Article Text

Abstract
Background There is an increasing demand for chimeric antigen receptor (CAR) T cell products from patients and care givers. Here, we established an automated manufacturing process for CAR T cells on the CliniMACS Prodigy platform that is scaled to provide therapeutic doses and achieves gene-transfer with virus-free Sleeping Beauty (SB) transposition.
Methods We used an advanced CliniMACS Prodigy that is connected to an electroporator unit and performed a series of small-scale development and large-scale confirmation runs with primary human T cells. Transposition was accomplished with minicircle (MC) DNA-encoded SB100X transposase and pT2 transposon encoding a CD19 CAR.
Results We defined a bi-pulse electroporation shock with bi-directional and unidirectional electric field, respectively, that permitted efficient MC insertion and maintained a high frequency of viable T cells. In three large scale runs, 2E8 T cells were enriched from leukapheresis product, activated, gene-engineered and expanded to yield up to 3.5E9 total T cells/1.4E9 CAR-modified T cells within 12 days (CAR-modified T cells: 28.8%±12.3%). The resulting cell product contained highly pure T cells (97.3±1.6%) with balanced CD4/CD8 ratio and a high frequency of T cells with central memory phenotype (87.5%±10.4%). The transposon copy number was 7.0, 9.4 and 6.8 in runs #1–3, respectively, and gene analyses showed a balanced expression of activation/exhaustion markers. The CD19 CAR T cell product conferred potent anti-lymphoma reactivity in pre-clinical models. Notably, the operator hands-on-time was substantially reduced compared with conventional non-automated CAR T cell manufacturing campaigns.
Conclusions We report on the first automated transposon-based manufacturing process for CAR T cells that is ready for formal validation and use in clinical manufacturing campaigns. This process and platform have the potential to facilitate access of patients to CAR T cell therapy and to accelerate scaled, multiplexed manufacturing both in the academic and industry setting.
- Cell Engineering
- Immunotherapy
- Receptors, Chimeric Antigen
- Translational Medical Research
Data availability statement
Data are available on reasonable request. All data directly relevant to the study are included in the article. Further data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Conventional manufacturing of chimeric antigen receptor (CAR) T cells is complex requiring multiple labor-intensive open handling steps and a dedicated infrastructure. Thus, automated, device-based processes will not only reduce potential operator errors and the risk for (cross-) contaminations but also associated expenses.
WHAT THIS STUDY ADDS
We developed the first automated, closed, cGMP-compliant, transposon-based process for the manufacture of CAR T cells. Our TCE process yields CAR-engineered T cells with a favorable early memory phenotype at clinical scale. CD19 CAR T cells conferred potent and specific anti-tumor reactivity in vitro and in vivo.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This process and platform have the potential to facilitate widespread patient access to CAR T cell therapy. It is now ready to be advanced to formal validation testing for clinical CAR T cell products and implemented in academic and industry manufacturing sites.
Background
Immunotherapy with chimeric antigen receptor (CAR) T cells has demonstrated therapeutic potential for treating B cell leukemia and lymphoma and multiple myeloma, resulting in a steadily increasing demand for CAR T cell products by caregivers and patients.1–4 At present, ‘conventional’ CAR T cell manufacturing takes place in labor-intensive production campaigns that entail a complex sequence of manual and ‘open’ process steps, with little to no automation.5–7 Automated manufacturing of advanced therapy medicinal products in closed-system devices has been shown to reduce operator errors and the risk for contamination and cross-contamination.8–10 Therefore, automation is an intuitive strategy to scale-up and standardize CAR T cell manufacturing with the ambition to increase patient access and improve outcome, as well as to reduce complexity and cost associated with this therapy.
Substantial effort and expense is currently invested in procuring viral vectors for CAR-modification and accordingly, there is significant interest to implement virus-free gene-transfer vectors in CAR T cell manufacturing.11 12 The Sleeping Beauty (SB) transposon system is virus-free and has been used successfully to accomplish CAR gene-transfer in human T cells in conventional pre-clinical and clinical manufacturing.5 6 SB transposase and transposon can be vectorized as mRNA and DNA, respectively, and handled with lower risk and in a lower biosafety level environment compared with viral vectors. Therefore, the implementation of SB-based gene-transfer on an automated CAR T cell manufacturing device is conducive to both cost–efficient point-of-care production at academic sites, as well as industry-scaled centralized CAR T cell manufacturing.
We and others have recently shown that the insertion of plasmid DNA that contains bacterial CpG motifs to encode SB transposase and transposon leads to an inflammatory response and consequently poor viability and a low percentage of target cells that stably express the transposon.6 13 14 As an alternative, we have therefore used mRNA or minicircle (MC) DNA to encode SB transposase and MC DNA to encode SB transposons. This strategy leads to reduced toxicity, augmented gene-transfer rate and increased absolute yield of CAR T cells that can be obtained within the time period that is typically available for CAR T cell manufacturing in the clinical setting.6 MCs are plasmid-derived circular DNA vectors without prokaryotic origin of replication and without antibiotic resistance gene and are derived from a parental plasmid by intramolecular recombination.15
In this study, we have manufactured SB transposon-based CAR T cells using the CliniMACS Prodigy T Cell Engineering (TCE) process. Electroporation and culture conditions have been optimized to maximize the yield of CAR T cells that can be obtained within twelve days of culture. The robustness and reproducibility of the process was assessed in manufacturing campaigns that were performed on different devices and controlled by different operators.
Material and methods
MC-encoded SB transposase and transposon
MC DNA encoding SB100X transposase16 and a pT2 CD19 CAR transposon were produced at PlasmidFactory and have been previously described. The CD19 CAR sequence encodes a FMC63-derived scFv linked to an IgG4 hinge spacer, a 4-1BB co-stimulatory and a CD3z signaling domain, a T2A sequence as well as a truncated epidermal growth factor receptor (EGFRt).17
Manual small-scale nucleofection and electroporation
T cells were isolated from healthy donor leukapheresis (LP) or buffy coats using either a Pan T cell isolation Kit or a CD8+ isolation Kit and activated using T Cell TransAct, human (all Miltenyi Biotec). Three days later, aliquots of 1E6 activated T cells were either nucleofected using a 4D-Nucleofector (Lonza) according to the manufacturer’s instructions (P3 Primary cell 4-D nucleofector Kit, program FI-115) or electroporated using a CliniMACS Prodigy connected to the CliniMACS Electroporator (Miltenyi Biotec). Electroporation was carried out in CliniMACS Electroporation Buffer (Miltenyi Biotec) using the following pulse combination: I. 950 V 104 µs burst/bi-polar and II. 400 V 2000 µs burst.
