Article Text

Abstract
Background The Food and Drug Administration recommends that people living with HIV (PWH) with a CD4+ T cell count (CD4) ≥350 cells/µL may be eligible for any cancer clinical trial, but there is reluctance to enter patients with lower CD4 counts into cancer studies, including immune checkpoint inhibitor (ICI) studies. Patients with relapsed or refractory cancers may have low CD4 due to prior cancer therapies, irrespective of HIV status. It is unclear how baseline CD4 prior to ICI impacts the proportion of treatment-emergent adverse events (TEAE) and whether it differs by HIV status in ICI treated patients.
Methods We conducted a pilot retrospective cohort study of participants eligible for ICI for advanced cancers from three phase 1/2 trials in the USA and Spain. We determined whether baseline CD4 counts differed by HIV status and whether the effect of CD4 counts on incidence of TEAE was modified by HIV status using a multivariable logistic regression model.
Results Of 122 participants, 66 (54%) were PWH who received either pembrolizumab or durvalumab and 56 (46%) were HIV-negative who received bintrafusp alfa. Median CD4 at baseline was 320 cells/µL (IQR 210–495) among PWH and 356 cells/µL (IQR 260–470) among HIV-negative participants (p=0.5). Grade 3 or worse TEAE were recorded among 7/66 (11%) PWH compared with 7/56 (13%) among HIV-negative participants. When adjusted for prior therapies, age, sex, and race, the effect of baseline CD4 on incidence of TEAE was not modified by HIV status for any TEAE (interaction term p=0.7), or any grade ≥3 TEAE (interaction term p=0.1).
Conclusions There was no significant difference in baseline CD4 or the proportions of any TEAE and grade ≥3 TEAE by HIV status. CD4 count thresholds for cancer clinical trials should be carefully reviewed to avoid unnecessarily excluding patients with HIV and cancer.
- Immunotherapy
- CD4-Positive T-Lymphocytes
- Clinical Trials as Topic
- Oncolytic Viruses
Data availability statement
No data are available. Data are from existing published data that are noted in ClinicalTrials.gov.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
People living with HIV (PWH) are at increased risk of cancers for which immune checkpoint inhibitors (ICI) are currently approved. Recent prospective trials have demonstrated safety of ICI in PWH with CD4+ T cell count (CD4) ≥100 cells/µL. However, nearly 75% of recent ICI clinical trials excluded PWH. HIV-negative participants with advanced cancers generally do not have a CD4 eligibility threshold to enter ICI trials.
WHAT THIS STUDY ADDS
Among participants with advanced cancers entering ICI clinical trials that were studied baseline CD4 counts do not differ by HIV status, low CD4 counts occur irrespective of HIV status, and CD4 counts <350 cells/µL do not appear to increase the risk of treatment-emergent adverse events or impact survival in both PWH and HIV-negative participants.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Findings from this study support recommendations to review CD4 thresholds for cancer clinical trials that will allow inclusion of PWH with cancer in current and future ICI clinical trials.
Introduction
The incidence and risk of cancer in people living with HIV (PWH) is rising globally.1 However, in many countries PWH have historically been marginalized from receiving cancer care. They are significantly more likely to be diagnosed with cancers at advanced stage,2 less likely to receive treatment for some cancers,3–5 and more likely to be excluded from cancer clinical trials.6 Among those who are required to receive curative-intent treatment based on treatment guidelines, being HIV-infected is associated with higher likelihood of not receiving standard cancer treatment modalities.4 Those with AIDS-defining cancers or low CD4+ T cell counts are even less likely to receive cancer treatment.4
Immune checkpoint inhibitors (ICI) have emerged as a cornerstone of treatment for many advanced cancers. In the USA, an estimated 39% of patients with cancer are eligible to receive ICI.7 Current Food and Drug Administration (FDA) approvals for ICI include their use in many cancers for which PWH are at increased risk, including cervical cancer,8 head and neck squamous cell carcinoma,9 10 cutaneous squamous cell cancer,11 hepatocellular carcinoma,12–14 Merkel cell carcinoma,15 16 Hodgkin’s lymphoma,17–19 and lung cancer.20 21 ICI therapy may be particularly promising in PWH because expression of immune checkpoints such as programmed cell death-1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4)—the targets of currently approved ICI—is increased in the setting of chronic HIV infection, leading to T-cell exhaustion which may persist despite antiretroviral therapy (ART).22 PWH are also at increased risk for virus-associated cancers,23 and emerging evidence shows high response rates to ICI therapy in certain virus-driven cancers such as non-Hodgkin’s lymphoma and natural killer/T cell lymphomas.24–26
Two prospective clinical trials have shown that ICI therapy is safe among PWH with advanced cancer who are receiving suppressive ART and with CD4+ T cell counts greater than 100 cells/µL or 200 cells/µL,27 28 a finding that buttresses observational and retrospective studies that have also shown safety of ICI in PWH.29 30 Despite these studies, translating these findings to widespread inclusion of PWH in cancer studies has required substantial effort. The Cancer Therapy Evaluation Program has initiated efforts to increase inclusion of PWH in ICI cancer clinical trials,31 and emerging guidelines provide recommendations for ICI use in these populations.32 Despite the strong evidence and advocacy efforts, 74% of clinical trials involving the use of ICI done between 2019 and 2020 excluded individuals with HIV infection and 7% conditionally included PWH only if they had adequate immune function.33 The HIV Working Group of the American Society of Clinical Oncology—Friends of Cancer Research Project for Modernizing Eligibility Criteria in Cancer Studies recommends that participants with CD4 >350 cells/µL should be eligible for cancer clinical trials and that lower thresholds are often appropriate.34 More recently, the FDA recommended that PWH with CD4+ T cell counts <350 cells/µL ‘should generally be eligible if the patient has a potentially curable malignancy or for interventions in a later stage of development that have demonstrated prior activity with a given cancer’.35
There are no data to definitively inform stratification of clinical trial eligibility by CD4+ T cell counts above or below 350 cells/µL. This arbitrary cut-off has been widely used in HIV research but has not been validated in cancer clinical trials. Many malignancies among PWH present at low CD4 counts.36 Moreover, in cases of relapsed or refractory disease, despite adherence to ART resulting in well-controlled HIV, systemic chemotherapy may contribute to decreases in CD4 counts which may not improve following cessation of treatment.37 Low CD4 counts are also seen in people without HIV after receiving cytotoxic cancer treatment38 but is not a condition for clinical trial eligibility for HIV-negative individuals.
