Article Text

Abstract
Background Activation of the programmed cell death protein 1/programmed death-ligand 1 (PD-1/PD-L1) pathway has been extensively described as a pivotal mechanism to escape immune surveillance and elicits suppressive effect on antitumor immunity. Blockade of the PD-1/PD-L1 interaction by checkpoint inhibitors has been shown to result in tumor shrinkage and prolong patient survival. However, regulatory machinery for PD-1/PD-L1 expression is largely unknown.
Methods We used bioinformatic tools and biochemical methods to investigate the significance of F-box and WD repeat domain containing 7 (FBW7) in regulating PD-1 protein stability. By generating a panel of FBW7 and PD-1 encoding plasmids, we expressed FBW7 and PD-1 or their mutants to performed immunoprecipitation and immunoblotting assays. The efficacy of cotargeting FBW7 to enhance antitumor immunity was evaluated in C57BL/6J mice. These laboratory findings were further validated in tumor samples obtained from patients with non-small cell lung cancer (NSCLC).
Results We identified FBW7 as a E3 ubiquitin ligase for PD-1 protein, in which FBW7 promotes the K48-linked polyubiquitination of PD-1 protein at Lys233 residue. Cotargeting FBW7 accelerates PD-1 protein degradation and enhances antitumor immunity in vivo. Moreover, we demonstrated that cyclin-dependent kinase 1-mediated phosphorylation of Ser261 residue primes PD-1 protein nucleus translocation and binding with FBW7. Higher expression of FBW7 characterizes a ‘hot’ tumor microenvironment and confers more favorable responses to PD-1 blockade therapy.
Conclusions This study highlights the critical role of FBW7 in determining PD-1 protein stability. FBW7 ubiquitinates PD-1 in a phosphorylation-dependent manner, as a consequence, leading to PD-1 protein degradation and cytotoxic lymphocytes infiltrating the tumor microenvironment. Screening FBW7 status would predict clinical response to anti-PD-1 immunotherapy in patients with NSCLC, and targeting FBW7 is a promising strategy to enhance antitumor immunity.
- Immunotherapy
- Lung Neoplasms
- Programmed Cell Death 1 Receptor
- Translational Medical Research
- Tumor Escape
Data availability statement
Data are available on reasonable request. Not applicable.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- Immunotherapy
- Lung Neoplasms
- Programmed Cell Death 1 Receptor
- Translational Medical Research
- Tumor Escape
WHAT IS ALREADY KNOWN ON THIS TOPIC
The expression of PD-1/PD-L1 has been shown to correlate with clinical response to PD-1/PD-L1 blockade therapy in patients with advanced cancer, while the regulatory machinery for PD-1/PD-L1 protein is not fully understood.
WHAT THIS STUDY ADDS
We demonstrated that the F-box and WD repeat domain containing 7 (FBW7) acts as an E3 ubiquitin ligase for PD-1 protein, which promotes K48-linked polyubiquitination of PD-1 protein and accelerates its destruction. The cyclin-dependent kinase 1-mediated phosphorylation is required for PD-1 protein trafficking into the nucleus and binding with FBW7. High expression of FBW7 in the tumor microenvironment confers increased sensitivity to anti-PD-1 blockade therapy in non-small cell lung cancer (NSCLC) patients receiving immunotherapy.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The FBW7-PD-1 pathway is a novel mechanism governing PD-1 protein abundance in the tumor microenvironment. Targeting FBW7 would be a promising strategy to enhance antitumor immunity in NSCLC.
Background
The tumor microenvironment is highly immunosuppressive in patients with advanced cancer. Cancer cell uses this immunosuppressive microenvironment to evade immunologic surveillance.1 Over time, increasing strengths and efforts have led to the discovery of immune checkpoints that facilitate immune escape and promote tumor outgrowth. The programmed cell death protein 1/programmed death-ligand 1 (PD-1/PD-L1) pathway is a paradigm of ‘immune shield’ to protect against immune elimination in the setting of advanced cancer.2 3 Upregulation of PD-L1 protein on cancer cells binds PD-1 protein on T cells, which leads to T cell dysfunction and exhaustion.4 Although it has been well established that blockade of the sustained PD-1/PD-L1 inhibitory signaling invigorates active T cells and results in robust clinical antitumor activity.5 6 There are considerable interests of using immunotherapy in non-small cell lung cancer (NSCLC), for example, in patients with treatment-naïve metastatic NSCLC, concurrent chemotherapy and pembrolizumab (anti-PD-1) treatment yielded an overall survival rate of 69.2% at 1 year.7 The IMPOWER150 trial also demonstrated that atezolizumab (anti-PD-L1) treatment resulted in a significantly longer overall survival than platinum-based chemotherapy among patients with NSCLC.8 Thus, the anti-PD-1/PD-L1 immunotherapy exerts exciting clinical benefits not only in achieving tumor shrinkage, but also in generating long-term disease remission.
Despite these inspiring results in clinical practice, therapeutic outcomes of PD-1/PD-L1 blockade are highly variable and the PD-1/PD-L1-positive patients seem to preferentially benefit from PD-1/PD-L1 blockade immunotherapy.9 However, only a fraction of PD-1/PD-L1-positive tumors are responsive to PD-1/PD-L1 blockade and Immunohistochemistry (IHC)-based detection of PD-1/PD-L1 has been proven problematic since their expression is dynamic and readily inducible by interferon gamma (IFN-γ).10 The cellular abundance of PD-1/PD-L1 is tightly controlled by various factors that are still under intensive investigation. Transcriptional activation of PD-1/PD-L1 could be driven by the amplification of the encoding gene locus and cytokines within the tumor microenvironment. The importance of post-translational modification of PD-L1 protein has attracted increasing interest and there are compelling evidence showing ubiquitination of PD-L1 by the E3 ubiquitin ligase Cullin3-speckle-type POZ protein (SPOP) promotes proteasome-mediated degradation, whereas the COP9 signalosome 5 (CSN5) deubiquitinase stabilizes PD-L1 protein in cancer cells.11 12 Although these findings reveal preliminary clues indicating ubiquitination/deubiquitination serves as a critical modification of PD-L1 protein, there is still a big gap regarding the biochemical basis for the regulation of PD-1 protein. We, therefore, sought to decipher the machinery associated with PD-1 protein expression and evaluate their potential implications in antitumor immunotherapy.
The F-box and WD repeat domain containing 7 (FBW7) protein is the F-box component of the Skp1-Cul1-F-box (SCF) E3 ubiquitin ligase, in which it determines the substrate recognition specificity of the SCF complex.13 Most substrates of FBW7 are oncogenic proteins, including c-Myc, c-Jun, mTOR, cyclin E, Notch and MCL-1, implying that FBW7 may serve as a putative tumor suppressor.14–16 In agreement with this notion, mutation of FBW7 gene has been detected in various cancers, with an overall frequency of 6%. Notably, 43% of the identified mutations are found to be missense mutations (Arg465 and Arg479) within the WD40 domain that are responsible for substrate recognition.17 We and others have reported the significance of FBW7 in determining sensitivity to chemotherapy and targeted therapy in multiple cancer cell lines by the stabilization of anti-apoptotic MCL-1 protein, and targeting FBW7 overcomes resistance to anticancer treatment in solid tumors.18–20 These findings may add FBW7 into a rapidly growing list of genomic alterations that are frequently tested in the clinical settings. However, whether FBW7 confers sensitivity to anti-PD-1/PD-L1 immunotherapy remains unclear.
Here, we describe a critical finding in the rapidly evolving field of cancer immunotherapy. By using biochemistry-based approaches, we identify the FBW7 E3 ligase whose activation potentiates response to anti-PD-1 immunotherapy in NSCLC. Specifically, FBW7 mediates the K48-linked polyubiquitination of PD-1 protein at Lys233 residue, which depends on the phosphorylation of Ser261 residue by cyclin-dependent kinase 1 (CDK1). More favorable clinical benefit and longer duration of response are found in NSCLC patients with higher expression of FBW7. We characterize the immunological features underlying this enhancement of antitumor efficacy as follows: higher expression of FBW7 results in PD-1 protein ubiquitination and destruction, which in return facilitates the blockade of PD-1/PD-L1 immune evasion pathway. Our findings thus indicate that the FBW7-PD-1 signaling plays a pivotal role in regulating the efficacy of anti-PD-1 immunotherapy in NSCLC. Screening FBW7 status may help to predict the responsiveness to anti-PD-1 immunotherapy and cotargeting FBW7 would provide more clinical benefits for patients with advanced NSCLC.
