalent to intratumoral (i.t.) PXL-NP administration. In all, these results demonstrate a role of the TdLN, and in particular CD8+ stem-like T cells, in mediating successful antitumor PXL/aPD-1 chemoimmunotherapy. Our findings have the potential to improve understanding of how cancer therapies influence the antitumor immune landscape in secondary lymphoid tissues, which has been underinvestigated in the literature, and may inform future clinical investigations into combination chemoimmunotherapy for TNBC and/or delivery strategies and technologies to target drugs to TdLNs to achieve a successful patient response.
Results
PPS-NPs enhance the delivery and therapeutic benefit of chemoimmunotherapy in E0771 TNBC
To investigate the effects of combination chemoimmunotherapy within TdLNs in the context of TNBC, we used the ER, PR, and HER2 negative murine mammary carcinoma model E0771 which readily forms tumors after orthotopic implantation into the mammary fat pad of C57Bl/6 mice32 33 (figure 1A,B). E0771 tumors develop a progressively expanded and disordered vasculature,32 a property which has been postulated to permit enhanced permeability and retention (EPR effect) of nanomaterials in the TME.21 We first sought to determine if a nanoparticle platform previously established in our laboratory (PPS-NP) improved TME accumulation of encapsulated small molecule. PPS-NP feature a cross-linked PPS core surface-stabilized with a Pluronic F-127 (PEG-bl-PPG-bl-PEG) block copolymer corona and are synthesized via emulsion polymerization.29 By virtue of their hydrophobic core, PPS-NP are able to efficiently encapsulate a variety of hydrophobic small molecules via a simple mixing process30 (figure 1C). Cyanine5.5-carboxylic acid (Cy5.5), a hydrophobic small molecule fluorescent dye, was used to model drug release from PPS-NPs in vivo.31 Cy5.5 was encapsulated into PPS-NP, yielding Cy5.5-NP which were administered intravenously via the jugular vein of mice bearing d10 or d25 E0771 tumors to recapitulate various stages of disease (figure 1D). PPS-NP demonstrated a modest delivery benefit over Cy5.5 administered as a free small molecule, 24 hours post-injection, but only in the context of late stage disease where vasculature is more disordered and presumably more permeable32 but not in early stages of disease (figure 1E,F).
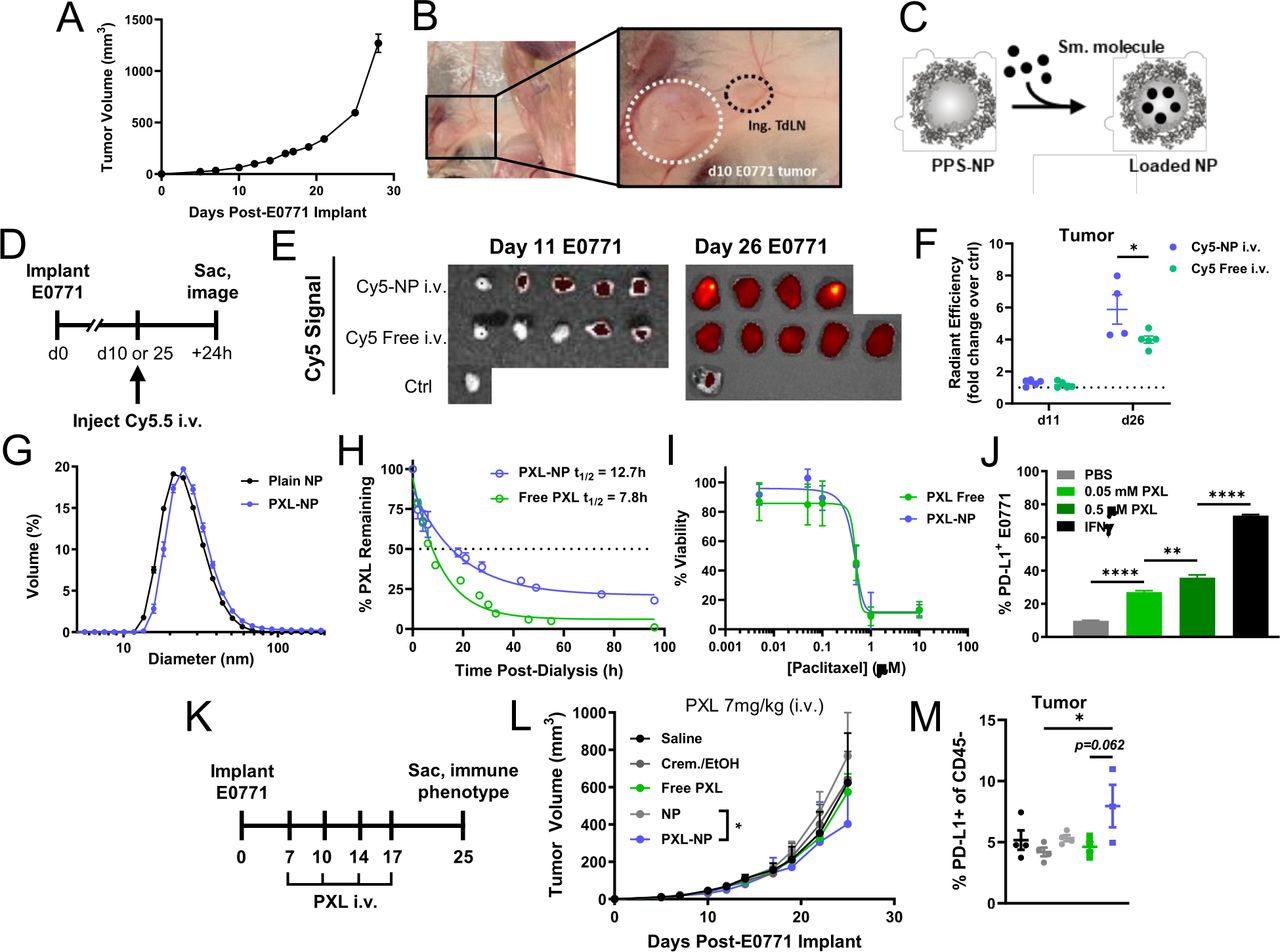
The 30 nm poly(propylene sulfide) NPs increase accumulation of encapsulated small molecule in late but not early stage E0771 TNBC tumors, facilitate sustained release of PXL with sustained cytotoxic and immune checkpoint activation effects, and improve tumor control from systemic chemotherapy. (A) E0771 tumor growth following implantation in the fourth mammary fat pad. (B) Ex vivo image of E0771 tumor microenvironment and proximal (tumor-draining) inguinal LN in the fourth MFP on day 11 post-implant. (C) Schematic of PPS-NP structure and loading of small molecules into hydrophobic NP core. (D) Fluorescent Cy5.5 model cargo-loaded NP (Cy5.5-NP) were administered intravenously into mice bearing d10 or d25 E0771 tumors and mice were sacrificed 24 hours later for tissue imaging. (E) IVIS images and (F) quantification of Cy5.5 content in tumors 24 hours post-intravenous injection of Cy5.5-NP. (G) Hydrodynamic diameter of unloaded (24.9±6.6 nm) or PXL-loaded (26.9±6.9 nm) NP analyzed via DLS. (H) Per cent of initial loaded PXL (starting concentration 1 mg/mL) amount remaining in NP or free during in vitro release with dialysis at 37°C. (I) Viability of E0771 tumor cells in vitro following 48 hours treatment with various concentrations of PXL in NP or free as determined via alamarBlue assay. (J) PD-L1 expression on E0771 cells treated with indicated concentrations of free PXL for 24 hours in vitro as determined using flow cytometry. (K) E0771 tumor-bearing mice (5×105 E0771 cells implanted s.c. in the fourth MFP) were treated intravenously on days 7, 10, 14, and 17 post-tumor implantation with PXL (7 mg/kg (140 μg) total dose) formulated in 35 mg/mL PPS-NP (PXL-NP) or Cremophor/Ethanol (free PXL), or corresponding saline, Cremophor/ethanol, and unloaded NP controls. (L) E0771 tumor growth in response to treatment. (M) Mice from (L) were euthanized on day 25 post-implant and tumors processed and analyzed using flow cytometry for PD-L1 expression on CD45– tumor cells. n=3–4 mice per treatment group. Statistical analyses by unpaired two-tailed t-test or one-way or two-way analysis of variance with Tukey’s test. Data represented as mean±SEM. *p<0.05, **p<0.01, ****p<0.0001. Cy5.5, Cyanine5.5-carboxylic acid; DLS, dynamic light scattering; i.v., intravenous; IVIS, in vivo imaging system; MFP, mammary fat pad; PD-L1, programmed death ligand 1; PPS-NPs, poly(propylene sulfide) nanoparticles; PXL, paclitaxel; s.c., subcutaneous; TdLN, tumor-draining lymph node; TNBC, triple negative breast cancer.
