Article Text

Abstract
The broad activity of agents blocking the programmed cell death protein 1 and its ligand (the PD-(L)1 axis) revolutionized oncology, offering long-term benefit to patients and even curative responses for tumors that were once associated with dismal prognosis. However, only a minority of patients experience durable clinical benefit with immune checkpoint inhibitor monotherapy in most disease settings. Spurred by preclinical and correlative studies to understand mechanisms of non-response to the PD-(L)1 antagonists and by combination studies in animal tumor models, many drug development programs were designed to combine anti-PD-(L)1 with a variety of approved and investigational chemotherapies, tumor-targeted therapies, antiangiogenic therapies, and other immunotherapies. Several immunotherapy combinations improved survival outcomes in a variety of indications including melanoma, lung, kidney, and liver cancer, among others. This immunotherapy renaissance, however, has led to many combinations being advanced to late-stage development without definitive predictive biomarkers, limited phase I and phase II data, or clinical trial designs that are not optimized for demonstrating the unique attributes of immune-related antitumor activity—for example, landmark progression-free survival and overall survival. The decision to activate a study at an individual site is investigator-driven, and generalized frameworks to evaluate the potential for phase III trials in immuno-oncology to yield positive data, particularly to increase the number of curative responses or otherwise advance the field have thus far been lacking. To assist in evaluating the potential value to patients and the immunotherapy field of phase III trials, the Society for Immunotherapy of Cancer (SITC) has developed a checklist for investigators, described in this manuscript. Although the checklist focuses on anti-PD-(L)1-based combinations, it may be applied to any regimen in which immune modulation is an important component of the antitumor effect.
- clinical trials, phase III as topic
- clinical trials as topic
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immunotherapy, specifically immune checkpoint inhibitors (ICIs) directed against programmed cell death protein 1 and its ligand (PD-(L)1), is now the backbone of the standard of care for many tumors. For patients with tumors that respond to anti-PD-(L)1 therapy, prolonged duration of response (DOR) or even delayed tumor shrinkage is possible even after discontinuation of therapy,1–5 visualized as long event-free plateaus on Kaplan-Meier curves.6 7 The long-term responses seen with immunotherapy contrast markedly with the other conventional therapeutic pillars of medical oncology: chemotherapy and targeted therapy, both of which nearly universally lead to the eventual emergence of resistant tumors.8 9
Not all tumors respond to ICI monotherapy, and the currently available assays for patient selection (eg, mutation burden/microsatellite instability and PD-L1 expression) are indirect measures of the immune susceptibility of tumors.10 A substantial unmet need exists in multiple malignancies to enhance overall response rates (ORRs) to ICIs and to develop and validate predictive biomarkers of long-term benefit. Combination approaches involving anti-PD-(L)1 added to cytotoxic, targeted, antiangiogenic, radiation, or other immune therapy have been put forward as a key strategy to expand the population of patients that benefit from immunotherapy.11–13 Anti-PD-(L)1-based combinations have demonstrated improved median overall survival (OS) and gained regulatory approval for the treatment of several tumors including hepatocellular carcinoma (HCC),14 melanoma,15 non-small cell lung cancer (NSCLC),16 renal cell carcinoma (RCC),17 triple-negative breast cancer (TNBC),18 and others. This immunotherapy renaissance has also generated renewed interest in the immune mechanisms of action of conventional anticancer therapies, revealing substantial potential contributions of adaptive and innate immunity to the effects of chemotherapies,19–21 tumor-targeted therapies,22–24 and antiangiogenic therapies.25 Unfortunately, experience has shown that preclinical evidence for combination synergy must be confirmed in treated patients, given the inadequacy of most murine models to recapitulate the organ-specific immune contexture as well as the heterogeneity, immune editing, and immune defenses of human disease.10 26 27
No uniform method has been established to predict the probability of success for individual clinical trials. Aggregate strategies to forecast probability of success based on historical attrition rates from phase to phase for specific indications28 or sponsors29 have been described, but these approaches provide incomplete information for future prediction and fail to account for unique aspects of immunotherapy drug development. Machine learning approaches have been put forward that can identify aspects of trial protocols and sponsor track-records as important features in predicting success.30–32 These tools are currently strictly exploratory, and in the absence of validated predictive algorithms, the go/no-go decision for phase III trials relies on expert opinion.
Except for a small number of combination regimens in development, most will require randomized phase III trials to demonstrate patient benefit and establish their place in the treatment paradigm. Phase III trials are time- and resource-intensive, and the cost associated with late-stage studies has steadily increased. Although pivotal trials for agents with substantial clinical benefit can be conducted at lower costs compared with studies where the effect sizes are marginal,33 the overall number of new approvals from the US Food and Drug Administration (FDA) per billion inflation-adjusted dollars of research and development spending has steadily decreased by 50% approximately every 9 years since 1950.34 Today, phase III trials are conducted by both pharmaceutical sponsors and academic institutions. The studies designed by pharmaceutical sponsors are most commonly designed with short-term endpoints35 to support primary or supplementary regulatory approval. The imperative to bring drugs to market more rapidly, which benefits both the sponsor and patients, may compromise efforts to answer important questions, including accurate biomarkers for patient selection, optimal biologic dose,35 determination of the optimal duration of therapy, and assessments to determine durability of clinical benefit. For agents that are already approved, phase III studies conducted by pharmaceutical sponsors are often designed to expand understanding of the clinical utility of a given agent (eg, different lines of therapy, alternative combination regimens) to further improve patient outcomes and broaden the number of patients with cancer who can benefit from the therapy. Studies designed by academic clinicians and researchers, on the other hand, generally focus on the discovery and/or evaluation of biomarker-based patient selection, investigation of alternative schedules, optimization of the duration of therapy, and characterization of the durability of clinical benefit. The complimentary roles that industry and academic teams play, respectively, are essential, and all within the cancer clinical research community must work together to develop immunotherapy regimens in a way that maximizes not only their short-term benefits, but also their curative potential for patients with cancer.
The Society for Immunotherapy of Cancer (SITC) recognizes the challenges in developing immunotherapy combinations and has initiated various efforts to work collaboratively with industry and academia to improve the benefit to patients. These include initiatives to promote the development of predictive biomarkers, and to develop criteria for designing and interpreting phase II and phase III immunotherapy-based clinical trials. In this manuscript, SITC seeks to provide investigators with a framework for selecting and prioritizing phase III trials with immuno-oncology agents, focusing primarily on anti-PD-(L)1 combinations, in order to optimally use resources, improve patient outcomes, and more rapidly bring the unique benefits of immunotherapy to a broader population of patients with cancer.
Definition of value in phase III clinical trials
The value of a clinical trial may have multiple definitions, depending on the stakeholder using the term. The financial value of a trial is typically defined as the expected revenue for an investigational regimen minus the costs for research and development, all multiplied by the probability of successful regulatory approval. As many as 50% of phase III oncology trials are unsuccessful,36 37 with lack of efficacy being the major reason for failure in late-stage development.38 Oncology trials are some of the most expensive phase III studies, with estimated per-patient costs of more than $100,000 USD.33 The cost of a failed phase III study includes not only the resources invested in the trial itself (including numbers of patients) but also the time that could have been spent pursuing a more effective strategy to improve outcomes for patients.