Data on the effect of CD4+ T cell counts on immune-related adverse events (irAE) are conflicting. Some studies suggest that falling CD4+ T cell counts among patients receiving ICI therapy may be associated with higher risk of irAE.39 Other studies suggest that patients with higher lymphocyte counts (and therefore likely higher CD4+ T cell counts) prior to receiving ICI therapy may have higher risk for irAE.40 41 Additionally, it is unclear whether there is a difference in the proportion of overall treatment-emergent adverse events (TEAE) and whether this is affected by pre-ICI CD4+ T cell counts among participants with and without HIV receiving ICI for advanced cancer.
We aimed to determine whether baseline CD4+ T cell counts differ by HIV status among patients in the USA and Spain receiving ICI for advanced cancer in prospective clinical trials, and whether the effect of CD4+ T cell counts on incidence of TEAE was modified by HIV status. Determining the association between baseline CD4+ T cell counts, the risk of TEAE and impact of CD4+ T cell count on survival could help to stratify treatment decisions with ICI for patients with cancer with and without HIV.
Methods
Study design and participants
We conducted a retrospective cohort study of participants who received ICI for advanced cancers in three contemporaneous phase 1/2 trials that enrolled patients between 2016 and 2021. The first trial (bintrafusp alfa) was a phase 2 single-center trial at the National Cancer Institute in the USA which included participants with metastatic or advanced human papillomavirus (HPV)-associated malignancies for which no effective therapy existed or where standard therapy had failed (NCT02517398 and NCT03427411).42 Participants were enrolled between January 2016 and July 2019 and received bintrafusp alfa, a bifunctional fusion protein composed of the extracellular domain of the human transforming growth factor-beta (TGF-β) receptor II linked to an anti-programmed death-ligand 1 (PD-L1) monoclonal antibody. All eligible participants were tested for HIV infection prior to enrollment. Participants with HIV were allowed if they were on stable ART with CD4+ T cell counts above 300 cells/µL; however, no such patients were entered. There was no CD4 count requirement for HIV-uninfected patients. The second trial, Cancer Immunotherapy Trials Network (CITN)−12, is an ongoing multicenter phase 1 trial at seven sites across the USA (NCT02595866). This trial included only participants with HIV and metastatic or locally advanced cancer for which no standard therapy existed or where standard therapy had failed.27 Participants included in this analysis were enrolled between April 2016 and June 2021 and received pembrolizumab, a monoclonal antibody targeting PD-1. All participants were required to have a CD4+ T cell count 100 cells/µL or greater. Participants with CD4+ T cell counts <200 cells/µL received prophylactic antibiotics against opportunistic infections. The third trial, DURVAST, was a multicenter phase 2 trial conducted in eight sites across Spain from the Spanish Lung Cancer Group (NCT03094286). This trial included only participants with HIV and metastatic or locally advanced cancers in which anti-PD-1 or anti-PD-L1 antibodies have demonstrated activity, or other cancers refractory to standard treatment or for which no standard treatment existed.28 Participants were enrolled between May 2017 and June 2018 and received durvalumab, a monoclonal antibody targeting PD-L1. There was no cut-off for CD4+ T cell counts, but all participants were required to have undetectable HIV viral load. Participants with CD4+ T cell counts <200 cells/µL received prophylactic antibiotics against opportunistic infections.
Statistical analysis
The primary objectives were to compare baseline CD4+ T cell counts between PWH and HIV-negative participants, to compare the proportion of TEAE between PWH and HIV-negative participants, and to determine whether the effect of baseline CD4+ T cell counts on incidence of grade 3 or higher TEAE was modified by HIV status. All participants enrolled in the three trials were included in the analysis. Adverse events included those that occurred from baseline to end of treatment (the first occurrence of each TEAE) and were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) V.4.03 for the DURVAST trial and CTCAE V.5.0 for the bintrafusp alfa trial and CITN-12.
