Article Text

Abstract
Background Galectin-9 is a member of the family of lectin proteins and crucially regulates human immune responses, particularly because of its ability to suppress the anticancer activities of T lymphocytes and natural killer cells. Recent evidence demonstrated that galectin-9 is highly expressed in a wide range of human malignancies including the most aggressive tumors, such as high-grade glioblastomas and pancreatic ductal adenocarcinomas, as well as common malignancies such as breast, lung and colorectal cancers. However, solid tumor cells at rest are known to secrete either very low amounts of galectin-9 or, in most of the cases, do not secrete it at all. Our aims were to elucidate whether T cells can induce galectin-9 secretion in human cancer cells derived from solid malignant tumors and whether this soluble form displays higher systemic immunosuppressive activity compared with the cell surface-based protein.
Methods A wide range of human cancer cell lines derived from solid tumours, keratinocytes and primary embryonic cells were employed, together with helper and cytotoxic T cell lines and human as well as mouse primary T cells. Western blot analysis, ELISA, quantitative reverse transcriptase-PCR, on-cell Western and other measurement techniques were used to conduct the study. Results were validated using in vivo mouse model.
Results We discovered that T lymphocytes induce galectin-9 secretion in various types of human cancer cells derived from solid malignant tumors. This was demonstrated to occur via two differential mechanisms: first by translocation of galectin-9 onto the cell surface followed by its proteolytic shedding and second due to autophagy followed by lysosomal secretion. For both mechanisms a protein carrier/trafficker was required, since galectin-9 lacks a secretion sequence. Secreted galectin-9 pre-opsonised T cells and, following interaction with other immune checkpoint proteins, their activity was completely attenuated. As an example, we studied the cooperation of galectin-9 and V-domain Ig-containing suppressor of T cell activation (VISTA) proteins in human cancer cells.
Conclusion Our results underline a crucial role of galectin-9 in anticancer immune evasion. As such, galectin-9 and regulatory pathways controlling its production should be considered as key targets for immunotherapy in a large number of cancers.
- Tumor Microenvironment
- T-Lymphocytes
- Receptors, Immunologic
- Immune Evation
Data availability statement
Data are available on reasonable request. The datasets used and/or analysed during the current study are available from the corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Disclaimer: this video summarises a scientific article published by BMJ Publishing Group Limited (BMJ). The content of this video has not been peer-reviewed and does not constitute medical advice. Any opinions expressed are solely those of the contributors. Viewers should be aware that professionals in the field may have different opinions. BMJ does not endorse any opinions expressed or recommendations discussed. Viewers should not use the content of the video as the basis for any medical treatment. BMJ disclaims all liability and responsibility arising from any reliance placed on the content.
WHAT IS ALREADY KNOWN ON THIS TOPIC
Recent evidence demonstrated that the immune checkpoint protein called galectin-9 is highly expressed in a wide range of human malignancies including the most aggressive tumors, such as high-grade glioblastomas and pancreatic ductal adenocarcinomas. This is often associated with elevated blood plasma levels of soluble galectin-9. However, solid tumor cells at rest are known to secrete either very low amounts of galectin-9 or, in most of the cases, do not secrete it at all. Therefore, it is crucially important to understand whether: (1) immune cells attacking the tumor, like T cells, can induce galectin-9 secretion in human cancer cells derived from solid malignant tumors;and whether (2) this soluble form displays higher systemic immunosuppressive activity compared with the cell surface-based protein.
WHAT THIS STUDY ADDS
We discovered for the very first time that T lymphocytes induce galectin-9 secretion in various types of human cancer cells derived from solid malignant tumors via differential biochemical mechanisms. Cross-links between secreted galectin-9 and another immune checkpoint protein called VISTA were demonstrated for the first time.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Based on our work, galectin-9 itself and the expression systems associated with its production could be considered as specific and highly efficient targets for immunotherapy of a wide range of human cancers.
Background
Galectin-9 is a member of the family of proteins that were first identified as a group of lectins which specifically bind to carbohydrates containing β-galactosides.1–5 Galectin-9 has a so-called tandem structure and contains two distinct carbohydrate recognition domains (CRDs) within one molecule.1–5 These CRDs are fused together by a polypeptide linker. As such, galectin-9 may be present in three main isoforms characterized by the length of their linker peptide which can be long (49 amino acids), medium (27 amino acids), and short (15 amino acids).1–5
Galectin-9 is known as a major contributor to the regulation of human immune responses,6 7 particularly because of its ability to suppress the cytotoxic activities of T lymphocytes and natural killer (NK) cells.6 7 In cytotoxic T cells galectin-9 acts through receptors such as Tim-3 (T cell immunoglobulin and mucin-containing protein 3), VISTA (V-domain Ig-containing suppressor of T cell activation)7 and programmed cell death protein 1 (PD-1).8 Galectin-9 can induce leakage of the proteolytic enzyme granzyme B from the intracellular granules of cytotoxic T cells thus leading to their programmed death, primarily by acting through VISTA and Tim-3.7 In NK cells, galectin-9 operates mostly through Tim-3 and impairs their cytotoxic activities.6 As such, galectin-9 is used by cancer cells to escape immune surveillance. It is also employed by foetal cells where it protects the embryo against rejection by the mother’s immune system.9
Recent evidence demonstrated that galectin-9 is highly expressed in a large number of human malignancies including very aggressive tumors such as high-grade glioblastomas10 11 and pancreatic ductal adenocarcinomas11 as well as more common malignancies like breast, lung and colorectal cancers.9 10 Galectin-9 plays a crucial role in the immune escape of hematological malignancies such as acute myeloid leukemia (AML), a very aggressive blood/bone marrow cancer.6 Importantly, we found that AML cells secrete high levels of galectin-9.6 However, solid tumor cells secreted either very low amounts of galectin-9 or, in most cases, did not secrete it at all,10 in which case cell surface-based galectin-9 nonetheless appeared to still play a role against T cells.
In contrast to the above, recent evidence demonstrated that galectin-9 can also participate in the opsonization of Gram-negative bacteria,12 13 thus promoting their phagocytosis by macrophages12 and neutrophils.13 Gram-negative bacteria actually upregulate galectin-9 secretion by myeloid cells, and this enhances the process of bacterial opsonization.12 It would be much more efficient for malignant tumors to secrete galectin-9 since it upregulates apoptotic processes in cytotoxic T cells and facilitate their destruction through phagocytosis by tumor-associated macrophages.12 We, therefore, hypothesized that interactions with T cells induce galectin-9 secretion in human solid tumor cells and as such preopsonise T lymphocytes to minimize attack on tumor cells.