Other conceptions of value exist. From a patient perspective, value may include more difficult-to-quantify interactions between clinically meaningful benefit, out-of-pocket healthcare costs, and effects on quality of life (QOL).39 40 Effective anticancer treatments also benefit society as a whole by allowing patients to return to being productive contributors rather than having prolonged chronic illness ultimately resulting in death. An added facet of value that may be considered by investigators is the potential for a trial to provide informative data on biomarkers of response and resistance or for patient selection in order to maximize likelihood of success for future studies.41
For the purposes of this manuscript, the primary consideration for determining the value of phase III trials is potential benefit to patients. Arguably, the greatest benefit for an individual patient is durable disease control and ideally a cure without the need for further treatment. While short-term endpoints such as progression-free survival (PFS) may be attractive to sponsors and investigators because of the ability to shorten the duration of registrational trials and control for the impact of additional therapy or off-trial crossover, patients, when asked, place little value on treatments that delay disease progression on imaging without helping them live longer or improving their QOL.42 Immunotherapy may also prolong OS in the absence of, or in excess of, clinically evident disease control (eg, ipilimumab,43 sipuleucel-T,44 tebentafusp-tebn45), which could represent a meaningful advance in a particular disease state depending on the magnitude of effect.
Assumptions on immunotherapy combinations
The definition of an immune therapy or immune modulatory agent remains a matter of debate. For the purposes of this manuscript, immunotherapy agents are defined as any modality where the primary mechanism of antitumor effect is mediated by an immune effector cell. In colloquial terms, immunotherapies treat the immune system to kill cancer as opposed to treating the tumor itself. As such, the secondary immune-mediated antitumor activity for several anticancer agents (eg, antibody-dependent cellular cytotoxicity in the case of trastuzumab24 46 or potential enhanced T cell recognition of tumors mediated by BRAF inhibition22), is insufficient to consider these modalities as immunotherapies in and of themselves.
The rationale for anti-PD-(L)1-based immunotherapy is to tip the balance of the cancer-immunity interface47 in favor of detection and elimination of cancer cells by the immune system, despite ongoing immunoediting.48 ICI therapy with anti-PD-(L)1 is thought to contribute to tumor control via the reinvigoration and expansion of tumor antigen-specific T cells, especially a memory stem-like population expressing PD-1, SLAMF6, and TCF1.49–54 Despite intensive investigations, the mechanisms responsible for the antitumor effects of anti-PD-(L)1-based ICI therapy remain incompletely defined. Certain biological effects, for example, the PD-L1-mediated anti-apoptotic ‘molecular shield’ function and direct regulation of metabolism in tumor cells, may play a role in the activity of pathway blockade in combination with direct tumor cytotoxic agents.55–57 These T cell-independent effects of PD-(L)1 blockade are poorly understood, and their importance to clinical benefit is not known.
Clinical activity of anti-PD-(L)1 has been shown to correspond with high tumor mutational load in several settings, (particularly insertion/deletions, presumably due to the likelihood of neoantigen production from frameshifting mutations),58 59 expression of PD-L1 by tumor and immune cells within the tumor microenvironment (TME),60 61 an interferon gamma (IFNγ) gene signature within the TME, the presence of intratumoral lymph node-like structures62 and a high frequency of pre-existing clonally expanded tumor-specific CD8+ T cells relative to disease burden.52 63 64 The mechanisms by which tumors avoid elimination after ICI treatment—both intrinsic and acquired—are also incompletely understood. Some features associated with non-response to PD-(L)1 blockade include a lack of neoantigens, upregulation of alternative checkpoints, silencing of antigen presentation machinery, aberrant IFNγ signaling in the microenvironment, and exclusion of cytotoxic T cells or all immune cells.63 65–68 Notably, features of responsiveness and non-responsiveness may be disease-specific.
Combination approaches may enhance tumor responses to anti-PD-(L)1 by augmenting neoantigen production and/or presentation,19 increasing CD8+ T cell infiltration, proliferation, and survival,23 inhibiting alternative checkpoints or other immunosuppressive factors within the TME,15 69 70 and several other mechanisms that are still actively being elucidated.11 13 71 72 When combining immune therapies with cytotoxic, antiangiogenic or molecular-targeted agents, both direct antitumor effects and secondary immune modulatory effects of the latter agents may contribute to the overall clinical antitumor effect, but the relative contribution of each component cannot be distinguished clinically or with laboratory correlative studies.12 13 73 74 Responses to combination treatment approaches that do not include additional immunotherapies may be less durable, as was the case with anti-PD-1 plus BRAF/MEK inhibition in melanoma.75 76 The most evident readout for approaches that induce tumor antigen-specific T cells in the advanced disease setting is the appearance of long rightward plateaus in the tails of Kaplan-Meier PFS and OS curves,77–79 indicative of prolonged patient benefit even after most patients have stopped therapy.