Materials and methods
Reagents
Oridonin (S2335), MG132 (S2619), alvocidib (S1230), Ro-3306 (S7747), palbociclib (S1116) were purchased from Selleck Chemicals. Phytohemagglutinin (PHA, HY-N7038A) was purchased from MedChemExpress. Cycloheximide (2112S) was purchased from cell signaling Technology. λ-PPase (P2316S) was purchased from Beyotime. Puromycin (P8833) was purchased from Sigma. The anti-mouse PD-1 antibody (clone J43) (BE0033-2) was purchased from BioxCell. Matrigel matrix (356234) was purchased from Corning.
Cell lines and cell culture
All the cell lines were obtained between 2018 and 2020, and they were validated by short tandem repeat analysis, tested for mycoplasma contamination within the last 6 months and used at passage numbers <10. HEK293 cells were cultured in DMEM medium (Gibco) supplemented with 1% penicillin/streptomycin (Gibco) and 10% FBS (Gibco). Jurkat, THP-1, MOLT-4 and Lewis lung cancer (LLC) cells were cultured in RPMI 1640 (Gibco) medium. Human peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors by gradient centrifugation, stimulated with 5 µg/mL plate-bound anti-CD3/CD28 (Biolegend) and cultured in RPMI 1640 medium supplemented with 10 ng/mL IL-2 (Biolegend). Primary human T cells were isolated from PBMCs using a negative selection kit (STEM CELL Technologies). PHA at a final concentration of 150 ng/mL was added in the culture medium to stimulate the expression of PD-1 protein in MOLT-4 cells. All cells were cultured at 37°C in an atmosphere of 5% CO2.
Constructs, mutants and lentivirus production
Overexpression plasmids for Myc-FBW7, Myc-FBW7ΔF, Myc-FBW7 ΔWD40, HA-Ub, His-Ub, His Ub K48, His Ub K63, and shRNAs targeting FBW7 were preserved in our in-house construct bank as we have described.20 All the constructs used in our study were thoroughly sequenced. The Flag tagged PD-1 and its mutants (K210R, K233R, K210R/K233R, S261A, S261D, ΔPEST, and ΔNLS) were generated using the mutagenesis kit following manufacturer’s instructions. To generate the EGFP tagged PD-1 fusion constructs, PD-1 cDNA sequence was in-frame fused to EGFP sequence at the C-terminal. The shFBW7 lentivirus was produced by transfecting HEK293 cells with pLKO.1 shRNAs together with the psPAX2 and pMD2.G packaging plasmids using Lipofectamine 3000 (Invitrogen). Virus-containing supernatants were collected 48 hours after transfection and stored at −80°C. Cells were infected with the virus supernatants for 48 hours supplemented with polybrene, and resistant clones were selected by 2 µg /mL puromycin. siRNA targeting CDK1 (sense: 5’-UAUUUUGGUAUAAUCUUCCAU-3’; anti-sense: 5’-GGAAGAUUAUACCAAAAUAGA-3’) was synthesized by RIBOBIO (Guangzhou,China). Primer sequences for mutagenesis were listed in online supplemental table S1.
Supplemental material
RNA isolation and real-time PCR
Total RNA was extracted with TRIzol (Takara) from the indicated cells and transcribed into cDNA using a reverse transcription kit (Takara). Real-time PCR was performed following the standard protocol (Bio-Rad). The primer sequences for real-time PCR assay were shown as follows: PD-1 forward: 5’-ACGAGGGACAATAGGAGCCA-3’, PD-1 reverse: 5’-GGCATACTCCGTCTGCTCAG-3’, β-actin forward: 5’-ACGCCAACACAGTGCTGTCTG-3’, β-actin reverse: 5’-GGCCGGACTCGTCATACTCC-3’.
Plasmid transfection
Cells were seeded in 6-well plates or 6 cm dishes and incubated overnight to allow attachment. Cells were transfected with plasmids targeting indicated genes using Lipofectamine 3000 for 48 hours according to the manufacturer’s instructions.
Cellular fractionation assay
Subcellular protein fractionation was performed by using a commercially available kit (Thermo). α-tubulin and Lamin A/C were used as equal loading controls for non-nucleus and nucleus proteins, respectively.
Flow cytometry
Cell surface expression of PD-1 was measured by Alexa Fluor 647-conjugated anti-PD-1 antibody (Abcam) using flow cytometry. Zombie Red Fixable Viability Kit (Biolegend) was used to distinguish live cells and dead cells.
Immunoprecipitation and Western blot analysis
After indicated treatment, cells were harvested and lysed with RIPA buffer (Beyotime) containing protease inhibitor cocktail (Roche). The protein concentration was measured with a BCA protein quantification kit (Thermo). The cell extracts were incubated with Flag tag antibody, PD-1 antibody or FBW7 antibody with gentle rotation overnight at 4°C. The immunocomplex was pelleted with protein A/G agarose beads (Santa Cruz) for 2 hours and then washed with PBS for three times, resuspended, boiled with 2×SDS loading buffer and subjected to Western blot analysis.
Equal amounts of proteins (20–60 µg) were separated by SDS–PAGE and transferred onto a nitrocellulose membrane (Millipore). The membrane was blocked with 5% skim milk and incubated with the indicated primary antibodies overnight at 4°C. Protein bands were detected with horseradish peroxidase (HRP)-conjugated secondary antibodies and visualized by a chemiluminescence analysis system. Detailed antibody information was listed in online supplemental table S2.
Immunofluorescent staining
Cells were fixed with 4% paraformaldehyde, permeabilized with Triton X-100 and then incubated with primary antibodies overnight at 4°C. Cells were then incubated with Alexa Fluor 594-conjugated secondary antibodies and counterstained with DAPI the next day. Fluorescence signals were detected using a fluorescence microscope (Zeiss).
IHC and TUNEL assays
IHC and TUNEL staining were performed on slides from formalin-fixed and paraffin-embedded tissues. After antigen retrieval and blocking, slides were incubated with the indicated primary antibodies overnight at 4°C, and then incubated with HRP-conjugated secondary antibodies. The expression of indicated protein was visualized by DAB (Dako) and hematoxylin counter staining. The IHC density was scored as negative (score 0), weakly positive (score 1), moderately positive (score 2) and strongly positive (score 3). The percentage of positive cells was also scored (<5%, score 0; 6%–25%, score 1; 26%–50%, score 2; 51%–75%, score 3; and >75%, score 4). The intensity score was multiplied by the proportion score to yield the IHC H-score as we have described previously.
For the TUNEL staining assay (Roche), slides were manipulated following standard protocols and the results were visualized under a fluorescent microscope.
Animal models
C57BL/6J mice and thymus-deficient nude mice (male, 6–8 weeks old) were purchased from Qinglongshan Animal Breeding Field (Nanjing, China) and GemPharmatech (Nanjing, China), respectively. Approximately 2×106 LLC cells (for immune competent C57BL/6 J mice) or 1×106 LLC cells (for thymus-deficient nude mice) were suspended in 80 µL of a mix of Matrigel and PBS (3:1), and were subcutaneously injected into the flanks of mice. When the tumors reached the appropriate size, oridonin (5 mg/kg) or Ro-3306 (4 mg/kg) was injected intraperitoneally every 3 days, and anti-PD-1 antibody (200 µg per mouse) was injected intraperitoneally once per week. Tumor volume and body weight were measured and recorded every 3 days. At the end of the experiment, the mice were humanely sacrificed, and tumors were carefully isolated. The tumor volume was calculated using the following formula: tumor volume=L (length)×W(width)2/2. Body weight change rate was calculated using the following formula: body weight change rate= (body weight after tumor removal – body weight before tumor inoculation)/body weight before tumor inoculation.
Bioinformatics analysis and patient samples
To assess putative association between FBW7 and PD-1, we performed bioinformatics analysis using UniProt (https://www.UniProt.org/), GEPIA (http://gepia.cancer-pku.cn/), cBioPortal (http://www.cbioportal.org/), nuclear localization signal (NLS) Mapper (https://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi), KinasePhos V.2.0 (http://kinasephos2.mbc.nctu.edu.tw/), Scansite4 (https://scansite4.mit.edu/%23home) and TIMER V.2.0 (http://timer.cistrome.org/) online tools.