We report for the first time that T lymphocytes induce galectin-9 secretion in various human solid malignant tumors. This occurs via two mechanisms: first, by the translocation of galectin-9 onto the cell surface followed by its proteolytic shedding and, second, by lysosomal secretion. For both mechanisms a protein carrier/trafficker is required since galectin-9 lacks a secretion sequence. Secreted galectin-9 preopsonises T cells and when they interact with other immune checkpoint proteins, their activity is completely attenuated. As an example, we studied the immunosuppressive cooperation of galectin-9 and VISTA in human cancer cells. Our results indicate that galectin-9 and its regulatory pathways may be considered as key targets for highly efficient immunotherapy of a large number of cancers.
Methods
Descriptions of materials used, cell lines and primary human cells/samples as well as animals are given in online supplemental file 1.
Supplemental material
Western blot analysis
Galectin-9, VISTA, granzyme B, and Tim-3 were measured by Western blot and compared with the amounts of β-actin (protein loading control), as previously described.14–16
ELISAs
Levels of galectin-9, VISTA, IL-2 and TGF-β were measured in cell culture medium, human and mouse blood plasma and some of the cell lysates by ELISA using R&D Systems kits (see the Materials section) according to manufacturer’s protocols.
Quantitative reverse transcriptase-PCR analysis
To detect VISTA mRNA levels, we used quantitative reverse transcriptase PCR (qRT-PCR).16 Total RNA was isolated using a GenElute mammalian total RNA preparation kit (Sigma-Aldrich) according to the manufacturer’s protocol, followed by RT-PCR of a target protein mRNA (also performed according to the manufacturer’s protocol). This was followed by qRT-PCR. The following primers were used. Galectin-9: 5′-CTTTCATCACCACCATTCTG-3′, 5′-ATGTGGAACCTCTGAGCACTG-3′, Tim-3: 5′-CATGTTTTCACATCTTCCC-3′, 5′-CTATGGCATTGCAAACGCACA-3′, VISTA: forward – 5’-GATGCACCATCCAACTGTGT-3’, reverse – 5’- GCAGAGGATTCCTACGATGC-3’; actin: forward – 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′, reverse – 5′-CTAGAAGCATTTGCGGTCGACGATGGAGGG-3′. Reactions were performed using a LightCycler 480 qRT-PCR machine and SYBR Green I Master kit (Roche, Burgess Hill, UK). The assay was performed according to the manufacturer’s protocol. Values representing VISTA mRNA levels were normalized against those of β-actin.
Characterization of galectin-9 and Tim-3/VISTA interactions
An ELISA-based analysis was used as described before.17 Plates were first coated with anti-galectin-9 antibody followed by blocking with 2% bovine serum albumin (BSA). Cell culture media were then applied and incubated for at least 2 hours at room temperature after which plates were extensively washed with TBST. A glycine-HCl pH-lowering buffer (pH 2.0) was then applied to extract the bound proteins. Extracts were mixed with equal volumes of lysis buffer (pH 7.5) and with 4×sample buffer for SDS-PAGE at a ratio of 1:3. Samples were then subjected to Western blot analysis using rabbit anti-galectin-9, mouse anti-Tim-3 and rabbit anti-VISTA primary antibodies, as described above.
Detection of PI-3K activity
PI-3K activity was detected using spectrophotometric method based on detection of substrate (PI-4,5-diphosphate) phosphorylation, as previously described.18
Intracellular calcium measurement
MCF-7 cells were plated and cultured as described above. Cells were washed once with the recording buffer (RB) containing the following components: NaCl—145 mM, KCl—5.6 mM, glucose—5.6 mM, MgCl2—1 mM, HEPES—15 mM, pH 7.4, as well as 0.5 mg/mL BSA. The buffer was supplemented with 0.25 mM sulfinpyrazone to prevent the removal of the dye from the cells. Subsequently, cells were mixed with 2 µM acetoxymethyl (AM) ester of Fluo-4 (Ca2+-chelating dye) and incubated at room temperature under dark conditions for 30 min. Cells were then washed twice with RB and incubated for another 30 min in RB to allow degradation of AM esters.
Fluorescence measurements were performed in a microplate fluorometer at the following wavelengths: excitation—485 nm and emission—538 nm.
On-cell Western
VISTA levels on the cell surface as well as binding of anti-CD4 and anti-CD8 antibodies to T cells were detected using on-cell Western analysis with Li-Cor imaging system as described before.19
Cell viability assay
Cell viability was measured using an MTS assay kit (Promega) according to the manufacturer’s protocol.
Statistical analysis
Each experiment was performed at least three times and statistical analysis, was conducted using a two-tailed Student’s t-test. Statistical probabilities (p) were expressed as * when p<0.05; **, p<0.01 and *** when p<0.001.
Results
T lymphocytes activate secretion of galectin-9 in human cancer cells derived from solid malignant tumors
We investigated whether T cells could induce the secretion of galectin-9 in human cancer cells derived from solid malignant tumors. We studied those cells which do not secrete galectin-9 on their own and those which secrete very small amounts of the protein. In addition, we tested non-malignant rapidly proliferating human cells including HaCaT keratinocytes and primary human embryonic cells. All the cells tested expressed clearly detectable and often high amounts of galectin-9.10 For cancer cell models we selected MCF-7 breast cancer cells, WT3ab Wilms tumor cells, G401 kidney rhabdoid tumor cells and LN-18 high grade glioblastoma cells. All cells were cocultured with Jurkat T cells for 16 hours at a ratio of 1:1. Jurkat T cells were then separated, washed 2–3 times with fresh medium followed by incubation of the cocultured cells using respective cell-specific media (see online supplemental figure 1 for details). Cells were cultured then for 4 hour and galectin-9 secretion was measured at 2, 3 and 4 hours. Following separation, Jurkat T cells secreted barely detectable amounts of galectin-9. Human cancer cells secreted high levels of galectin-9, HaCaT keratinocytes secreted less and human fetal cells produced almost undetectable amounts of the protein (figure 1A). Galectin-9 expression in MCF-7,10 WT3ab,16 HaCaT9 and primary human embryonic cells9 12 was confirmed in our earlier studies. The expression of this protein in LN-18 and G401 cells was determined in this study (online supplemental figures 2, 3).