Assessing potential value in phase III trials
For the purposes of this paper, the value of a trial is primarily dependent on the potential benefit for patients within the context of the particular patient population. Also important is the possibility for the study to improve outcomes for future patients. As such, the framework for evaluating value includes assessment of the mechanism and biology of the combination, phase I and phase II data, trial design, and potential for impact. The framework for assessing value is illustrated in figure 1.
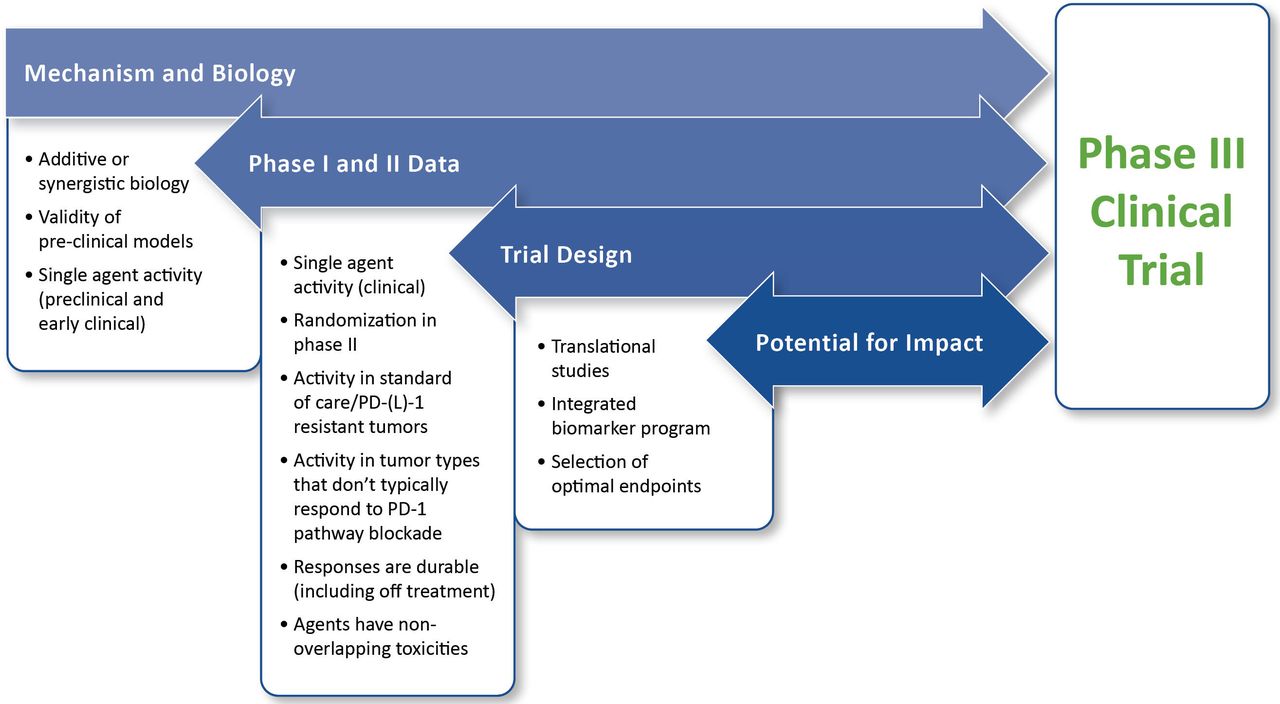
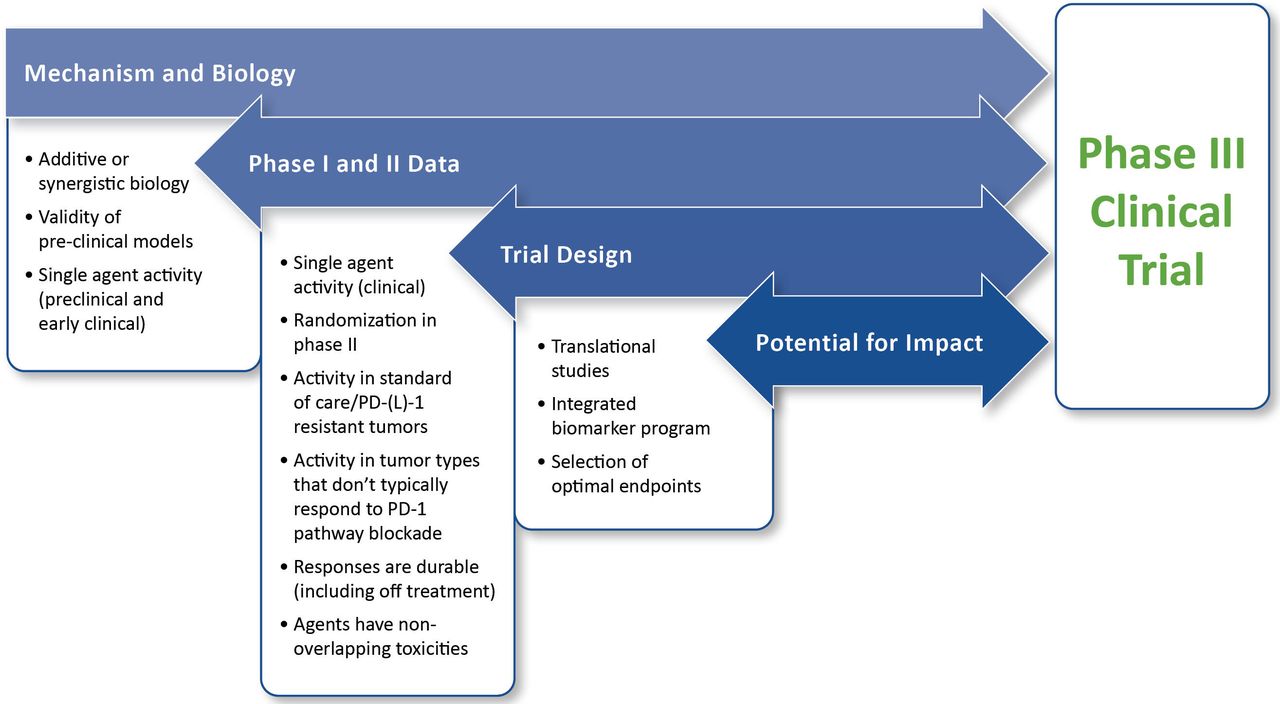{kind=link}
Framework for evaluating planned phase III trials in immuno-oncology. This framework assumes that fundamental practical and institutional considerations for a study are satisfied and is intended to assess the immuno-oncology aspects of a planned trial. The highest priority is placed on clinical data. If the preclinical and early clinical data are both unconvincing, extreme caution is warranted before moving forward. Detailed descriptions for the evaluation and prioritization of individual components of the framework are provided in the text.Abbreviation: PD-(L)1, programmed cell death protein 1 and its ligand
Mechanism and biology
Preclinical data serve many purposes, including characterization of pharmacokinetics and pharmacodynamics of individual drugs and combinations, identification of optimal doses of each drug in combination and their potential schedule dependencies, exploration of biomarkers, and identification of potential mechanisms of resistance. When evaluating the preclinical data supporting a planned phase III trial of an anti-PD-(L)1-based combination regimen, activity as monotherapy, interactions between the agents, and validity of the models used are three key factors for consideration, as summarized in box 1.
Key considerations for preclinical and early clinical data supporting planned phase III studies of immunotherapy-based combinations
Mechanism and biology for immunotherapy combinations
Additive or synergistic biology.
Validity of preclinical models.
Single-agent activity (preclinical and early clinical).
In the years after the 2009 publication of the International Conference for Harmonization guideline on non-clinical requirements for oncology therapeutics development (ICH S9), the average number of non-clinical studies performed and number of animals used per development program has consistently trended downward.80 A 2021 systematic review of immunotherapy combination trials found that 72% of studies lacked published data in more than one model system supporting efficacy of the specific combination in the target indication.13 Even though the FDA’s guidance document on development of combinatorial regimens states that preclinical data including appropriate animal models and toxicology studies are required to assess the potential of a multidrug indication,81 many combination trials of oncology drugs are initiated to empirically evaluate an agent in combination with a standard of care, and often without scientific or preclinical evidence to support the combination regimen.
Combination approaches may enhance response rates via additivity or synergy or by independent action, where benefit at the population level occurs because each patient has more than one opportunity for exposure to one drug that will have meaningful antitumor activity for them.74 Preclinical data should ideally show that an investigational combination addresses a specific mechanism responsible for non-response in the clinic, which would ultimately enable a therapeutic antitumor immune response, such as the observation of expansion of effector CD8+ T cells following combination ipilimumab plus nivolumab not seen for either single agent.70 It is further important to validate that the combination does not restrain the activity of anti-PD-(L)1, which is a distinct possibility for some T cell-ablative chemotherapies or small molecule inhibitors that block or restrain T cell activation, signaling, and/or proliferation.
A major challenge for immunotherapy is the paucity of preclinical models that faithfully recapitulate the ongoing interplay between tumor and immune system during human oncogenesis, progression, and metastasis.10 47 Transplantable tumor models lack tissue-specific immune contexture82 and genetically engineered inducible tumor models have been shown to carry distinct mutational signatures with lower neoantigen burdens compared with human disease.83 84 New models such as the NINJA mouse85 with inducible expression of neoantigens, may overcome some of these shortcomings. Also promising are humanized mouse models co-engrafted with human CD34+ hematopoietic stem and precursor cells and human leukocyte antigen (HLA)-matched cell line-derived or patient-derived tumors.86 87 However, genetically homogenous inbred mice that were raised in sterile environments will never adequately account for the profound variability in human patients nor the effects of the microbiome on immunotherapy efficacy.88 The inclusion of veterinary models89 in the preclinical portfolio enhances the potential value for a planned phase III trial; however, these studies are currently almost as difficult to conduct and interpret as human trials.