We used a multivariable logistic regression model adjusted for sex, ethnicity, age, CD4+ T cell count, number of prior systemic therapies, prior radiotherapy, and HIV status. Variables for inclusion in the model were selected a priori based on being known or suspected risk factors for TEAE. We included an interaction term for CD4+ T cell count and HIV status.
Secondary analyses included the assessment of 3-year survival stratified by CD4+ T cell counts. Survival was measured from the time of study enrollment to time of death from any cause. The Kaplan-Meier method and log-rank tests were used to estimate and compare survival rates, respectively.
The distribution of baseline clinical and demographic characteristics between PWH and HIV-negative participants was compared using descriptive statistics including χ2 tests for categorical variables and Wilcoxon rank-sum tests for continuous variables.
All tests were two-sided with a significance level of p<0.05. Stata V.13 was used to perform all analyses (StataCorp, College Station, Texas, USA).
Results
All 122 participants from the three trials were included. Of these, 66 (54%) were PWH who received either pembrolizumab (CITN-12) or durvalumab (DURVAST), and 56 (46%) were HIV-negative who received bintrafusp alfa (table 1).
Participant characteristics at baseline*
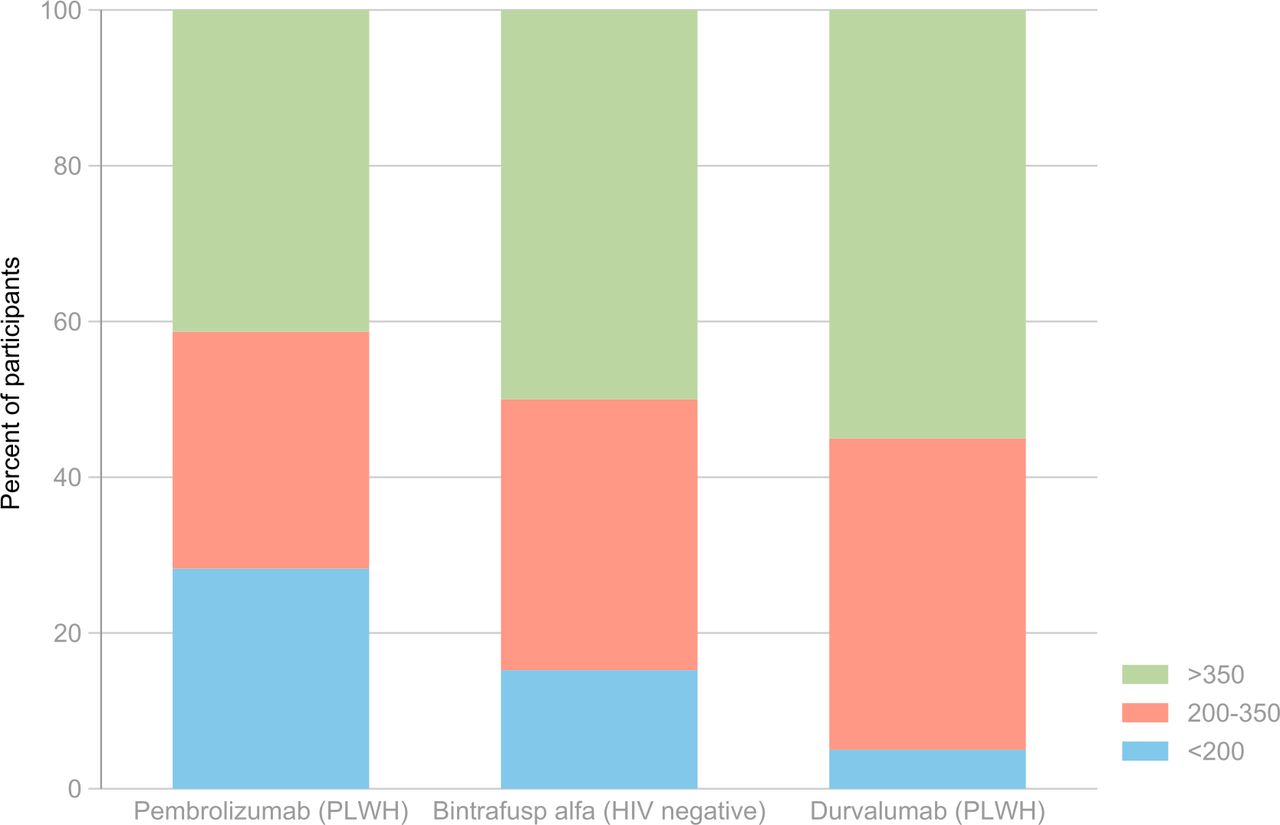
Distribution of baseline CD4+ T cell counts by clinical trial. PLWH, people living with HIV.