Given the demonstrated benefit of PPS-NPs on encapsulated dye delivery the E0771 TME, we sought to apply the PPS-NP platform to deliver the small molecule chemotherapeutic PXL. PXL was dissolved in dimethyl sulfoxide (DMSO) and added at 5%–10% v/v to PPS-NP and mixed by inversion for 5 min, yielding PXL-NPs30 which retained their ~30 nm size as determined via dynamic light scattering (DLS) (figure 1G). Encapsulated PXL was released from NP in an extended manner over free formulated PXL (t1/2 =~13 vs ~8 hour) without need for external stimuli (figure 1H) and PXL released from NP retained its tumoricidal activity as determined by an in vitro E0771 cell viability assay (figure 1I). In addition to inducing cell death, PXL also upregulated expression of PD-L1 on E0771 cells in vitro (figure 1J), a feature which may sensitize this model to disruption of the PD-1:PD-L1 signaling axis via function blocking mAbs, and is consistent with previous reports of chemotherapeutics causing PD-L1 induction in a manner similar to interferons.34 PXL-NPs were administered intravenously at 7 mg/kg to E0771 tumor bearing mice throughout the course of tumor growth (figure 1K), which resulted in modestly reduced tumor growth over treatment with non-drug loaded NPs as control in vivo (figure 1L, online supplemental figure S1). Flow cytometry analysis of CD45− cells in the TME revealed an increase in PD-L1 expression in vivo with PXL-NP only, which may also result from improved delivery via NP (figure 1M). To further probe the effect of PXL dose on E0771 tumor growth, PXL-NPs were administered intravenously at various concentrations using the same treatment schedule. Antitumor effects were only modestly enhanced at a dose of 10 mg/kg PXL that was implemented in further studies (online supplemental figure S2).
Supplemental material
Given the success of PXL-NP as a monotherapy following systemic administration, the therapeutic efficacy of combination PXL/aPD-1 chemoimmunotherapy in the E0771 model was evaluated (figure 2A). PXL in its free or NP-formulated form was administered intravenously at a 10 mg/kg dose (PXL dose) while aPD-1 mAb was delivered intraperitoneally (i.p., the route used in the vast majority of all preclinical studies of ICB13 17 18 33) at a dose of 5 mg/kg (mAb dose). As a monotherapy, PXL-NP treatment reduced tumor growth more substantially compared with free PXL (formulated in 20% DMSO/saline vehicle) and saline, whereas treatment with vehicle control or unloaded NP intravenously had no effect (figure 2B,C, online supplemental figure S3). In the context of combination therapy, PXL-NP/aPD-1 resulted in the greatest inhibition of tumor growth, surpassing the effect of free PXL/aPD-1, but surprisingly not aPD-1 mAb alone (figure 2D, online supplemental figure S3). Immune profiling on day 18 of tumor growth revealed increased levels of tumor infiltrating CD45+ immune cells with PXL-NP/aPD-1 treatment compared with free PXL/aPD-1 and aPD-1 alone (expressed as % of total tumor events), which was not attributed to the CD11c+ DC compartment (figure 2E,F, online supplemental figure S4). However, PXL-NP/aPD-1 did result in an increase in the T cells within the TME over free PXL/aPD-1, including CD3+ and CD8+ T cells, as well as levels of cycling Ki67+ CD8+ T cells (figure 2G, online supplemental figure S5). However, levels of immune suppressive FoxP3+ TREG cells responsible for dampening antitumor immune responses were also increased over aPD-1 alone (figure 2G), and the ratio of CD8 to TREG cells in the TME was increased for aPD-1 monotherapy over PXL-NP/aPD-1, possibly explaining why tumor growth was similar between these groups (figure 2H).

NP encapsulation improves chemotherapeutic efficacy of PXL, and in combination with systemically administered aPD-1 immunotherapy augments the expansion of tumor infiltrating T cells and maintains immunotherapy effects on immune cell activation and CD8 T lymphocyte expansion in TdLNs. (A) E0771 tumor-bearing mice treated intravenously on days 7, 10, 14, and 17 post-tumor implantation of PXL (10 mg/kg (200 µg) total dose) formulated in 35 mg/mL PPS-NP (PXL-NP) or free drug formulated in 20% DMSO/saline (free PXL), alone or in combination with i.p. administration on days 10, 14, and 17 of aPD-1 (5 mg/kg mAb dose) formulated in saline. (B) E0771 tumor growth in response to controls, (C) PXL monotherapy and (D) PXL/aPD-1 combination therapy or aPD-1 monotherapy treatment. Mice were sacrificed on day 18 and tissues processed and analyzed using flow cytometry (E–K). Levels (% of total tumor events) of tumor-infiltrating (E) CD45+ immune cells, (F) CD11c+ and mature CD86+CD11c+DCs, (G) CD3+, CD8+, cycling Ki67+CD8+, and CD4+FoxP3+ TREG T cells, and (H) ratio of intratumoral CD8+/TREG cells. Cell counts in the TdLN of (I) CD45+ immune cells, (J) CD11c+ and mature CD86+CD11c+DCs, and (K) CD3+, CD8+, cycling Ki67+CD8+, and CD4+FoxP3+ TREG T cells. n=5–6 mice per treatment group. Statistical analyses by one-way or two-way analysis of variance with Tukey’s test. Data represented as mean±SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. aPD-1, anti-PD-1, DC, dendritic cell; DMSO, dimethyl sulfoxide; i.p., intraperitoneal; i.v., intravenous; mAb, monoclonal antibodies; n.s., not significant; PBS, phosphate buffered saline; PD-1, programmed death-1; PPS-NP, poly(propylene sulfide) nanoparticles; PXL, paclitaxel; TdLN, tumor-draining lymph node.
Because of the role of LNs in antitumor immunity and in breast cancer staging and progression35, we wanted to understand how the immune landscape changed in the TNBC TdLN. As was seen in the TME, similar increases in immune cell levels in the TdLN with systemic PXL-NP/aPD-1 treatment were measured (figure 2I). Unlike the TME, however, increased counts of CD11c+ DCs and mature DCs expressing the T cell costimulatory molecule CD86 were reflected in the TdLN with PXL-NP/aPD-1 treatment (figure 2J), though this was not significantly increased over aPD-1 alone. Combination therapy also increased counts in the T cell compartment, most notably of proliferating Ki67+ CD8+ T cells but also of TREGs as in the TME (figure 2K). That we have observed pleiotropic effects of PXL on immune stimulation and tumor growth is in line with literature reports: PXL elicits both proinflammatory and anti-inflammatory effects on tumor-resident immune cells as well as the induces secretion of i.t. lymphangiogenic factors that may contribute to a more highly suppressive immune milieu in the TME in some murine models.36–39. In the E0771 model, the presence of tumor-resident TREGs have been observed previously wherein i.t. administration of TREG depleting anti-cytotoxic T-lymphocytes-associated protein 4 mAb clone provided enhanced tumor protection compared with non-depleting clones.18 Increased presence of TREGs in tumors, TdLNs, and blood was observed with PXL-NP/aPD-1-treated mice compared with aPD-1 monotherapy (figure 2), suggesting that the pleiotropic effects of PXL on enhancing activity of TREGs in this model may be significant and explain the lack of additional efficacy afforded by combination therapy compared with aPD-1 alone. Nevertheless, the observation of antitumor immune cell stimulation in the TdLN was surprizing given that systemic drug administration is typically thought to result in poor lymphatic accumulation13 21 25 and may suggest a role for the TdLN in the antitumor immune response following chemoimmunotherapy.
Biodistribution reveals LN accumulation of PPS-NP from various routes of administration
To understand the observation of increased immune expansion in TdLNs of mice treated with systemic PXL/aPD-1 chemoimmunotherapy, the biodistribution of PPS-NP in non-draining and TdLNs from systemic administration was explored. PPS-NPs covalently labeled with Alexa Fluor (AF) 555 to allow NP tracking were loaded with Cy5.5 as a model for encapsulated drug that can be passively released from the NP. Biodistribution profiles of NP-encapsulated Cy5.5 to LNs 24 hours post-injection was assessed via in vivo imaging system (IVIS) imaging after administration i.d. in the flank ipsilateral to the inguinal LN co-draining d10 E0771 tumors, an administration route well established to result in NP accumulation within TdLNs18 and compared with those achieved by intravenous administration (figure 3A). Cy5.5-NP injection i.d. resulted in high levels of NP (AF555) signal within tumor draining inguinal and axial LNs, but lower levels in non-TdLNs (nTdLNs) as expected (figure 3B,C). While intravenous administration resulted in lower NP signal to TdLNs compared with i.d., NP accumulation in all LNs was appreciably higher than background and exceeded that of i.d. NP in nTdLNs. Notably, while i.d. administration of Cy5.5-NP resulted in enhanced accumulation of Cy5.5 cargo in TdLNs over free Cy5.5 i.d., neither formulation resulted in appreciable accumulation of Cy5.5 in LNs after intravenous injection (figure 3D–F). This could be due to blood circulation representing a larger concentration sink for intravenous administered materials, which can drive small molecule diffusion from the NP core. In contrast, levels of Cy5.5 in the TdLN may be more detectable following i.d. administration given that materials are more concentrated in lymph. Similarly, AF610 covalently-labeled aPD-1 was administered i.d. or i.p. (5 mg/kg mAb dose) and LNs analyzed 24 hours later for AF610 mAb content, revealing higher levels of accumulation in TdLNs with i.d. administration but appreciable accumulation of mAb in all LNs with systemic i.p. administration (figure 3G,H). In other tissues of interest, notably the spleen and tumor, NP (AF555) and aPD-1 (AF610) accumulation is equivalent for both i.d. and intravenous (NP) or i.p. (mAb) administration routes, and Cy5.5 accumulation in spleens and tumors is equivalent regardless of formulation with NP or free (online supplemental figure S6). Thus, while the TdLN can be specifically targeted via locoregional skin injection of NP or mAb, systemic administration results in both NP and mAb delivery to all LNs. Thus, systemic administration of NP-formulated PXL and aPD-1 likely results in co-delivery to LNs, meaning that the elevated levels of immune cells in TdLNs of PXL-NP/aPD-1 treated mice (figure 2) may result from direct interaction of chemoimmunotherapy with LN immune cells.