With improved mechanistic understanding of the pathways by which tumors exclude cytotoxic effector cells or evade recognition and elimination, additional rational combination approaches may be explored that include agents without activity as monotherapies. In these cases, it will be important to establish preclinical evidence that the inactive agent modulates the immune microenvironment toward blockade or reversal of a critical and non-redundant pathway of immune suppression or infiltration by tumor-antigen-specific T cells, with or without the presence of an IFNγ gene expression signature.59 65–68
Phase I and phase II data
Data from early-phase trials provides the first evidence that the mechanistic rationale from preclinical studies translates into safe and effective treatments for human patients. Several characteristics of ICIs are mismatched to the traditional paradigm for phase I and phase II development including a non-linear relationship between dose and efficacy49 50 60 and the potential for a poor concordance between short-term objectives such as extending median PFS, and the impact on long-term survival for the entire population or a significant subset of patients.77 78 90 Effective assessment of immunotherapy agents in early-phase studies is a major obstacle for the field10 and combinations are even more complicated to evaluate in phase I due to patient heterogeneity, potential for selection biases that exist at institutions with large phase I programs, strong and variable monotherapy response rates to PD-(L)1 inhibitors, and lack of prospective randomization.
The design of the phase I and phase II studies follows from the indication in which the investigational combination is being evaluated. With respect to likelihood of success in subsequent phase III trials and overall impact, priority should be given to combinations that demonstrate activity in anti-PD-(L)1-resistant tumors or tumors that do not typically respond to anti-PD-(L)1-based therapy. Assessing activity in populations progressing on prior anti-PD-(L)1 can be confounded by several factors including late responses, lack of confirmed progression, and variable mechanisms of resistance—selection of patients for these trials has been addressed in a recently published SITC manuscript.91 Finally, toxicity data should indicate that the combination will be tolerable, especially in settings where the benefit of the combination relative to the individual drugs given in sequence is merely additive. The key considerations for evaluating phase I and phase II data are summarized in box 2.
Key considerations for phase I/II data in planned phase III studies of immunotherapy-based combinations
Phase I and phase II data for immunotherapy combinations
Single-agent activity (clinical).
Randomization in phase II.
Activity in standard of care/programmed cell death protein 1 and its ligand (PD-(L)1)-resistant tumors.
Activity in tumor types that do not typically respond to PD-1 pathway blockade.
Responses are durable (including off-treatment).
Agents have non-overlapping toxicities.
The combination approaches that have been successfully developed to date have mostly included agents that have activity as monotherapies. Examples of agents that have been approved in combination with anti-PD-(L)1 include the anti-cytotoxic T lymphocyte antigen-4 (CTLA-4) checkpoint inhibitor ipilimumab,92 antiangiogenic agents,14 tumor cell-targeted small molecules,17 93 and chemotherapies.16 18
This paradigm may not be absolute, however. The anti-LAG3 checkpoint inhibitor relatlimab, which has minimal to no activity as a single agent, was approved by the FDA based on improved PFS for patients with previously untreated metastatic or unresectable melanoma who were treated with relatlimab in combination with nivolumab compared with those receiving nivolumab alone in RELATIVITY-047.94 Compelling rationale for the clinical development of relatlimab plus nivolumab was provided by durable responses seen with the combination in patients with melanoma whose disease had progressed on prior anti-PD-(L)1 treatment.95 This example stands in contrast to the phase III ECHO-301/KEYNOTE-252 trial96 evaluating the combination of pembrolizumab plus the inhibitor of indoleamine 2,3-dioxygenase-1 (IDO-1), epacadostat and the phase III PIVOT IO 001 trial97 evaluating the combination of nivolumab plus the second-generation interleukin-2, bempegaldesleukin—other agents without activity as monotherapy. Importantly, no biomarkers indicative of response to the combination were described in early studies, nor was evidence of efficacy in a population with tumors resistant to anti-PD-1 established before proceeding to phase III in either case.
In evaluating the early-phase data supporting a planned phase III trial, particularly when evaluating combinations in PD-(L)1 antagonist-naïve populations, randomized phase II studies may provide important information on the likelihood of success in later stages. Historically, phase II trials in oncology have been significantly less likely to be designed with randomization and placebo controls compared with other medical and surgical disciplines.98 Non-controlled phase II studies are at risk of leading to phase III trials with little likelihood of demonstrating benefit if data from earlier trials does not adequately estimate the outcomes that patients would expect without receiving the investigational intervention and if the endpoints selected do not definitively measure antitumor activity as compared with disease biology.99 Both are true for immunotherapy combinations.
Randomization should be encouraged in phase II not only because it allows for more precise estimates of the required effect size and sample size for definitive trials,100 but also because it reduces the potential for errors introduced due to intertrial variability and non-representative patient selection. A multi-arm, randomized phase II trial with appropriate statistical assessment also provides the opportunity to test for bona-fide interaction between the components of a combination as opposed to independent drug action, which could potentially be recapitulated by giving the agents in sequence, or biomarker-based selection to identify the populations that derive benefit with monotherapy.74 Because of the small size of randomized phase II trials, it is important to make sure there are no major imbalances in the prognostic factors of the two arms and to carefully assess the performance of the control arm relative to the historical expectation. In the randomized phase II CITYSCAPE study evaluating the combination of atezolizumab plus the anti-TIGIT tiragolumab in advanced NSCLC, the performance of the control arm was much worse than what was seen in the phase III IMpower110 study that led to regulatory approval for atezolizumab monotherapy.101 The phase III SKYSCRAPER trial subsequently failed to meet its PFS endpoint.
Endpoints evaluated in phase II should also be relevant to the immune mechanism of action of anti-PD-(L)1 combinations, with consideration given to evaluation of longer-term milestone survival102 or biomarker-based measures of tumor response. Notably, the phase III IMspire150 trial evaluating the combination of atezolizumab with vemurafenib and cobimetinib76 for the treatment of BRAF-mutant melanoma was not preceded by a randomized phase II study—although this regimen gained FDA approval based on an investigator-assessed PFS endpoint, it has not gained acceptance in practice due primarily to weak evidence of benefit by immune-based clinical endpoints (ie, milestone PFS, durability of response, and OS) relative to immunotherapy alone. A similar triplet, spartalizumab with dabrafenib and trametinib, did not demonstrate evidence of an immune effect in a randomized phase II study and the phase III COMBI-I trial evaluating the regimen was negative.75
Randomization in phase II may not be necessary if the combination is being evaluated in tumors that are resistant to the standard of care or for tumor types that typically do not respond to anti-PD-(L)1 such as microsatellite stable colorectal cancer, soft tissue sarcomas, or well-differentiated neuroendocrine tumors. The pivotal study for atezolizumab in combination with bevacizumab for the treatment of advanced HCC, IMbrave150, for example, was initiated based on response rates in the phase I setting.103 For combination targeted and cytotoxic therapies, proof of concept in the form of a ≥20% increase above the median historical response rate without the experimental agent in phase II has been shown to enrich for success in phase III.6 Consideration should be given to adaptive, biomarker-driven designs in phase II to stratify patients based on known determinants of response to anti-PD-(L)1 such as tumor mutation burden (TMB) and PD-L1 status to determine if an investigational agent has immune-enhancing effects in ‘cold’ tumors. Examples of this strategy include the BIONIKK and INFORM2 trials.104 105
Master protocols or platform trials incorporating adaptive design features to evaluate single drugs across multiple populations, multiple drugs on a single population, or complex multi-arm, multi-stage designs including multiple treatments simultaneously are increasingly being advanced. These trials are information-rich and often include biomarker components for patient selection. Although the statistical power for these trials may be reduced because of small sample size and frequent crossover, the likelihood of exposing individual patients to the best therapy may be increased because crossover allows patients to receive subsequent treatments based on initial responses.106 Although these novel designs may provide interesting exploratory evidence—especially on biomarkers—several combinations that advanced through adaptive trials have failed to demonstrate benefit in phase III.