HIV-negative participants had HPV-related malignancies (anal cancer, cervical cancer, and head and neck cancer). PWH had both virus-associated (anal cancer, head and neck cancer, Kaposi sarcoma, hepatocellular carcinoma, and non-Hodgkin’s lymphoma) and non-virus-associated cancers (eg, bladder cancer, breast cancer, cholangiocarcinoma, lung cancer, and melanoma). Overall, 36% of participants were cisgender females and the median age at study entry was 54.5 years (IQR 47–62). Participants with HIV were predominantly cisgender male (88%) and white (65%), while participants without HIV were predominantly cisgender female (64%) and white (82%). All participants without HIV had received at least one prior systemic cancer therapy while 2% of participants with HIV had not received any prior systemic cancer therapy. The median number of systemic cancer therapies received prior to trial enrollment was highest in CITN-12 (2.5) followed by the bintrafusp alfa study (2.0), and the DURVAST study (1.0). Most (85%) participants with HIV had not received prior radiation therapy while 86% of participants without HIV had received prior radiation therapy. Nearly half of the participants overall had CD4+ T cell counts >350 cells/µL at baseline (figure 1). Among HIV-negative participants, one had a CD4+ T cell count <100 cells/µL and six had CD4+ T cell counts between 100 and 200 cells/µL. All PWH had CD4+ T cell counts >100 cells/µL, with 14 having CD4+T cell count between 100 and 200 cells/µL.
Median CD4+ T cell count at baseline was 320 cells/µL (IQR 210–495) among PWH and 356 cells/µL (IQR 260–470) among HIV-negative participants (p=0.5). When restricted to the two studies that did not have an eligibility cut-off for CD4+ T cell counts (bintrafusp alfa and DURVAST studies), the median CD4+ T cell count at baseline was 397 cells/µL (IQR 294–513) among PWH and 356 cells/µL (IQR 260–470) among HIV-negative participants (p=0.5). The proportion of participants with CD4+ T cell count <350 cells/µL did not differ significantly between PWH and HIV-negative participants (p=0.6). The baseline median absolute lymphocyte count was significantly higher among PWH (1400 cells/µL, IQR 1000–1900) than HIV-negative participants (900 cells/µL, IQR 600–1400) (p=0.001).