Matched NSCLC and adjacent normal lung tissues were acquired from 23 patients who underwent primary surgical resection in our institution between January 1, 2016 and September 30, 2016. In another cohort, a total number of 48 cases of NSCLC who received anti-PD-1 therapy in our department between January 1, 2017 and February 18, 2021 were enrolled. Response Evaluation Criteria In Solid Tumors (V.1.1) was used to assess the clinical efficacy of anti-PD-1 therapy. The clinical benefits of anti-PD-1 therapy were defined as durable clinical benefit (DCB: complete response, partial response, or stable disease lasting >6 months) and no durable clinical benefit (NDB: progressive disease or stable disease that lasted <6 months). The expression of FBW7 and PD-1 was determined by IHC, respectively.
Statistical analysis
All experiments were repeated at least in triplicate, and the data were calculated as the means±SEM. Statistical analysis was performed using SPSS V.10.0. Student’s t-test and one-way analysis of variance were applied to determine statistically significant differences (significance was set to p<0.05 and represented as *p<0.05, **p<0.01 and ***p<0.001) between different groups.
Results
Identification of the FBW7-PD-1 interaction
FBW7 recognizes and binds to the CDC4 phosphodegron (CPD) sequence (S/T-x-x-x-S/T) of its substrates, including c-Jun, c-Myc, cyclin-E, and MCL-1 (figure 1A). By using online bioinformatic tools, it is predicted that the PD-1 protein has two putative CPD sequences that could be recognized by FBW7. In addition, proteins that could be ubiquitinated are rich in proline (P), glutamic acid (E), serine (S) and threonine (T) (PEST domain). Alignment analysis of PD-1 protein sequence indicated a conserved PEST domain that overlapped with the C-terminal CPD (online supplemental figure S1A). Furthermore, the PD-1 protein level in patients with FBW7 mutations tended to be higher than that in patients without FBW7 mutations in a lung adenocarcinoma cohort from The Cancer Genome Atlas (TCGA) database (online supplemental figure S1B). These findings may suggest a putative interaction between FBW7 and PD-1.
Supplemental material

FBW7 interacts with PD-1 protein. (A) Sequence alignment of the phosphodegron sequences recognized by FBW7 in c-Jun, c-Myc, cyclin E, MCL-1 and PD-1. Conserved degron sequences are shown in red. (B, C) Western blot analysis with antibodies specific for the indicated proteins of HEK293 cells transfected with Flag-tagged PD-1 and Myc-tagged FBW7 constructs. β-actin was used as equal loading control. (D) The primary PBMCs, THP-1 cells and MOLT-4 cells, which express endogenous PD-1 and FBW7, were precipitated with anti-PD-1/anti-FBW7 antibody or isotype IgG, and the resultant immunoprecipitates were subjected to immunoblotting. (E) The MOLT-4 and Jurkat cells were treated with increasing concentrations of oridonin and analyzed for PD-1 protein abundance by Western blot. (F) The THP-1 cells were transfected with indicated constructs or treated with increasing concentrations of oridonin. The expression of exogenous and endogenous FBW7 and PD-1 protein was determined by immunoblotting. (G) Primary T cells and MOLT-4 cells were treated with oridonin at final concentrations of 5 µM and 10 µM for 24 hours. After indicated treatment, cells were incubated with Alexa Fluor 647-conjugated anti-PD-1 antibody. Cell surface expression of PD-1 was assessed by flow cytometry. Representative flow cytometry images were shown. FBW7, F-box and WD repeat domain containing 7; PBMCs, peripheral blood mononuclear cells;DMSO,dimethyl sulfoxide.
To verify the results of bioinformatic analysis, we first assessed the effect of FBW7 on PD-1 mRNA transcription. The results of real-time PCR assay showed that FBW7 increased the mRNA level of PD-1, whereas its ΔF inactive mutant minimally affected PD-1 mRNA expression (online supplemental figure S1C). Blockade of the proteasome activity by MG132 entirely abolished FBW7-induced PD-1 transcription, suggesting that FBW7 primarily regulated PD-1 expression at protein level. Next, we transfected plasmids encoding FBW7 and PD-1 in HEK293 cells. Overexpression of Myc-tagged full-length FBW7 (FBW7 WT) readily reduced the protein abundance of Flag-tagged PD-1. In contrast, the FBW7 ΔF mutant restored the abundance of Flag-tagged PD-1 protein (figure 1B). To explore whether PD-1 directly bind to FBW7, pull-down assay with an anti-Flag antibody was performed and the result showed the Myc-tagged FBW7 protein coprecipitated with the Flag-tagged PD-1 protein. However, the removal of the WD40 domain (FBW7 ΔWD40), which is required for substrate binding, abolished the interaction between FBW7 and PD-1 (figure 1C). To collaborate with these notions, we investigated the FBW7-PD-1 interaction in physical conditions by using primary PBMCs. Fresh cell lysates of primary PBMCs were precipitated with anti-FBW7 or anti-PD-1 antibody and the resultant precipitates were subjected to immunoblotting. The immunoprecipitation experiment also confirmed that FBW7 and PD-1 mutually bind to each other (figure 1D). Monocytes and lymphocytes are two major components of PBMCs, as such, we investigated whether these cells were responsible for mediating the FBW7-PD-1 interaction. The human acute monocytic leukemia THP-1 and human T lymphoblast leukemia MOLT-4 cell lines express endogenous PD-1 and FBW7. Immunoprecipitation assay done in the two cell lines further confirmed an endogenous FBW7-PD-1 interaction in both monocyte-derived and T cell-derived cell lines (figure 1D). While FBW7 is frequently mutated in T cell leukemia, it was not surprizing to notice activation of FBW7 by a natural terpenoids oridonin readily suppressed PD-1 protein expression in the FBW7 WT MOLT-4 cells, it failed to do this in the FBW7 R505C mutant acute T cell leukemia Jurkat cells (figure 1E). In contrast, activation of FBW7 by pharmacological and molecular approaches in the FBW7 WT THP-1 cells resulted in decreased PD-1 protein abundance (figure 1F). Since PD-1 is a cell surface biomarker of immune cells, we assessed the significance of FBW7 on cell surface expression of PD-1. The primary human T cells and MOLT-4 cells were incubated with a fluorescence-conjugated anti-PD-1 antibody and analyzed by flow cytometry. The results suggested an immediate reduction in membrane PD-1 protein following 24 hours treatment with oridonin (figure 1G), whereas knockdown of endogenous FBW7 promoted membrane PD-1 expression (online supplemental figure S1D).
To investigate whether FBW7-mediated downregulation of PD-1 protein was due to the effects on protein turnover and stability, we expressed Flag-tagged PD-1 together with FBW7 WT or its mutant in HEK293 cells and measured the half-time of PD-1 protein. The Flag-tagged PD-1 protein became unstable when Myc-tagged FBW7 WT was introduced into HEK293 cells. However, the Myc-tagged FBW7 ΔF mutant failed to do this and the half-time of Flag-tagged PD-1 was largely restored (figure 2A). Pharmacological activation of FBW7 by oridonin also suppressed PD-1 protein stability and promoted its turnover in THP-1 and MOLT-4 cells (figure 2B), whereas inhibition of FBW7 by shRNAs led to an opposite effect (figure 2C,D). The protein–protein interaction was specific to PD-1 because we did not find a casual effect between FBW7 and PD-L1. As illustrated in figure 2E, overexpression of FBW7 did not affect the protein level of PD-L1. Consistently, there were no significant changes in PD-L1 protein half-time, even in the presence of FBW7 plasmid (figure 2F). Taken together, the FBW7 selectively interacts with PD-1 protein and promotes its degradation.