With long-term follow-up results emerging for the registration trials for immunotherapy combinations, it is now apparent that many patients achieve years-long disease control (remission or even cure) after cessation of treatment. Among the 12 patients who completed 2 years of pembrolizumab treatment for advanced non-squamous NSCLC in KEYNOTE-021, for example, all had durable response beyond 2 years, as reported by an estimated 3-year DOR rate of 100%.3 Analysis of treatment-free survival (TFS, defined as the area between Kaplan-Meier curves for time to protocol therapy cessation and time to subsequent systemic therapy initiation or death) for the patients with metastatic melanoma treated in CheckMate 067 demonstrated superiority for combination nivolumab plus ipilimumab (11.1 months) versus either monotherapy (4.6 months with nivolumab and 8.7 months with ipilimumab).107 Similar results were seen for patients with RCC treated with nivolumab and ipilimumab versus sunitinib in CheckMate 214 including in the International Metastatic RCC Database Consortium (IMDC) favorable risk population.108 Durable responses (lasting >1 year) in phase I and phase II trials may provide evidence that a combination contributes to the establishment of immune memory leading to tumor rejection.
Finally, the toxicity data from phase I and phase II trials should be carefully scrutinized. While synergistic toxicity of combinations is undesirable, the increase in toxicity must be weighed against the potential benefit. The paradigm for dose selection for cytotoxic chemotherapies all-too often leads to inadequately characterized toxicities in registrational trials for immunotherapies and molecularly targeted agents.35 Toxicities not predicted by preclinical models may emerge in early-phase trials, such as the unexpected hepatotoxicity seen with the combination of vemurafenib and ipilimumab109 or the combination of pazopanib with either pembrolizumab or nivolumab.110 111 The tolerability of immunotherapy agents administered as combinations may be radically distinct from the respective monotherapies. For example, standard doses of nivolumab and ipilimumab could not be given concomitantly in the phase I trial of the dual regimen for advanced melanoma,15 although overall activity with dual immunotherapy exceeded that expected of either single agent across dosing schedules. Active combinations may also have distinct safety profiles in different patient populations, as was seen in the phase I CheckMate 012 trial evaluating nivolumab plus ipilimumab as first-line treatment for advanced NSCLC112 in which decreasing the dose and frequency of administration of anti-CTLA-4 compared with the melanoma indication was necessary to reduce adverse events. Complicating matters further, reducing the dose of a more active agent in order to accommodate the toxicity of another agent in a combination may lead to diminished benefit, as was seen in IMspire150 where vemurafenib was given at 720 mg in the atezolizumab arm instead of the standard 960 mg dose used in the control arm. No improvement was seen in PFS assessed by independent review for the triplet regimen compared with BRAF/MEK inhibition alone, possibly due to submaximal activity of the immunotherapy component in the context of the combination in addition to dose-dependent tyrosine kinase inhibitor (TKI) activity being worse than the standard of care.76
Trial design
Robust preclinical and phase I/II data are necessary but not sufficient for the success of later stage trials. In order to maximize value to patients and the field, phase III trials evaluating immunotherapy combinations should be designed to account for the unique characteristics of agents with immune mechanisms of action and to include integrated translational studies that will provide informative data even if desired endpoints are not met. The key considerations for trial design are summarized in box 3.
Key considerations for trial design in planned phase III studies of immunotherapy-based combinations
Trial design for immunotherapy combinations
Translational studies.
Integrated biomarker program.
Selection of optimal endpoints.
As discussed in the Assumptions on immunotherapy combinations section, treatment with anti-PD-(L)1 is expected to lead to atypical patterns of radiographic response, delayed separation of OS curves, and long-term event-free survival (EFS) in a subset of patients.2 4 Because of these characteristics, traditional clinical trial design schemes used for cytotoxic and tumor or vasculature-targeted therapies may fail to adequately demonstrate patient benefit for combinations involving immunotherapies.
For most phase III trials in the advanced cancer setting, evidence of benefit is based on a time-to-event primary endpoint using a statistical design that assumes an exponential distribution in which the hazard of an endpoint event remains constant over time—thus also proportional between two treatment groups over time—with patients continuing to experience events. The delayed clinical benefit and durable responses expected with anti-PD-(L)1 can result in non-proportional hazards in the treatment arms (with hazards for the treatment arms actually crossing in some cases).113 114 As a result, typical statistical models may be inappropriate or at least have a loss of statistical power to demonstrate difference between immunotherapy and control arms, and thus studies may take considerably longer to reach the targeted number of endpoint events for analysis.78 79 115
The challenges for statistical design in immuno-oncology underscore the importance of randomized phase II data to inform phase III trial design, as discussed in the Phase I and phase II data section. Although requiring several years of follow-up in every phase II immunotherapy trial to detect the emergence of a tail in the survival curve would be an unreasonable prerequisite for moving to phase III, milestone OS and PFS analyses at shorter-term time points (eg, 12 and 18 months) and the identification of patients with TFS can inform whether the combination is displaying characteristics of immune-mediated tumor control. With information from phase II, appropriate statistical plans may be used to accommodate non-proportionality for immunotherapy trials, such as weighted log-rank tests, piecewise exponential distributions, or cure rate models.78 79 115–117 Statistical methods to estimate treatment effects in the presence of the experimental and control arms crossing have also been published.118–120 Finally, results from randomized biomarker-driven phase II trials also inform phase III decisions between an enrichment/targeted, biomarker stratified, or unselected design.121 Randomized phase II endpoints, however, must be interpreted in the context of the agents being used in the combination and the disease setting being treated—short-term endpoints such as ORR may be less informative for combinations involving highly active cytotoxic chemotherapies, for example.
Successful phase III oncology trials are more likely to employ biomarker-driven patient selection.37 As mentioned in previous sections, the identification and validation of reliable biomarkers to predict clinical benefit with anti-PD-(L)1-based combinations remains an unsolved challenge for the field. Validated surrogate markers for benefit with immunotherapy and surrogate endpoints for OS for drug development would greatly streamline phase III trials.
The discovery and validation of immunotherapy biomarkers is an ongoing priority for the field. Several key considerations must be taken into account for biomarker discovery trial design. Predictive biomarkers may be identified in secondary analyses in randomized clinical trials via interaction test between the treatment and the biomarker in an appropriate statistical model.122 It is important to differentiate between predictive and prognostic biomarkers either by the inclusion of a control arm in biomarker discovery or confirmation within a single-arm study of a clear effect of the therapy. The tissue source for analysis must also be taken into account (ie, metastatic lesion rather than primary) as well as the timing for obtaining samples (ideally within 1 year of treatment). In order to isolate the contribution of the immunotherapy component, it is important that biomarker analyses exclude samples that have been exposed to intervening therapy. Furthermore, the biomarker preferably should be confirmed for an immunotherapy-only regimen to rule out effects of a cytoablative or T cell-restraining component of a combination. Finally, it is important to analyze the interaction test between the biomarker and an appropriate immunotherapy endpoint, such as TFS, rather than short-term outcome measures.