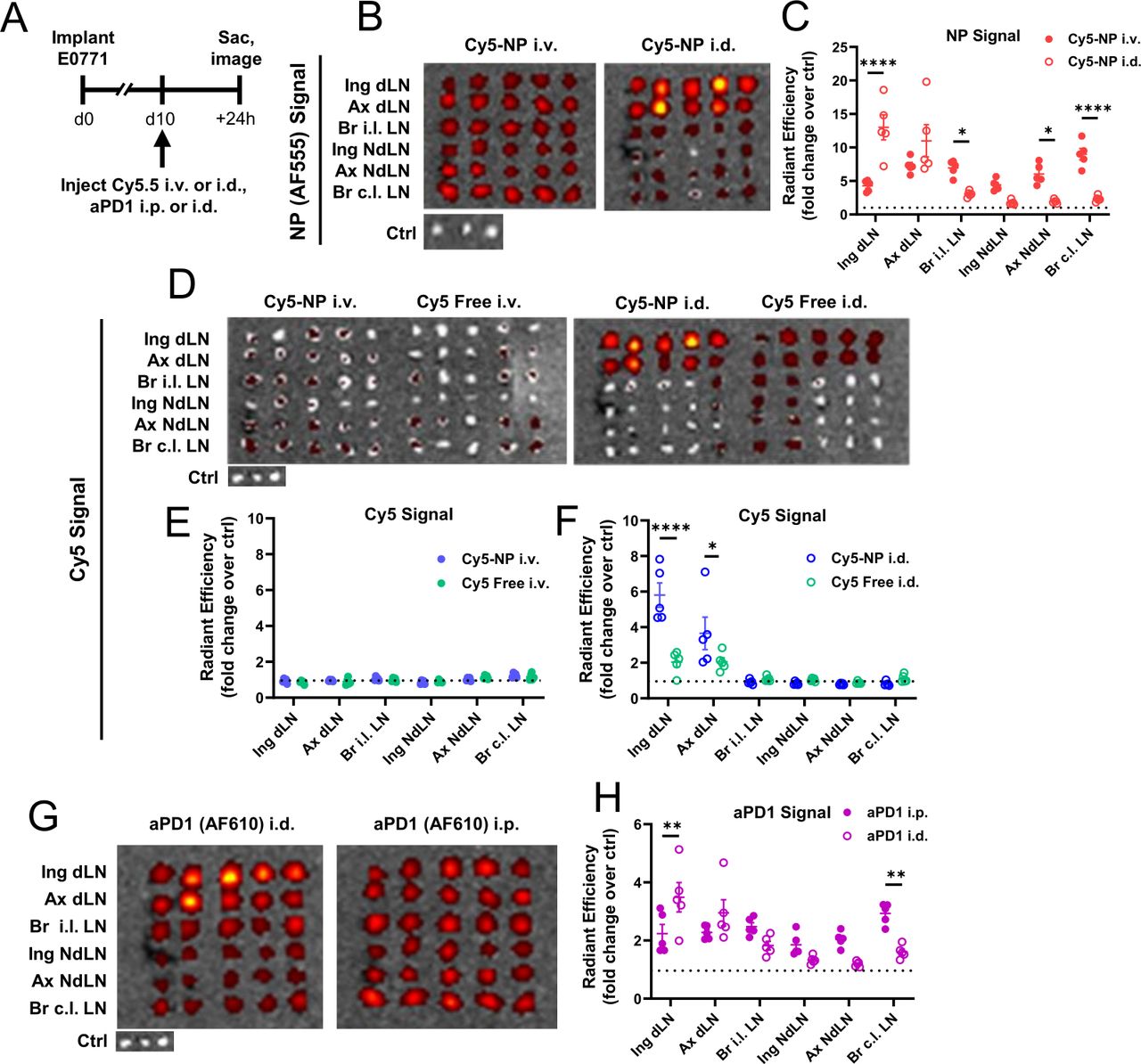
LN accumulation is enhanced by NP encapsulation for model small molecular cargo Cy5.5 and locoregional compared with systemic administration for both Cy5.5 and aPD-1-AF610 mAb. (A) Schematic of biodistribution experiment in which Cy5.5 encapsulated in 35 mg/mL PPS-NP (AF555-labeled) or free (in 20% DMSO/saline), or aPD-1 mAb labeled with AF610, was injected intravenously (NP)/i.p. (mAb), or i.d. in the ipsilateral flank co-draining to d11 E0771 TdLNs, and 24 hours later TdLNs analyzed for NP (AF555), Cy5.5, or aPD-1 (AF610) fluorescent signal. (B) Representative IVIS images and (C) quantification of NP (AF555) fluorescent signal in tumor-draining and non-draining LNs from intravenous or i.d. administration. (D) Representative IVIS images and quantification of Cy5.5 fluorescent signal in tumor-draining and non-draining LNs of free or NP-formulated Cy5.5 24 hours after (E) intravenous and (F) i.d. administration. (G) Representative IVIS images and (H) quantification of aPD-1 (AF610) levels in tumor-draining and non-draining LNs of d11 E0771-bearing mice 24 hours after i.p. or i.d. administration. n=4–5 mice per treatment group. Statistical analyses by two-way analysis of variance with Sidak’s test. Data represented as mean±SEM. *p<0.05, **p<0.01, ****p<0.0001. AF, Alexa Fluor; aPD-1, anti-programmed death-1; Cy5.5, Cyanine5.5-carboxylic acid; DMSO, dimethyl sulfoxide; i.d., intradermal; i.p., intraperitoneal; i.v., intravenous; IVIS, in vivo imaging system; mAb, monoclonal antibodies; nTdLN, non-tumor draining lymph node; PPS-NP, poly(propylene sulfide) nanoparticles; TdLN, tumor-draining lymph nodes.
Locoregional chemoimmunotherapy delivery to TdLN unleashes drug synergies
Given results that intravenous PPS-NP-formulated PXL, but not free PXL, in combination with aPD-1 causes APC and T cell expansion within the TdLN (figure 2) and that PPS-NPs administered intravenously accumulate within LNs (figure 3C), the effect of locoregional PXL/aPD-1 therapy resulting in localized drug delivery to the TdLN via PPS-NPs was tested. Mice bearing E0771 tumors were treated locoregionally i.d. in the flank ipsilateral to the tumor co-draining the inguinal TdLN with NP-formulated or free-formulated PXL as a monotherapy or in combination with aPD-1, and tumor size and animal survival monitored. As a proof of concept, 2 mg/kg PXL dose (5× reduction from 10 mg/kg dose) was used due to i.d. administration resulting in ~5× higher delivery of NP to TdLNs compared with intravenous, thus roughly approximating the dose accumulating in the TdLN in our systemic therapy results from figure 2 (figure 3C). During the course of treatment, NP formulated PXL resulted in improved tumor reduction in the context of locoregional monotherapy and combination therapy (online supplemental figure S7 A-C,G-H). Despite this early inhibition of tumor growth, no impact on overall survival was observed with any formulation (online supplemental S7 D-F). Nevertheless, these data indicate that chemoimmunotherapy accumulation within LNs may have a direct antitumor effect.