Automated large-scale electroporation and CAR T cell manufacturing
Manufacturing campaigns were done on a CliniMACS Prodigy connected to the CliniMACS Electroporator. Technical details and handling of the CliniMACS Prodigy have been described.9 18 19 Unless mentioned to the contrary, all materials were obtained from Miltenyi Biotec. In brief, a CliniMACS Prodigy tubing set TS520 was sterile welded to a CliniMACS Prodigy EP-2 electroporation accessory and installed on the Prodigy and electroporator unit. A LP was connected to the tubing set, the white blood cell count determined and CD4 and CD8 T cells isolated using CliniMACS CD4 Reagent and CliniMACS CD8 Reagent. Then, 2E8 T cells were resuspended in TexMACS GMP medium supplemented with 12.5 ng/mL MACS GMP recombinant human IL-7 (rhIL-7) and MACS GMP recombinant human IL-15 (rhIL-15), and activated with 4 mL MACS GMP T cell TransAct for 3 days. During these 3 days, T cells were maintained in agitated culture. For electroporation, T cells were automatically rebuffered in 20 mL CliniMACS Electroporation Buffer and transferred into the Cell Bag of the EP-2. The Nucleic Acid Bag of the EP-2 was pre-loaded with 495 µg MC DNA (PlasmidFactory) (ie, 270 µg pT2 CD19 CAR transposon MC and 225 µg SB100X transposase MC). Electroporations were done sequentially on aliquots of about 600 µL (cell/DNA ratio 5.7:1) for 33 times using the same pulse as in the small-scale electroporation. Post-electroporation, T cells were recovered in 76 mL TexMACS GMP medium supplemented with 3% heat-inactivated human AB serum (Capricorn Scientific) as well as 12.5 ng/mL of rhIL-7 and rhIL-15, respectively, and cultured according to a pre-defined activity matrix that specifies the chronological order of all process steps (including washing and media exchange procedures). The complete TexMACS GMP medium did not contain serum after day 6.
Phenotypic analyses of CAR T cells
Cellular composition, CD4/CD8 ratio and CAR-expression was determined by flow cytometric analysis after staining with one or several of the following antibodies and reagents: anti-CD45, anti-CD4, anti-CD3, anti-CD16, anti-CD56, anti-CD19, anti-CD14, anti-CD8, anti-CD45RO, anti-CD95, anti-CD62L and 7-AAD (all Miltenyi Biotec). CAR-modified T cells were identified using the EGFRt marker and staining with an anti-EGFRt antibody (clone: C225, ImClone Systems) conjugated in-house to AlexaFluor 647. Data were acquired on a MACSQuant Analyzer 10 and analyzed using MACSQuantify software (both Miltenyi Biotec).
Functional analyses of CAR T cells
Cytolytic activity was assessed by co-culturing 1E4 CAR-modified or non-CAR-modified control T cells (mock) and GFP-labeled target cells at an effector-to-target ratio of 1:1 for 24 hours before analyzing the frequency of residual viable target cells by flow cytometry. Cytokine production was analyzed from the same co-culture by harvesting 100 µL supernatant from the 24 hours co-culture and quantification using the human MACSPlex Cytokine 12 Kit (Miltenyi Biotec) according to the manufacturer’s instructions.
All in vivo studies were approved by the Institutional Animal Care and Use Committee of the University of Würzburg. In brief, NSG mice (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ, Charles River) were inoculated with 1E6 firefly luciferase expressing Raji tumor cells by intravenous (i.v.) injection, and 7 days later treated with 4.7E6 T cells from the final cell product (from run #3, containing 2E6 CAR T cells) or 4.7E6 mock T cells (also i.v.). Serial bioluminescence imaging on an in vivo imaging system Lumina imager (Perkin Elmer) was performed to assess tumor burden and distribution.
Transposon copy number analyses by droplet digital PCR
Genomic DNA (gDNA) of CD19-CAR T cell products was isolated at the end of each large scale run using PureLink gDNA Mini Kit (Invitrogen, California, USA). Droplet digital PCR (ddPCR) was set up and analyzed in technical duplicates. Each ddPCR reaction contained ready-to-use ddPCR Supermix for Probes No dUTP (Bio-Rad, California, USA), 20 ng of the respective fragmented gDNA, 0.1 µL of CviQI (10 000 U/mL) (NEB, Frankfurt am Main, Germany), 0.1 µL of DpnI (20,000 U/mL) (NEB, Frankfurt am Main, Germany), 200 nM of each respective ddPCR FAM/HEX probe, and 600 nM of each respective forward and reverse primer in a final volume of 25 µL. CviQI was used to fragment the gDNA to ensure single-copies per droplet. DPNI was used to specifically digest non-integrated vectors. The ddPCR CAR probe (FAM-agcagcatggtggcggcgct-BQH1), and primer CAR forward (5′- atctggatgtcggggatcag −3′) and primer CAR reverse (5′- gcttgctcaactctacgtct −3′) were designed to bind in the CAR sequence to amplify integrations resulting from CAR transposition events. Ribonuclease P/MRP 30 subunit (RPP30) gene was used as the copy number reference (two copies per genome) with ddPCR RPP30 probe (HEX-tggacctgcgagcgggttctgacc-BHQ1), RPP30 forward (5′- ggttaactacagctcccagc −3′) and RPP30 reverse (5′- ctgtctccacaagtccgc −3′). QX200 droplet generator (Bio-Rad, California, USA) and PX1 PCR Plate Sealer (Bio-Rad, California, USA) were used to generate droplets and seal the reactions. The subsequent PCR amplification was performed in C1000 Touch Thermal Cycler (Bio-Rad, California, USA) using the following program: 95°C 10 min; 40 cycles of 94°C 30 s, 50°C 30 s, 60°C 1 min; and final extension of 98°C 10 min. Droplet fluorescence measurements for each sample were performed by a QX200 Droplet Reader (Bio-Rad, California, USA). The results were analyzed using QuantaSoft (Bio-Rad, California, USA) with manual thresholds and Poisson distribution with 95% CI.
Gene expression analyses on CAR T cells
2E5 T cells from the final cell product (run #3) were lysed for 20 min in a 1:3 mix of RLT buffer (Qiagen, Hilden, Germany) and water. Native CD4 T cells and CD8 peripheral blood T cells were used as controls. Lysed samples were processed for gene expression analysis using the nCounter CAR T characterization panel from Nanostring (Nanostring, Seattle, Washington, USA.), following the manufacturer’s instructions. For hybridization, 200 µg/mL Proteinase K (Invitrogen, Waltham, Massachusetts, USA) was added to the master mix. Mastercycler epgradient (Eppendorf, Hamburg, Germany), nCounter Pro Prep Station and nCounter Digital Analyzer (Nanostring) were used for further sample processing. Counting was performed using the setting 555 fields of view.
