Article Text

Abstract
Background Oncolytic virus V937 showed activity and safety with intratumoral administration. This phase 1 study evaluated intravenous V937±pembrolizumab in patients with advanced solid tumors.
Methods Patients had advanced non-small cell lung cancer (NSCLC), urothelial cancer, metastatic castration-resistant prostate cancer, or melanoma in part A (V937 monotherapy), and metastatic NSCLC or urothelial cancer in part B (V937+pembrolizumab). Prior immunotherapy was permitted >28 days before study treatment. Patients received intravenous V937 on days 1, 3, and 5 (also on day 8 in part B) of the first 21-day cycle and on day 1 of subsequent cycles for eight cycles. Three ascending dose-escalation cohorts were studied. Dose-escalation proceeded if no dose-limiting toxicities (DLTs) occurred in cycle 1 of the previous cohort. In part B, patients also received pembrolizumab 200 mg every 3 weeks from day 8 for 2 years; dose-expansion occurred at the highest-dose cohort. Serial biopsies were performed.
Results No DLTs occurred in parts A (n=18) or B (n=85). Grade 3–5 treatment-related adverse events (AEs) were not observed in part A and were experienced by 10 (12%) patients in part B. The most frequent treatment-related AEs (any grade) in part B were fatigue (36%), pruritus (18%), myalgia (14%), diarrhea (13%), pyrexia (13%), influenza-like illness (12%), and nausea (12%). At the highest tested dose, median intratumoral V937 concentrations were 117,631 copies/mL on day 8, cycle 1 in part A (n=6) and below the detection limit for most patients (86% (19/22)) on day 15, cycle 1 in part B. Objective response rates were 6% (part A), 9% in the NSCLC dose-expansion cohort (n=43), and 20% in the urothelial cancer dose-expansion cohort (n=35).
Conclusions Intravenous V937+pembrolizumab had a manageable safety profile. Although V937 was detected in tumor tissue, in NSCLC and urothelial cancer, efficacy was not greater than that observed in previous studies with pembrolizumab monotherapy.
Trial registration number NCT02043665.
- Oncolytic Viruses
- Oncolytic Virotherapy
- Immunotherapy
- Lung Neoplasms
- Urinary Bladder Neoplasms
Data availability statement
Data are available upon reasonable request. Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, New Jersey, USA (MSD), is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company’s clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data-sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the USA and European Union or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country-specific or region-specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis-driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data-sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Intratumoral V937, an unmodified strain of Coxsackievirus A21, demonstrated antitumor activity and manageable safety when administered alone or in combination with immune checkpoint inhibitors in patients with advanced melanoma, and intravesical V937 induced changes in the tumor micro-environment (eg, upregulation of programmed cell death ligand 1) in patients with non-muscle-invasive urothelial cancer. Intravenous administration of V937 may be more feasible and may allow access of virus to otherwise inaccessible metastatic sites.
WHAT THIS STUDY ADDS
The phase 1 STORM study was the first to evaluate multiple intravenous doses of V937 as well as the combination of intravenous V937 plus the anti-programmed death 1 antibody pembrolizumab in patients with advanced solid tumors.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
V937 can be safely delivered by intravenous administration and was detected in tumor tissue. However, objective responses associated with intravenous V937+pembrolizumab in the expansion cohorts (non-small cell lung cancer and urothelial cancer) were not greater than those seen with pembrolizumab monotherapy in previous studies.
Background
Oncolytic viruses are being investigated for the treatment of various cancers and have at least two distinct mechanisms of action: direct lysis of tumor cells and induction of locoregional and/or systemic antitumor immunity.1 One oncolytic virus, the genetically modified herpes simplex virus 1 talimogene laherparepvec, received approval in 2015 for treatment of melanoma.1 2 Coxsackievirus A21, a naturally occurring human picornavirus, causes mild cold-like symptoms in humans.3 4 It enters cells via binding to intracellular adhesion molecule 1 (ICAM-1) and decay-accelerating factor receptors,3 both of which are overexpressed in a wide array of cancer cell types.5 Results of clinical studies with V937, an unmodified strain of Coxsackievirus A21, have been reported in advanced melanoma6–8 and non-muscle-invasive urothelial cancer.9 These studies assessed monotherapy and combination therapy with immune checkpoint inhibitors, the latter investigated because of the effects of oncolytic viruses on the tumor microenvironment (including increased CD8+ T-cell infiltration and programmed cell death ligand 1 (PD-L1) expression,10 as well as decreased regulatory and suppressor T cells).11
In advanced melanoma, intratumoral V937 with or without immune checkpoint inhibitors demonstrated manageable safety and systemic antitumor activity.6–8 Intratumoral V937 plus the anti-cytotoxic T-lymphocyte antigen 4 antibody ipilimumab6 or the anti-programmed death 1 antibody pembrolizumab7 appeared to provide an additive benefit. Objective response rates (ORRs) with these combinations (47%–50%)6 7 were greater than historic rates with the individual agents (12%–34%).8 12 13 In non-muscle-invasive urothelial cancer, first-line intravesical V937 was well tolerated and induced changes in the tumor microenvironment including upregulation of PD-L1 and lymphocyte activation gene-3 expression, supporting further studies in combination with immune checkpoint inhibitors.9
Compared with intratumoral injection, intravenous administration of oncolytic viruses may be more feasible (requiring less training and fewer logistical concerns) and may allow access of virus to otherwise inaccessible metastatic sites. Studies of intravenous V937 treatment are limited. In a first-in-human study, a single intravenous V937 dose was well tolerated in patients with advanced solid tumors, with some evidence of transient antitumor activity.14 Here, we report the primary outcomes of the phase 1 STORM study (Protocol VLA-009; NCT02043665) evaluating intravenous V937±pembrolizumab in patients with advanced solid tumors, including expansion cohorts in non-small cell lung cancer (NSCLC) and urothelial cancer. The rationale for assessing the combination of these agents in NSCLC and urothelial cancer in part B of the study was because these tumor types are known to have high expression of ICAM-1 and evidence of pembrolizumab activity.