We next sought to directly compare immunostimulatory effects of locoregional and systemic PXL-NP/aPD-1 chemoimmunotherapy. A 10 mg/kg PXL dose was used from both administration routes in order to make a direct comparison at the tested dose where the highest efficacy in intravenous therapy was observed. E0771 tumor-bearing mice were treated according to the same schedule as previously described (figure 2A), and mice were sacrificed on day 18 for immune phenotyping analysis. In the context of PXL-NP monotherapy, i.d. administration provided a modest reduction in tumor burden over intravenous (p=0.059; figure 4A, online supplemental figure S8), whereas route of administration had little effect on the efficacy of PXL-NP/aPD-1 combination therapy (p=0.967; Figure 4B, online supplemental figure S8). In the context of locoregional therapy administered i.d., slight but non-significant reduction in tumor volume was observed between PXL-NP monotherapy and PXL-NP/aPD-1 combination when administered i.d. (p=0.116), and little differences were observed between combination therapy and aPD-1 monotherapy (p=0.402; Figure 4C, online supplemental figure S8). In contrast to tumor growth, immune phenotyping revealed large expansion of DCs and T cells in TdLNs of mice treated with i.d. PXL-NP/aPD-1 compared with systemic combination therapy and i.d. administration of PXL-NP or aPD-1 monotherapies (figure 4D–I). Most strikingly, i.d. PXL-NP/aPD-1 treatment resulted in higher numbers (p=0.055) and frequency of mature CD86+ DCs over aPD-1 alone, demonstrating PXL’s adjuvant effect 30 40 (figure 4E,F). This coincided with an increase in the number (figure 4G) and frequency of stem-like Tcf1+Tim3– (of PD-1+CD8+) T cells, a population increasingly investigated for their crucial role in patient response to immunotherapy14 16 41 42, in the TdLN with PXL-NP/aPD-1 i.d. treatment compared with either monotherapy i.d. (figure 4I). Additionally, elevated levels of TREGs were also observed in the TdLN in line with our findings in the systemic therapy context (figure 4G). In the blood, while the number of CD8+ T cells remained unchanged with therapy (online supplemental figure S9), a higher proportion of circulating CD8+ T cells were Ki67+ with i.d. PXL-NP as monotherapy or combination therapy and PD-1+ with i.d. combination (figure 4J). Additionally, a higher number and frequency (of PD-1+CD8+) of stem-like Tcf1+ Tim3– cells were elevated in the blood with i.d. combination therapy over either monotherapy i.d. (figure 4K,L), suggesting that PXL-NP/aPD-1 accumulation in the TdLN may mobilize stem-like cells into circulation. As in the TdLN, modestly higher levels of TREGs were also observed in the blood with i.d. combination therapy compared with aPD-1 i.d. (online supplemental figure S9), and in the tumor, an increased ratio of CD8+ to TREG infiltration of aPD-1 i.d. relative to i.d. combination therapy was observed (online supplemental figure S9). Despite this, the viability of CD8+ tumor infiltrating lymphocytes (TILs) in tumors of mice treated with i.d. combination therapy was elevated (figure 4M), a property which is beneficial in the clinical context.43 44 Together with our data in the systemic context (figure 2), these results suggest that TdLN accumulation of chemoimmunotherapy, enabled via use of PPS-NPs and further enhanced using locoregional injection, causes significant improvements in T cell quantity and quality, most notably that of the Tcf1+Tim3– stem-like cell population, in TdLNs which is also observed in blood circulation. Given that Tcf1+Tim3– stem-like cells have been increasingly emphasized in the literature as being critical in mediating powerful antitumor immunity, and successful immunotherapy has been correlated with the presence of these cells,14 16 these results suggest that directing chemoimmunotherapy to LNs may be a promising approach to improving clinical therapeutic responses. While combination chemoimmunotherapy may increase expansion of TREGs in any tissue regardless of administration route which detracts from therapeutic efficacy brought on by immune stimulation in this murine model, it is important to note that chemoimmunotherapy does improve overall survival of patients with TNBC compared with monotherapies in the clinic.6 45 Additionally, these clinical investigations commonly involve multi-agent chemotherapy regimens in combination with ICB,6 which offers an alternative explanation to the lack of improved efficacy of over ICB monotherapy in our simplified single-agent chemotherapy approach. Nevertheless, our results suggest an unexpected role of TdLN-mediated stimulation in the clinical success of chemoimmunotherapy.
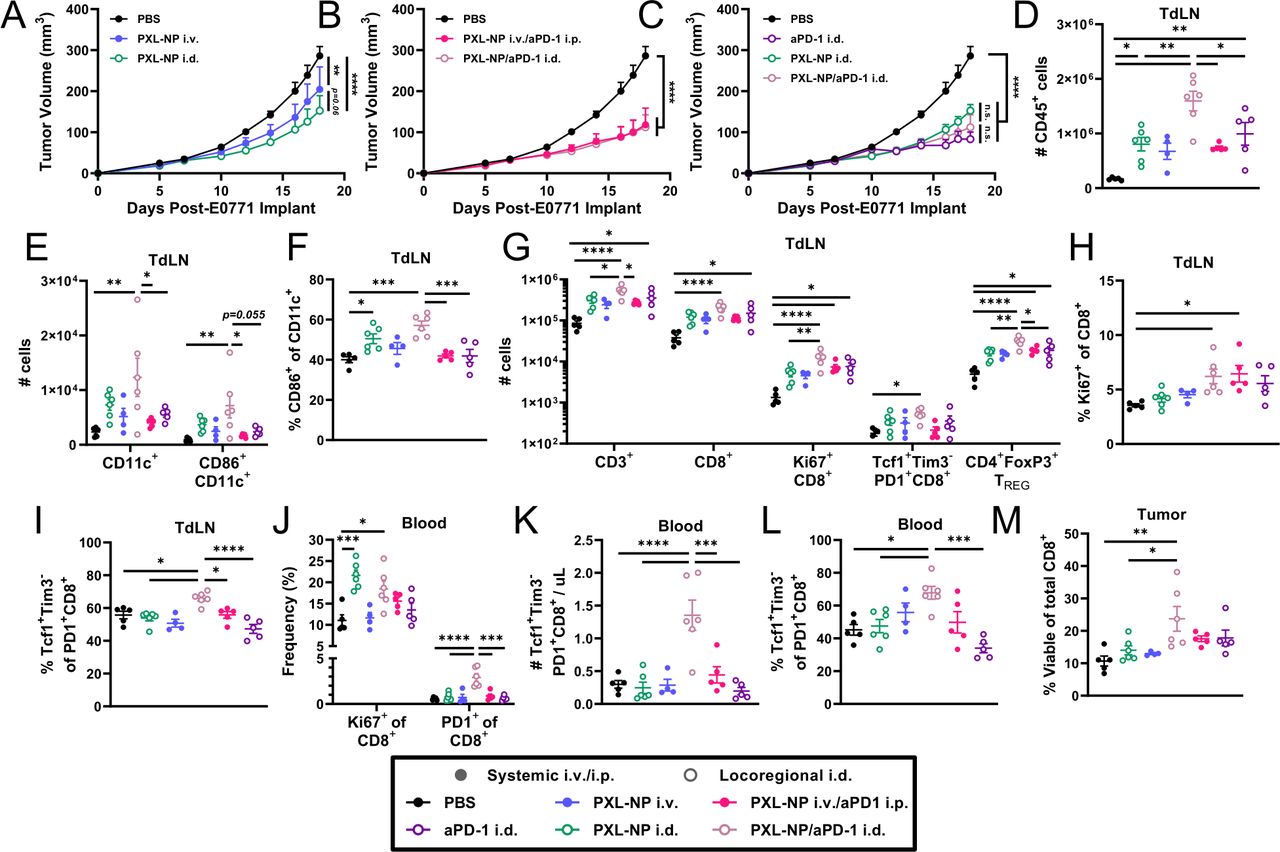
Locoregional administration i.d. that directs drug accumulation to TdLNs improves the therapeutic efficacy of PXL-NP chemotherapy but not in combination with aPD-1 immunotherapy and, when administered in combination locoregionally i.d., augments the activation and expansion of leukocytes in TdLNs, CD8 T cells in TdLNs and blood, and tumor infiltration of viable of CD8 T cells. E0771 tumor growth in response to i.d. or intravenous administration on days 7, 10, 14, and 17 post-tumor implantation of PXL (10 mg/kg total dose) formulated in 35 mg/mL PPS-NP (PXL-NP) as a monotherapy or in combination with i.d. or i.p. administration on days 10, 14, and 17 of aPD-1 (5 mg/kg total dose) formulated in saline. (A) Comparison of i.d. and intravenous PXL-NP monotherapy. (B) Comparison of i.d. and intravenous/i.p. PXL-NP/aPD-1 combination therapy. (C) Comparison of i.d. administered therapies. Mice from (A-C) were sacrificed on day 18 and tissues processed and analyzed using flow cytometry (D–M). Cell count in the TdLN of (D) CD45+ immune cells and (E) CD11c+ and mature CD86+CD11c+DCs. (F) CD86+frequency of CD11c+ DCs in the TdLN. Cell count in the TdLN of (G) CD3+, CD8+, cycling Ki67+CD8+, Tcf1+Tim3– PD1+CD8+ stem-like, and CD4+FoxP3+ TREG T cells. (H) Ki67+ frequency of CD8+T cells in the TdLN. (I) Tcf1+Tim3– frequency of PD-1+CD8+ T cells in the TdLN. (J) Ki67+ and PD-1+ frequencies of CD8+ T cells in the blood. (K) Number of Tcf1+Tim3– PD-1+CD8+ stem-like cells per µL blood. (L) Tcf1+Tim3– frequency of PD-1+CD8+ T cells in the blood. (M) Viability (% live) of tumor-infiltrating CD8+T cells. n=4–6 mice per treatment group. Statistical analyses were done using one-way or two-way analysis of variance with Tukey’s test. Data represented as mean±SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. aPD-1, anti-PD-1; i.d., intradermal; i.p., intraperitoneal; i.v., intravenous; PBS, phosphate buffered saline; PD-1, programmed death-1; PPS-NP, poly(propylene sulfide) nanoparticles; PXL, paclitaxel; TdLN, tumor-draining lymph nodes.