Data were first analyzed with nSolver analysis software V.4.0 using default settings for quality control and for normalization on positive control, housekeeping genes (without UBB) and sample references. Data were analyzed with a HyperScale architecture developed by Rosalind Inc (San Diego, California, USA) using default settings for fold changes and p values. The threshold for the log2 fold change was set between 1.5 and −1.5 and the significance threshold (p-adj.) was set at 0.01.
Results
Validation of bi-pulse electroporation for gene-transfer into T cells
MC-encoded SB100X transposase, as well as pT2 eGFP and pT2 CD19 CAR MC transposon donor vectors were generated from parental plasmid DNA and their size verified by gel electrophoresis (figure 1A). We used an electroporator unit with controlled pulse strength, length and mode and performed bi-pulse electroporations with activated T cells where we applied a first pulse that comprised a change in the electric field direction and a second pulse that comprised an unidirectional electric field (figure 1B). We performed electroporations with MC-encoded SB100X transposase either in combination with GFP MC or CD19 CAR MC transposon into T cells from N=3 donors and assessed T cell viability and gene-transfer rate. On day 3 after electroporation, T cell viability and GFP expression were >66% and >86%, respectively, indicating that the bi-pulse shock was appropriate and well tolerated by activated human T cells (figure 1C). Consistent with our prior work, we did not observe CAR-expression in T cells that had only been electroporated with the CD19 CAR MC transposon in the absence of SB100X transposase6 (figure 1D).

Small scale transfection of T cells using bi-pulse electroporation. (A) Restriction digestion analysis of conventional plasmids and MCS. Lane 1: SB100X plasmid digested with Sac I; Lane 2: SB100X MC digested with Pac I; Lane 3: PT2 CD19 CAR plasmid digested with Nhe I; Lane 4: PT2 CD19 CAR MC digested with Pac I, Lane 5: PT2 EGFP plasmid digested with Nhe I; Lane 6: PT2 EGFP MC digested with Pme I; lanes M: 1 kb DNA ladder (NE). (B) Schematic representation of applied bi-pulse system (red=first pulse, green=second pulse). (C) Viability and transfection efficiency (TE) of GFP-MC electroporated (Pulse) or non-electroporated (No pulse) T cells using the established bi-pulse system. (D) Episomal CAR-expression from MC vectors. T cells were electroporated with CD19 CAR transposon donor MC vector (CAR TP) in the presence or absence of transposase (SB100X), CAR-expression was assessed on day 6. (E–G) CD8 T cells were (co-)electroporated with mock (m, no DNA) or CD19 CAR and SB100X either encoded on plasmid (P) or MC in equimolar amounts using the two different electroporation systems. (E) viability, (F) transfection efficiency and (G) a representative flow cytometry analysis of anti-EGFRt staining 12 days after electroporation are shown. (C), (E, F) show mean+SD from N=3 donors. Statistical analysis was performed using paired t-test.
We sought to ascertain our prior observation of markedly increased T cell viability and gene-transfer rate when MC DNA instead of plasmid DNA was used to encode transposase and transposon. To provide a point of reference, we performed nucleofections using an optimized protocol that we established in previous work.6 T cells were electroporated vs. nucleofected with MC DNA encoding SB100X transposase and CD19 CAR MC transposon (transposase to transposon at 1:1.2 weight ratio, total amount 5.5 µg) or an equimolar amount of plasmid DNA. Immediately after nucleofection vs. electroporation, there was a loss of viable T cells, however, there was rapid recovery and by day 12, all T cell cultures contained a high percentage of viable T cells. In particular, there was a higher percentage of viability after electroporation of MC DNA vs. plasmid DNA and nucleofection of MC DNA vs. plasmid DNA (figure 1E). We also observed significantly higher stable CAR gene-transfer after transposition from MC DNA vs. plasmid DNA, both after electroporation and nucleofection (figure 1F,G). Taken together, these data show that electro-transfer using a bi-pulse shock leads to efficient insertion of MC DNA transposase and transposon vectors and a high frequency of T cells that express an SB transposon.
Electroporation of MC transposase and transposon results in high-level CAR gene-transfer
In order to facilitate scale-up and implementation on the CliniMACS Prodigy automated manufacturing platform, we performed titration experiments to determine the amount of MC DNA in relation to volume and T cell number during electroporation that results in the maximum yield of CAR-modified T cells. We maintained the ratio of transposase and transposon at the 1:1.2 weight ratio as before and varied the total amount of MC DNA that was added to the electroporation reaction (range: 3.3–48.3 µg in 600 µL and 5E6 activated T cells). As anticipated, there was a steady increase in stable gene-transfer rate determined at day 6 after electroporation with <5% CAR-expressing T cells at the lowest and >80% at the highest MC DNA amount (online supplemental figure 1A). However, we also observed gradually increasing toxicity when higher amounts of MC DNA were used for electroporation. Accordingly, the highest yield of CAR-modified T cells was obtained when 33 µg of MC DNA (i.e., 15 µg MC SB100X transposase and 18 µg MC CD19 CAR transposon) were used for the electroporation reaction (online supplemental figure 1B). Together, these data show that the amount of MC DNA for electroporation into T cells ought to be titrated to balance gene-transfer rate vs. toxicity in order to obtain a maximum yield of CAR-modified T cells.
Supplemental material
Automated, scaled manufacturing process consistently yields therapeutic CAR T cell doses
We proceeded to perform N=3 automated manufacturing runs at large scale. The runs were done on different CliniMACS Prodigy devices and executed by different operators. An overview on the essential steps of the automated manufacturing process is provided in figure 2A. At the start of the process on day 0, 2E8 T cells were isolated from LP and activated using TransAct. On day 3, T cells were re-buffered and sequentially electroporated in aliquots until all T cells had been processed using the bi-pulse electroporation program. Immediately after electroporation, T cells were moved into the cultivation chamber that had been pre-loaded with complete TexMACS medium for recovery and maintained for propagation and serial in-process and end-of-process controls until day 12 (figure 2B). We determined T cell viability and, as expected, noted a drop after electroporation but also rapid recovery with viability rates consistently above 95% (mean: 97.5%±0.7%) in each of the three runs on and after day 6 of the process (figure 2C). At the end of the process on day 12, there was a total of 8E8, 3.5E9 and 3.3E9 T cells in runs #1, #2 and #3, respectively (mean: 2.5E9±1.5E9), accounting for an 12.7±7.5-fold expansion in all three runs relative to the input of 2E8 T cells on day 0 (online supplemental figure 2). The percentage of T cells that was modified with the CAR_EGFRt transposon at the end of the process on day 12 was on average 28.8%±12.3% (range: 20%–43%), and was similar in the CD8 and CD4 T cell subset (online supplemental figure 3). We noted a higher percentage of CAR-expressing T cells at in-process controls in some of the runs, potentially due to T cells with less active transposon-expression entering a resting state toward the end of the process (figure 2D). The yield of CAR-modified T cells was 2.6E8, 7.0E8, and 1.4E9 in runs #1–3, respectively (figure 2E). Taken together, these data show that the automated process has reached a clinical scale and provides therapeutic doses of CAR T cells within a 12-day period.