Methods
Study design and patients
This phase 1, multicenter, open-label, dose-escalation study followed a 3+3 cohort design (online supplemental figure S1). The study contained two parts: part A explored escalating doses of V937 alone, and part B tested V937 in combination with pembrolizumab. In part A, eligible patients had histologically-confirmed metastatic NSCLC, urothelial cancer, castration-resistant prostate cancer, or stage IIIC/IV melanoma; part B was restricted to patients with histologically-confirmed metastatic NSCLC or urothelial cancer. The cancer types studied were based on preclinical data5 and results from the aforementioned clinical trials. Other key eligibility criteria in both parts included measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) V.1.1; Eastern Cooperative Oncology Group (ECOG) performance status of 0–2 (0/1 in part B); no significant V937 neutralizing antibodies (≤1:16 titer); adequate hematologic, renal, and hepatic function; and no chemotherapy, radiation therapy, hormonal therapy, or immunotherapy within 28 days of treatment initiation (within 21 days of treatment initiation in part B expansion and also with no more than one prior PD-(L)1 inhibitor regimen). In part A, patients in the highest-dose cohort (see Treatment for description of cohorts and dosing) must have had a lesion accessible for biopsy on day 8, cycle 1. In part B, patients in the highest-dose cohort (dose-expansion cohort) must have had a lesion accessible for biopsy before treatment and on day 15, cycle 1. Further details on inclusion/exclusion criteria are in the protocol (online supplemental material).
Supplemental material
Supplemental material

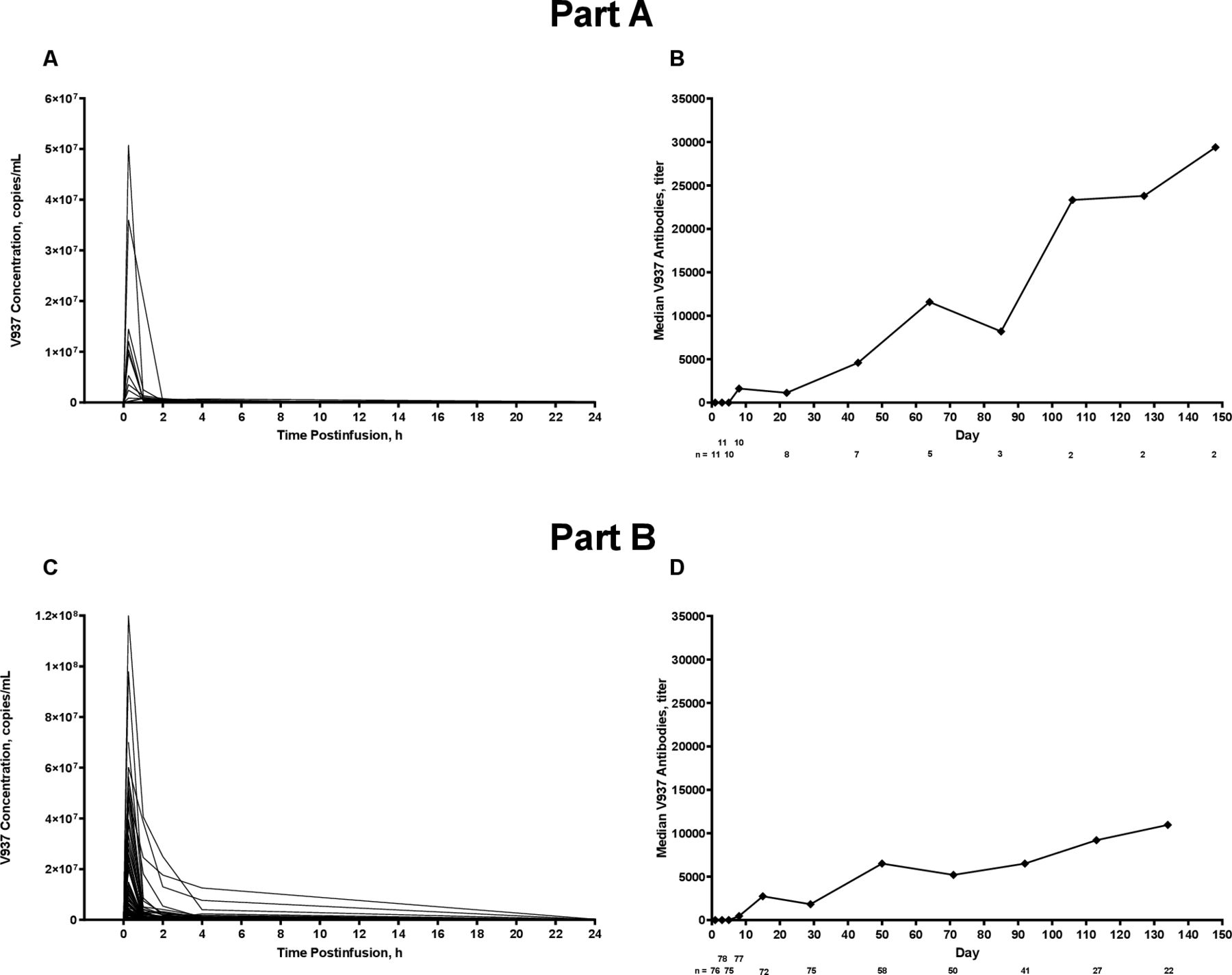
Individual serum concentrations of V937 on day 1, cycle 1, in cohort 3 and median serum concentrations of V937 antibodies over time in cohort 3. Part A (V937 monotherapy) is shown in panels A and B; part B (V937+pembrolizumab) is shown in panels C and D.