Next, to determine whether the effect of chemoimmunotherapy accumulation within TdLNs enhancing stem-like T cell mobilization can be recapitulated in a more physiologically relevant spontaneous tumor model, we utilized the MMTV-PyMT transgenic mouse model which reproduces many hallmarks of human breast cancer including more heterogeneity compared with implantable models.46 PD1-expressing CD8+ and CD4+ TH cells can be found in the TdLN, tumor, and systemic tissues of PyMT+ mice, indicating that immune checkpoints are upregulated in this model and thus chemoimmunotherapy may augment the anti-tumor immune response (figure 5A). We monitored PyMT+ females for spontaneous tumor growth and began treating once the primary tumor reached 50–100 mm3 in size (‘d0’). Mice were randomly assigned into groups and treated with either saline, aPD-1 mAb as a monotherapy (5 mg/kg), or PXL-NP (10 mg/kg PXL dose) plus aPD-1 mAb i.d. in the skin ipsilateral (i.l.) to the primary tumor co-draining to the TdLN. As before, PXL treatments were given on days 0, 3, 7, and 10, and aPD-1 treatments were administered on days 3, 7, and 10 (figure 5B). Tumor measurements revealed a modest but non-significant reduction with locoregional PXL-NP/aPD-1 treatment compared with saline control (p=0.066) over the course of treatment (figure 5C). On day 11, blood was drawn from mice for immune phenotyping (online supplemental figure S10), revealing little (Ki67+) to modest (PD-1+, p=0.09) differences in the frequency of functional markers on CD8 T cells (figure 5D). Nevertheless, an increase in the number of circulating Tcf1+Tim3– stem-like T cells with locoregional chemoimmunotherapy treatment compared with saline and aPD-1 monotherapy was observed (figure 5E). Combination therapy resulted in an increase in the overall survival of PyMT+ mice compared with saline (figure 5F). Overall, this indicates that secondary lymphoid tissues play a role in mediating the response to chemoimmunotherapy, and that chemoimmunotherapy accumulation in the TdLN elicits a mobilization effect on stem-like cells, in a physiologically relevant spontaneous breast tumor model.
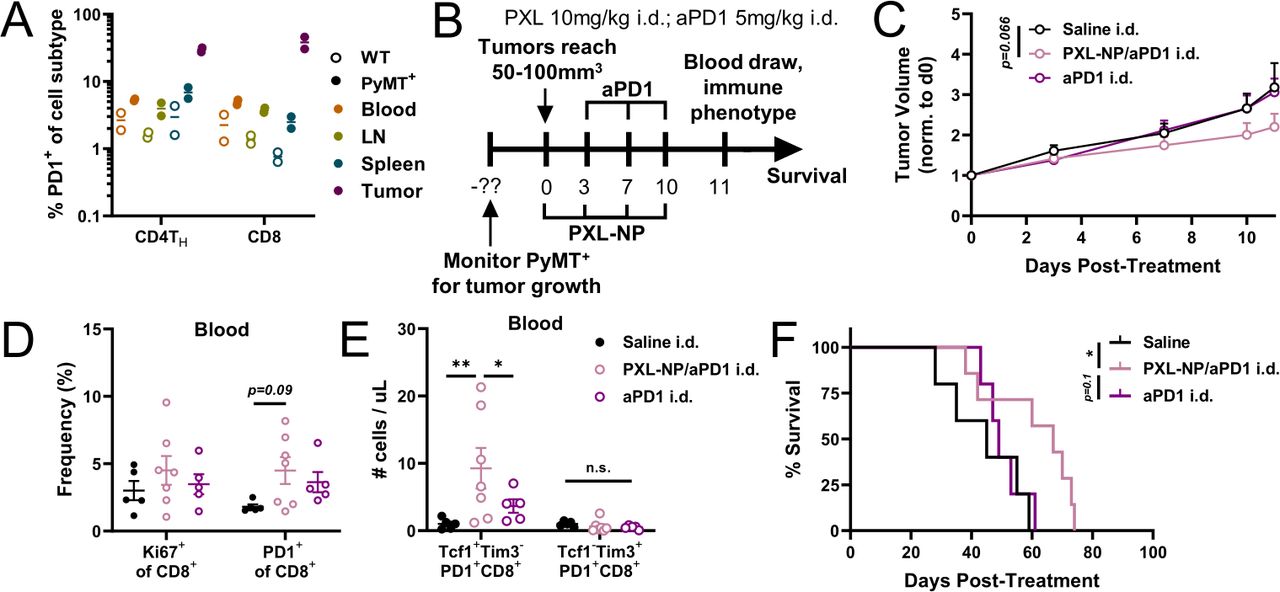
Locoregional administration i.d. of PXL-NP/aPD-1 combination therapy that directs therapeutic agents to TdLNs enhances CD8 stem-like cell mobilization into circulation in spontaneous MMTV-PyMT breast tumor model. (A) PD-1 expression in the CD4TH and CD8 T cell compartments in blood, TdLN, spleen, and tumor tissues of PyMT+ females (12 weeks old; solid circles) or PyMT– (WT; open circles) littermates. (B) Offspring genotyped at 6–8 weeks old and PyMT+ females monitored for tumor growth weekly from 8 weeks of age. On the largest tumor reaching 50–100 mm3 (day 0), PyMT+ mice were treated as before with PXL (10 mg/kg (200 µg) total dose) formulated in 35 mg/mL PPS-NP on days 0, 3, 7, and 10 i.d. in the flank co-draining the primary TdLN, and/or aPD-1 (5 mg/kg dose) on days 3, 7, and 10 i.d. (C) Average tumor curves throughout the course of treatment. Blood was drawn from mice on day 11 and analyzed via flow cytometry. (D) Ki67+ and PD1+ frequencies of CD8+ cells in the blood and (E) number of Tcf1+Tim3– or Tcf1–Tim3+ CD8+PD1+ stem-like or effector-like cells per µL blood. (F) Survival of mice from (C-E). n=5–7 mice per treatment group. Statistical analysis was done using two-way analysis of variance with Tukey’s test and log-rank (Mantel-Cox) test for survival analysis. Data represented as mean±SEM. *p<0.05, **p<0.01. aPD-1, anti-PD-1; i.d., intradermal; PD-1, programmed death-1; PXL, paclitaxel; TdLN, tumor-draining lymph nodes.