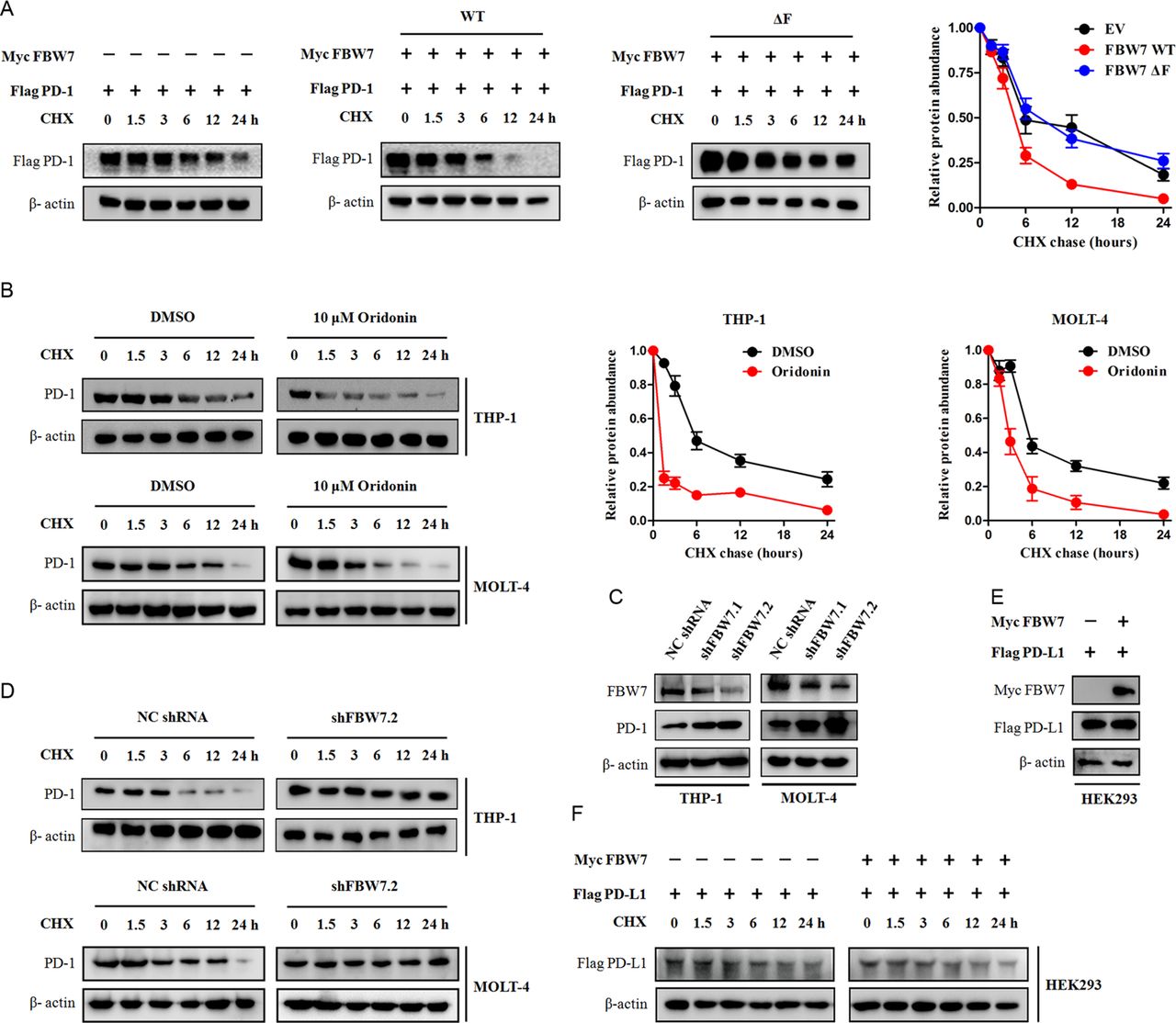
FBW7 determines PD-1 protein stability. (A) HEK293 cells were transfected with Flag-tagged PD-1 together with indicated Myc-tagged FBW7 constructs. Twenty hours after transfection, cells were split into 6 cm dishes and treated with 25 µg/mL CHX. At the indicated time points, cell lysates were prepared and probed with the Flag-tag antibody. β-actin was used as equal loading control. (B) The THP-1 cells and MOLT-4 cells were treated with 10 µM oridonin or DMSO and evaluated for PD-1 protein half-time by CHX chase assay. (C) The THP-1 cells and MOLT-4 cells were infected with shRNA lentivirus against FBW7 or negative control (NC), and selected with puromycin to eliminate non-infected cells. The resultant cells were analyzed for FBW7 and PD-1 expression by Western blot. (D) The THP-1 cells and MOLT-4 cells stably expressing shRNA targeting FBW7 were treated with CHX and evaluated for PD-1 protein half-time. (E, F) The Flag-tagged PD-L1 and Myc-tagged FBW7 constructs were expressed in HEK293 cells and evaluated for PD-L1 protein half-time by CHX chase assay. CHX, cycloheximide; FBW7, F-box and WD repeat domain containing 7;DMSO,dimethyl sulfoxide.
FBW7 E3 ligase ubiquitinates PD-1 protein
On the basis of FBW7 acting as a E3 ubiquitin ligase, we next investigated whether the binding to FBW7 resulted in PD-1 protein ubiquitination. The THP-1 and MOLT-4 cells were treated with oridonin and the endogenous PD-1 protein was immunoprecipitated and probed with anti-Ub antibody. The immunoprecipitation assay showed that oridonin enhanced PD-1 protein binding to FBW7 protein, as a consequence, leading to increased PD-1 protein ubiquitination (figure 3A). In consistent with this notion, the binding to exogenous FBW7 also markedly increased PD-1 protein ubiquitination. As shown in figure 3B, cotransfection with Flag-PD-1, Myc-FBW7, and HA-Ub plasmids in HEK293 cells resulted in a significant ubiquitination of Flag-PD-1 protein. In contrast, the Flag-PD-1 protein ubiquitination was abolished when the Myc-FBW7 plasmid was replaced by its catalytically inactive ΔF mutant. To map whether the PEST region of PD-1 protein is required for interacting with FBW7, an immunoprecipitation experiment was performed, and the result showed a dramatic reduction in FBW7 binding and ubiquitination on PEST region removal (figure 3C), indicating that the PEST region is essential and sufficient for interaction with FBW7.

FBW7 as a E3 uibiquitin ligase for PD-1 protein. (A) Western blot analysis of whole cell lysates (WCL) and immunoprecipitates derived from THP-1 and MOLT-4 cells treated with DMSO or oridonin (10 µM) for 24 hours, and cells were treated with 20 μM MG132 for additional 5 hours before harvesting. The ubiquitination status of endogenous PD-1 protein was determined by immunoprecipitation (IP). β-actin was used as equal loading control. (B) HEK293 cells were transfected with Flag PD-1 together with indicated Myc FBW7 construct. Thirty hours after transfection, cells were treated with 20 μM MG132 for additional 8 hours to block the proteasome pathway before cell collection. The cell lysates were subjected to IP and immunoblotting, respectively. (C) Immunoblotting analysis of Flag tag pull-down precipitates and WCL derived from HEK293 cells transfected with HA Ub, Myc FBW7 and indicated Flag PD-1 construct. Thirty hours after transfection, cells were treated with 20 μM MG132 for 8 hours before cell collection. (D) Myc-tagged FBW7, Flag-tagged PD-1, and His-tagged Ub WT, or Ub K48, or Ub K63 plasmids were expressed in HEK293 cells. PD-1 protein ubiquitination status was determined by Flag tag pull-down and anti-His immunoblotting. (E) Western blot analysis of WCL and immunoprecipitates derived from HEK293 cells transfected with Myc FBW7, HA Ub together with indicated Flag PD-1 construct. The ubiquitination status of Flag-tagged PD-1 protein was determined in the Flag tag pull-down precipitates by using anti-HA primary antibody. (F) The Flag-tagged PD-1 WT and its KR mutants were expressed in THP-1 cells. Thirty hours after transfection, cells were treated with or without MG132 before cell collection. The expression of Flag PD-1 protein was assessed by Western blot. FBW7, F-box and WD repeat domain containing 7; HA, hemagglutinin;DMSO,dimethyl sulfoxide.
The FBW7 primarily leads to K48 polyubiquitin linkage of its substrates, we therefore determined whether FBW7 promotes this kind of polyubiquitin linkage in PD-1 protein. To test this hypothesis, we generated two His-tagged Ub mutants that harbored only a single lysine residue (K48 only or K63 only) with all other lysines mutated to arginines. The immunoprecipitation assay indicated that the PD-1 ubiquitination status in HEK293 cells transfected with His-Ub K48 mutant was comparable to that in cells transfected with His-Ub WT plasmid. However, the ubiquitination level of PD-1 protein was significantly decreased when transfected with the His-Ub K63 mutant (figure 3D). As such, it was concluded that FBW7 predominantly promotes the K48 polyubiquitin linkage-mediated proteasomal degradation but not the K63-mediated signaling activation.
Since the K48 polyubiquitin linkage also occurs within the lysine residue of substrate protein, we determined which lysine residue is the ubiquitination site of FBW7 E3 ligase. The bioinformatic sequence alignment analysis suggested the K210 and K233 residues were highly conserved across a panel of species (online supplemental figure S2), raising the possibility that these two lysine residues might serve as potential ubiquitination sites. To verify the importance of K210 and K233 residues for FBW7-mediated PD-1 protein ubiquitination, we substituted each lysine residue or both with arginine and performed immunoprecipitation assay. Strikingly, amino acid substitution of the K210 residue had minimal effect on PD-1 protein ubiquitination, whereas mutation of the K233 residue sharply suppressed PD-1 protein ubiquitination (figure 3E). The substitution of both K210 and K233 residues did not further suppressed PD-1 protein ubiquitination, indicating that the K233 residue is crucial for FBW7 E3 ligase-mediated PD-1 protein ubiquitination. Ectopic expression of the KR mutant in THP-1 cells also suggested an increase in protein abundance in cells receiving K233R and K210/233R mutants (figure 3F). Collectively, these findings suggests that FBW7 E3 ligase ubiquitinates PD-1 protein at K233 residue, leading to proteasome-dependent degradation of PD-1 protein.