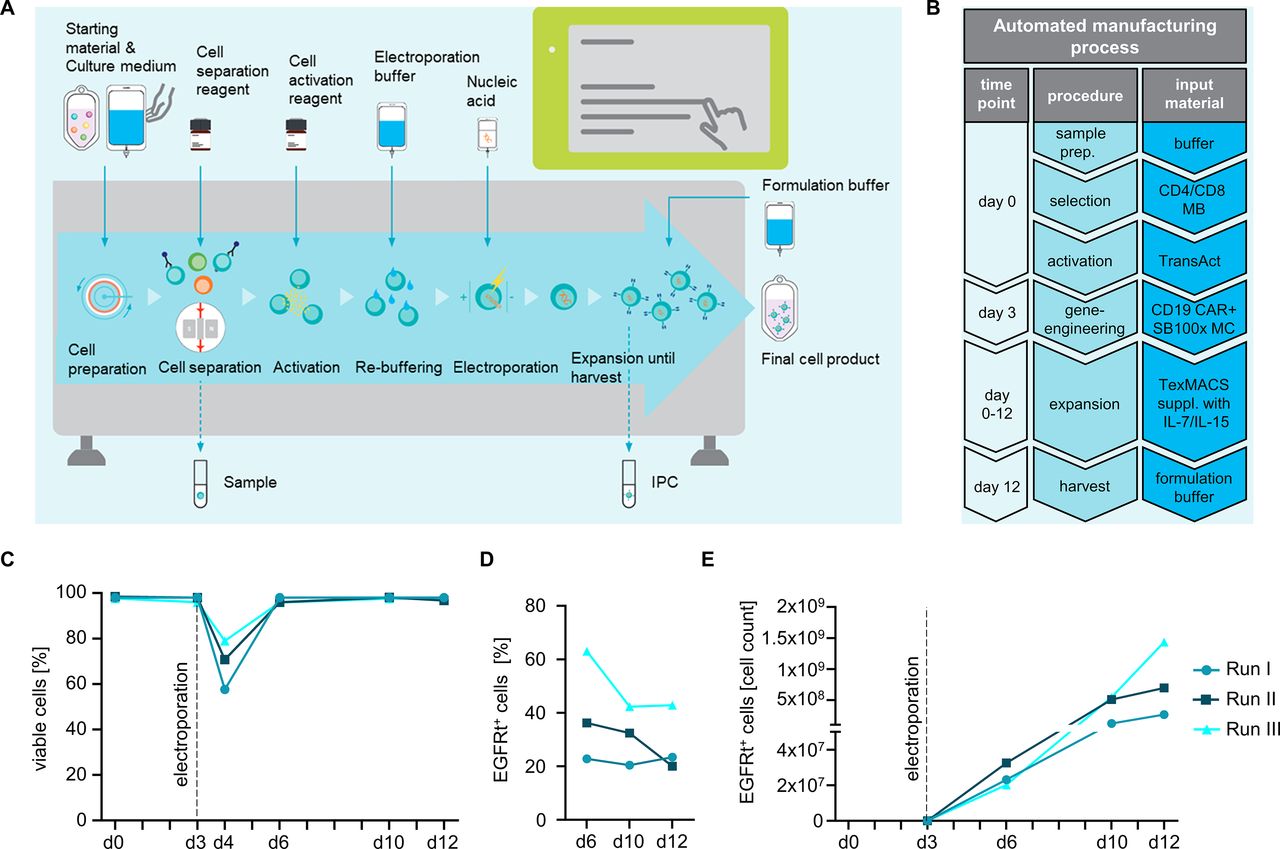
Large-scale automated manufacturing of CAR T cells. (A, B) Workflow of the automated manufacturing process. (A) After installing the single-use disposable tubing set (TS520 in combination with the EP-2 accessory), starting material was sterile connected and automatically processed including CD4/CD8 separation, activation, electroporation, expansion and final formulation with minimal hands-on time according to a pre-defined activity matrix. In-process controls (IPC) allow tracking of viability, cellular expansion, pH value, and glucose consumption as required. (B) Flow chart of the manufacturing process and materials required. (C) Viability of in-process control samples before and after electroporation, during the process and at day of harvest. (D) Percentage of gene-modified T cells and (E) CAR T cell expansion was tracked during the entire process.
CAR T cell products show balanced CD4/8 composition and a high frequency of Tcm cells
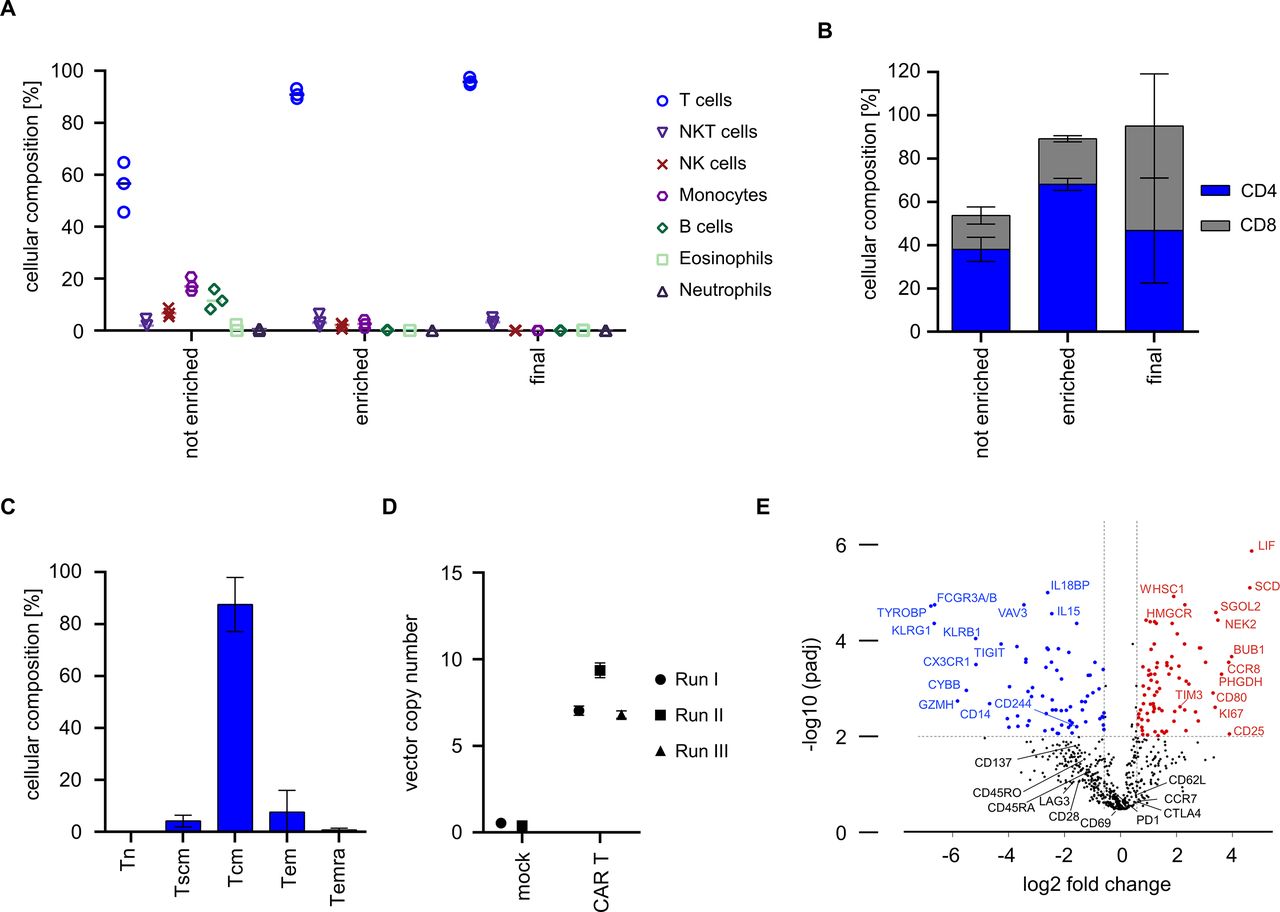
We were interested in determining the purity and cellular composition of the end-of-process product relative to input T cells that had been isolated from LP. The data show that enrichment from LP on day 0 had increased the percentage of T cells from 55.6%±9.5% to 91.1%±2.0% (figure 3A). Consistent with prior studies, the enriched T cell population also contained a fraction of NK cells, NKT cells and monocytes due to their low expression of CD8 and CD49. The final cell product contained highly pure T cells (mean: 96.0%±1.4%) with only a minor fraction of NKT cells (mean: 3.5%±1.4%) (figure 3A and online supplemental figure 4). There was a balanced ratio of CD4 and CD8 CAR T cells in the final cell product, even though there was a higher percentage of CD4 compared with CD8 T cells at process start (figure 3B). Of interest, there was a high percentage of CAR T cells with CD45RO+ and CD62L+expression, consistent with a central memory T cell phenotype (Tcm) (figure 3C). We determined the transposon copy number on the final cell product to be 7.0, 9.4 and 6.8 in runs #1, #2 and #3, respectively (figure 3D). We also performed gene expression analyses on the final drug product and found higher expression of distinct activation/proliferation markers (eg, CD25, CD80, Ki67) and balanced expression of activation/exhaustion markers (eg, PD-1, CTLA4, TIM-3, LAG-3, TIGIT) relative to native non-cultured T cells (figure 3E). Taken together, these data show that the automated process yields highly purified T cells that comprise an equal proportion of CAR-modified CD8 and CD4 T cells with desirable attributes in phenotype and genotype.
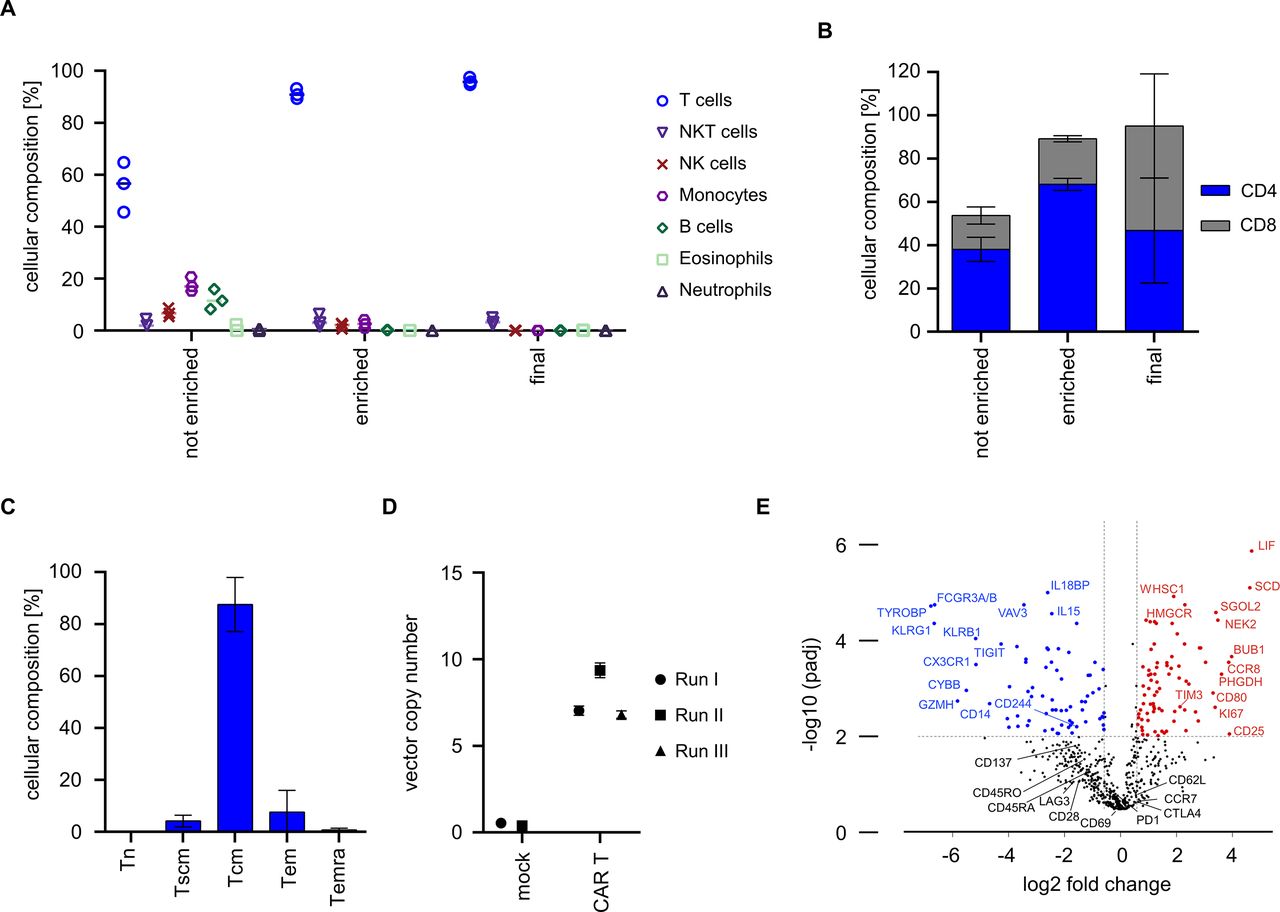
Composition, phenotype and genotype of CAR T cells from automated manufacturing runs. (A) Cellular composition was analyzed before and after enrichment on day 0 as well as in the final cell product using flow cytometry. (B) CD4/CD8 ratio was analyzed before and after enrichment as well as after harvest. (C) T cell immunophenotype in final cell product. T cells were stained with antibodies against CD45RO, CD62L and CD95 and frequency of naïve T cells (Tn), stem-cell memory T cells (Tscm), central memory T cells (Tcm), effector memory T cells (Tem), and CD45-RA+ effector memory T cells (Temra) was assessed in the final cell product by flow cytometric analysis. (D) Vector copy numbers were analyzed from final cell products (CAR T, N=3) or mock T cells (control, N=2). (E) Gene expression profiles of cells from final cell product (N=4) were compared by Nanostring analysis to that of freshly isolated T cells (N=10). (B, C) Show mean±SD from N=3 donors.