Treatment
In parts A and B, patients received intravenous V937 (manufactured by Nova Laboratories, Gloucester Crescent, Leicester, UK) in 100 mL of saline on days 1, 3, and 5 (also on day 8 in part B) of the first 21-day cycle and on day 1 of subsequent cycles for up to eight cycles. Three 50% tissue culture infectious dose (TCID50; dilution of virus required to infect 50% of a given cell culture) cohorts were studied at ascending doses based on dose-limiting toxicities (DLTs): 1×108 TCID50 (cohort 1), 3×108 TCID50 (cohort 2), and 1×109 TCID50 (cohort 3). The maximum dose of V937 administered was based on the titer that could be manufactured. Dose escalation occurred if no cycle 1 DLTs were observed at the previous dose. If DLTs occurred, additional rules regarding dose expansion and identification of the phase 2 dose were followed (see protocol in online supplemental material).
In part B, patients received pembrolizumab 200 mg every 3 weeks from day 8 for up to 2 years in addition to V937 by the same dose-escalation schema. If no cycle 1 DLTs were observed at the highest dose of V937+pembrolizumab, dose expansion of this cohort occurred. Treatment continued for the planned treatment duration or until confirmed complete response (CR) or disease progression, based on immune-related RECIST (irRECIST), or intolerance to study drug.
Assessments and endpoints
In parts A and B, adverse events (AEs) and DLTs were assessed using National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), V.4.03. AEs were evaluated from enrollment through 30 days (90 days for serious AEs) after cessation of study treatment. DLTs included most grade ≥3 non-hematologic or hematologic toxicities that were considered related to V937 and/or pembrolizumab and that occurred during the 21-day period of cycle 1 (see online supplemental table S1). A cohort committee reviewed safety data and DLTs before proceeding to the next V937 dose level and before dose expansion.
Tumor biopsy samples were obtained from cohort 3 on day 8, cycle 1 in part A and at screening and on day 15, cycle 1 in part B. V937 concentrations in tumor samples were assessed using quantitative PCR. Paired tumor samples in part B were assessed for PD-L1 expression using PD-L1 IHC 22C3 pharmDx (Agilent Technologies, Carpinteria, California, USA). Serum samples were taken at multiple time points (see protocol in online supplemental material) before and after V937 infusion in parts A and B for evaluation of V937 concentrations (assessed by PCR) and V937 neutralizing antibody titers. Samples were first run on the PCR assay, and if positive, further assessed using the tissue-culture based infectivity assay. For the PCR assay, viral RNA samples were extracted using Qiagen viral RNA mini kit and real-time (RT)-PCR was run using the QuantiFast Pathogen RT-PCR+IC kit. Samples were run on an Applied Biosystems StepOnePlus Real-Time PCR system and analyzed using StepOne V.2.1 software. The lower limit of quantification for this assay was 15,000 copies/mL. Samples below this level were reported as below the level of detection. The infectivity assay assessed the TCID50 of the samples on SK-MEL-28 cells for the determination of viral titer in serum, urine, sputum, fecal, throat swab, injection site, and outer dressing swab samples. Results were reported as TCID50/mL. If no cytopathic effect was detected in any well, the results were reported as <32 TCID50/mL. Tumor and serum samples were analyzed at central laboratories.
In parts A and B, contrast-enhanced CT or MRI scans were performed at screening and at cycles 3, 5, and 7 and month 6 in part A and every 12 weeks starting at cycle 5 in part B. Tumor response was assessed at each scan visit using irRECIST and RECIST V.1.1 criteria.15 16 Bone scans were performed in part A for patients with prostate cancer (see protocol in online supplemental material).
Primary endpoints were safety (AEs and DLTs) and to determine if V937 was capable of tracking to malignant tumors (as assessed by culturing of virus from biopsy samples). Secondary endpoints included V937 pharmacokinetics, serum V937 neutralizing antibody titers, biomarkers, and preliminary efficacy assessment based on ORR, time to response, duration of response, progression-free survival (PFS), and overall survival (OS).
Statistical analyses
Outcomes were summarized descriptively in part A. For the part B NSCLC dose-escalation cohort, it was estimated that the ORR associated with pembrolizumab monotherapy would be ~20% based on historic data from a heavily pretreated NSCLC cohort.17 A sample size of 43 patients provided ~80% power for the null hypothesis that the ORR is 20% versus the alternative ORR of 30% using a two-sided, one-sample binomial test at a significance level of 0.05. For the part B urothelial cancer dose-escalation cohort, it was estimated that the ORR associated with pembrolizumab monotherapy would be ~24% based on historic data from a heavily pretreated urothelial cancer cohort.18 A sample size of 35 patients provided >80% power for the null hypothesis that the ORR is 24% versus the alternative ORR of 37% using a two-sided, one-sample binomial test at a significance level of 0.05.