TdLNs, not mammary tumors, contain a pool of T cells that respond to chemoimmunotherapy
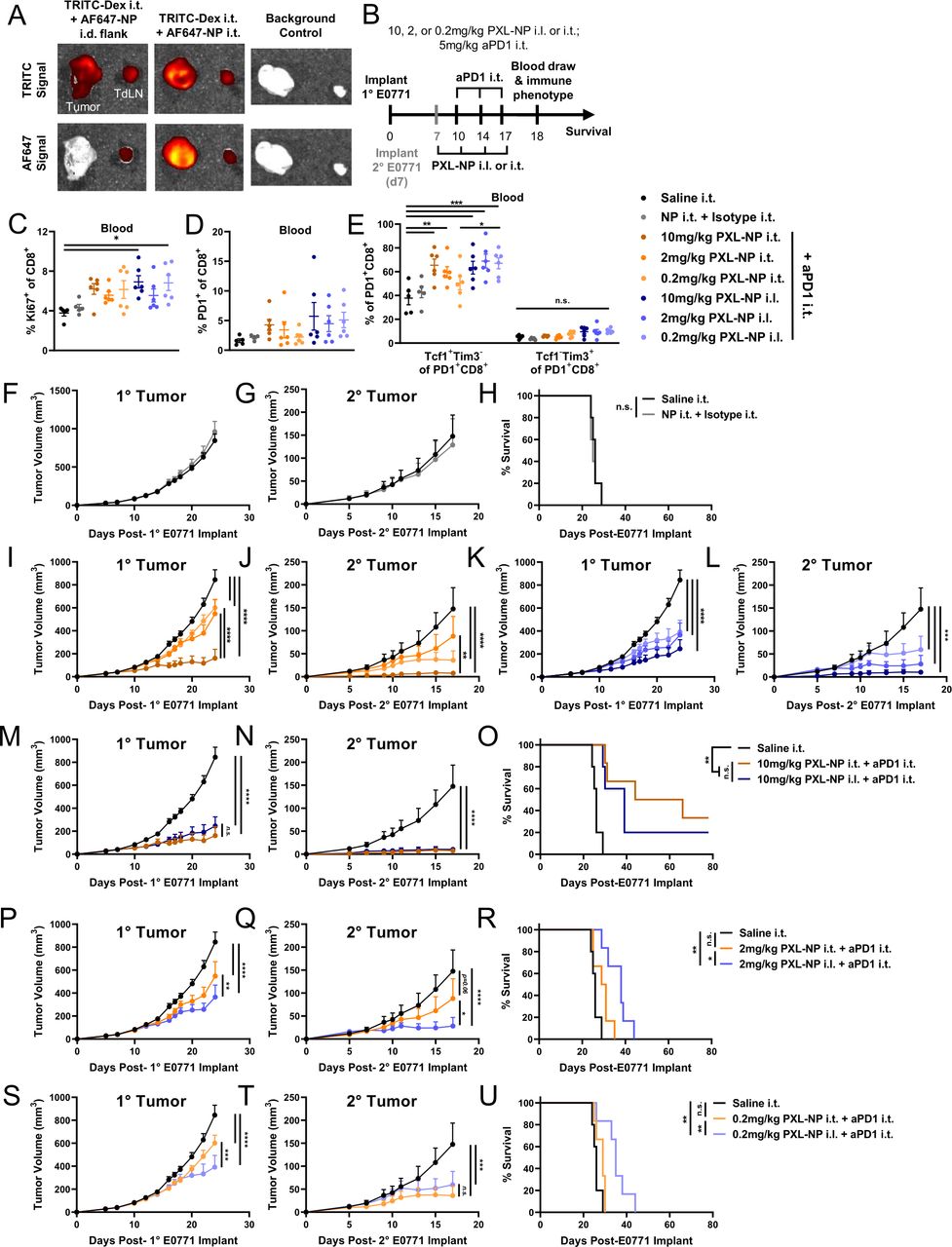
To rule out the possibility that the observed effect of stem-like cell mobilization is a direct or indirect result of PXL adjuvant accumulation in the TME, we next sought to investigate the source of the T cells that expand and mobilize in response to treatment, which we hypothesize is due to the adjuvant effects of PXL and originates in the LN. It should be noted that in the previous experiment, despite locally administered PXL-NP/aPD-1 being equivalent to aPD-1 alone in terms of tumor control, combination therapy did enhance local and systemic immune cell activation over aPD-1 alone (figure 4). We thus next wanted to investigate whether changing PXL-NP administration route augmented the effects of our previously established locoregional PXL-NP/aPD-1 approach from figure 4. To test this hypothesis, PXL-NP in varying doses (10, 2, or 0.2 mg/kg PXL) was administered either i.t. or i.l. in E0771 tumor bearing animals in order to compare the effects of PXL-NP accumulation in the tumor and TdLN (i.t.) versus TdLN alone (i.l.), the route used previously in figure 4. aPD-1 (5 mg/kg mAb dose) was administered i.t. in order to allow mAb delivery to both the tumor18 and TdLN in order to maximally inhibit ICB pathways in both tissues that might otherwise be limited by conventional systemic routes of drug administration.18 While delivery of NP in the flank i.l. to the tumor enables NP delivery to the TdLN, injection i.t. allows delivery to both the TME and TdLN (figures 3 and 6A, online supplemental figure S6 A).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dose reduction study reveals benefit of PXL-NP/aPD-1 combination chemoimmunotherapy directed to TdLNs by locoregional administration i.d. compared with i.t. on improving systemic tumor control and animal survival associated with increased mobilization of Tcf1+ stem like cells into the circulation. (A) On day 11 post E0771 tumor implant, mice were injected with 30 nm TRITC-dextran (0.05 mg in 10 µL saline) i.t. and AF647-labeled PPS-NPs (20 µL, 40 mg/mL) either i.t. or in the i.l. flank and sacrificed 4 hours post-injection for tissue imaging via IVIS CT. (B) Schedule for the therapeutic study, primary E0771 tumors were implanted as previously on day 0 and secondary tumors implanted in the contralateral fat pad on day 7. Mice were treated with PXL-NP i.l. or i.t. on days 7, 10, 14, and 17 post-tumor implantation (10, 2, 0.2 mg/kg total PXL dose) formulated in 35 mg/mL PPS-NP (PXL-NP) in combination with i.t. administration on days 10, 14, and 17 of aPD-1 (5 mg/kg mAb dose) formulated in saline. Blood was drawn on day 18 post-primary tumor implant and analyzed via immune phenotyping. (C) Ki67+ and (D) PD-1+ frequency of circulating CD8+T cells. (E) Tcf1+Tim3– stem-like and Tcf1–Tim3+ terminally differentiated cell frequencies of circulating PD-1+ CD8+ T cells. (F–G) E0771 primary or secondary tumor growth in response to saline or combination of unloaded NP and isotype mAb until first animal reaching humane endpoint. (H) Survival of mice treated in (F–G). (I–J) Primary and secondary tumor volumes of PXL-NP i.t. treated groups. (K–L) Primary and secondary tumor volumes of PXL-NP i.l. treated groups. (M–N) Comparison of primary and secondary tumor volumes of mice treated in (B–L) i.l. or i.t. with 10 mg/kg (P–Q) 2 mg/kg or (S–T) 0.2 mg/kg PXL-NP in combination with aPD-1 i.t. (O,R,U) Survival of mice treated in (B–L) i.l. or i.t. with (O) 10 mg/kg (R) 2 mg/kg or (U) 0.2 mg/kg PXL-NP in combination with aPD-1 i.t. n=5–6 mice per treatment group. Statistical analysis by one-way or two-way analysis of variance with Tukey’s test and log-rank (Mantel-Cox) test for survival analysis. Data represented as mean±SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. aPD-1, anti-PD-1; i.l., ipsilateral; i.t., intratumoral; IVIS, in vivo imaging system; mAb, monoclonal antibodies; PD-1, programmed death-1; PPS-NP, poly(propylene sulfide) nanoparticles; PXL, paclitaxel; TdLN, tumor-draining lymph nodes.
Mice were implanted with primary E0771 tumors and treated with the same schedule as previously described (figure 6B), and secondary tumors were implanted on day 7 post-primary tumor inoculation to test the ability of locoregional combination therapy to elicit tumor control on disseminated disease. Blood was drawn on day 18 for immune phenotyping of circulating T cells (online supplemental figure S11), which revealed an increase in the frequency of Ki67+ cycling CD8+ T cells with i.l. PXL-NP treatment and a non-significant increase in PD-1+ frequency of CD8+ T cells (figure 6C,D, online supplemental figure S11). Additionally, we once again observed an increase in the frequency of circulating Tcf1+Tim3– stem-like CD8+ T cells which was most pronounced in 10 mg/kg PXL-NP i.t. and all doses of PXL-NP i.l. in combination with aPD-1 i.t., while no significant changes in the frequency of Tim3+Tcf1– terminally differentiated CD8 cells were observed (figure 6E, online supplemental figure S11).
In the context of both PXL-NP/aPD-1 i.t. and PXL-NP i.l./aPD-1 i.t. treatment, primary and secondary tumor growth was demonstrated to be dose dependent, with 10 mg/kg PXL dose resulting in the strongest tumor inhibition while the combination of plain, unloaded NP plus isotype IgG mAb had no effect on tumor growth (figure 6, online supplemental figure S12). Remarkably, PXL-NP injected i.l. or i.t. resulted in similar levels of primary tumor reduction and overall survival (p=0.4099) at 10 mg/kg (figure 6M–O), whereas at lower doses 2 and 0.2 mg/kg, i.l. PXL-NP led to reduced primary tumor volume and improved survival compared with i.t. delivery (figure 6P–U). In line with results from other experimental configurations (figures 2 and 4), this may be due to the reported pleiotropic effects of PXL on tumor-resident immune cells, particularly TREGs, which were increased in tumors, TdLNs, and blood with combination therapy in the E0771 model (figures 2 and 4, online supplemental figure S11).
Similar to effects on primary tumor size, at a dose of 10 mg/kg both i.l. and i.t. PXL-NP treatment elicited strong inhibition of the secondary, untreated tumor to a similar extent (figure 6N). This was consistent between i.l. and i.t. administration at 0.2 mg/kg (figure 6T), though at the 2 mg/kg dose i.l. treatment caused more potent slowing of secondary tumor growth than i.t. administration (figure 6Q). Notably, the relatively sustained efficacy of i.l. PXL-NP at low PXL doses (figure 6) demonstrate the potential of this strategy to allow dose sparing. In all, this data suggests that the TdLN plays a critical role in mediating systemic antitumor immunity with PXL/aPD-1 combination therapy, and that the TdLN, likely not the tumor, is the primary source of T cells that are expanding in response to combination chemoimmunotherapy as PXL-NP directed to the TdLN alone (i.l.) had a remarkably similar effect on treated primary and untreated secondary tumors compared with PXL-NP directed to the tumor and TdLN via i.t. administration.