Automatically manufactured CAR T cells confer potent anti-tumor function in vitro and in vivo
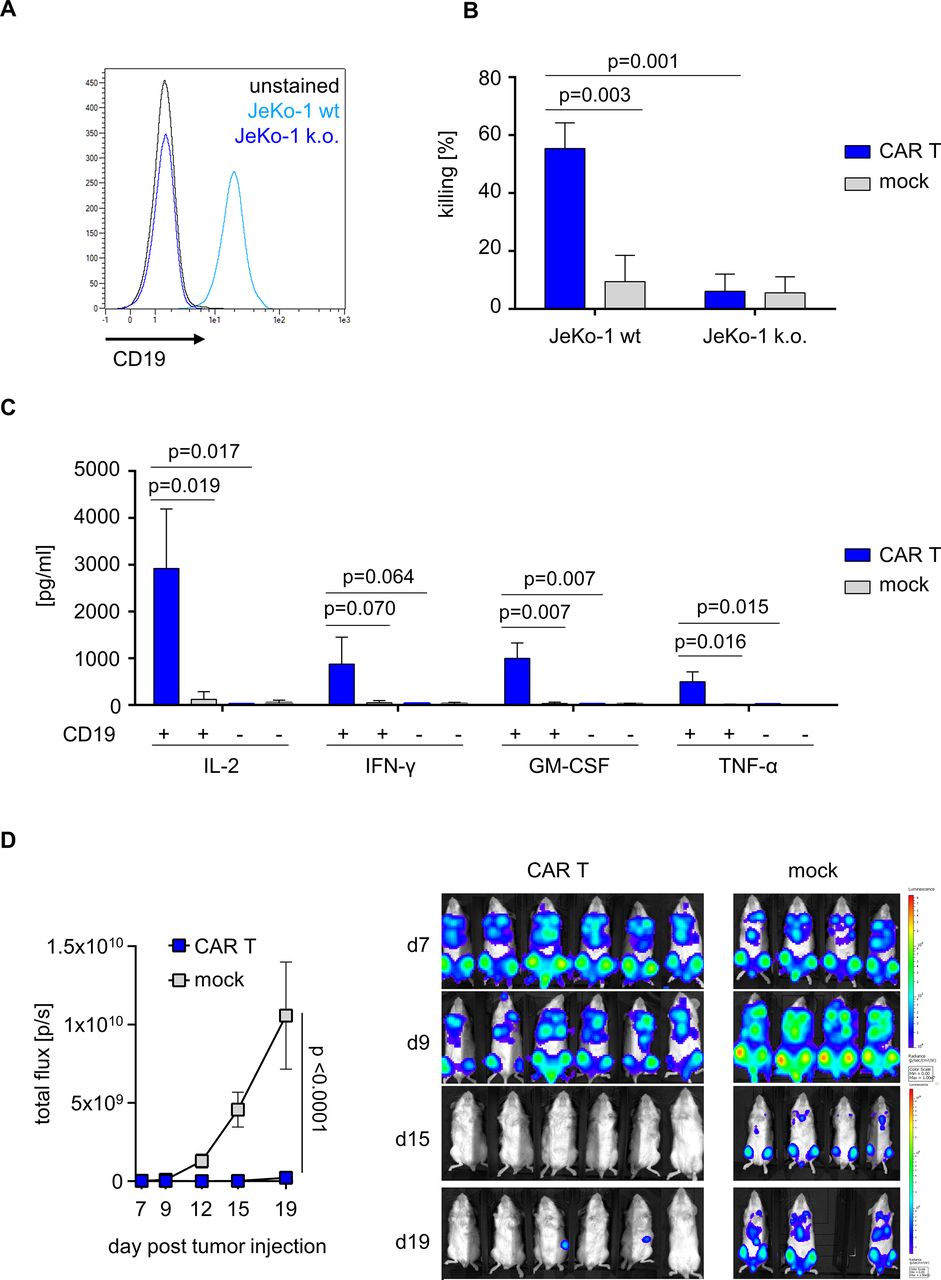
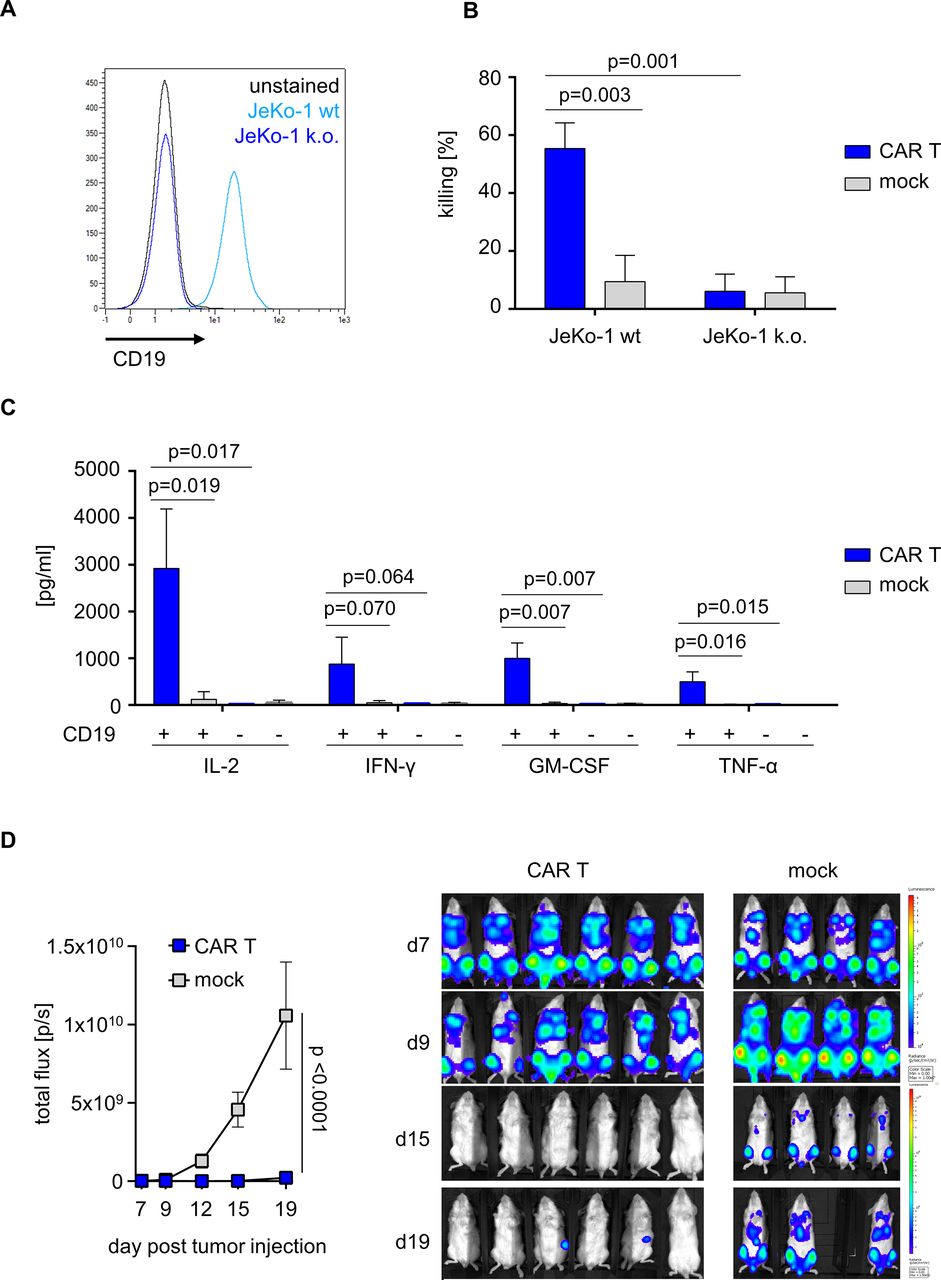
We sought to assess the anti-tumor reactivity of CD19 CAR T cells that had been prepared on the automated, scaled manufacturing process in runs #1–3. To demonstrate specificity and selectivity for CD19-expressing target cells, we generated isogenic CD19 knockout variants from the native JeKo-1 mantle cell lymphoma cell line (figure 4A). CD19 CAR T cells but not mock T cells conferred potent and specific cytolysis of native CD19-positive JeKo-1 target cells in a flow cytometry-based lysis assay. There was no specific cytolytic activity of CD19 CAR T cells compared with mock T cells against CD19-negative target cells (figure 4B). CD19 CAR T cells produced several effector and helper cytokines including IL-2, IFN-γ, GM-CSF and TNF-α in response to stimulation with CD19-positive target cells, whereas cytokine production in response to stimulation with CD19-negative target cells was similar to background and not different from mock T cells (figure 4C). The anti-tumor potency of CD19 CAR T cells that had been produced on the automated and scaled manufacturing process was assessed in a xenograft in vivo model of aggressive lymphoma (NSG/Raji). Following lymphoma inoculation, the adoptive transfer of 2E6 CD19 CAR T cells conferred a significant anti-tumor effect compared with the transfer of mock T cells (BLI signal at study endpoint on day 12, p<0.001) (figure 4D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Anti-tumor reactivity of CD19 CAR T cells from automated manufacturing runs. (A) Histogram of CD19-expressing JeKo-1 wild-type (WT) or CD19 knockout (k.o.) JeKo-1 cells. (B) Effector and target cells were co-cultured for 24 hours at an effector-to-target ratio (E:T) of 1:1. Killing was analyzed using flow cytometry. (C) Cytokine release (interleukin-2 (IL-2), interferon-gamma (IFN-γ), granulocyte-macrophage colony-stimulating factor (GM-CSF), and tumor-necrosis factor-alpha (TNF-α)) was measured after co-culturing mock or CD19 CAR T cells either in the presence or absence of CD19-positive target cells. (D) After engrafting luciferase expressing Rajiffluc tumor cells in NSG mice for 7 days, non-electroporated mock or CD19 CAR engineered T cells were i.v. injected. Tumor burden was tracked using an in vivo tumor imaging system (IVIS). (B, C) Show mean+SD from N=3 donors. (D) Represents mean±SD from N=6 mice (CAR T) and N=4 mice (mock). Statistical analysis was performed using parametric unpaired t-test with 95% CI.
Collectively, these data show that CAR T cells produced on the automated, scaled, transposed-based manufacturing platform confer potent and specific anti-tumor activity in vitro and in vivo, suggesting they will be effective in a clinical setting in humans.
Discussion
CAR T cell therapy is a transformative treatment and has shown clinical efficacy in selected hematological malignancies.1 20 Because manufacturing of CAR T cells is complex, their dissemination to a broader range of patients remains challenging.9 In order to cover the growing demand, advanced manufacturing concepts characterized by robustness and reproducibility, scalability and exportability are required.4 We and others have previously shown that CAR T cells can be manufactured in an automated, closed and GMP-compliant manufacturing process with viral gene-transfer.9 19 21–23 In this project, we developed an alternative process that is based on virus-free SB transposon gene-transfer. Though virus-free SB transposon-engineered CAR T cells have been evaluated in clinical trials, all previously published manufacturing protocols included manual and multiple ‘open’ handling steps that pose a risk for operator errors, contamination and cross-contamination.5 6 These risks can be alleviated through the use of device-based, closed and automated manufacturing processes. We provide the first demonstration that virus-free SB transposon-based gene-transfer can be implemented on an automated process platform to yield therapeutic doses of CAR T cells.