Safety and efficacy analyses were performed in all patients who received study drug. For ORR, 95% CIs were based on the exact method for binomial data. PFS and OS were estimated using the Kaplan-Meier method. Analyses were conducted using SAS statistical software V.9.3 and V.9.4 (SAS Institute, Cary, North Carolina, USA).
Results—part A
Patients
Part A was conducted between March 2014 and August 2016. At the cut-off date (April 12, 2017), 18 patients were enrolled and treated with V937 monotherapy (urothelial cancer, n=5; NSCLC, n=5; castration-resistant prostate cancer, n=4; melanoma, n=4). Median time from initial diagnosis was 47.5 months (range, 12–196). Median age was 65.9 years (range, 32–81). Most patients (83%) had an ECOG performance status of 0 or 1. All patients received previous anticancer therapy; 94% received prior immunotherapy. Median number of prior treatments was 4 (range, 2–10; table 1). Median duration of V937 treatment was 6.3 weeks (range, 0.1–21.3); patients received a median of 5 infusions (range, 1–10). The most common reason for study drug discontinuation was disease progression (44%; online supplemental figure S2).
Demographics and baseline characteristics by cohort (parts A and B)

Kaplan-Meier analysis of progression-free survival (PFS) (A) and overall survival (OS) (B) in the non-small cell lung cancer (NSCLC) and urothelial cancer dose-expansion cohorts (part B, V937+pembrolizumab).
Safety
No DLTs occurred with V937 monotherapy; dose escalation proceeded as planned. Treatment-related AEs were experienced by 11 (61%) patients (table 2 and by CTCAE grade in online supplemental table S2). No treatment-related AEs were grade 3–5 in severity or led to V937 discontinuation or dose modification. The most common treatment-related AEs were fatigue (28%), pyrexia (28%), influenza-like illness (17%), and lethargy (17%).
Treatment-related adverse events by cohort (parts A and B)
Efficacy
Efficacy was determined primarily by investigator-assessed irRECIST. Efficacy according to RECIST V.1.1 was similar (online supplemental table S3).
Best percentage changes from baseline in target lesions are shown in online supplemental figure S3. There was one confirmed partial response (PR; cohort 3, prostate cancer) to V937 monotherapy (1×109 TCID50), for an ORR of 6% (table 3). Nine (50%) patients had stable disease. Time to response for the patient with a PR was 1.2 months; the patient was censored in the assessment of duration of response (because of death >84 days after the last assessment). Sixteen (89%) patients experienced a PFS event (disease progression or death). Median PFS was 2.6 months (95% CI, 1.2 to 3.5), and the 13-week PFS rate was 39%. Seventeen (94%) patients died. Median OS was 6.4 months (95% CI, 3.0 to 11.1), and the 52-week OS rate was 22%.
Efficacy results by cohort (part A and B)
{kind=link}
{kind=link}
{kind=link}
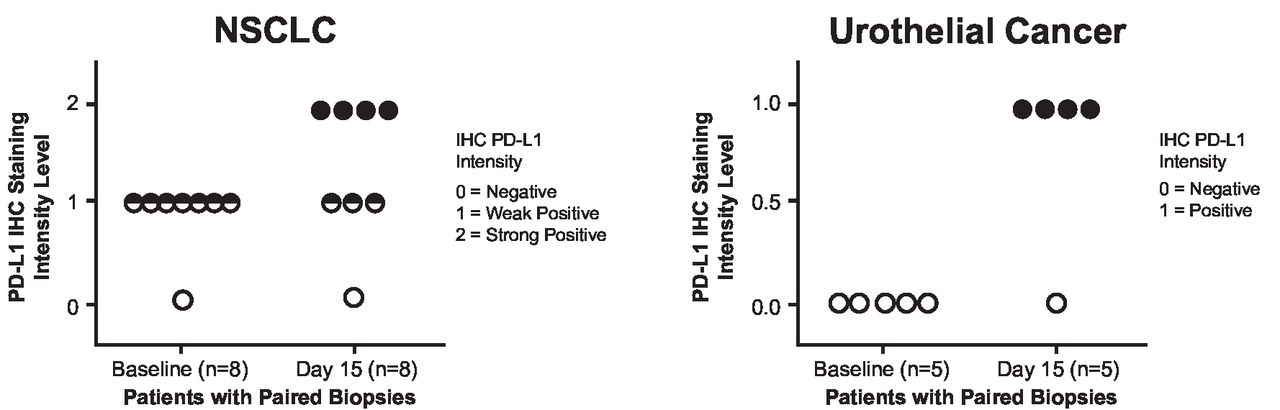
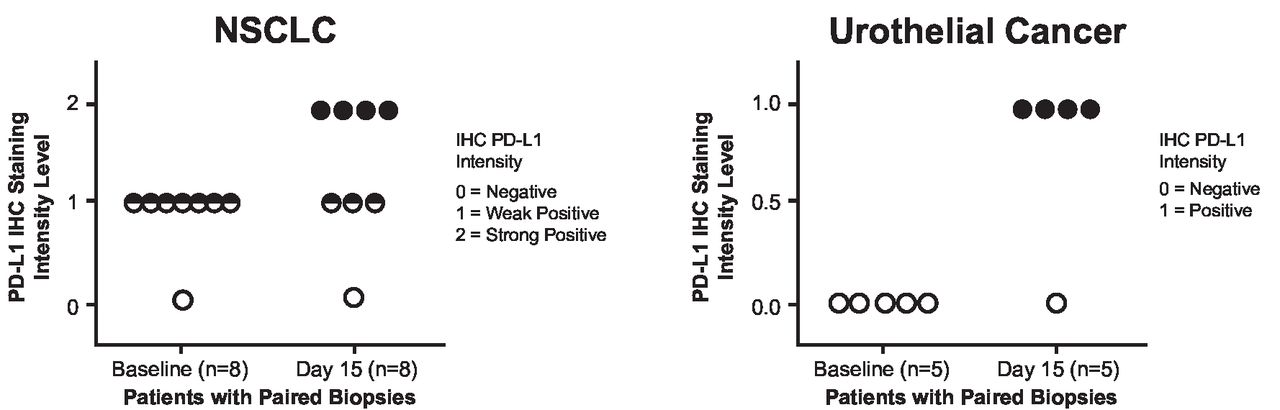
Programmed cell death ligand 1 (PD-L1) expression levels (immunohistochemistry) in tumor cells from paired biopsies in the non-small cell lung cancer (NSCLC) and urothelial cancer dose-expansion cohorts (part B, V937+pembrolizumab). Please contact C M Rudin at rudinc@mskcc.org for access to the biomarker data presented in this figure.