Static and dynamic measures of tumor immune status and response to treatment are needed and trial protocols should specify when and how samples should be collected as well as analyzed (ie, fresh or archival tissue; primary or metastatic disease, including disease site; core biopsy or resection, etc). Promising future areas for development include molecular profiling to understand the tumor-intrinsic microenvironment,59 68 artificial intelligence-based algorithms for analyzing fixed or even live tissue specimens (eg, Graticule or Elephas platforms),123–126 blood-based biomarkers of disease burden such as circulating tumor DNA (ctDNA),127 128 and functional imaging to assess immune responses.129–131 An example of this approach is the exploratory analyses of the IMvigor 010 phase III study evaluating adjuvant atezolizumab compared with observation for patients with operable urothelial cancer that demonstrated positive ctDNA after resection could serve as a prognostic marker to identify the subgroup of patients most likely to have a chance of benefit from an effective adjuvant therapy.132
Durability of response after stopping therapy offers clinical evidence that either all tumor was eliminated, or an immunotherapy combination elicited immune memory, as discussed in the Phase I and phase II data section. Most clinical trials of immunotherapies mandate treatment for 2 years or until disease progression, but some patients may be able to stop therapy earlier if antitumor immune memory has been established. In CheckMate 153, patients with previously treated advanced NSCLC still receiving treatment at 1 year were randomly assigned to continue nivolumab until disease progression or unacceptable toxicity or to stop therapy with the option of on-study retreatment after disease progression.5 In exploratory analyses, median PFS was longer in the population that continued nivolumab beyond 1 year compared with those that discontinued therapy. However, this study did not formally investigate or stratify by response status at random assignment and therefore patients who were likely cured may have been included in the continuation of therapy group. It will be important for future trials to identify sensitive biomarkers for minimal residual disease to identify patients who may safely discontinue treatment. Patients who have elected to discontinue therapy after 12 months when no active disease is observed on CT scan, positron emission tomography (PET)/CT scan, or tumor biopsy had a post-discontinuation 3-year EFS rate of 95% in a retrospective study,133 and the prospective ECOG EA6192 PET-Stop trial to validate this approach is ongoing. These biomarker-based strategies should be prioritized to identify regimens which produce lasting antitumor immunity in the absence of ongoing therapy, enabling survivors to stop oncology treatment and return more fully to their precancer lives.
Neoadjuvant trials also offer a unique opportunity to evaluate the potential efficacy of an investigational regimen directly in the resection specimen. ICIs administered before surgery may induce a robust immune response against a broad repertoire of tumor antigens while the lesion is still in situ. Even with limited cycles, ICIs administered in the neoadjuvant setting have been demonstrated to elicit tumor antigen-specific T cells and major pathologic responses.134 Lymphocytic infiltration, necrotic tumor, and antigen-specific T cells in the resection specimen for a combination administered in the neoadjuvant setting can offer direct evidence for immune activity of a combination on a much more rapid timeline than required for the emergence of a tail on the survival curve in the metastatic setting. Caution is warranted, however, in applying pathologic complete response criteria developed for cytotoxic therapies to combinations with an anti-PD-(L)1 backbone, as distinct histopathologic features are observed in resection specimens after neoadjuvant treatment with immune agents.135 Furthermore, although neoadjuvant pembrolizumab plus chemotherapy did demonstrate improvements in pathologic complete response rate and event-free survival for patients with high-risk TNBC in KEYNOTE-522,136,137 the correlation between pathologic responses and OS has not yet been established for immuno-oncology based approaches, and may be confounded by the chemotherapy component.
Potential for impact
Provided the mechanistic rationale for a combination is solid, the early-phase data supports safety and efficacy, and the planned phase III trial is adequately designed, the final feature that determines potential value is the impact that the new regimen would have for patients and the field should it gain approval. Several of the considerations described in the previous sections also factor into the assessment of potential for impact, including the inclusion of correlative and translational studies to help understand the biology and early integration of the biomarker program.
The measure of potential impact that seems most straightforward to assess is unmet medical need, however, definitions of this term vary substantially among stakeholders and are inconsistently applied.138 Historically, unmet medical need has been used to describe a clinical setting with few treatment options and poor survival outcomes. An analysis of 237 indications described by investigators in publications as ‘unmet medical need’ included nearly 100 instances where the disease incidence was 1,000–10,000 cases per year and 5–10 National Comprehensive Cancer Network (NCCN)-recommended regimens were available with 5-year survival rates of 50% or greater.139 For the immunotherapy field, substantial unmet medical need exists for effective options to treat tumors that either do not respond or respond initially and then progress after first-line anti-PD-(L)1-based regimens.10
The design of second-line immunotherapy combination trials is further complicated when the first-line therapy includes anti-PD-(L)1, as it is difficult to differentiate bona-fide activity of a new agent from a ‘long tail’ from prior therapy. Combinations that render immune-excluded and immune desert tumors susceptible to infiltration and elimination by cytotoxic T cells would also fulfill an unmet need. As an example, the SWOG1616 trial is evaluating the addition of ipilimumab to nivolumab versus ipilimumab alone for patients with melanoma who have exhibited progressive disease on anti-PD-(L)1 monotherapy based on the hypothesis that anti-CTLA-4 will augment inadequate T cell infiltration and that the tumor-specific infiltrating T cells will require PD-1 blockade in order to stay active.140 Similar rationales are provided for combinations exploring intratumoral injections of oncolytic virus or toll receptor agonists together with PD-(L)1 pathway blockade.
In evaluating the landscape of competing trials to assess unmet need, it is important to consider the magnitude and type of efficacy expected for an investigational combination. Additionally, patient-centered concepts of value39 40 should be taken into account such as the potential for an investigational combination to offer disease control and even treatment-free remissions, with fewer detrimental impacts on QOL compared with the standard of care option or to reduce direct and indirect aspects of the financial burden of cancer care.
A tool for investigators to evaluate phase III immunotherapy combination trials
Based on the principal components for assessing potential value of phase III immunotherapy clinical trials described in the preceding section, a checklist for prioritizing planned phase III trials evaluating immunotherapy combinations is provided in table 1. Importantly, this checklist assumes that fundamental practical and institutional considerations for a study are satisfied (eg, the study is adequately powered, ethical approval is granted, funding is available, etc). The main purpose of the checklist is to enhance the likelihood of success based on the immuno-oncology aspects of a trial. Individual items on the checklist are categorized based on the application to preclinical, early clinical, or trial design considerations and assigned a level of priority to assist investigators in determining if the portfolio of supporting data and planned design are adequate to move forward to phase III.
A checklist for assessing planned phase III immunotherapy combination trials
The highest priority (level I) is placed on clinical data. Evidence for efficacy in a well-designed randomized phase II study where the contribution of individual components to activity is clearly defined may be sufficient to move forward to phase III even if the preclinical portfolio is lacking. This is especially true for combinations involving dual immunotherapies. By contrast, a combination lacking data from a randomized phase II would be more compelling with evidence of antitumor activity in multiple preclinical models, a well-defined immune mechanism of action, and a predictive biomarker to allow for selection of patients. If the preclinical and early clinical data are both unconvincing, extreme caution is warranted in moving forward to phase III.