Among PWH, 49/66 (74%) experienced any TEAE compared with 43/56 (77%) among HIV-negative participants. Grade 3 or worse TEAE were recorded among 7/66 (11%) PWH compared with 7/56 (13%) among HIV-negative participants. There were two infection-related adverse events among PWH (one grade 2 and one grade 3 soft tissue infections) and one (grade 1 papulopustular rash) among HIV-negative participants (online supplemental table 1–3). Tables 2 and 3 show the results of the multivariable analysis of the effect of baseline CD4+ T cell counts on proportion of TEAE adjusted for prior therapies, age, sex, and race, with an interaction term between HIV status and baseline CD4+ T cell count category. The effect of baseline CD4+ T cell count on incidence of TEAE was not modified by HIV status for any TEAE (interaction term p=0.7), or any grade 3 or higher TEAE (interaction term p=0.1). The unadjusted estimate of the risk of TEAE with CD4+ T cell count <350 cells/µL was 5.19 (95% CI 1.35 to 19.96; p=0.02). When adjusted for prior systemic therapies, prior radiotherapy, age, sex, ethnicity, and HIV status, CD4+ T cell count <350 cells/µL was not significantly associated with increased risk of any TEAE (adjusted OR (aOR) 7.74; 95% CI 0.75 to 79.56; p=0.09) or any grade ≥3 TEAE (aOR 0.67; 95% CI 0.15 to 3.05; p=0.6).
Supplemental material
Effect of baseline CD4+ T cell counts on any treatment-emergent adverse effects
Effect of baseline CD4+ T cell counts on grade ≥3 treatment-emergent adverse effects
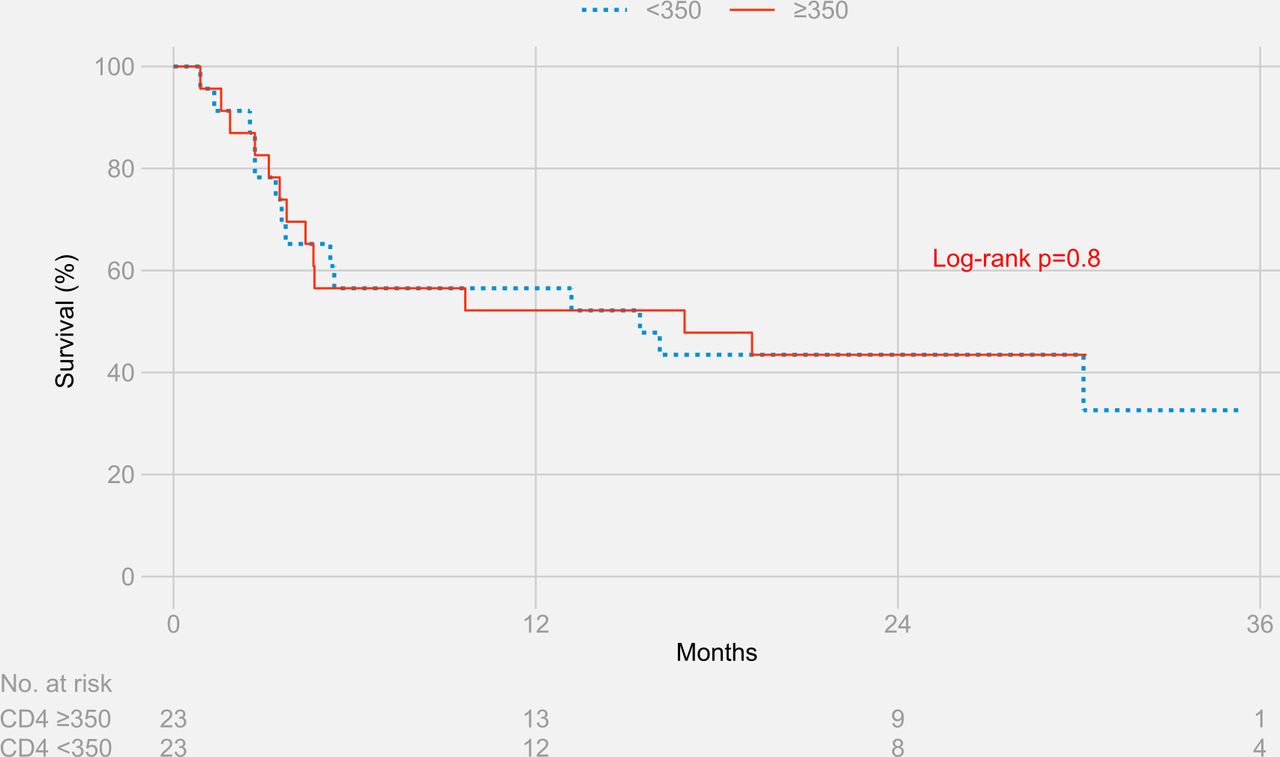
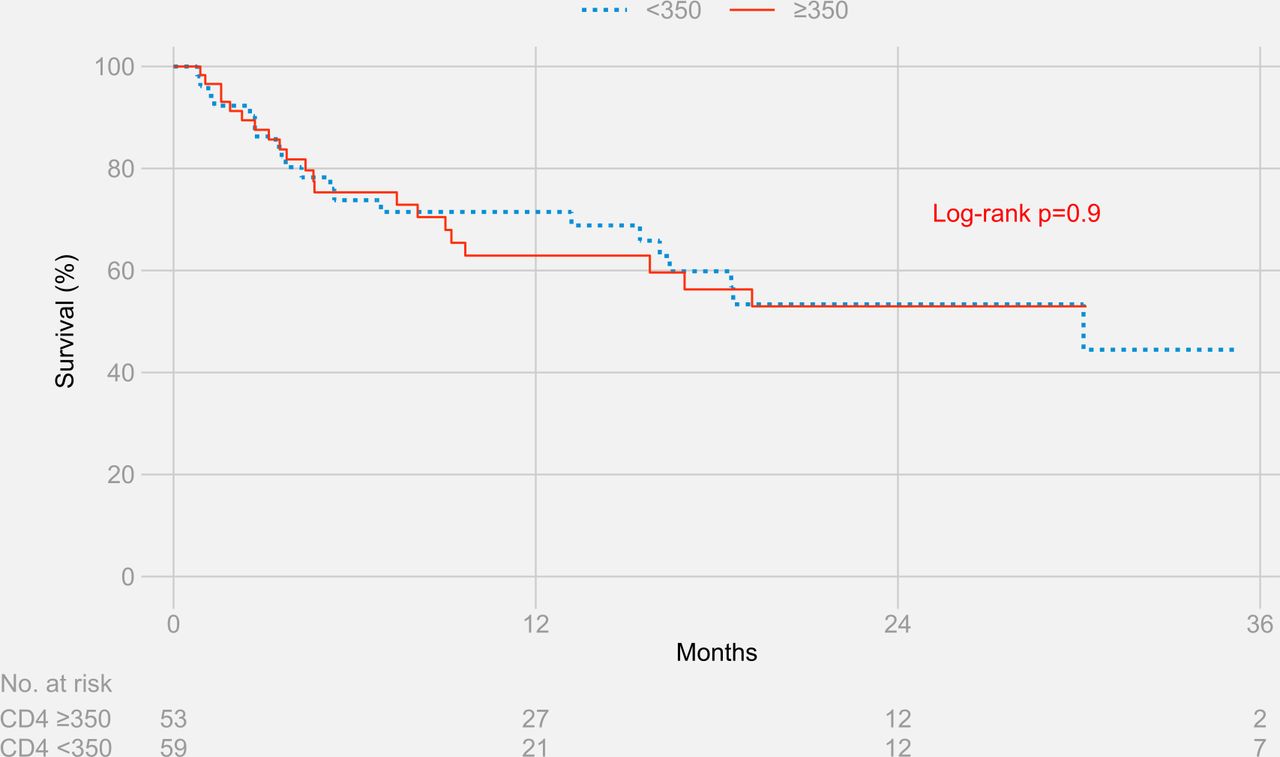
The Kaplan-Meier estimates for survival at 3 years were 44.5% for participants with CD4+ T cell counts ≥350 cells/µL and 53.0% for participants with CD4+ T cell counts <350 cells/µL (log-rank p value=0.9) (figure 2). Results remained similar when stratified separately by HIV status (figure 3 and figure 4)

Three-year survival stratified by CD4+ T cell counts in people living with HIV and HIV-negative participants.