Correlative studies in highest-dose cohort (cohort 3)
On day 8, cycle 1, median V937 concentration in tumor samples (n=6) was 117,631 copies/mL (range, 12,749–262,946). None of the six patients experienced a radiological response.
Median maximum observed serum concentration of V937 on day 1, cycle 1 was 75 copies×105/mL (range, 8–508), and median time of maximum observed concentration was 0.25 hours (range, 0.25–1.00; figure 1A). Median serum concentrations of V937 neutralizing antibodies tended to increase over time, although the sample size was limited beyond cycle 1 (figure 1B). Median V937 infectivity in serum on day 1, cycle 1 was 320 TCID50/mL within 15 min post-infusion and decreased to 32 TCID50/mL at all time points from 1 to 24 hours post-infusion. In most patients, serum concentrations of V937 were detected up to 2 hours after infusion.
Results—part B
Patients
Part B was conducted between May 2016 and December 2019. As of the cut-off date (January 10, 2020), 85 patients were enrolled and treated with V937 and pembrolizumab (NSCLC, n=46 and urothelial cancer, n=39). Median time since initial diagnosis was 18.3 months (range, 1–175). Median age was 66 years (range, 40–83). Most patients (54%) had an ECOG performance status of 1. Nine (11%) patients were EGFR mutation-positive. The majority of the patients (96%) received previous anticancer therapy; median number of prior treatments was 5 (range, 1–40; table 1). Previous immune checkpoint inhibitor therapy was received by 20% of the patients. Median duration of V937 treatment was 10.3 weeks (range, 0.4–25.1), with patients receiving a median of 7 infusions (range, 2–12). Patients also received pembrolizumab for a median duration of 9.1 weeks (range, 0.1–102.3), with patients receiving a median of 4 doses (range, 1–34). The most common reason for study drug discontinuation was disease progression (69%; online supplemental figure S2).
Safety
No DLTs occurred with V937+pembrolizumab. Treatment-related AEs were experienced by 68 (80%) patients (table 2 and by CTCAE grade in online supplemental table S2). There were no grade 4 or 5 treatment-related AEs. Ten (12%) patients experienced grade 3 treatment-related AEs; colitis was the only event to occur in >1 patient (n=2 (2%)). Of these 10 patients, events were considered related to pembrolizumab in 8 patients and related to V937 in 2 patients. Five (6%) patients had treatment-related AEs leading to V937 and/or pembrolizumab discontinuation: acute myocardial infarction, fatigue, myalgia, immune-mediated pneumonitis, and colitis/diarrhea (n=1 each (1%)). Eleven (13%) patients had treatment-related AEs (all grades ≤3) resulting in dose modification (V937 and/or pembrolizumab dose delayed or interrupted); fatigue and increased blood creatine phosphokinase were the only events to occur in >1 patient (both n=2 (2%)).
Efficacy in NSCLC and urothelial cancer dose-expansion cohorts
Best percentage changes from baseline in target lesions are shown in online supplemental figure S3. In the NSCLC cohort (n=43), there were three confirmed CRs and one confirmed PR to V937+pembrolizumab, for an ORR of 9% (95% CI, 2.6% to 22.1%; table 3). All four patients had a durable response (CR or PR lasting ≥6 months); none had received previous treatment with immune checkpoint inhibitors. Median time to response was 3.0 months (range, 3.0–5.5), and duration of response ranged from 2.8 to 25.5+ months. Forty-one (95%) patients experienced a PFS event (figure 2A). Median PFS was 2.9 months (95% CI, 2.4 to 3.0), and the 26-week and 52-week PFS rates were 23% and 7%, respectively. Thirty-one (72%) patients died (figure 2B). Median OS was 11.5 months (95% CI, 7.2 to 15.9), and the 26-week and 52-week OS rates were 72% and 46%, respectively.