Supplemental material
Subcellular translocation of PD-1 protein underlies its interaction with FBW7
The FBW7 E3 ligase predominantly expressed in the nucleus of eukaryotes, and substrate proteins need to undergo nucleus translocation in order to interact with FBW7.20 Given that the PD-1 protein primarily localized in cell surface of immune cells, it is estimated that nucleus translocation primes PD-1 binding to FBW7. The removal of FBS in the culture medium efficiently resulted in enhanced binding to FBW7 and ubiquitination of PD-1 protein in THP-1 and MOLT-4 cells, respectively (figure 4A). Cell fraction analysis of endogenous PD-1 and exogenous Flag-tagged PD-1 indicated that a notable proportion of PD-1 protein being detected in the nucleus following FBS deprivation (figure 4B). In agreement with these notions, immunofluorescent staining of endogenous PD-1 protein in THP-1 and MOLT-4 cells showed the membrane-anchored protein gradually condensed in the nucleus in the absence of FBS (figure 4C).
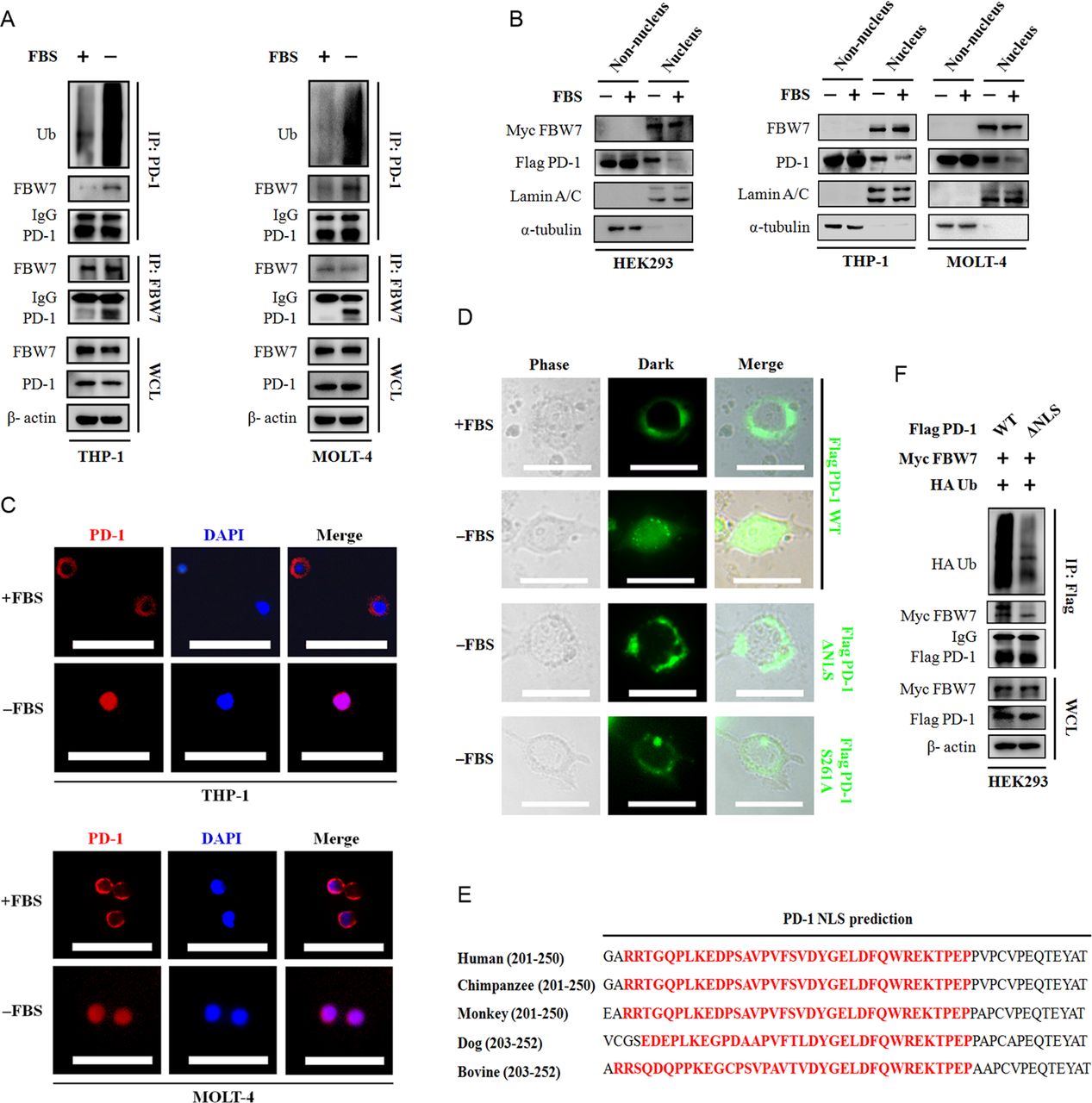
The nucleus translocation of PD-1 protein is required for its interaction with FBW7. (A) Immunoblotting of WCL and immunoprecipitates from THP-1 and MOLT-4 cells treated with or without FBS for 6 hours. MG132 was added into the culture medium 5 hours before harvesting. The ubiquitination status of PD-1 was assessed by using an antibody against Ub. β-actin was used as equal loading control. (B) HEK293 cells were transfected with Myc-tagged FBW7 and Flg-tagged PD-1 constructs.The resultant cells, THP-1 and MOLT-4 cells were cultured with or without FBS for 6 hours. After indicated treatment, cell fraction assay was performed and the expression of FBW7 and PD-1 in the non-nucleus lysates and nucleus lysates was determined by immunoblotting. α-tubulin and Lamin A/C were used as equal loading control for non-nucleus protein and nucleus protein, respectively. (C) Representative immunofluorescent images of the PD-1 protein localization in THP-1 and MOLT-4 cells cultured with or without FBS. Scale bar=100 µm. (D) The EGFP-tagged PD-1 constructs were transiently expressed in HEK293 cells and the distribution of PD-1 protein was visualized under a fluorescent microscope. Representative fluorescent images were shown. Scale bar=20 µm. (E) Alignment analysis of putative NLS region of PD-1 protein across a panel species. The conserved NLS region is highlighted in red. (F) Immunoblotting with the anti-Flag tag precipitates and WCL derived from HEK293 cells expressing Myc FBW7, HA Ub and Flag PD-1 WT or Flag PD-1 ΔNLS constructs. MG132 was added for 8 hour before cell collection. FBW7, F-box and WD repeat domain containing 7; HA, hemagglutinin; NLS, nuclear localization signal; WCL, whole cell lysates; FBS, fetal bovine serum.
To directly visualize the dynamic subcellular localization, PD-1 cDNA was in frame fused to EGFP at the C-terminal and transiently expressed in HEK293 cells. Consistent with our previous observations, FBS deprivation readily induced a striking movement of PD-1 protein from membrane to nucleus. The removal of NLS abolished PD-1 imported to the nucleus and the ΔNLS mutant was restricted to the cell membrane (figure 4D,E). As a consequence, PD-1 protein lacking NLS became stabilized (online supplemental figure S3) and was resistant to FBW7-mediated ubiquitination (figure 4F).