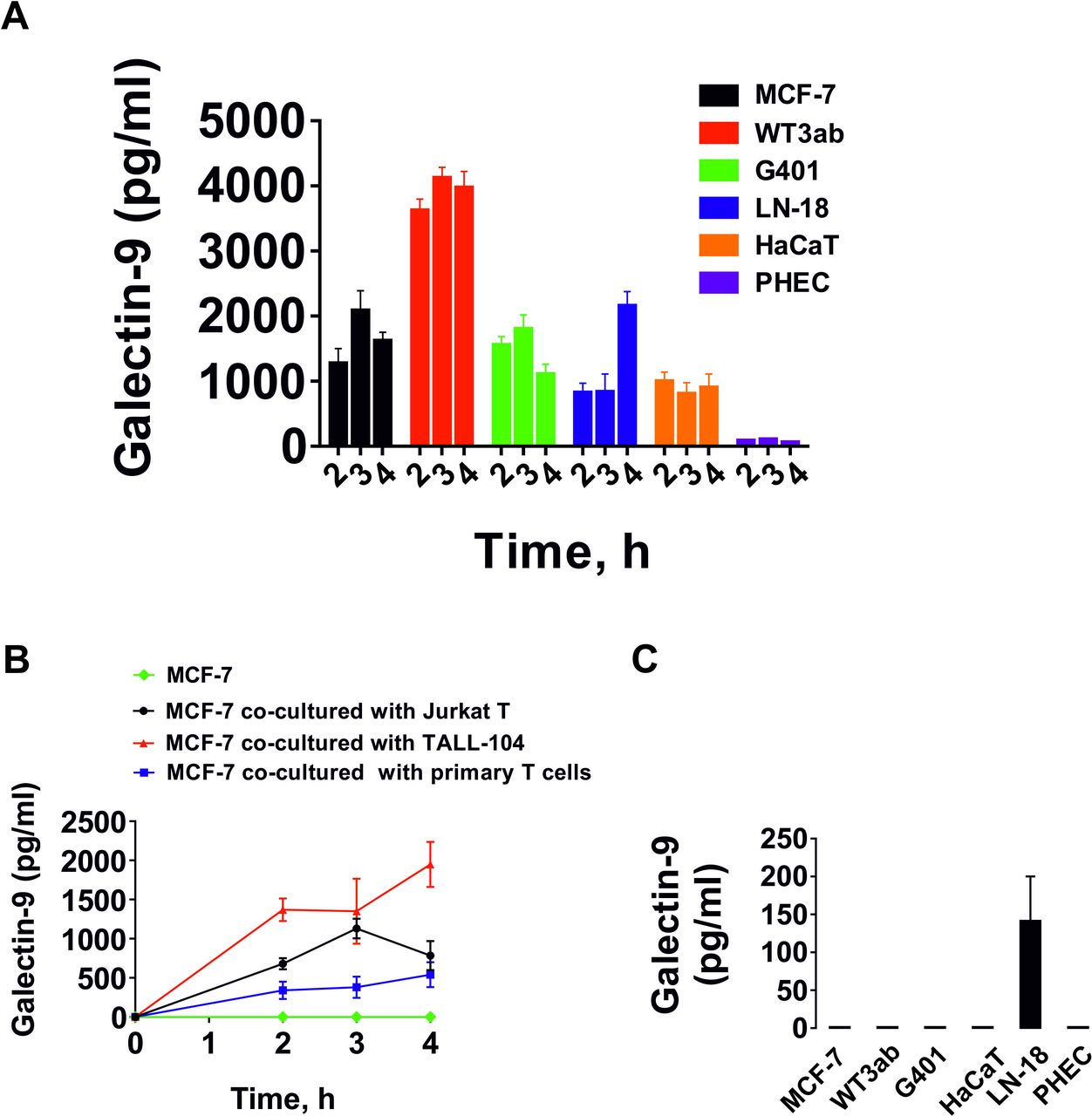
T cells induce galectin-9 secretion in human cancer cells derived from solid malignant tumors and keratinocytes. (A) Human cancer cells derived from solid malignant tumors, HaCaT keratinocytes and primary human embryonic cells were cocultured for 16 hours with Jurkat T cells at a ratio of 1:1. Cells were then separated and washed with fresh culture medium followed by culture for 4 hours. Galectin-9 release was measured at 2, 3 and 4 hours. Jurkat T cells did not release detectable amounts of galectin-9. (B) MCF-7 human breast cancer cells were cocultured for 16 hours with Jurkat T cells, TALL-104 or primary CD3-positive human T cells at a ratio of 1:1. The experiment was performed as described in section (A). T cells did not release detectable amounts of galectin-9. As a control we used MCF-7 cells which were not cocultured with T cells. (C) Galectin-9 secretion measured in resting cells studied in section A, which were cultured for 24 hours as outlined in the Materials and methods section. Quantitative data represent mean values±SEM of four independent experiments.
We then studied whether various T cell types can induce galectin-9 secretion. For comparison, we cocultured MCF-7 human breast cancer cells with Jurkat T cells (CD4-positive), TALL-104 cytotoxic (CD8-positive) lymphocytes as well as primary human CD3-positive T cells. All cocultures were done at a 1:1 ratio for 16 hours and then processed as described above. None of the T cells studied secreted any galectin-9 after separation (except Jurkat T cells which secreted barely detectable amounts (<100 pg/mL)). However, MCF-7 cells secreted high amounts of galectin-9 in all three cases (figure 1B). As mentioned above, among the studied adherent cells, only LN-18 were able to secrete detectable (but low) amounts of galectin-9 at rest in the absence of T lymphocytes (figure 1C).
We then investigated whether CD4 or CD8 proteins are involved in T cell-induced galectin-9 secretion. We cocultured primary human T cells with MCF-7 cells in the presence or absence of CD4 or CD8 neutralizing antibodies for 16 hours (online supplemental figure 4A). T cells were then, as described above, separated and galectin-9 levels released from cultured MCF-7 cells were then measured in the conditioned media after 2, 3 and 4 hours (online supplemental figure 4B). Galectin-9 was also measured in the medium where MCF-7 and primary human T cells were cocultured for 16 hours (online supplemental figure 4C). Binding of anti-CD4 and anti-CD8 to primary T cells was confirmed by on-cell Western analysis (online supplemental figure 4D).
We found that neither of the antibodies were able to significantly reduce T-cell induced galectin-9 secretion in MCF-7 cells, suggesting that CD4 and CD8 are unlikely to be involved in this process. However, since we saw a minor reduction in the effect caused by the anti-CD8 antibody, we cocultured highly active LN-18 glioblastoma cells with Jurkat T (which express only CD4 and not CD8 protein) cells for 16 hours, as described above. Jurkat T cells were then separated and LN-18 cells were cultured for the next 4 hour where galectin-9 levels were measured in the conditioned media after 2, 3 and 4 hours (online supplemental figure 4E). Galectin-9 was also measured in the medium where LN-18 and Jurkat T cells were cocultured for 16 hours (online supplemental figure 4F). We confirmed that anti-CD4 was highly expressed on Jurkat T cells and anti-CD8 was barely detectable using on-cell Western analysis (online supplemental figure 4G). Also, we measured both CD4 and CD8 in Jurkat T cells after coculture by flow cytometry and found that they were CD4 positive, while CD8 levels were barely detectable (online supplemental figure 4H). CD4 and CD8 neutralizing antibodies had no effect on T cell-induced galectin-9 secretion in LN-18 cells. A very minor (non-significant) reduction of galectin-9 secretion was observed in the presence of anti-CD8 antibody, similar to that seen with MCF-7 cells (online supplemental figure 4 B,E). However, this is unlikely to be specific due to the absence of CD8 expressions in Jurkat T cells. Importantly, FACS analysis confirmed the absence of any traces of CD4 in LN18 cells after coculture (this is another confirmation of a complete separation of adherent cells from suspension T cells; online supplemental figure 4H).