Discussion
TNBC remains an aggressive disease despite the modest clinical benefit achieved by the combination of traditional chemotherapy with ICB mAbs, generating interest in studying how to improve these therapies. As of late, ICB mAbs have been shown to act within secondary lymphoid tissues, most importantly TdLNs, and directing mAbs toward the TdLN via locoregional administration has been demonstrated as an effective strategy to improve ICB immunotherapy and simultaneously reduce toxic side effects.18 47 Thus, how combination chemoimmunotherapy interacts with TdLNs and the cells within remains an important question and obstacle to improving the efficacy of these combinations against TNBC. Using 30 nm polymeric PPS-NPs and a variety of administration routes, the accumulation of the small chemotherapeutic molecule PXL was directed to LNs, enabling its co-delivery with the ICB mAb aPD-1. This revealed strong stimulation of antitumor immunity within TdLNs, which resulted in mobilization of T cells into circulation and improved control of local and disseminated tumors and survival in a syngeneic implantable and transgenic spontaneous model of BC. Our results demonstrate that combination chemoimmunotherapy has a direct, stimulatory effect within secondary lymphoid tissues, particularly TdLNs, and supplement the current prevailing hypothesis that therapeutic effects are mediated predominantly through the TME via immunogenic cell death and reinvigoration of TILs.7 21 Appreciation for the role of TdLNs in the ongoing antitumor immune response has rapidly accumulated in recent years. Due to the presence of tumor antigen in lymph, TdLNs are an immune privileged site where tumor-specific T cells are primed and activated, including in sentinel LNs in the context of human breast cancer.48 49 Moreover, some have shown antitumor T cells in TdLNs to have a clonal relationship to TILs in the TME,19 48 50 and studies have demonstrated that when LN cell egress is blocked or LNs irradiated, a distinct lack of TILs is observed.19 51 Additionally, stem-like PD-1+Tcf1+ CD8 T cells, the population that proliferates and differentiates to give rise to antitumor effector-like T cells following ICB,14 16 52 have been observed in secondary lymphoid tissues18–20 51–53 including TdLNs where they are maintained by DCs as a reservoir to sustain antitumor immunity.19 20 Furthermore, stem-like T cells infiltrate the TME where they reside in APC niches and differentiate into potent effectors,14 highlighting the importance of CD8 T cell mobilization from TdLNs into circulation and tumors. When ICB mAb accumulation was directed to TdLNs via locoregional administration, an increase in mature DCs was observed in lymphoid tissues and proliferating CD8 T cells in lymphoid tissues and in circulation which was similar to that of i.t. administration directing accumulation to both the TdLN and tumor, suggesting that accumulation of chemoimmunotherapy in TdLNs enables local and systemic antitumor T cell immunity.18 The E0771 mammary TdLN is a unique survival niche containing antitumor T cells capable of expanding in response to ICB, whereas TME-resident T cells are exhausted and suppressed and thus lack this ability.32 Taken all together, the TdLN is a potent source of APCs and antitumor T cells capable of maturing, expanding, and mobilizing in response to chemoimmunotherapy. By directing chemoimmunotherapy to TdLNs via locoregional administration and NP-formulation of PXL, we demonstrate not only that the resulting T cell expansion and mobilization into circulation is a direct effect of therapy accumulating in the TdLN, but also that combination chemoimmunotherapy synergizes to exert an immune adjuvant effect greater than either monotherapy. Though our results did not include an analysis of T cell antigen specificity, we nevertheless observed strong tumor inhibition alongside expansion of TdLN-resident CD8 T cells expressing PD-1, which is upregulated following antigen experience and activation.54 Taken together, our results support that the TdLN is a potent target tissue for immunotherapeutic drug combinations. Our results also highlight a role for nanotechnology in enabling drug accumulation within TdLNs, which we achieved through both systemic (intravenous) and locoregional (i.d.) administration routes. The current paradigm of anticancer nanotechnology efficacy focuses on the EPR of delivered drugs into tumor tissue.21 However, the veracity of the EPR effect as a clinical paradigm associated with therapeutic responses to nanomedicines is in debate due to the lack of translational success by the vast majority of TME-targeted nanotechnologies.55–57 In the context of BC, where the primary tumor is often resected, the use of nanomaterials to achieve TdLN targeting of chemoimmunotherapy may hold potential to leverage the benefits of nanotechnology on drug delivery and release but also circumvent the potential absence of EPR effect in human solid tumors. Moreover, because of the intrinsic tumoricidal properties of chemotherapy, sentinel LN-targeted adjuvant or neoadjuvant regimens could provide the additional benefit of preventing lymph-borne metastases,25 which are particularly prevalent in BC and contribute greatly to disease progression and mortality. Lastly, the benefits classically ascribed to be afforded by nanotechnology in the context of tumor targeting via EPR, such as extended release, dose reduction, and reduction in toxic side effects, have also been demonstrated in the context of LN-targeting.25 26 Use of an NP platform that offers these benefits, such as PPS-NPs described herein,30 58 is thus particularly promising for improving LN targeting of encapsulated therapeutics.
Overall, our findings suggest a previously unappreciated role of secondary lymphoid tissues in mediating effects of chemoimmunotherapy and the benefit of nanotechnologies in unleashing drug synergies, resulting in enhancement of systemic antitumor immunity that led to the control of both local and disseminated TNBC and improved overall survival. Future studies into the mechanisms underlying PXL/aPD-1 chemoimmunotherapy mobilization of PD1+Tcf1+ stem-like cells from TdLNs have the potential to reveal pathways by which other potent combination therapies that enhance systemic T cell response and durable patient response may be designed. Such studies include a deeper investigation into the antigen specificity of expanded T cell populations using an exogenous model antigen system, such as OVA257–264, in the E0771 model or endogenous antigen in an alternative BC model.
By leveraging existing drug delivery technology to achieve directed delivery of chemoimmunotherapy to TdLNs, we demonstrate the potential of this approach to elicit potent antitumor immunity and achieve dose sparing. Thus, our results provide rationale to utilize innovations in drug delivery technologies that achieve LN targeting of therapeutics to achieve enhanced immune responsiveness and clinical management of TNBC.
Materials and methods
Cells
E0771 murine breast adenocarcinoma cells were cultured under sterile conditions in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum and 1% penicillin-streptomycin-amphotericin B (Gibco, Thermo Fisher Scientific). For in vitro experimentation, 104 E0771 cells were plated in 24-well plates (VWR) in complete DMEM and allowed to adhere for 4–6 hours. Cells were treated for 24 hours with PXL (LC Labs) at a final well concentration of 0.05 or 0.5 µM, recombinant mouse interferon-gamma (Invitrogen) at 100 ng/mL, or phosphate buffered saline (PBS) in triplicate. After 24 hours treatment, cells were removed from the plate via 0.05% Trypsin-EDTA (Thermo Fisher), neutralized with complete DMEM, and washed twice with PBS. Cells were then stained with Live/Dead and BV786 anti-PD-L1 (BioLegend) according to the flow cytometry protocol below.
Mice
Female C57BL/6 mice (The Jackson Laboratory) of 8–10 weeks old were used. Isoflurane was used as anesthesia and animal sacrifice was performed using CO2 asphyxiation or cervical dislocation. All procedures were completed in compliance with protocols approved by the Georgia Tech Institute for Animal Care and Use Committee (IACUC). To implant E0771 tumors, 5×105 E0771 cells were injected subcutaneously into the fourth mammary fat pad in sterile PBS. E0771 tumors were measured using calipers in the x, y, and z directions starting at day 5 post-implantation, and tumor volume was calculated as the volume of an ellipsoid ( ×xyz). For therapeutic experiments, mice were randomly assigned into treatment groups without blinding across all cages to minimize cage effects.
×xyz). For therapeutic experiments, mice were randomly assigned into treatment groups without blinding across all cages to minimize cage effects.
PyMT breeding and genotyping
Male MMTV-PyMT+/– mice backcrossed onto a C57Bl/6 background (kind gift from the laboratory of Dr Douglas K. Graham, Emory University and Children’s Healthcare of Atlanta) were bred in a sterile environment. At 6–8 weeks of age, sterile scissors were used to excise a 2–5 mm tail biopsy from each mouse. Offspring genotypes were determined using real-time PCR via a commercial vendor (Transnetyx, Cordova, Tennessee, USA). All procedures were completed in compliance with protocols approved by the Georgia Tech IACUC.