To apply the checklist, an investigator considers the gestalt of high-priority and low-priority items satisfied by the combination. Generally, compelling data from a randomized phase II trial alone may be sufficient, whereas multiple trial design or preclinical parameters are necessary to support moving forward to phase III.
Considerations for hematologic malignancies
Anti-PD-(L)1 therapy is a relatively minor component of the armamentarium for hematologic malignancies, although pembrolizumab and nivolumab have both demonstrated efficacy for the treatment of relapsed and refractory Hodgkin lymphoma.141 142 The checklist or a modified version may still be applied to evaluate potential value of phase III trials for hematologic malignancies, even when the backbone agent is not an ICI. Furthermore, the immunotherapies that are most commonly deployed in the treatment of hematologic malignancies may represent the next frontier for immune-unresponsive solid tumors. Multispecific antibodies143 and chimeric antigen receptor T cell therapies144 145 are actively being developed for the treatment of solid tumors. The ability to sample hematologic cancers at the single-cell level has also allowed resistance mechanisms for these modalities to be studied more directly.146 147 Lessons learned from hematologic malignancies will be paramount as these approaches advance through development for solid tumors, for example, the paradigm of a shorter intense induction treatment plan leading to off-therapy control as well as sensitive methods for evaluating minimal residual disease.
Future directions to maximize value in phase III immunotherapy combination trials
Several anti-PD-(L)1-based immunotherapy combinations are now integrated into the standard of care for solid tumors, and the pace of development is not expected to slow. However, it must be emphasized that a positive result in phase III does not guarantee meaningful benefit to patients.39 148 149 For example, statistical significance does not always correlate with clinical significance. Furthermore, for studies designed with PFS as the primary endpoint, successful prolongation of PFS is not always associated with prolonged survival,42 nor does not necessarily improve QOL.150 151 The optimal phase III trial is well-sized for the analysis of OS and captures measures that are particularly important for immunotherapies, such as DOR and TFS.1 Because continuous treatment may not be necessary to affect a long-lasting immune response, limited durations of therapy should also be studied to evaluate the durability of immune-mediated tumor control. To this end, it is recommended that clinical definitions should be established to define when a treatment effect is considered durable and when therapy can safely be stopped.
Trials evaluating immunotherapy combinations should ideally include comparisons to arms in which the agents are given in sequence to assist in ruling out the possibility for independent drug action,74 especially when one component of a combination may suppress antitumor T cell responses. The phase III DREAMseq trial demonstrated superior OS and ORR when patients with stage III-IV BRAF-mutant melanoma received ipilimumab plus nivolumab followed by dabrafenib plus trametinib at disease progression compared with the inverse sequence,113 and the phase II SECOMBIT trial also found favorable survival outcomes with an immunotherapy-first sequence.152 The INSIGNIA trial (EA5163/S1709) is studying whether pembrolizumab alone as a first-line treatment followed by pemetrexed and carboplatin with or without pembrolizumab after disease progression is superior to induction with pembrolizumab, pemetrexed and carboplatin followed by pembrolizumab and pemetrexed maintenance for the treatment of stage IV NSCLC. Future studies should continue to address the effect of sequencing.
The identification, validation, and integration of biomarkers for response prediction and evaluation are also high priorities for future combination trials. The inclusion of biomarker-based patient selection is not only predictive of phase III studies leading to FDA approval,28 36 37 41 but also may provide value for patients by helping select treatments with the greatest likelihood of benefit. Furthermore, biomarkers may also assist in ‘right-sizing’ therapy by identifying patients who have been cured and may safely discontinue treatment. As discussed in the Trial design section, patients with no evidence of active disease by PET/CT and tumor biopsy have been demonstrated to achieve durable EFS after discontinuation of therapy.133 Further biomarker validation is needed to identify patients who may benefit from immunotherapy—such as the use of ctDNA132 as well as sensitive surrogate markers for early response, for example.129 130
Future iterations of the checklist may include numerical values associated with each item, enabling investigators to weigh potential value and directly compare planned phase III trials based on a score. In order to objectively weigh each criterion, it will be necessary to perform retrospective and prospective analyses with multiple variations of experimental combinations of values. Simulation-based approaches may accelerate this undertaking. Similar to biomarker validation, receiver operating characteristics analysis will be necessary to evaluate the predictive power of the checklist, if converted into a quantitative instrument. Such an undertaking, although important, is beyond the scope of this manuscript, which is intended to provide the conceptual and theoretical framework for the checklist.
Conclusion
The advent of anti-PD-(L)1 ushered in a new era for oncology and transformed the outlook for tumors that were once almost universally fatal. Combinations involving anti-PD-(L)1 plus chemotherapies, tumor-targeted therapies, and blockade of additional immune checkpoints have all demonstrated superior response rates and survival outcomes compared with ICI monotherapy in some disease indications, expanding the population of patients who derive benefit with immunotherapy. Not all agents offer enhanced benefit when administered in combination with anti-PD-(L)1 and a surfeit of combination trials with shaky mechanistic rationale, weak data from phase I and phase II, and inadequate design to evaluate agents with immune mechanisms of action is of low value to the immunotherapy field and to patients. To maximize the return on investment for phase III trials and increase the likelihood that patients will benefit, go/no-go decisions should be based on a rigorous framework. SITC urges widespread adoption of the checklist tool for assessing potential value in phase III studies to maximize the return on investment in terms of financial, time, and human costs for late-stage oncology trials and ultimately offer improved outcomes to patients with cancer.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
The authors acknowledge SITC staff for their contributions including Sam Million-Weaver, PhD, for medical writing; Angela Kilbert and Emily Gronseth, PhD for editorial support; and Matthew Erickson and Nichol Larson for project management, editorial support and assistance. The authors thank Martin "Mac" Cheever for conversations during initial manuscript development, prior to his passing. Additionally, the authors wish to thank SITC for supporting the manuscript development.