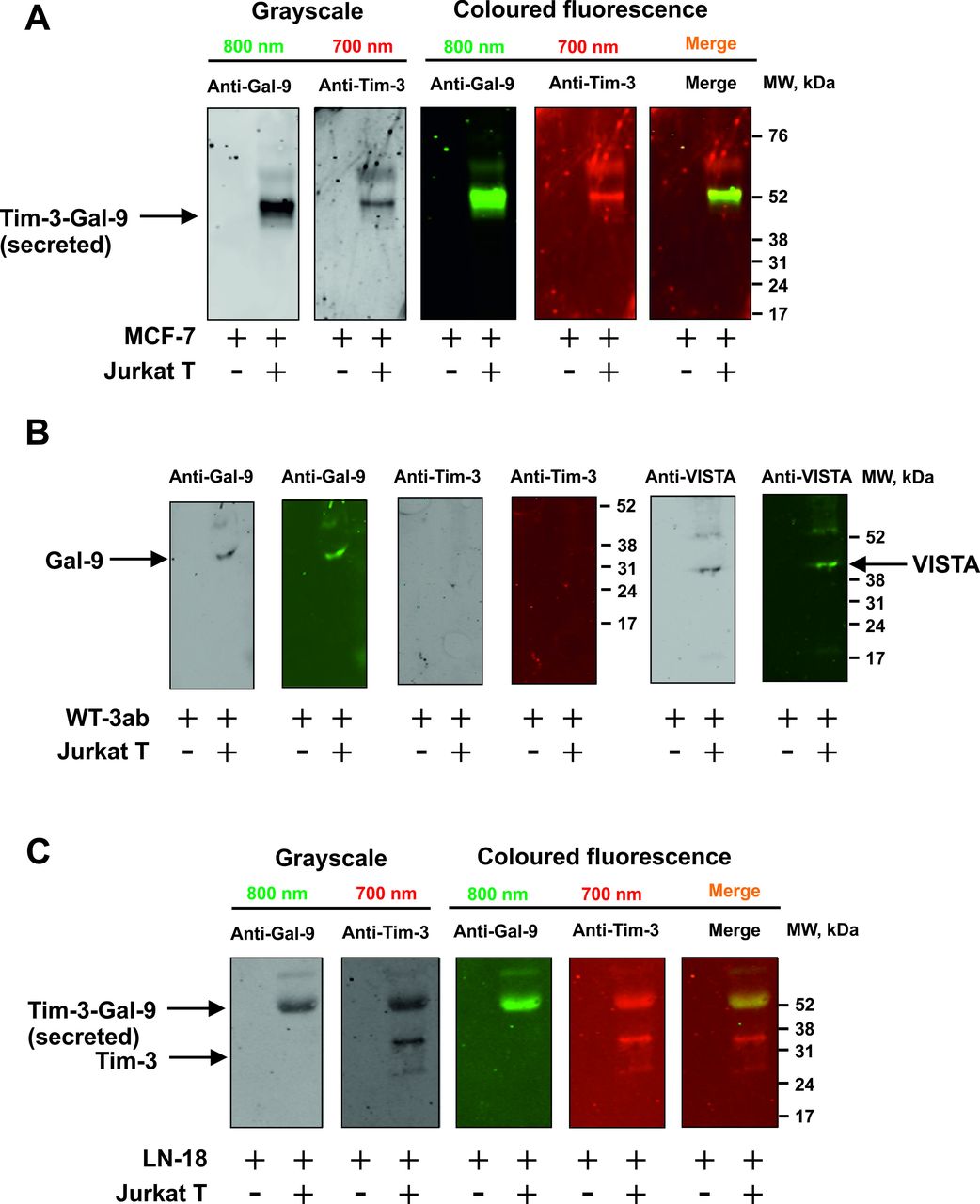
Importantly, MCF-7 cells express only one galectin-9 binding receptor—Tim-3,10 16 WT3ab express only VISTA and traces of Tim-3,16 while LN-18 express both Tim-3 and VISTA (online supplemental figure 3). We assessed whether galectin-9 is released in a complex together with fragments of its receptors, which can act as carriers/traffickers for this protein. We immunoprecipitated galectin-9 as described before6 7 17 and detected it using Western blot analysis without boiling the samples. When MCF-7 cells were cocultured with Jurkat T cells, this resulted in galectin-9 to be secreted in a complex with Tim-3 fragment (figure 2A). However, in WT3ab cells it was secreted in a complex with VISTA fragment (figure 2B). VISTA was dissociated from galectin-9 during SDS-PAGE electrophoresis, unlike Tim-3, confirming our findings reported earlier that Tim-3 has higher affinity to galectin-914 compared with VISTA.7

Tim-3 or VISTA traffic galectin-9 during the process of secretion. MCF-7 (A), WT3ab (B) or LN-18 (C) cells were cocultured with Jurkat T cells at a ratio of 1:1 for 16 hours. Cells were then separated and washed with fresh culture medium. Solid cancer cells were then cultured for 2 hours and galectin-9 was immunoprecipitated as described in the Materials and Methods. Eluted proteins were analyzed by Western blot in order to detect galectin-9, Tim-3 and VISTA. Images are from one experiment representative of four which gave similar results.