Supplemental material
CDK1-mediated PD-1 phosphorylation is required for ubiquitination
Protein phosphorylation within the CPD motif is required for the recognition by FBW7, such as GSK3-mediated phosphorylation of c-Myc primes its interaction with FBW7.21 In order to determine whether PD-1 protein also follows this dogma, HEK293 cells were transfected with the Myc-FBW7 and Flag-PD-1 plasmids and treated with a non-selective λ-phosphotase (λ-PP) before immunoprecipitation assay. Strikingly, dephosphorylation by λ-PP entirely abolished PD-1 binding to FBW7 (online supplemental figure S4A). Thus, phosphorylation of PD-1 would be essential for earmarking PD-1 protein nucleus translocation and degradation. An integrative alignment analysis of functional domains in PD-1 sequence converged at the Ser261 residue, which is predicted to be phosphorylated by CDK1 (online supplemental figure S4B). To characterize potential significance of CDK1 for PD-1 protein phosphorylation and degradation, we inhibited the CDK1 activity by the pan-CDKs inhibitor Alvocidib or the selective CDK1 inhibitor Ro-3306. As shown in figure 5A, the interaction between Flag-PD-1 and Myc-FBW7 was compromised when cells treated with both inhibitors. Of noted, treatment with Ro-3306 produced more profound suppressive effect on PD-1 protein ubiquitination and this treatment disrupted PD-1 protein nucleus translocation (figure 5A, online supplemental figure S4C). Using small interference RNA (siRNA) to knockdown CDK1 expression in THP-1 and using Ro-3306 to block CDK1 activity in MOLT-4 cells rescued PD-1 protein from being ubiquitinated (figure 5B). To verify the CDK1-mediated phosphorylation site in PD-1, we generated the phosphorylation resistant Flag-PD-1 S261A mutant and found that defects of Ser261 phosphorylation resulted in almost exclusively nucleus staining for PD-1 protein (figure 4D), which also impaired the PD-1 protein ubiquitination. In contrast, the phosphorylation mimicking S261D mutant was more fragile to FBW7-mediated ubiquitination (figure 5C). Thus, it was assumed that CDK1-mediated Ser261 phosphorylation was required and sufficient for the nucleus import of PD-1 protein. To support this notion, the PD-1 protein was precipitated with the anti-Flag antibody and probed with an antibody against importin α. As shown in figure 5D, the PD-1 S261A mutant failed to interact with importin α whereas the PD-1 S261D mutant elicited enhanced affinity to this nucleus translocator. Thus, we propose a model in which phosphorylation of PD-1 by CDK1 enables PD-1 trafficking and translocates into the nucleus where it interacts with FBW7 to undergo ubiquitination.
Supplemental material

Phosphorylation of PD-1 prompts FBW7-mediated ubiquitination and protein destruction. (A) HEK293 cells were transfected with Myc FBW7, Flag PD-1 and HA Ub constructs. Thirty hours after transfection, cells were treated with the pan-CDK inhibitor Alvocidib (10 µM) or the selective CDK1 inhibitor Ro-3306 (10 µM), together with 20 μM MG132 for another 8 hours. The ubiquitination status and the binding with FBW7 were determined with anti-Flag immunoprecipitates. β-actin was used as equal loading control. (B) The THP-1 cells and MOLT-4 cells were transfected with indicated plasmids and treated with either siRNA targeting CDK1 or Ro-3306 (10 µM). The ubiquitination status of PD-1 and its binding with FBW7 were assessed as described. (C) The Flag-tagged PD-1 and its S261A/S261D mutant was expressed in HEK293 cells. Phosphorylation status of the serine residue was determined in the anti-Flag immunoprecipitates by using an anti-pSer antibody. The ubiquitination status of PD-1 and its binding with FBW7 were assessed as described. (D) The effect of Ser261 phosphorylation on PD-1 ubiquitination was evaluated in HEK293 cells. Cells were transfected with indicated plasmids and cultured in the presence or absence of FBS for 6 hours. The binding to nucleus cotranslocator importin α was assessed in the anti-Flag immunoprecipitates. FBW7, F-box and WD repeat domain containing 7; WCL, whole cell lysates;FBS,fetal bovine serum.
Manipulating the PD-1-FBW7 pathway to enhance the antitumor efficacy of PD-1 blockade therapy
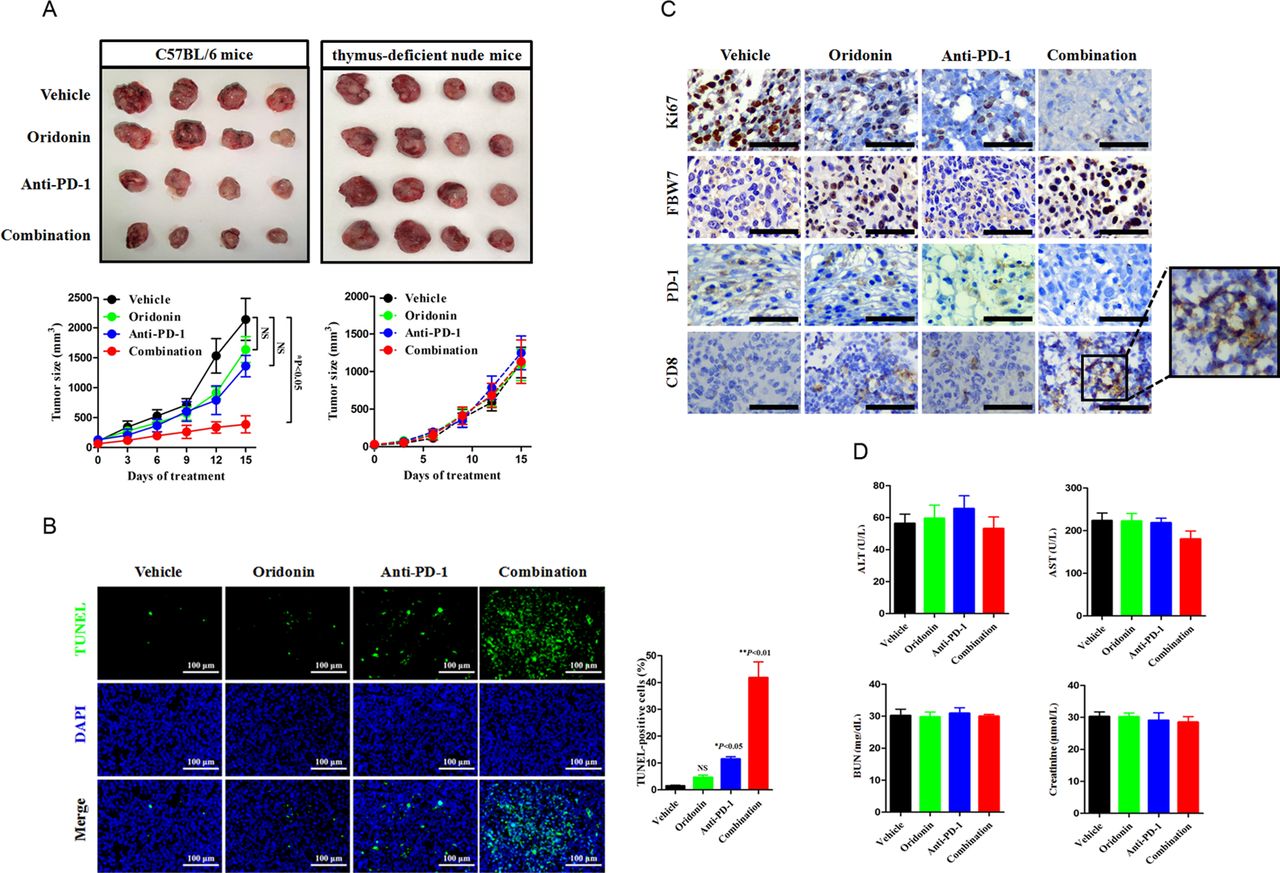
Given the observations in biochemical studies, we wondered whether FBW7 regulates PD-1 expression in primary tumors setting. The LLC cell injection into the syngeneic C57BL/6 mouse model enables perturbation of immunotherapy in immune competent host and provides an opportunity to assess whether a combinational strategy might be therapeutically efficacious. The anti-mouse PD-1 monoclonal antibody (200 µg per week) was administrated with or without oridonin (5 mg/kg every 3 days) by intraperitoneal injection, and the tumor size was routinely recorded and calculated. It was not surprizing that the combinational treatment yielded more profound antitumor efficacy, as the addition of a FBW7 agonist reduced the tumor burden over the use of PD-1 blockade agent alone (figure 6A). The combinational treatment also yielded a higher magnitude of cancer cell apoptosis, which was supported by increased proportion of TUNEL-positive cells (figure 6B). IHC staining of Ki67 revealed a sharp decrease in mice receiving the combinational treatment, and such reaction was accompanied by increased infiltration of CD8+cytotoxic lymphocytes (CTLs) (figure 6C). The combinational strategy was safe and did not increase the incidence of treatment associated toxicity because it did not further disrupted the hepatic and renal functions (figure 6D). To demonstrate that this enhancement of antitumor efficacy required the presence of T cells, we treated the immunocompromised mice with the same treatment and found that oridonin failed to synergize with PD-1 blockade therapy in thymus-deficient nude mice (figure 6A). Thus, a tumor microenvironment contexture with the enrichment of T cells dictates immunological response to anti-PD-1-based combinational therapy.