References
Footnotes
Twitter @PAscierto, @EmensLeisha, @robertferrismd, @gulleyj1, @kmargolin, @matsadoc, @brian_rini
Contributors MBA originated the concept of the checklist. MBA and MS led the conception and design of the work as the Chairs of the manuscript development group. All other authors are listed alphabetically. KAM, RSH, and RWH provided input on the direction and scope of the project as members of a steering committee formed prior to beginning the manuscript development. All authors contributed equally during the manuscript development, providing input on the outline, contributing to the writing, and offering critical review and editing during the manuscript development.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests MBA—Consulting Fees: Aveo, BMS, Eisai, Exelixis, Genentech, Iovance, Merck, Novartis, Pfizer, Roche, Pyxis, Werewolf, Asher Bio, Calithera, Idera, Agenus, Apexigen, Neoleukin, Adagene, AstraZeneca, Elpis, ScholarRock, Surface, ValoHealth, Sanofi, Fathom; Ownership Interest (less than 5%): Werewolf, Pyxis, Elpis. HA-S—Nothing to disclose. PAA—Consulting Fees: BMS, Roche, Array, Novartis, Merck Serono, Pierre Fabre, Incyte, Medimmune, AstraZeneca, Sun Pharma, Sanofi, Idera, Ultimovacs, Sandoz, Immunocore, 4SC, Italfarmaco, Nektar, Boehringer-Ingelheim, Eisai, Regeneron, Daiichi Sankyo, Pfizer, Oncosec, Nouscom, Lunaphore, Seagen, iTEOS, Medicenna; Contracted Research: BMS, Roche-Genentech, Array, Sanofi. MRB—Consulting Fees: Novartis, Kite/Gilead, CRISPR Therapeutics, Autolus, Takeda, Agios, Iovance, Bluebird Bio, BMS; Fees for Non-CE Services: Agio, BMS, Incyte, Sanofi, Kite/Gilead; Contracted Research: Immatics, Novartis, Kite/Gilead, CRISPR Therapeutics, Autolus, TMUNITY. DSC—Ownership Interest (less than 5%): IGM Biosciences, Corvus. MD—Consulting Fees: Lava Therapeutics, Amgen, Janssen, Roche/Genentech. LAE—Consulting Fees: Genentech, F. Hoffmann-La Roche, Chugai, GPCR, Gilead, Immune Onc, Immutep, Shionogi, Mersana; Contracted Research: AbbVie, AstraZeneca, Bolt Therapeutics, Bristol Myers Squibb, Compugen, Corvus, CytomX, EMD Serono, Genentech, F. Hoffmann-La Roche, ImmuneOnc, Maxcyte, Merck, Next Cure, Silverback, Takeda, Tempest; Grants from Non-Industry Entities: HeritX Incorporated, NSABP Foundation, Translational Breast Cancer Research Consortium, Breast Cancer Research Foundation, National Cancer Institute, Department of Defense, Johns Hopkins University, University of California San Francisco, Cornell University, Dana Farber Cancer Institute. Royalties: Elsevier; IP Rights: Aduro Biotech. MSE—Nothing to Disclose. RLF—Consulting Fees: Achilles Therapeutics, Aduro Biotech, Bicara Therapeutics, BMS, Brooklyn Immunotherapeutic, Everest Clinical Research Corporation, F. Hoffman-La Roche, Genocea Biosciences, Hookipa Biotech GmbH, Instill Bio, Kowa Research Institute, Lifescience Dynamics Limited, MacroGenics, Merck, Mirati Therapeutics, Nanobiotix, Novasenta, Numab Therapeutics AG, OncoCyte Corporation, Pfizer, PPD Development, L.P., Rakuten Medical, Sanofi, Seagen, Vir Biotechnology, Zymeworks; Contracted Research: AstraZeneca/MedImmune, BMS, Merck, Novasenta, Tesaro; Stock: Novasenta. TFG—Contracted Research: AstraZeneca, BMS, Merck, Sillajen, Vascular Biogenics. JLG—Nothing to disclose. RSH—Consulting Fees: AbbVie Pharmaceuticals, ARMO Biosciences, AstraZeneca, Bayer HealthCare Pharmaceuticals, Bolt Biotherapeutics, BMS, Candel Therapeutics, Cybrexa Therapeutics, eFFECTOR Therapeutics, Eli Lilly and Company, EMD Serono, Foundation Medicine, Genentech/Roche, Genmab, Gilead, Halozyme Therapeutics, Heat Biologics, I-Mab Biopharma, Immunocore, Infinity Pharmaceuticals, Loxo Oncology, Merck and Company, Mirati Therapeutics, Nektar, Neon Therapeutics, NextCure, Novartis, Ocean Biomedical, Oncternal Therapeutics, Pfizer, Ribbon Therapeutics, Ventana Medical Systems, Sanofi, Seattle Genetics, Shire PLC, Spectrum Pharmaceuticals, STCube Pharmaceuticals, Symphogen, Takeda, Tesaro, Tocagen, WindMIL Therapeutics, Xencor; Contracted Research: AstraZeneca, Eli Lilly and Company, Genentech/Roche, Merck and Company; Board Member (non-executive/independent): Immunocore Holdings Limited, Junshi Pharmaceuticals. RWH—Stock: Black Diamond, Xilio. JL—Consulting Fees: Iovance, Boston Biomedical, Pfizer, BMS, GSK, Novartis, Incyte, Immunocore, YKT Global, iOnctura, Apple Tree, Roche, Pierre Fabre, AstraZeneca, EUSA Pharma, MSD, Ervaxx, Merck, Ipsen, Aptitude, Eisai, Calithera, Ultmovacs, Seagen; Contracted Research: BMS, MSD, Novartis, Pfizer, Achilles, Roche, Nektar, Covance, Immunocore, Pharmacyclics, Aveo. KAM—Consulting Fees: Iovance DMC, ImaginAb SAB, Checkmate Pharma DSMB, Xilio advisor, Werewolf advisor, all except ImaginAb less than 10k/year;Temporary higher consulting fees: ImaginAb (to provide one time extra consulting up to 25k). LM—Consulting Fees: Tethis spa; Fees for Non-CE Services: Roche. SSR—Consulting Fees: Amgen, BMS, Genentech/Roch, Merck, AstraZeneca, Takeda, Eisai, Daiichi Sankyo, Sanofi, GSK, Lilly; Contracted Research: Tesaro, Merck, AstraZeneca, Advaxis, BMS, Amgen, Takeda, Genmab, GSK. MMR—Consulting Fees: BMS, Tolmar, WebMD; Contracted Research: Novartis, Pfizer, Ipsen, TerSera, Merck, Pierre Fabre, Roche, AstraZeneca, BMS, Bayer, DebioPharm. BIR—Consulting Fees: BMS, Pfizer, GNE/Roche, Aveo, Synthorx, Compugen, Merck, Corvus, Surface Oncology, 3DMedicines, Aravive, Alkermes, Arrowhead, GSK, Shionogi, Eisai; Contracted Research: Pfizer, F. Hoffmann-La Roche, Incyte, AstraZeneca, Taris, Seattle Genetics, Arrowhead Pharmaceuticals, Immunomedics, Bristol Myers Squibb, Mirati Therapeutics, Merck, Surface Oncology, Dragonfly Therapeutics, Aravive, Exelixis; Ownership Interest (less than 5%): PTC therapeutics. MS—Consulting Fees: Adaptimmune, Pfizer, Kadmon, Pierre-Fabre, Biond, Nextcure, Incyte, Alligator, Bristol Myers, Ocellaris, Simcha, Rootpath, Numab, Evolveimmune, Biontech, Immunocore, Glaxo Smith Kline, Adagene, Asher, Kanaph, iTEOS, Genocea, Trillium, Sapience, Targovax, Molecular Partners, Ontario Institute for Cancer Research, Jazz Pharmaceuticals, Gilead, Innate pharma, Tessa, Stcube, Oncosec, Regeneron, AstraZeneca, Agenus, Idera, Apexigen, Verastem, Rubius, Genentech-Roche, Boston Pharmaceutical, Servier, Dragonfly, Boehringer Ingelheim, Nektar, Pieris, AbbVie, Zelluna, Seattle Genetics; Stock options: Adaptive Biotechnologies, Amphivena, Intensity, Actym, Evolveimmune, Nextcure, Repertoire, Oncohost, Rootpath, Asher; Stock: Johnson and Johnson, Glaxo-Smith Kline. SITC staff: SMW—Shares owned: Pacific Biosciences, Editas Medicine. ME, AK—Nothing to disclose.
Provenance and peer review Not commissioned; externally peer reviewed.