Both receptors were present in LN-18, where Tim-3 was the carrier of galectin-9 (figure 2C), which is in line with our previous findings for primary human embryonic cells which also express both Tim-3 and VISTA receptors.12
We then wished to assess the interactions between MCF-7 and cytotoxic TALL-104 cells in order to discover if MCF-7-derived galectin-9 is capable of opsonising cytotoxic T cells. We cocultured MCF-7 and TALL-104 cells at a ratio of 1:1 and then separated them as described above. TALL-104 cells were lysed and around 50–60 µg/well of total cellular protein from resting TALL-104 lysates was loaded for Western blot detection of galectin-9 (these cells express low amounts of galectin-9 and Tim-316). We then loaded less (ca. 15 µg/well) protein of TALL-104 cells lysed after coculture with T cells, so that their own galectin-9 and Tim-3 were undetectable. Samples were not boiled in both cases. As a result, in lysates of TALL-104 cells cocultured with MCF-7 cells, we observed a band at ca. 52 kDa detectable by both anti-Tim-3 and anti-galectin-9 antibodies (figure 3A). This corresponds to the galectin-9 secreted by MCF-7 cells (figure 3B), which was detected in the medium following the immunoprecipitation of galectin-9. Importantly, this complex had lower affinity to the TALL-104 cell surface receptors than to Tim-3 trafficking it, since the band corresponded to galectin-9 and a single Tim-3 fragment.6 7 We could rule out that this was not an internal complex made by TALL-104 cells since the internal complex is known to have a molecular weight of ca. 70 kDa before Tim-3 is shed.6 Importantly, we found that galectin-9 mRNA levels were significantly downregulated in TALL-104 after coculture with MCF-7 cells. The same observation was made regarding VISTA mRNA levels, while Tim-3 mRNA levels were significantly upregulated (see figure 3C for details). In MCF-7 cells both Tim-3 and galectin-9 mRNA levels were significantly upregulated (figure 3D). In parallel, TALL-104 cells were relatively unable to deliver granzyme B into MCF-7 cells (figure 3E, F), which is in line with our previous observations.10
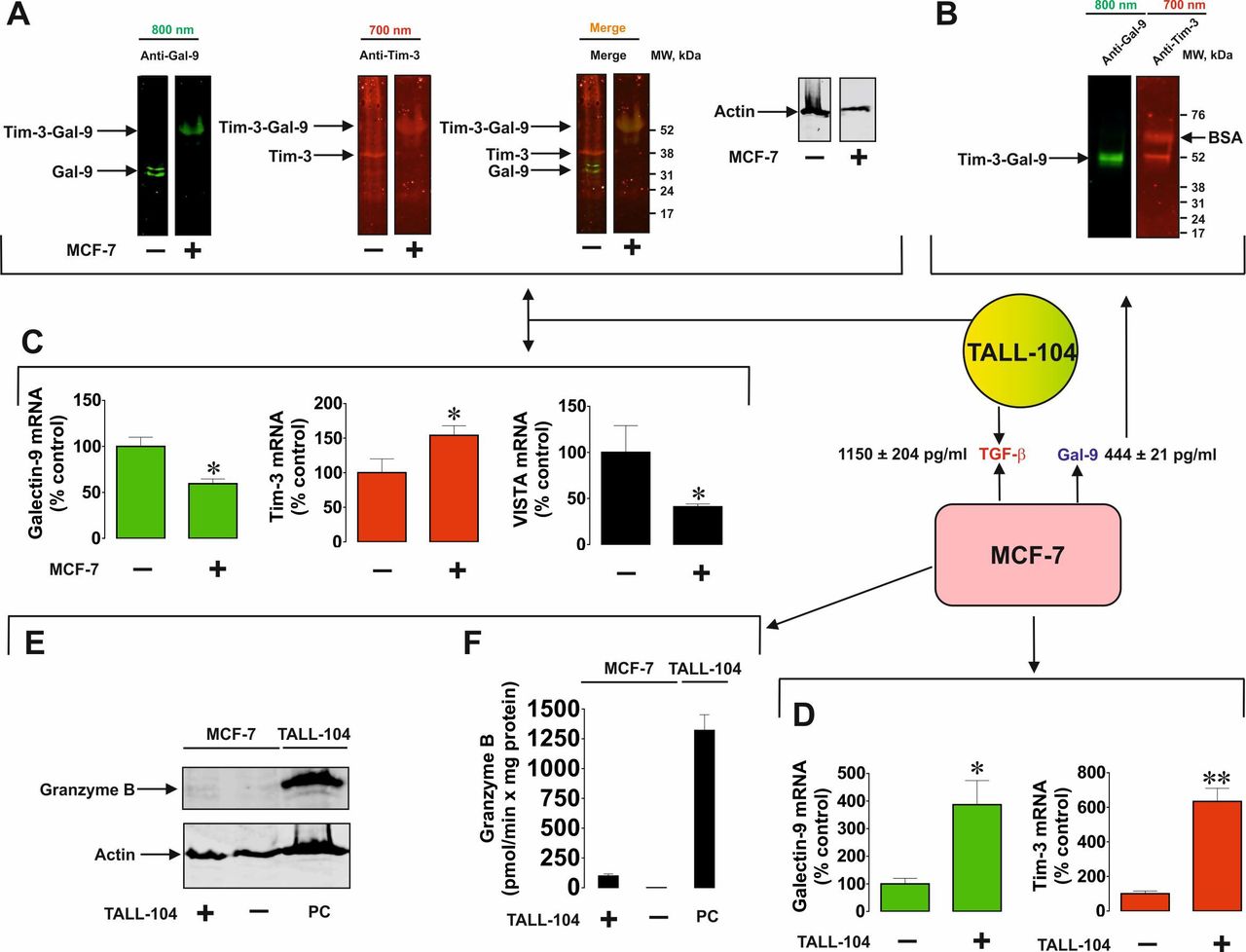
Solid tumor cell-derived galectin-9 interacts with cytotoxic T cells. MCF-7 human breast cancer cells were cocultured with TALL-104 for 16 hours at a ratio of 1:1. (A) Resting TALL-104 cells and those cocultured with MCF-7 were lysed and subjected to Western blot analysis (without boiling the samples) in order to detect Tim-3 and galectin-9. (B) Galectin-9 and Tim-3 were detected following immunoprecipitation of galectin-9 from the medium collected from the cocultured cells. Levels of galectin-9 and TGF-β were analyzed in the medium by ELISA as described in the Materials and methods section. mRNA levels of galectin-9, Tim-3 and VISTA were analyzed in TALL-104 (C) and MCF-7 (D) cells by qRT-PCR as described in the Materials and methods section. Granzyme B quantities (E) and activities (F) were analyzed in both MCF-7 and TALL-104 cells. Images are from one experiment representative of four which gave similar results. Quantitative data represent mean values±SEM of four independent experiments. *p<0.05, **p<0.01 vs control. qRT-PCR, quantitative reverse transcriptase PCR.