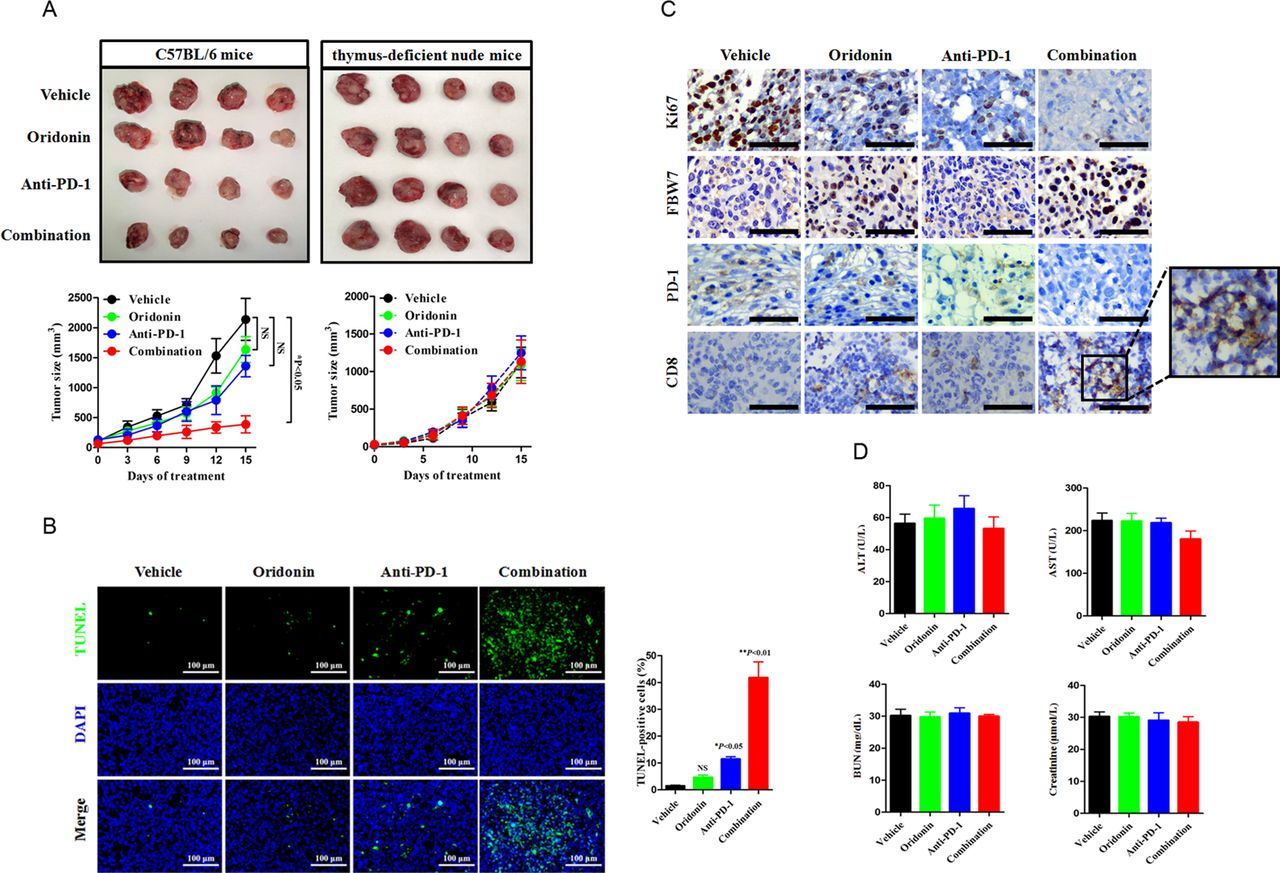
Cotargeting FBW7 enhances antitumor immunity in vivo. (A) The immune competent C57BL/6 J mice and the immune compromised nude mice were subcutaneously injected with LLC cells. The mice were treated with vehicle, oridonin, anti-PD-1 antibody or their combination. The tumor volume was routinely monitored. At the end of the experiment, the syngeneic tumor was carefully removed and photographed. NS, not significant; *p<0.05 vs vehicle. (B) Evaluation of cell apoptosis in the syngeneic tumor by TUNEL assay. Representative images were shown. Scale bar=100 µm. NS, not significant; *p<0.05 vs vehicle, **p<0.01 vs vehicle. (C) Representative images of IHC staining of Ki67, FBW7, PD-1 and CD8 in the syngeneic tumor. The enlarged window indicated the infiltration of CD8+CTLs in the tumor microenviroment. Scale bar=50 µm. (D) Measurement of hepatic and renal function in tumor bearing mice receiving indicated treatment. CTLs, cytotoxic lymphocytes; FBW7, F-box and WD repeat domain containing 7; LLC, Lewis lung cancer; AST, aspartate transaminase; ALT, alanine transaminase; BUN, blood urea nitrogen.
FBW7 status predicts clinical response to anti-PD-1 therapy in patients with NSCLC
To extend these results and determine their applications in patients treated with anti-PD-1 immunotherapy, we validated FBW7 as a biomarker predicting response to immunotherapy in patients with NSCLC. The correlation between FBW7 and PD-1 was verified in surgical resected samples from 23 NSCLC patients (stages I–IIIA) by IHC staining. It was noted that higher expression of PD-1 protein tended to be detected in NSCLC patients with relatively lower level of endogenous FBW7 protein, whereas higher level of FBW7 expression elicited an opposite outcome (figure 7A). The linear regression analysis of IHC score in the 23 cases of NSCLC also confirmed a negative correlation between FBW7 and PD-1 (figure 7B). These results indicated the FBW7-PD-1 interaction also existed in clinical setting, and highlighted that FBW7 status would be a novel biomarker implying response to antitumor immunity. For example, higher expression of FBW7 protein in the tumor microenvironment would accelerate PD-1 protein ubiquitination and degradation, which in return ameliorated the PD-1/PD-L1 immune suppressive pathway.

Screening FBW7 to predict response to anti-PD-1 immunotherapy in NSCLC. (A, B) A total number of 23 cases of surgical resected NSCLC were analyzed for FBW7 and PD-1 expression by IHC staining. Representative IHC images were shown. Scale bar=50 µm. Each NSCLC sample yielded an IHC H-score for FBW7 and PD-1, respectively. The correlation between FBW7 and PD-1 was determined by linear regression analysis.**p<0.01. (C) Evaluation of the tumor microenvironment in DCB (n=27) and NDB (n=21) patients. The panCK was used to distinguish cancer cells and non-cancerous cells. Representative IHC images for tumor nests (positive for panCK), FBW7, and infiltrating CD8+CTLs were shown. Scale bar=50 µm. ***p<0.001. CTLs, cytotoxic lymphocytes; DCB, durable clinical benefit; FBW7, F-box and WD repeat domain containing 7; NBD, no durable clinical benefit; NSCLC, non-small cell lung cancer; IHC, immunohistochemistry.
To distinguish the expression pattern of FBW7 and its association with the clinical response to immunotherapy, we retrospectively examined FBW7 status in 48 cases of patients with advanced NSCLC who received anti-PD-1 immunotherapy. These patients were divided into DCB group and NDB group based on their response to PD-1 blockade therapy. Pretreatment samples obtained from the DCB group who experienced a tumor response tended to have relatively higher level of FBW7, whereas the IHC result of FBW7 of the NDB group was opposite to that in the DCB group (figure 7C). While both cancer cells and non-cancerous cells in the tumor microenvironment express FBW7, we thus assess which component is the major source of FBW7. We found that cells positive for FBW7 minimally overlapped with pan cytokeratin (panCK), and a notable proportion of CD8+CTLs infiltrated the FBW7-positive tumor microenvironment in the DCB group, In contrast, the NDB group manifested as an immune desert microenvironment since there was no detectable CTLs. As such, it is reasonable to believe the FBW7-positive tumor microenvironment could imply ‘hot tumors’, whereas tumors negative for FBW7 would be ‘cold tumors’.
Discussion
Our study highlighted a novel regulatory machinery of PD-1 protein stability, in which we identified the FBW7 E3 ligase promotes its ubiquitination and proteasomal degradation, thereby facilitates the antitumor response of PD-1 blockade agents in NSCLC. By characterizing FBW7 status in NSCLC patients, we present a clinically relevant finding that higher expression of FBW7 confers increased sensitivity to anti-PD-1 immunotherapy. In this context, screening FBW7 status would facilitate the discrimination of responders and nonresponders before anti-PD-1 immunotherapy initiation may direct the application of immunotherapy to patients who are more likely to benefit from it, while it also provides the addition of FBW7 agonist strategy to FBW7low patients that are unlikely to show a response. Deciphering the molecular mechanism underlying the FBW7-PD-1 interaction would definitely accelerate our understanding of the PD-1/PD-L1 immune suppressive signaling.