CAR T cell therapy is associated with significant effort and expense in supply and clinical use. A recent cost analysis of conventional CAR T cell manufacturing has identified GMP-trained personnel as the most expensive item.7 The use of automated manufacturing has the potential to substantially reduce personnel effort, and to facilitate scaled manufacturing in order to increase throughput in production facilities. We have refrained from calculating operator hands-on-minutes when performing the manufacturing runs on our automated platform, as this may still vary when the process is exported to other facilities that have a different setup and infrastructure and is performed by other operators with distinct experience and training. We are confident to state, however, that the operator hands-on-minutes (excluding analyses of in-process and end-of-process controls) have been considerably reduced in comparison to a ‘conventional’ non-automated manufacturing process for CAR T cells in one of our ongoing clinical trials.5
We have recently shown that CAR T cells can be efficiently modified by SB transposition when using MC-encoded transposase and transposon.6 In order to scale up our process, we initially focused on the identification of transfection conditions that permit high gene-transfer rates and low electroporation-associated toxicities. In accordance with prior studies reported by Alzubi et al10 and Schwarze et al,24 we modified pulse strength, length, mode and direction to arrive at a bi-pulse shock that permitted high frequencies of stable gene-transfer and high viability in activated CD8 and CD4 T cells. We then performed titration experiments to determine the optimal amount of MC DNA for electroporation and defined a sweet spot with maximum yield of CAR-expressing T cells.6 This enabled subsequent scale-up experiments to demonstrate automated enrichment, activation, electroporation, expansion and formulation of the CAR T cell product. Distinct from our previously reported viral CAR T cell manufacturing process,9 we initiated manufacturing from a higher input number of 2E8 T cells to compensate for the expected drop in viability post-electroporation. Accordingly, the recovery volume post-electroporation was adapted to reach at least a concentration of 5E5 T cells/mL. Another critical parameter that we found affected T cell expansion was the frequency of dead and apoptotic T cells in the cultivation chamber. Although we found electroporation of MC to be associated with lower toxicity compared with plasmid DNA,6 a relevant proportion of T cells still died during electroporation. Therefore, we optimized culture conditions and maintained T cells in the presence of 3% serum post-electroporation between day 3 and day 6, which resulted in rapid expansion and T cell viability rates consistently >95% until the end-of-process. Future efforts will be directed at refining the culture medium to develop a serum-free process.
We performed phenotypic, genomic and functional analyses with the CD19 CAR T cell product obtained with our automated manufacturing process. At the end of the process, we were pleased to find almost exclusively T cells and only a minor fraction of NKT cells, a balanced ratio of CD8 CAR-modified and CD4 CAR-modified T cells and a very high percentage of T cells with Tcm phenotype. Tcm have been associated with a consistent ability for engraftment, expansion and persistence in vivo and superior outcome in clinical trials.25–27 The development and preservation of a high frequency of T cells with Tcm phenotype may have been favored by a high percentage of naïve T cells in the input population and our use of rhIL-7 and rhIL-15 during the manufacturing process.28 The CD19 CAR construct used in our study has been evaluated in previous preclinical and clinical studies and shown to confer specific and potent anti-lymphoma reactivity.17 27 In the NSG/Raji model we administered 2E6 CD19 CAR T cells that we obtained from our automated manufacturing process. The cell product reached the pre-clinical benchmark of significant anti-lymphoma efficacy set by T cells expressing the same CD19 CAR but engineered with lentiviral gene-transfer in a manual manufacturing process.17
There are several conceptual advantages with using virus-free transposition rather than viral gene-transfer for CAR T cell engineering. First, while viral vectors are limited to a payload<10 kb, SB and other transposons have a ‘yet-to-be-determined’ upper cargo limit,29 making them more attractive to next generation CAR T cell applications combining, for example, bispecific and tandem CARs, safety switches, selection markers,30 additional co-stimulatory moieties, releasable cytokines or logic gate circuits.31 Second, SB and other transposons can be used under biosafety level 1, thereby reducing regulatory and infrastructure requirements on manufacturing facilities in comparison to viral vectors that have to be handled under biosafety level 2 conditions. Micklethwaite et al32 recently reported on the development of T cell lymphoma in two patients that had been treated with CD19 CAR T cells engineered with the piggyBac transposon system. Whether the manufacturing process and electroporation protocol potentially affected the genomic insertion of the piggyBac transposon and the propensity of T cells for transformation remains unclear. In contrast to viral vectors and piggyBac transposons, that are biased towards integration in transcriptionally active genes,33 SB transposons integrate randomly in the genome.34 We have determined the SB transposon copy number in our final CAR T cell product to be within the range that has been deemed acceptable for advancing to evaluation in clinical trials.5 Furthermore, we have annotated the genomic insertion profile of the CD19 CAR transposon used in this study in previous work and found a high frequency of insertions into genomic safe harbors without preference for transcriptionally active genes or genic regions.6
In conclusion, we report the first automated, scaled transposon-based manufacturing process for CAR T cells that is ready for formal validation and use in clinical manufacturing campaigns. This process and platform have the potential to facilitate the more widespread access of patient to CAR T cell therapy and to accelerate scaled, multiplexed manufacturing both in the academic and industry setting.
Data availability statement
Data are available on reasonable request. All data directly relevant to the study are included in the article. Further data are available on reasonable request.
Ethics statements
Patient consent for publication
Acknowledgments
The authors acknowledge the support of Nadine Mockel-Tenbrinck, Carola Barth, Sabrina Schmitz and Stefan Wild at Miltenyi Biotec for their scientific advice, as well as the technical assistance of Silke Frenz at Universitätsklinikum Würzburg in in vivo studies. The authors also thank Brian Webster (Miltenyi Biotec) and N. Christine Øien (Universitätsklinikum Würzburg) for revising and editing the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
DL and RM contributed equally.
SP and TS contributed equally.
Contributors DL, RM, TS and MH conceptualized the study; DL, RM, CB, SB, SL, RK-S, KT, NW, LG, SP and TL generated and analyzed data; MA, AK, MSchm, MSchl and ME supervised defined aspects of the project; DL, RM and CB wrote a first draft of the manuscript. SP, TS and MH reviewed and edited the manuscript. TS and MH were responsible for the overall content as guarantor. All authors read and approved the final manuscript.
Funding The authors were supported by the patient advocacy group 'Hilfe im Kampf gegen den Krebs e.V.', Würzburg, Germany and 'Forschung hilft' - Stiftung zur Förderung der Krebsforschung an der Universität Würzburg (SP and MH). Further, the authors were supported by grants from the Deutsche Forschungsgemeinschaft, project number 324392634, TRR 221, SP and MH and project number SFB- TRR 338/1 2021 –452881907-TP A2 (MH), and the Bayerisches Zentrum für Krebsforschung BZKF, Leuchtturm Zelluläre Immuntherapie (SP and MH). The authors were also supported by the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 116026 (T2EVOLVE). This Joint Undertaking receives support from the European Union's Horizon 2020 research and innovation program and EFPIA.
Competing interests DL, CB, SL, KT, NW, MA, AK and TS are employees of Miltenyi Biotec. MH is listed as an inventor on patent applications and granted patents that have been filed by the Fred Hutchinson Cancer Research Center, Seattle, WA and the University of Würzburg that are related to CAR technologies and the use of MC DNA for genetransfer into lymphocytes and that have been licensed—in part—to industry. MH is a cofounder and equity owner of T-CURX. MSchm and MSchl are listed as inventors on granted patents of PlasmidFactory that cover the use of transposons in combination with Minicircle technology for cell transfection. No competing financial interests exist for the remaining authors.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.