Nanoparticle synthesis and characterization
PPS-NPs were prepared as previously described via emulsion polymerization.28–30 59 Pluronic-F127 (poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol); PEG-bl-PPG-bl-PEG) (Sigma-Aldrich) 500 mg was added to Milli-Q water degassed by three argon purge cycles under inert conditions. The resulting Pluronic mixture dissolved for 30 min stirring at 1500 revolutions per minute (RPM), after which it degassed again via three argon purge cycles. Propylene sulfide monomer (Tokyo Chemical Industry) 400 µL was added and the mixture stirred for 30 min to form an emulsion. Separately, the four-arm polymerization initiator (synthesized in-house as previously described60) was deprotected by reacting with sodium methoxide (Sigma-Aldrich) in methanol for 15 min, then added to the NP emulsion and allowed to stir for another 15 min. The reaction was finalized via addition of 1,8-Diazabicyclo(5.4.0)undec-7-ene and allowed to react for 24 hours under inert conditions. The NPs were then exposed to air for a minimum of 2 hours in order to allow cross-linking of PPS chains in the NP core, leading to formation of disulfide bonds. PPS-NPs were then transferred to 100 kDa molecular weight cutoff (MWCO) dialysis tubing (Spectrum Laboratories) and dialyzed against 5L of de-ionized (DI) water for 3 days at a minimum of six water changes to allow removal of unreacted reagents. PPS-NPs were removed from dialysis tubing and sterile-filtered using 0.22 µm syringe filters in a sterile hood. NP concentration was determined by placing a sample of known volume into pre-weighed tubes, flash freezing, and lyophilizing. NP size was determined by DLS using a Malvern Zetasizer (Malvern Panalytical). PPS-NPs were stored at 4°C until use.
PXL-NP in vitro release
PXL-encapsulating NPs (PXL-NP) were prepared similarly to previous work.30 PXL (LC Labs) was dissolved at 10 mg/mL in DMSO (VWR) and added at 10% v/v to a 40 mg/mL aqueous solution of PPS-NPs. PXL-NPs were mixed by inversion for 5 min to allow encapsulation, then added to 100 kDa MWCO dialysis cups (Spectrum Laboratories) in triplicate and dialyzed against 37°C DI water. Alternatively, 10 mg/mL PXL in DMSO was added to water as a free drug control, and dialyzed in the same manner in triplicate. Samples were taken before dialysis and at varying time points, lyophilized, resuspended in acetonitrile to dissolve encapsulated PXL, and filtered to remove residual NP materials. PXL concentration was determined using High Performance Liquid Chromatography (Agilent) with a reverse phase column in 80% acetonitrile: 20% water as the mobile phase. Data is presented as % remaining, which is the absolute concentration per time point divided by the initial concentration at time t=0 hour.
In vivo biodistribution
PPS-NPs containing residual core thiols were reacted with AF555 maleimide (Thermo Fisher) in PBS overnight, then separated from free, unreacted dye using a Sepharose CL-6B size exclusion column (Sigma-Aldrich). NP fractions were combined, concentrated using a 30 kDa spin filter (Millipore Sigma), and stored at 4°C until use. Cy5.5 (Lumiprobe), a hydrophobic fluorophore, was used to model PXL. Cy5.5 was dissolved in DMSO at 5 mg/mL and encapsulated into AF555-labeled NPs as described above. Free dye was cleaned from Cy5.5-NPs using two successive 7 kDa MWCO Zeba spin columns (Thermo Fisher) immediately prior to injection. Encapsulation efficiency was determined by measuring fluorescence before and after cleaning with Zeba columns using a BioTek Synergy H4 plate reader at ex/em 685/720 nm, which was used to concentration-match the Free Cy5.5 control group. Alternatively, aPD-1 (clone RMP1-14, BioXCell) was reacted for ~4 hours with AF610 NHS ester and cleaned of free dye on a Sepharose CL-4B size exclusion column (Sigma-Aldrich), after which fractions were combined and concentrated using a 10 kDa spin filter (Millipore Sigma). Mice bearing E0771 tumors ~100 mm3 were injected with Cy5.5-NP or Free Cy5.5 either i.d. in the flank or intravenous via the jugular vein, or 100 µg AF610-aPD-1 either i.d. in the flank or i.p. similar to therapeutic experiments. Mice were euthanized 24 hours post-injection, and tissues imaged using the IVIS Spectrum CT (PerkinElmer). Primary (inguinal) and secondary (axillary) draining and non-draining LNs, tumor, and spleen were homogenized using bead-filled tubes (OPS Diagnostics) and a FastPrep-24 homogenizer (MP Biomedical). Samples were analyzed on a BioTek Synergy H4 plate reader at ex/em 555/575 (AF555, NPs), 605/625 (AF610 aPD-1), and 685/720 (Cy5.5 cargo). Per cent injection was determined using a standard curve of injectate in tissue homogenate from naïve mice. Living Image software (PerkinElmer) was used to analyze images.
Therapeutic PXL-NP/aPD-1 studies
For all studies, 5×105 E0771 cells were injected into the fourth mammary fat pad in sterile PBS (day 0). E0771 tumor bearing mice were treated with doses indicated in each figure. PXL-NP or controls (free PXL, solvent vehicle, plain NP, or PBS) were administered either i.t. in the tumor, i.d. in the flank draining to the inguinal TdLN, or intravenous in the jugular vein on days 7, 10, 14, and 17 post-tumor implant. aPD-1 (clone RMP1-14, BioXCell) or control (PBS or Rat IgG2a isotype control, BioXCell) was administered i.t. in the tumor, i.d. in the flank, or i.p on days 10, 14, and 17 post-implant. Tumors were measured every 2–3 days and mice were sacrificed when tumors exceeded 15 mm in any direction, or on day 18 or 25 post-implant (1 or 8 days after the last treatment) for flow cytometry analysis. In select experiments, secondary tumors were implanted in the fourth fat pad contralateral to the primary tumor (5×105 E0771 cells injected subcutaneously in sterile PBS) on day 7 following primary tumor implantation and monitored as previously described.
For treatment of MMTV-PyMT mice, females carrying the PyMT transgene were monitored weekly for tumor growth. When the largest tumor reached 50–100 mm3 in size, PXL-NP and aPD-1 treatments were prepared as described and administered i.d. to allow accumulation in the LN co-draining the largest tumor (TdLN). Tumor measurements were obtained every 2–3 days until day 11 post-treatment when 100 µL of blood was taken via facial vein laceration into a tube containing 10 µL EDTA (0.5M; Invitrogen). Mice were monitored weekly and euthanized when the primary tumor reached 15 mm in any direction.
Flow cytometry
Tumor, spleen, blood, and TdLN tissues were harvested post sacrifice. TdLN and tumor tissues were incubated with 1 mg/mL collagenase-D for 1 and 4 hours (respectively). Tissues were then manually dissociated and pushed through a 70 µm mesh strainer (Grenier Bio One) to obtain a single cell suspension in PBS +/+ (Corning). Splenocytes and whole blood were treated with red blood cell lysis buffer (Sigma-Aldrich) according to manufacturer protocol. Cell suspensions were centrifuged for 5 min at 400 g, washed, and plated into 96 well U-bottom plates (Corning) for staining. Next, cells were incubated with 100 µL of PBS containing 2.4G2 Fc block (Tonbo Biosciences), incubated for 5 min on ice, and washed with PBS. Cells were then treated with Zombie Aqua live/dead fixable viability kit (BioLegend) in PBS for 30 min at room temperature, then washed with fluorescence activated cell sorting (FACS) buffer (1% bovine serum albumin in PBS 1× −/−). Cells were then stained for the following surface markers (flow antibodies listed in table 1) in FACS buffer on ice for 30 min. Cells were washed in FACS buffer. Cells were then stained for intracellular markers (flow antibodies listed in table 1) using the FoxP3 intracellular fixation/permeabilization kit (eBioscience) according to manufacturers’ protocols, washed, and resuspended in FACS buffer, and transferred into flow cytometry tubes. Samples were run on an LSRFortessa flow cytometer (BD Biosciences) and data was analyzed via FlowJo software.
Statistical analyses
Statistical significance was calculated with Prism V.9 software (GraphPad). Data is presented as mean±SEM. ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05 by unpaired two-tailed t-tests or one-way or two-way analysis of variance followed by Tukey post hoc test for multiple comparisons. For survival curves, log-rank (Mantel-Cox) test was performed. Tumor volume curves and immune profiling plots from in vivo therapeutic experiments included n=4–6 mice per group, standard practice for murine tumor studies.
List of flow cytometry antibodies used
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MPM and SNT designed research studies. MPM conducted experiments and analyzed data. MPM, MJO’M, and SNT interpreted results. MPM and SNT prepared figures and wrote the manuscript. SNT is the guarantor.
Funding This work was supported by National Institutes of Health (NIH) grants R01CA207619 (SNT), R01CA247484 (SNT), T32GM008433 (MJO’M), and S10OD016264, and a CCR15330478 grant from Susan G Komen (SNT). MPM was supported by a National Science Foundation Graduate Research Fellowship.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.