The highly suppressive property of tumor microenvironment is a landmark of cancer and numerous efforts aiming to remove the immune suppressive executors are emerging.22 Recent studies have showed that ubiquitination, glycosylation, acetylation and palmitoylation are critical post-translational modifications of the immune suppressive PD-1/PD-L1 signaling. These modifications contribute to PD-1/PD-L1 protein trafficking, stabilization, subcellular localization, signaling transduction and dictate the efficacy of PD-1/PD-L1 blockade immunotherapy.23–26 Among these modifications, ubiquitination attracts increasing interest since a panel of agonists/antagonists for E3 ligases/deubiquitinases has been commercially available. Although there is a pioneering preclinical report in colon cancer indicating FBXO38 as a E3 ligase for PD-1 protein,27 it is yet not clear which E3 ligases are responsible for PD-1 ubiquitination in NSCLC. Our findings are not reminiscent of the FBXO38 experiments, in contrast, we report that FBW7 is a key determinant in directing PD-1 protein abundance and stability in NSCLC. This is a striking finding because the cell membrane-anchored PD-1 protein could be degraded by a nucleus-located E3 ligase. The most likely explanation for such disorientated pattern of regulation is that PD-1 protein traffics from cell membrane to nucleus. This presumed theory could be supported by the identification of nucleus PD-L1 protein in circulating tumor cells and doxorubicin-treated breast cancer cells.28 29 The PD-L1 protein moves to the nucleus in response to hypoxia, where it acts as a transcriptional factor and switches cancer cell apoptosis to pyroptosis.30 31 In this study, we identified nucleus localization of PD-1 protein, which is triggered by CDK1-mediated phosphorylation at Ser261 residue. The nucleus PD-1 protein binds to FBW7 E3 ligase and undergoes ubiquitination, which results in the termination of PD-1/PD-L1 immune suppressive signaling. This argues a possibility that kinases potentiate post-translational modification of PD-1/PD-L1 protein and correlate with the antitumor immune responses. In line with this notion, the CDK4/6 has been implicated to maintain PD-L1 protein stabilization (online supplemental figure S5) and selective CDK4/6 inhibitors have been demonstrated to cooperate with anti-PD-L1 immunotherapy in a doxycyclin-inducible animal model of ErbB2-positive breast cancer.32 However, we reported an intriguingly opposite result that the combination with CDK1 inhibitor antagonizes the antitumor efficacy of PD-1 blockade therapy (online supplemental figure S6). This paradox may attribute to distinct protein-protein interaction patterns in which the CDK4/6-PD-L1 interaction primarily occurs in cancer cells, whereas the CDK1-PD-1 crosstalk predominantly occurs in immune cells. Inhibition of CDK4/6 activity in cancer cells leads to cell cycle arrest and increases tumor immunogenicity. As such, the addition of CDK4/6 inhibitors to PD-L1 blocking agents elicits more favorable therapeutic outcomes. In our study, suppression of CDK1 would impair Ser261 phosphorylation of PD-1 protein in immune cells, thereby preventing its binding to FBW7 E3 ligase and stabilizing PD-1 protein. Thus, CDK1 inhibitors maintain the PD-1/PD-L1 immune suppressive signaling, which antagonizes the antitumor activity of PD-1 inhibitors. Taken together, the discriminations between CDK4/6 and CDK1, and between PD-L1 and PD-1, would therefore warrant increasing clinical cautions in the setting of their combinations.
Supplemental material
Supplemental material
Our study also highlighted the FBW7 E3 ligase as an inspiring target for remodeling the tumor microenvironment. Our preclinical LLC syngenic tumor model suggests that targeting FBW7 alongside anti-PD-1 therapy improves the antitumor efficacy, which may provide additional treatment options for patients unlikely to have durable response to anti-PD-1 immunotherapy alone. We found that expression of FBW7 is closely related to the infiltration of CD8+CTLs. Higher level of FBW7 expression would accelerate the removal of PD-1 protein, thus turning PD-1high/CTLslow tumors (cold tumors) into PD-1low/CTLshigh tumors (hot tumors) (figure 8). In our cohort, tumors from the DCB group were enriched in FBW7 expression and manifested as FBW7-positive ‘hot tumors’, whereas patients in the NDB group were FBW7-negative ‘cold tumors’. To the best of our knowledge, this study, for the first time, establishes a possible link that connects the FBW7 status in the tumor microenvironment with the clinical benefits of anti-PD-1 immunotherapy. Screening FBW7 status would serve as a biomarker predicting patient’s response to anti-PD-1 immunotherapy in NSCLC. More importantly, cotargeting FBW7 sharply increased CD8+CTLs infiltration and suppressed tumor outgrowth in the immunocompetent mouse model. Therefore, the FBW7 status could mirror the tumor microenvironment and screening its expression would predict response to anti-PD-1 immunotherapy in NSCLC. Therapeutic reactivation of FBW7 in ‘cold tumors’ may reconstruct the tumor microenvironment, and therefore, improve the efficacy of PD-1 blockade agents.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A schematic diagram of CDK1-mediated PD-1 and FBW7 interaction and their underlying implications for antitumor immunity. The PD-1 protein is primarily expressed in the cell membrane and FBW7 is expressed in the nucleus of immune cells. In the FBW7 OFF condition, PD-1 phosphorylation is suppressed by the CDK1 inhibitor and it could not undergo nucleus translocation. Thus, the PD-1 protein constitutively expresses in immune cells, which in return activates the PD-1/PD-L1 immune suppressive pathway and inhibits CTLs infiltration. Tumors harboring the FBW7low/PD-1high feature manifest as ‘cold tumor’ and are not sensitive to PD-1 blockade immunotherapy. In contrast, the CDK1-mediated phosphorylation of PD-1 protein at Ser261 residue triggers a rapid movement into the nucleus, where PD-1 binds to FBW7 and undergoes ubiquitination and destruction. Overexpression of FBW7 could enhance this interaction, as a consequence, leading to the suppression of PD-1/PD-L1 signaling (FBW7 ON). The CD8+CTLs infiltrates the FBW7high/PD-1low ‘hot tumor’, which confers increased responsiveness to anti-PD-1 immunotherapy. CTLs, cytotoxic lymphocytes; FBW7, F-box and WD repeat domain containing 7.
Collectively, this study suggests a pivotal role of FBW7 E3 ligase in regulating antitumor immunity. FBW7 serves as a novel biomarker reflecting the landscape of tumor microenvironment, and it also acts as a therapeutic target for remodeling the PD-1high/CTLslow cold tumors. The FBW7-PD-1 interaction elucidates a post-translational machinery for the immune suppressor PD-1 and establishes a new combinational strategy to sensitize anti-PD-1 immunotherapy.
Data availability statement
Data are available on reasonable request. Not applicable.
Ethics statements
Patient consent for publication
Ethics approval
The experiment using human specimens was approved by the Institutional Ethics Committee of Jinling Hospital (2018NZGKJ-60). All animal experiments were conducted in compliance with institutional guidelines and approved by the Ethical Review Committee of Jinling Hospital (2018GKJDWLS-03-106). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We would like to thank laboratories of Wenyi Wei (Beth Israel Deaconess Medical Center, Harvard Medical School) and Mien-Chie Hung (University of Texas MD Anderson Cancer Center/China Medical University) for helpful suggestions. We also express gratitude to all the patients and their families who participated in our study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
JL, LW and NH contributed equally.
Contributors JL, MY and YS wrote the manuscript, generated figures, designed experiments, performed experiments and analyzed data; LW and NH performed experiments, provided resources, analyzed data and edited the manuscript; YS and MY supervised the study, designed experiments, provided resources and edited the manuscript; DW, JN and SZ performed experiments, analyzed data and edited the manuscript; HL, TL and JY verified data and edited the manuscript. YS accepted full responsibility for the work and the conduct of the study. All coauthors read, edited and approved the manuscript.
Funding This study was sponsored by grants from National Natural Science Foundation of China (#81802301 to MY, #82002456 to DW), Natural Science Foundation of Jiangsu Province (#BK20180290 to MY,#BE2019719 to YS), and Science Foundation of China Post-doctoral Research (#2020M670093ZX to MY).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.