Human cancer cells derived from solid malignant tumors operate two mechanisms of galectin-9 secretion induced by T cells
We considered the possibility of employment of two differential mechanisms of T cell-induced galectin-9 secretion from human cancer cells derived from solid malignant tumors. We cocultured MCF-7 and Jurkat T cells at a ratio of 1:1 for 16 hours and then separated them, as described above, subsequently culturing MCF-7 cells in fresh medium for up to 4 hour, measuring galectin-9 secretions at 2, 3 and 4 hour. Cells were cocultured in the absence and presence of various inhibitors. This included U73122 (PLC inhibitor), Gö6983 (PKC inhibitor), vacuolin-1 (inhibitor of lysosomal exocytosis), AZD2014 (mTOR inhibitor), EDTA (matrix metalloproteinase inhibitor) and protease inhibitor cocktail (PIC) containing blockers of lysosomal proteases and matrix metalloproteinases (figure 4A).
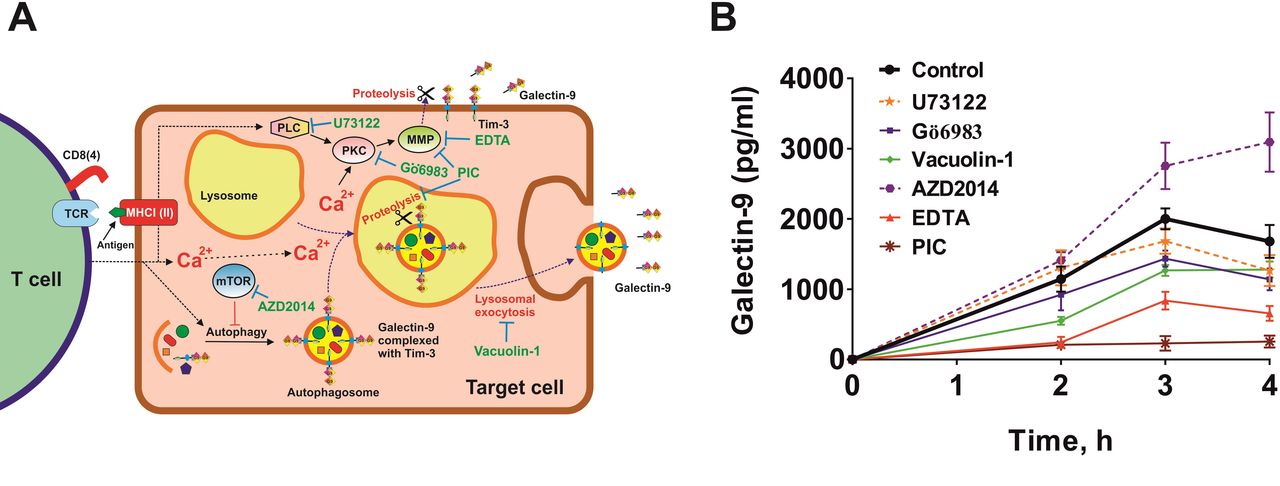
Galectin-9 secretion induced by T cells in MCF-7 human breast cancer cells can occur through two mechanisms. (A) Possible biochemical mechanisms underlying T cell-induced galectin-9 secretion in human solid cancer cells. Two potential mechanisms were investigated. First, Ca2+/PKC-dependent translocation of galectin-9 onto the cell surface followed by proteolytic shedding by matrix metalloproteinases (MMPs), which can also be activated by PKC. Another mechanism could possibly be associated with galectin-9 in complex with a carrier protein joining autophagosomes, which merge with lysosomes, where galectin-9 is shed and secreted via lysosomal exocytosis. (B) MCF-7 cells (adherent) were cocultured with Jurkat T cells (suspension) for 16 hours at a ratio of 1:1. Jurkat T cells were then removed and washed away from MCF-7 cells. After replacing the culture medium, MCF-7 cells were incubated for 4 hour and galectin-9 release was measured after 2, 3 and 4 hour. To investigate the mechanisms, replacement medium contained respective pharmacological inhibitors (PIC—protease inhibitor cocktail)—U73122—30 µM, Gö6983–70 nM, AZD2014—10 µM, EDTA—200 µM. Data are the mean values±SEM of 5 independent experiments.
We found that secretion was downregulated by Gö6983, vacuolin-1, EDTA and PIC (figure 4B). AZD2014 upregulated the secretion, which suggests that it was autophagy-dependent lysosomal secretion (confirmed by the inhibitory effect of vacuolin-1), unlike in AML cells where galectin-9 secretion is autophagy-independent.6 These results suggest that there are two mechanisms involved in secretion of galectin-9. The first one is translocation of this protein onto the cell surface (by PLC-independent, PKC-dependent processes) followed by its shedding off the cell surface by matrix metalloproteinases (as in AML cells6). The other one is autophagosome formation followed by lysosomal exocytosis, where galectin-9 is also shed off. This conclusion can be made based on the fact that secretion of galectin-9 was attenuated by PIC blocking both lysosomal proteases and matrix metalloproteinases.
PKC involved in the process is most likely to be calcium-dependent and diacyl glycerol (DAG)-independent (PLC had no influence on the process of secretion). We checked whether intracellular calcium levels were upregulated in MCF-7 by coculturing them with Jurkat T cells at a ratio of 1:1 for 2 hours followed by detection of intracellular calcium levels using Fluo4 reactive dye as described in the Materials and methods section. We found that intracellular calcium levels were significantly upregulated (online supplemental figure 5) in MCF-7 cells cocultured with Jurkat T cells compared with resting MCF-7 cells. This suggests that the environment was supportive of activating Ca2+-dependent PKC isoforms, which are inhibited by Gö6983 together with DAG/Ca2+-dependent isoforms of this enzyme.