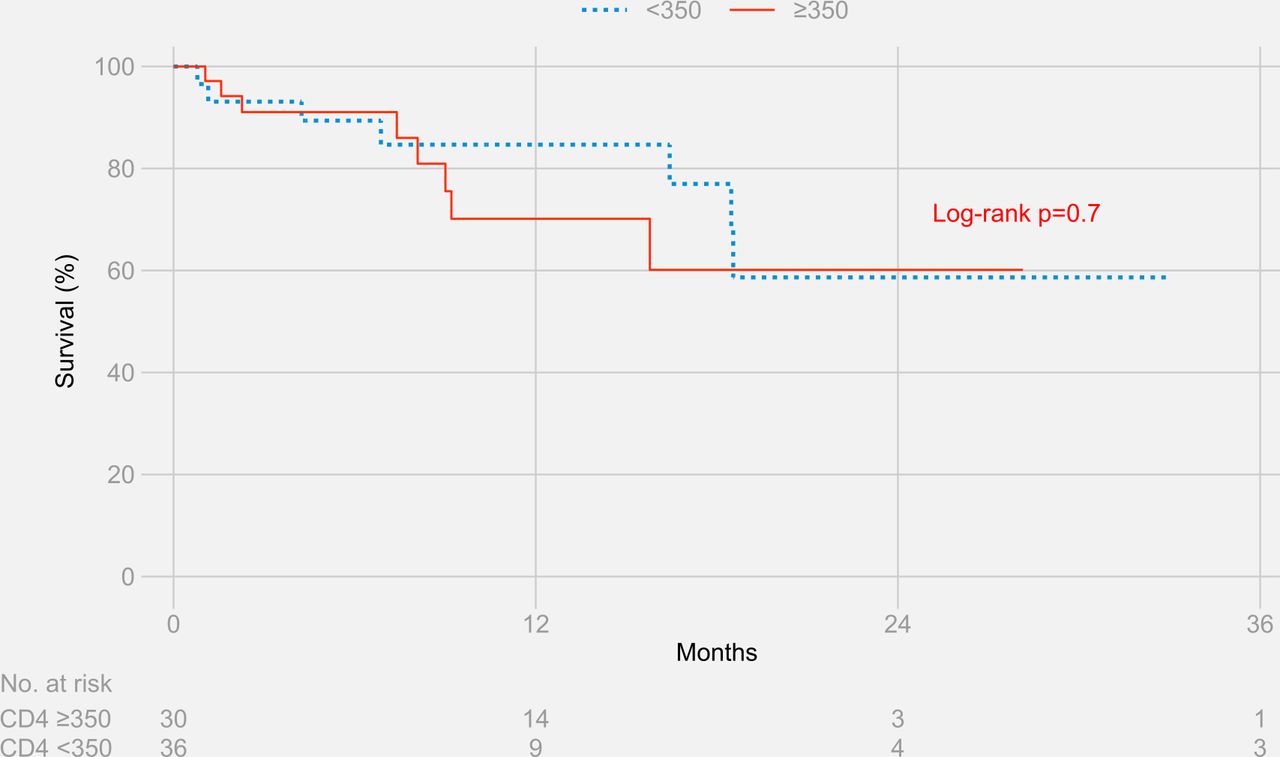
Three-year survival stratified by CD4+ T cell counts in people living with HIV.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Three-year survival stratified by CD4+ T cell counts in HIV-negative participants.
Discussion
Despite the demonstrated safety of ICI in PWH and cancer, only a quarter of recent ICI clinical trials included PWH, and the few trials that included PWH only enrolled a small number.33 Within the literature, PWH who have low CD4+ T cell counts are less likely to receive cancer treatment compared with HIV-negative individuals,4 which further marginalizes a population that is already at high risk of mortality from cancer.
In this study, we found that there was no significant difference in baseline CD4+ T cell counts comparing participants with and without HIV who had received prior cancer treatment and were subsequently enrolled in clinical trials to receive ICI therapy for advanced cancers. This finding did not change when comparing only studies that did not have an eligibility cut-off for CD4+ T cell counts (bintrafusp alfa and DURVAST studies). Importantly, low baseline CD4+ T cell counts were observed irrespective of HIV status among trial participants eligible for immunotherapy. Half of HIV-negative participants within these analyses had CD4+ T cell counts <350 cells/µL at baseline. The lowest CD4+T cell count in this population was 88 cells/µL. We found that the proportions of any TEAE and grade 3 or worse TEAE did not differ significantly between participants with and without HIV. In addition, we found that the association between baseline CD4+ T cell counts and the proportion of TEAE was not modified by HIV status. In CITN-12 and DURVAST which demonstrated safety of pembrolizumab and durvalumab in PWH with cancer, an important finding was that HIV viral load among study participants remained <400 copies/mL and that CD4+ T cell counts did not decline over the course of the trials.27 28 These findings support previous data on the safety of ICI by showing that being HIV-positive does not increase the risk of TEAE among participants receiving ICI,27 28 regardless of the CD4+ T cell counts at start of ICI therapy. Notably, there were only two recorded infection-related TEAE among PWH and one in HIV-negative participants. Our findings provide new evidence that low CD4+ T cell counts do not appear to increase incidence of TEAE among patients with and without HIV receiving ICI therapy for advanced cancers.