In the urothelial cancer cohort (n=35), there were three confirmed CRs and four confirmed PRs, for an ORR of 20% (95% CI, 8.4% to 36.9%). A durable response was seen in 6 (17%) patients. Median time to response was 3.0 months (range, 2.8–9.0), and duration of response ranged from 2.3 to 24.3+ months. None of the seven patients with a response had received previous treatment with immune checkpoint inhibitors. Twenty-seven (77%) patients experienced a PFS event (figure 2A). Median PFS was 3.0 months (95% CI, 2.2 to 5.8), and the 26-week and 52-week PFS rates were 34% and 24%, respectively. Twenty-five (71%) patients died (figure 2B). Median OS was 9.4 months (95% CI, 4.6 to 16.4), and the 26-week and 52-week OS rates were 60% and 43%, respectively.
Correlative studies in NSCLC and urothelial cancer dose-expansion cohorts
On day 15, cycle 1, median V937 concentration in tumor samples from 22 patients with data available was 101,280 copies/mL (range, 15,366–198,000) among the three patients with detectable levels. None of the three patients was a responder. The remaining 19 patients had V937 concentrations below the level of detection. For PD-L1 expression, immunohistochemistry analysis of paired tumor samples from 13 patients (NSCLC, n=8 and urothelial cancer, n=5) with negative or low baseline PD-L1 expression revealed a notable increase in PD-L1-positive tumor cells on day 15 in 8 patients (figure 3).
Results in serum were similar to those in part A (figure 1C and D). Median V937 infectivity in serum on day 1, cycle 1 was 441 TCID50/mL within 15 min post-infusion, which decreased to 32 TCID50/mL at all time points from 2 to 24 hours post-infusion.
Discussion
The phase 1 STORM study was the first to evaluate multiple intravenous doses of the oncolytic virus V937, as well as the combination of intravenous V937+pembrolizumab, in patients with advanced solid tumors. Intravenous V937 reached tumor tissue in approximately one-third of the patients tested. Safety was manageable and pharmacokinetics were similar with V937 alone and with V937 in combination with pembrolizumab. There were no DLTs, no deaths because of treatment-related AEs, and no unexpected safety concerns. Toxicity was as anticipated given prior experience with pembrolizumab and V937 monotherapy. In the combined NSCLC and urothelial cancer dose-expansion cohorts (part B), the types of treatment-related AEs reported were consistent with those previously described for intratumoral V937 in patients with melanoma8 and pembrolizumab in patients with NSCLC or urothelial cancer.19–21 Our results add to the growing list of oncolytic viruses that can be safely delivered by intravenous administration.22
Antitumor activity was limited with V937 alone (part A), with one patient experiencing a PR at the highest dose assessed. The primary rationale behind this study was to test the hypothesis that tumor infection with V937 could substantially stimulate immune response to subsequent programmed cell death 1 (PD-1) checkpoint inhibition with pembrolizumab. However, our results in NSCLC and urothelial cancer suggest a lack of additive benefit with the combination of intravenous V937+pembrolizumab. Interestingly, none of the nine patients (n=3 each of melanoma, NSCLC, and urothelial cancer) with detectable levels of V937 in tumor tissue were responders. The results from our study contrast with previous findings in which V937 was administered intratumorally as monotherapy8 or in combination with ipilimumab6 or pembrolizumab7 in patients with advanced melanoma. In those studies, V937 demonstrated antitumor activity alone and combination therapy appeared to have an additive benefit. There are several plausible explanations. It is possible that insufficient intratumoral V937 levels were reached by intravenous administration or that the viral titer achievable by manufacture was inadequate. Additionally, viral delivery to the tumor may have been limited by low levels of ICAM-1 (not assessed in our study), physiological dilution of V937 in the peripheral blood, and/or binding of V937 to serum proteins. It is also possible that induction of an inflammatory cascade also included acute PD-L1 upregulation (figure 3), inhibiting T-cell responses despite pembrolizumab. The observed upregulation of PD-L1 expression after V937 administration may suggest that the dosing schedule of pembrolizumab was not optimal. A chance of enhanced efficacy may also have been negatively influenced by the presence of neutralizing antibodies against V937, which increased over time in the study, EGFR mutations in 11% of patients, and the heavily pretreated study population. Finally, since early blockade of PD-1 may have enhanced antiviral immunity and resulted in more rapid viral clearance, starting pembrolizumab was delayed to day 8 in part B (combination therapy cohort). It is unknown whether an alternate dosing schedule (eg, more frequent V937 dosing) would have enhanced viral delivery to the tumor and potentially improved outcomes.
Although cross-study comparisons must be interpreted with caution, response rates in patients with advanced NSCLC were numerically lower with intravenous V937+pembrolizumab in STORM than with pembrolizumab monotherapy in previous studies. The ORR of 9% in our NSCLC expansion cohort (part B) was half the rate of 18% (95% CI, 14 to 23) achieved with pembrolizumab alone in patients with advanced NSCLC in KEYNOTE-010.19 Differences in PD-L1 expression and receipt of previous therapy may have contributed. All patients in KEYNOTE-010 were required to have PD-L1 tumor proportion score (TPS) ≥1% (42% had PD-L1 TPS ≥50%), and although all patients were previously treated in that study, most (69%) had received one line of previous therapy.19 Similarly, in KEYNOTE-042, all patients were required to have PD-L1 TPS ≥1% (31% with PD-L1 TPS ≥50%) and had not received previous therapy.23 In contrast, PD-L1 expression was not required for enrollment in STORM (patients with PD-L1 TPS <1% would not typically be candidates for pembrolizumab monotherapy), and the majority (72%) of the patients had received ≥2 lines of previous therapy. Prior findings from KEYNOTE-001 indicate that response rates to pembrolizumab monotherapy are expected to be greater in patients who are treatment naive compared with those who were previously treated.24
In the urothelial cancer expansion cohort (part B), an ORR of 20% was achieved with intravenous V937+pembrolizumab. This response rate was generally consistent with that in previous reports with pembrolizumab alone in patients with advanced urothelial cancer.20 21 The study most suitable for comparison is KEYNOTE-045, in which the ORR was 21%.20 Both STORM and KEYNOTE-045 enrolled previously treated patients regardless of PD-L1 status. A somewhat higher ORR of 26% was noted with pembrolizumab monotherapy in KEYNOTE-012,21 perhaps owing in part to nearly one-quarter of the patients receiving first-line therapy and/or the prerequisite of tumor PD-L1 expression at enrollment.21
Limitations to this study include the lack of comparator monotherapy arms for the dose-expansion NSCLC and urothelial cancer cohorts in part B. In addition, sample sizes for some of the analyses were relatively small (eg, V937 concentrations in tumor biopsies) and efficacy was not a primary objective. These results should therefore be interpreted with caution.