Galectin-9 can cooperate with VISTA to suppress the anticancer activities of T lymphocytes
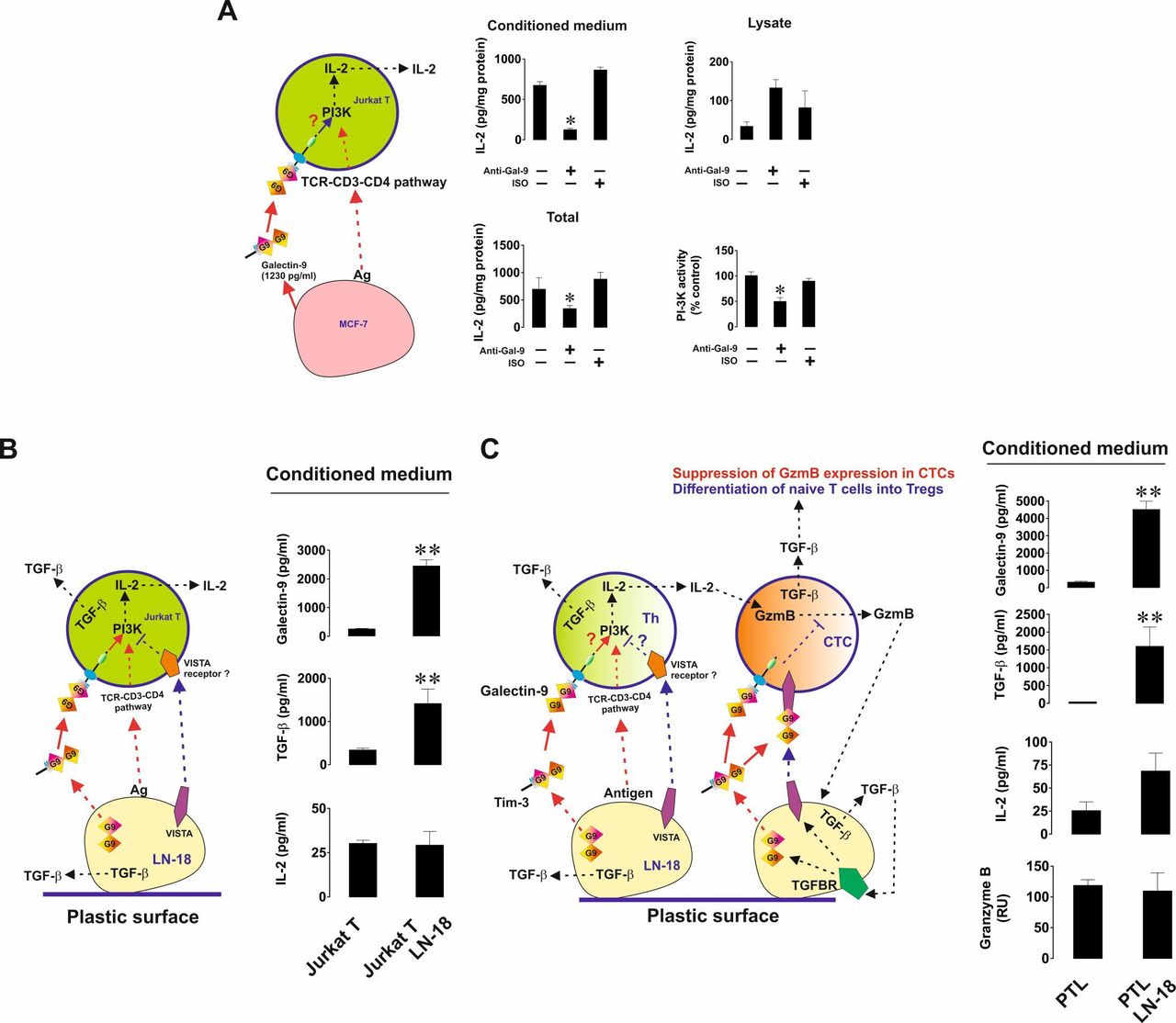
We then investigated the immunosuppressive activity of galectin-9 and how it can cooperate with other immune checkpoints in order to achieve T cell inactivation. First, we cocultured MCF-7 human breast cancer cells with Jurkat T cells (CD4 positive cells displaying T helper activity) at a ratio of 1:1 for 16 hours in either the absence or presence of 9S2-1 galectin-9 neutralizing (blocking) antibody12 or isotype control antibody. On completion of the incubation, we measured IL-2 (a T helper-derived cytokine inducing activity of cytotoxic T cells) in cell culture medium and Jurkat T cell lysates. We also analyzed phosphatidyl inositol 3-kinase (PI-3K) activity in Jurkat T cell lysates (PI-3K activity and its pathway contribute to IL-2 expression in helper T cells and indicate activation of TCR signaling20). Importantly, resting Jurkat T cells secreted barely detectable amounts of IL-2 and MCF-7 did not secrete any.
We found that secretion (but not the levels of cell-associated IL-2) as well as the total amounts of IL-2 (secreted+cell-associated levels detected in the lysates) produced by Jurkat T cells during the interaction with MCF-7 cells were significantly reduced in the presence of galectin-9 neutralizing antibody but not isotype control antibody (figure 5A). The same effect was observed for PI-3K. This means that galectin-9 supports PI-3K activation in Jurkat T cells and enhances IL-2 production by them. We then cocultured Jurkat T cells with LN-18 (which unlike MCF-7 cells also express VISTA), also at a ratio of 1:1 for 16 hours. We found that, in the presence of LN-18, Jurkat T cells did not secrete more IL-2 than resting Jurkat T cells. However, the secreted levels of transforming growth factor-β type 1 (TGF-β) and galectin-9 were highly upregulated (figure 5B). TGF-β, in line with our previous findings,9 16 was found to upregulate the expressions of both galectin-9 and VISTA in LN-18 cells on both mRNA and protein levels (online supplemental figure 3). We then cocultured LN-18 cells with primary human CD-3-positive T cells and assessed the release of IL-2, granzyme B, TGF-β and galectin-9 into the medium. We found that no increase was observed in the levels of released granzyme B and IL-2, however both TGF-β production and galectin-9 secretion were significantly upregulated (figure 5C).

Galectin-9 produced by human cancer cells derived from solid malignant tumors triggers PI-3K activation and IL-2 production in helper T cells. Human cancer cells expressing both galectin-9 and VISTA suppress both helper and cytotoxic T cell activities. (A) MCF-7 human breast cancer cells were cocultured with Jurkat T cells (CD4-positive helper T cells) and galectin-9 release was measured by ELISA. Cells were cocultured for 16 hours at a ratio of 1:1 in the absence or presence of galectin-9 neutralizing antibody or isotype control antibody. IL-2 was measured in the cell culture medium and T cell lysates. Total IL-2 levels were calculated as a sum of released and cell-associated (detected in lysates) IL-2. LN-18 high grade human glioblastoma cells were cocultured with Jurkat T lymphocytes (B) or primary human CD3-positive T cells (C) at a ratio of 1:1 for 16 hours. Release of galctin-9, TGF-β, IL-2 and, where applicable, granzyme B were analyzed as outlined in the Materials and methods section. Data are shown as mean values±SEM of 6 independent experiments. PI-3K activity was analyzed in T cell lysates. Quantitative data represent mean values±SEM of five independent experiments. *p<0.05, **p<0.01 vs control. In the schemes—Th represents T helpers and CTC—cytotoxic T cells.