We also found that overall survival did not differ by CD4+ T cell counts for all participants when stratified by counts greater or less than 350 cells/µL. Previous studies on the use of ICI in PWH have not reported survival outcomes. To our knowledge, this is the first study to compare baseline CD4+ T cell counts between cancer trial participants with and without HIV who have received cancer treatment with ICI, and to compare TEAE rates and survival outcomes by CD4+ T cell counts.
Given that current clinical trials of ICI are largely in patients with cancer, inclusion of participants with HIV would provide additional opportunities to learn about viral biology through correlative studies. Pembrolizumab has been shown to enhance activation of HIV-1 specific CD8+ T cell without adverse effects.43 Emerging correlative results from the CITN-12 study indicate that pembrolizumab can reverse the latency of HIV in vivo.44 This study showed no evidence of HIV clonal expansion despite latency reversal, which provides additional safety data.
Clinical studies that simultaneously advance both the HIV cure and cancer treatment fields are only feasible if more trials of ICI therapy explicitly include and enroll PWH and cancer. In addition, strong consideration should be given to include PWH in trials of other immune and cellular therapies, including chimeric antigen receptor T-cell therapies, that also have the potential to contribute to both the cancer and HIV cure agendas.45
This study had several limitations. First, participants received different types of ICI therapy which bind to different epitopes, and which have different safety profiles. Although all three treatments are humanized monoclonal antibodies, durvalumab is an IgG1 antibody, pembrolizumab is an IgG4 antibody, and bintrafusp alfa is an IgG1 antibody linked to the extracellular domain of the human TGF-β receptor II. These differences may confound the comparison of proportion of TEAE between participants with and without HIV who received different types of ICI. This limitation further highlights the issue of exclusion of PWH from cancer immunotherapy clinical trials. Increased inclusion of this marginalized group in future studies will enable similar analyses to be conducted with less inherent bias or confounding. Regarding the cohort of PWH who received either pembrolizumab or durvalumab, the studies included in our analyses did not reveal any new safety signal.27 28 As an illustrative example (online supplemental table 4), a review of the literature showed that among participants with metastatic head and neck cancers who received durvalumab monotherapy, any grade of TEAE occurred in 57.1% to 63.1% of participants, and grade ≥3 TEAE occurred in 9.7% to 12.3% of participants.46–49 In the DURVAST trial included in our manuscript, 50% of participants experienced any TEAE, and all were grade <3, suggesting that the toxicities experienced by the HIV-positive participants in the DURVAST trial were neither more frequent nor more severe than what would be expected. Similarly, studies that evaluated pembrolizumab in advanced or metastatic HPV-associated cancers in HIV-negative individuals reported any TEAE ranging between and 58% and 65.3%,10 50 and grade ≥3 TEAE ranging between 12.2% and 18%.50 51 In the CITN-12 study included in our manuscript, 97% of participants who received pembrolizumab experienced any TEAE, and 20% experienced grade 3 TEAE. There were no grade 4 or 5 TEAE. Second, CITN-12 restricted enrollment to participants with CD4+ T cell counts >100 cells/µL. This may have skewed the median CD4+ T cell count among PWH upward. Interestingly, DURVAST did not have a CD4 cut-off, yet all enrolled participants had CD4+ T cell count >100 cells/µL. When we restricted the baseline comparison to only those studies that did not have a CD4 cut-off (bintrafusp alfa and DURVAST studies), there was no significant difference in median CD4+ T cell counts between participants with and without HIV. Third, there was heterogeneity of tumor types in each cohort. Attempts to address this limitation by conducting tumor-specific subgroup analyses were precluded by the small sample sizes for each tumor type. However, we do not feel this is likely to affect interpretation of the data as our results point to the broad safety of ICI among people with and without HIV with at least CD4+ T cells over 100 cells/µL and a broad variety of cancer types. A retrospective series from our group provides some support for the use of ICI in lower CD4+ T cell counts as one patient had a baseline CD4+ T cell count of 43 cells/µL and achieved a partial response with no TEAE.22 Fourth, baseline CD4+ T cell counts may be greater in patients not treated with prior lymphocytotoxic modalities such as radiation and chemotherapy. This would limit the generalizability of this study. Fifth, 86% of participants without HIV received prior radiation which is expected to decrease CD4+ T cell counts. To address this limitation, we adjusted for prior therapies in the multivariable model. Finally, the bintrafusp alfa trial of HIV-negative participants was a single-site study in the USA, which might limit the generalizability of our findings for HIV-negative participants. However, this study provides an important comparison cohort because it specifically examined HPV-associated cancers which are highly prevalent in PWH and is representative of an important early phase ICI study. Moreover, despite eligibility for the bintrafusp alfa study allowing for inclusion of PWH per FDA guidance, no PWH were enrolled in the study.