In conclusion, intravenous V937+pembrolizumab had a manageable safety profile in patients with advanced solid tumors. Efficacy in the expansion cohorts (NSCLC and urothelial cancer) was not greater than that seen with pembrolizumab monotherapy in previous studies.
Data availability statement
Data are available upon reasonable request. Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, New Jersey, USA (MSD), is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company’s clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data-sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the USA and European Union or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country-specific or region-specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis-driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data-sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.
Ethics statements
Patient consent for publication
Ethics approval
This was a multicenter study. The study protocol and amendments were approved by institutional review boards (IRBs) or independent ethics committees at each study site. For example, for Dr Brendan Curti, the IRB Providence Health and Services (Oregon) (IRB ID: MODCR2017000057) reviewed and approved the study protocol on February 14, 2017. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank the patients and their families and caregivers for participating in this study, along with all investigators and site personnel. Medical writing assistance was provided by Michael S. McNamara, MS, of ICON plc (Blue Bell, PA, USA). This assistance was funded by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @charlesrudin, @mattzibelman
Contributors CMR: conception, design or planning of the study; acquisition of the data; analysis of the data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; provision of study materials/patients; final approval of the submitted manuscript; accountability for the work; and guarantor. HSP: conception, design or planning of the study; acquisition of the data; analysis of the data; interpretation of the results; drafting of the manuscript; reviewing or revising the manuscript for important intellectual content; provision of study materials/patients; final approval of the submitted manuscript; and accountability for the work. MZ: acquisition of the data; analysis of the data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; final approval of the submitted manuscript; and accountability for the work. WLA: acquisition of the data; analysis of the data; interpretation of the results; drafting of the manuscript; reviewing or revising the manuscript for important intellectual content; provision of study materials/patients; final approval of the submitted manuscript; and accountability for the work. KJH: conception, design or planning of the study; acquisition of the data; analysis of the data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; provision of study materials/patients; final approval of the submitted manuscript; and accountability for the work. DD: acquisition of the data; reviewing or revising the manuscript for important intellectual content; provision of study materials/patients; final approval of the submitted manuscript; and accountability for the work. AGH: acquisition of the data; reviewing or revising the manuscript for important intellectual content; final approval of the submitted manuscript; and accountability for the work. SJO: analysis of the data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; final approval of the submitted manuscript; and accountability for the work. TDC: acquisition of the data; reviewing or revising the manuscript for important intellectual content; final approval of the submitted manuscript; and accountability for the work. GMW: acquisition of the data; analysis of the data; interpretation of the results; drafting of the manuscript; reviewing or revising the manuscript for important intellectual content; final approval of the submitted manuscript; and accountability for the work. RRJ: acquisition of the data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; final approval of the submitted manuscript; and accountability for the work. DEG: acquisition of the data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; provision of study materials/patients; final approval of the submitted manuscript; and accountability for the work. JER: conception, design or planning of the study; acquisition of the data; analysis of data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; provision of study materials/patients; final approval of the submitted manuscript; and accountability for the work. CR: conception, design or planning of the study; acquisition of the data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; provision of study materials/patients; administrative, logistical or technical support; final approval of the submitted manuscript; and accountability for the work. DCC: acquisition of the data; analysis of the data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; final approval of the submitted manuscript; and accountability for the work. BDC: acquisition of the data; reviewing or revising the manuscript for important intellectual content; provision of study materials/patients; final approval of the submitted manuscript; and accountability for the work. JRM: interpretation of the results; reviewing or revising the manuscript for important intellectual content; final approval of the submitted manuscript; and accountability for the work. YR: analysis of the data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; final approval of the submitted manuscript; and accountability for the work. EVS: analysis of the data; interpretation of the results; reviewing or revising the manuscript for important intellectual content; final approval of the submitted manuscript; and accountability for the work. LG: conception, design or planning of the study; reviewing or revising the manuscript for important intellectual content; administrative, logistical or technical support; final approval of the submitted manuscript; and accountability for the work. SG: conception, design or planning of the study; analysis of the data; acquisition of the data; interpretation of the results; drafting of the manuscript; reviewing or revising the manuscript for important intellectual content; provision of study materials/patients; administrative, logistical or technical support; final approval of the submitted manuscript; and accountability for the work.
Funding This study was sponsored by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, New Jersey, USA. This work was supported in part by National Cancer Institute Cancer Center Support, grant P30 CA008748.
Competing interests CMR has consulted regarding oncology drug development with AbbVie, Amgen, AstraZeneca, Epizyme, Genentech/Roche, Ipsen, Jazz, Lilly, and Syros. He serves on the scientific advisory boards of Bridge Medicines, Earli, and Harpoon Therapeutics. HSP has received speaker and advisory board honoraria from Ipsen, Eisai, Pfizer, and Bristol Myers Squibb. MZ has received research support (to institution) from Bristol Myers Squibb and Exelixis. He has received advisory board honoraria from Aveo, Exelixis, Janssen, Pfizer, and EMD Serono. WLA is a data safety monitoring board member for Lilly. KJH has received research funding from AstraZeneca, Boehringer-Ingelheim, MSD, and Replimune. He serves on the scientific advisory boards of Arch Oncology, AstraZeneca, Bristol Myers Squibb, Boehringer-Ingelheim, Codiak BioSciences, Inzen Therapeutics, ISA Pharma, Merck-Serono, MSD, Pfizer, and Replimune. He serves on the speaker’s bureau for Bristol Myers Squibb, MSD, and Replimune. DD has received research support (to institution) from Ambrx Biopharma, Beigene, Bristol Myers Squibb, EpimAb, Harbour BioMed, Maxinovel, MSD, Olema Pharmaceuticals, Pfizer, PhamAbcine, Roche, and Haihe Biopharma. She serves on the scientific advisory board of MSD and has received travel support from Novartis. AGH has nothing to disclose. SJO has served on the scientific advisory boards of Biontech, Ultimovacs, Merck, ImaginAB, and Array. He was a consultant for Rad Immune, Bristol Myers Squibb, and Biontech; has served on the speaker’s board for Bristol Myers Squibb; and has received honoraria from Array, Ultimovacs, Merck, Rad Immune, ImaginAB, Biontech, and Bristol Myers Squibb. TDC has received honoraria from AstraZeneca, Merck USA, Novartis, Roche, and Specialized Therapeutics Australia; travel/accommodation support from Astellas; conference support from Novartis; and research support (to institution) from Daiichi Sankyo, Beigene, Janssen, Amgen, Exelixis, Merck, Immutep, and Clovis. He serves on the scientific advisory boards of AstraZeneca, Novartis, and Cipla. GMW has received speaker honoraria from AstraZeneca, Medtronic, Johnson & Johnson, and Device Technologies Australia and consulting fees (Expert Input Forum) from MSD. RRJ has nothing to disclose. DEG has received research funding from AstraZeneca, BerGenBio, and Karyopharm. He serves on the scientific advisory boards of Regeneron, Sanofi, Jansen, Mirati, and Catalyst and on the steering committee of Bristol Myers Squibb. He is a shareholder in Gilead. JER has received research funding (institutional) from Bayer, SeaGen, AstraZeneca, Roche/Genentech, Astellas, and QED Therapeutics. He has received consulting fees from IMVax, Century Tx, Aadi, Alligator, Hengrui, Bayer, SeaGen, AstraZeneca, Roche/Genentech, Astellas, QED Therapeutics, Bristol Myers Squibb, Merck, Pfizer, Pharmacyclics, Boehringer Ingelheim, GlaxoSmithKline, Infinity, Janssen, Mirati, EMD-Serono, Gilead, Lilly, Tyra Biosciences, and Pharmacyclics. He has received honoraria from EMD-Serono, Research to Practice, MJH LifeSciences, Medscape, Uptodate, Clinical Care Options, and OncLive. CR has received research funding (institutional) from Viralytics, Roche, Viralytics/Merck, and Eisai and has participated in a consulting/advisory role with Bristol Myers Squibb. She has received honoraria from Bristol Myers Squibb, Pfizer, Novartis, and Eisai and travel, accommodations, and expenses from Astellas Pharma, Roche, Janssen, GlaxoSmithKline, Bristol Myers Squibb, and Ipsen. DCC has nothing to disclose. BDC has received research funding from Viralytics, Galectin Therapeutics, Clinigen, AstraZeneca (research institution), and Bristol Myers Squibb (research institution). He has participated on the scientific advisory boards of Clinigen, Cullinan Oncology, and Nektar. He is a data safety monitoring board member for Merck USA. JRM has received research funding (institutional) from Sillagen, Vyriad, Replimune, Corvus Pharmaceuticals, Tizona, Trishula Therapeutics, Merck, Eisai, Genentech, Peloton Therapeutics, Seagen, Rubius Therapeutics, and BioNtech. He has participated on the scientific advisory board of Merck. YR is an employee of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, New Jersey, USA, and stockholder in Merck & Co., Inc., Rahway, New Jersey, USA. EVS is an employee of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, New Jersey, USA, and stockholder in Merck & Co. Inc., Rahway, New Jersey, USA. LG is a former contractor to Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, New Jersey, USA, and stockholder in Merck & Co., Inc., Rahway, New Jersey, USA. SG has received honoraria from SITC advances in Cancer Immunotherapy as the Salt Lake City Program Organizer and presenter, travel support from QED Biopharmaceutical, as well as funding for other clinical trials from Bristol Myers Squibb, Rexahn, Incyte, Novartis, LSK, Five Prime, Mirati, QED, Debiopharm, Merck, Pfizer, AstraZeneca, MedImmune, Clovis, Immunocore, Elevar Therapeutics, and Seattle Genetics.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.