An important strength of this study was that CITN-12 and DURVAST are multicenter trials in the USA and Spain and therefore included a geographically diverse pool of participants, potentially broadening the applicability of our findings. Yet, global geographical disparities remain in cancer clinical trials for PWH due to the exclusion of sub-Saharan African and Latin American countries—where the burden of HIV and HIV-associated cancers is highest—from cancer clinical trials.52 Notably, there was a low proportion of black participants in these studies, highlighting previously demonstrated disparities in clinical trial enrollment among racial and ethnic minorities even in high-income countries.53 54 Expanding ICI clinical trial enrollment for PWH is an important step in reducing barriers to immunotherapy access.55 Overall, our findings provide strong supporting evidence for the safety of ICI therapies for PWH with advanced cancer. Though well-intentioned from a safety perspective, CD4+ T cell count thresholds represent a limitation or barrier for PWH otherwise eligible for cancer trials. These data suggest that rather than specific thresholds for PWH, cancer investigators may wish to consider scientifically meaningful inclusion criteria that considers the safety profile of the study agent. Participants with lower CD4+ T cell counts will require initiation of opportunistic infection prophylaxis as outlined by guidelines.35 56
In conclusion, this study shows that in these three early phase studies, baseline CD4+ T cell counts did not differ by HIV status among participants receiving ICI for a variety of advanced cancers. In particular, we have shown that HIV-negative trial participants with advanced cancer may have low CD4+ T cell counts at baseline. Importantly, we found that CD4+ T cell counts less than 350 cells/µL do not affect the risk of TEAE in both PWH and HIV-negative participants. Finally, we found that survival outcomes do not differ by CD4+ T cell counts. These results underscore the need for investigators and regulatory bodies to avoid unjustified thresholds and revise cancer trial eligibility criteria that permit inclusion of PWH with cancer. Not only will this reduce the barriers of PWH to partake of new advances in cancer treatment, but it will also open the doors to advancing the fields of cancer and HIV cure with potent new immune and cellular therapies.
Data availability statement
No data are available. Data are from existing published data that are noted in ClinicalTrials.gov.
Ethics statements
Patient consent for publication
Ethics approval
All studies were conducted in accordance with the principles of the Declaration of Helsinki and followed international standards consistent with the International Council for Harmonization E6 Guideline for Good Clinical Practice and were approved by the respective regulatory bodies at participating study sites. The original studies were approved by separate institutional review board (IRB)—NCT03094286 from eight hospitals (ICO-Badalona, Consorci Sanitaria de Terrassa, H. Clinic I Provincial de Barcelona, H Universitario Quiron Dexeus, Hospital Puerta de Hierro, H. La Paz, Hospital Virgen del Rocio, Hospital La Fe), NCT03427411—National Cancer Institute and NCT02595866—Fred Hutchinson Cancer. All participants provided written informed consent. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank the individuals who volunteered for this study and their families. We thank the nurses, physicians, the patient care, and clinical research support staff at the participating sites, including the Cancer Immunotherapy Trials Network 12 group and the Spanish Lung Cancer Group.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @gulleyj1, @ThomasUldrick
Contributors TAO and RR designed this study. TSU, MG-C, JLG, JS, RY, KL, ES, SPF and AW designed and led the clinical trials. KL, RR, TAO, JS, JM-P, TM, JLG, MG-C, TSU and RY cared for patients. MG-C, ES, SPF, AW, JM-P, TM, KL, JLG, JS and ES provided trial oversight. TAO and RR collected and analyzed data. All authors contributed to writing and approving the manuscript. Merck KGaA, Darmstadt, Germany, reviewed the manuscript for medical accuracy only before journal submission. The authors are fully responsible for the content of this manuscript and the views and opinions described in the publication reflect solely those of the authors. RR acts as the guarantor.
Funding This work was supported, in part, by the Intramural Program of the National Cancer Institute, National Institutes of Health, Department of Health and Human Services. Funding for the Cancer Immunotherapy Trials Network 12 (CITN-12; NCT02595866) study was obtained in part from the NCI UM1CA154967. Additional funding in support of clinical trial NCT02595866 was provided by Merck and Co. Inc., Kenilworth, New Jersey, USA. The DURVAST study NCT03094286 was sponsored by the Spanish Lung Cancer Group and funded by AstraZeneca.
Competing interests RR, TSU, KL, and RY report receiving research support from Celgene (now Bristol Myers Squibb) through a CRADA at the NCI. RR, TSU, KL, and RY report receiving drug for a clinical trial from Merck through a CRADA at the NCI. RR, KL, and RY report receiving drug for a clinical trial from EMD-Serono through a CRADA at the NCI. RY reports receiving drug for preclinical studies from Janssen and CTI BioPharma. TSU reports receiving other commercial research support from Roche through a CTA with Fred Hutchinson Cancer Center. JM-P reports receiving research support from AstraZeneca (through the Spanish Lung Cancer Group) and Merck. TSU and RY are coinventors on US Patent 10001483 entitled ‘Methods for the treatment of Kaposi’s sarcoma or KSHV-induced lymphoma using immunomodulatory compounds, and uses of biomarkers’. RY is also a coinventor on patents on a peptide vaccine for HIV and on the treatment of Kaposi sarcoma with IL-12, and an immediate family member of RY is a coinventor on patents related to internalization of target receptors, on KSHV viral IL-6, and on the use of calreticulin and calreticulin fragments to inhibit angiogenesis. All rights, title, and interest to these patents have been or should by law be assigned to the US Department of Health and Human Services; the government conveys a portion of the royalties it receives to its employee inventors under the Federal Technology Transfer Act of 1986 (P.L. 99-502). No potential conflicts of interest were disclosed by the other authors. TSU is currently an employee of Regeneron.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.