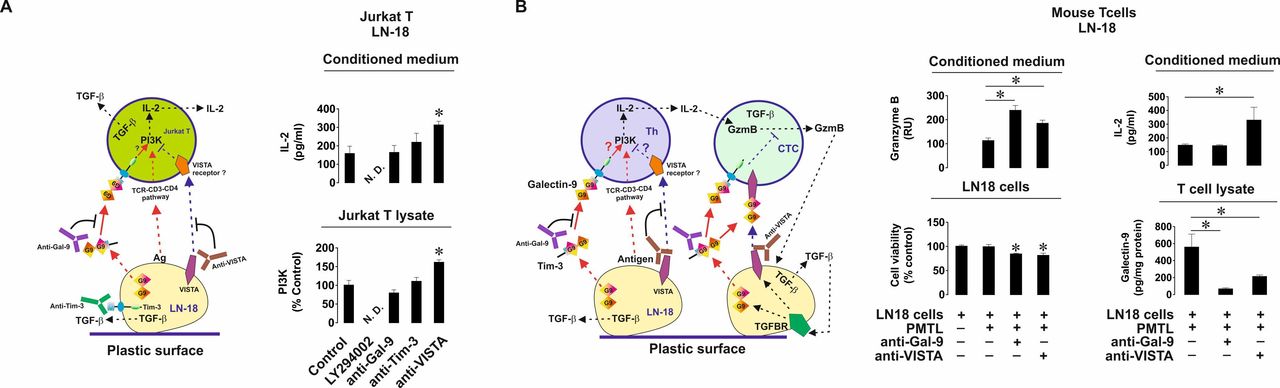
Importantly, when LN-18 cells were cocultured with Jurkat T cells or primary human T lymphocytes, cell surface levels of VISTA were significantly upregulated (online supplemental figure 6). No VISTA secretion from LN-18 was detected by ELISA. In order to assess the contribution of VISTA to the events triggered in T cells by galectin-9 we cocultured LN-18 and Jurkat T cells in the absence or presence of galectin-9 or VISTA-neutralizing antibodies.12 We found that IL-2 secretion (unlike in the presence of MCF-7 cells) was not changed when galectin-9 was neutralized but was upregulated on neutralization of VISTA (figure 6A). The same effect was observed with the PI-3K activity in Jurkat T cell lysates. This means that VISTA contributes to suppression of T helper activity. In order to confirm the role of PI-3K in IL-2 production in our experimental system, we added 30 µM LY294002 (PI-3K inhibitor) to the coculture and found that both PI-3K activity and IL-2 release were attenuated (figure 6A). Importantly, Tim-3 is present on the surface of LN-18 cells cocultured with Jurkat T cells as confirmed by FACS analysis, and expressed at almost equal levels compared with galectin-9 (online supplemental figure 7). To rule out the involvement of Tim-3 in the observed effects we cocultured the cells as described above in the presence or absence of Tim-3 neutralizing antibody (4BS12). Tim-3 neutralization had no effect on either PI-3K activity or IL-2 release (figure 6A).

VISTA cooperates with galectin-9 in order to suppress helper T cell and cytotoxic T cell activities. (A) LN-18 cells were cocultured with Jurkat T cells for 16 hours at a ratio of 1:1 in the absence or presence of either galectin-9, VISTA,Tim-3 neutralizing antibodies or 30 µM PI-3K inhibitor LY294002. PI-3K activity was measured in Jurkat T cell lysates and IL-2 release in the culture media. (B) LN-18 cells were cocultured with primary mouse T cells in the absence or presence of human galectin-9 or VISTA (which recognizes both mouse and human protein) neutralizing antibodies. Granzyme B activity and IL-2 levels were measured in the culture media. Viability of LN-18 cells was detected by MTS assay. Human galectin-9 levels were detected in mouse T cells by ELISA. Data are shown as mean values±SEM of 6 independent experiments. *p<0.05 vs control or indicated events. In the schemes—Th represents T helpers and CTC—cytotoxic T cells.
The next step was to confirm the observed effects in vivo using a xenograft model. First, in order to verify whether LN-18 cells are capable of suppressing mouse T cell activities we cocultured them for 16 hours with mouse T cells at a ratio of 1:1 in the absence or presence of galectin-9 or VISTA neutralizing antibodies. We found that coculture led to substantial activity of released granzyme B in the cell culture medium (figure 6B). This activity was further highly upregulated when galectin-9 was neutralized and also significantly upregulated when VISTA was neutralized (figure 6B). Respectively, the viability of LN-18 cells was reduced when either galectin-9 or VISTA were neutralized (figure 6B). Mouse IL-2 secretion was upregulated only when VISTA was neutralized, which is in line with the observation made when coculturing LN-18 with Jurkat T cells (figure 6A). Measurement of human galectin-9 in mouse T cell lysates (total cell-associated galectin-9) was conducted by ELISA (which had no cross-reactivity with mouse galectin-9) as outlined in the Materials and methods section. We found that when galectin-9 was neutralized, it was almost absent in T cells and when VISTA was neutralized, its levels were significantly downregulated in mouse T cells (figure 6B). This effect of VISTA neutralization may be also a result of the inability of galectin-9 to bind VISTA on mouse T cells.
Human cancer cells derived from solid malignant tumors are capable of secreting galectin-9 which suppresses cytotoxic T cell activities in vivo
In order to confirm our findings in vivo, we used C57 BL16 mice (wild type and not immunocompromised). All procedures complied with the UK Animals (Scientific Procedures) Act (1986) and were performed under a UK Home Office Project Licence in accordance with University of Kent Policy on the Use of Animals in Scientific Research (approved by the University of Kent animal committee). 5 mice were used as a control group and each of the other 5 mice subcutaneously received 2×106 LN-18 cells (which are highly tumorigenic) and blood of these mice was analyzed after 5 hour (since these mice are immunocompetent and engrafted human LN-18 are highly active, a 5-hour period was used to be able to observe maximal immune evasion responses of LN-18 cells before they were suppressed by mouse immunity).
First, we isolated the area where LN-18 cells were injected and subjected it to soft homogenization in cell lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA and 0.5% NP-40), in order to dissolve the proteins secreted into the microenvironment and to obtain lysed haematopoietic cells present within. We found that the obtained homogenates contained high levels of human galectin-9 (figure 7A) but did not contain any human VISTA. Both proteins were undetectable in the control group. In both, the control and LN-18-injected group. The injection microenvironment (subcutaneous area, where the LN-18 cells were injected) contained T cells since CD3 protein was clearly detectable in homogenates (figure 7A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Human cancer cells derived from solid malignant tumors secrete galectin-9 in vivo. C57 BL16 mice were subcutaneously injected with tumorigenic LN-18 cells (2 х 106 cells per mouse). Then CD3 protein, human galectin-9 and VISTA were measured in the microenvironment where the injection took place (A). We also measured human galectin-9 and TGF-β in blood plasma and human galectin-9 in primary mouse T cell lysates (B). Finally, K562 cells were cocultured with primary mouse T cells isolated from control and LN-18-injected mice. Images of K562 cells were then taken and their viability was analyzed by MTS assay (C). Images are from one experiment representative of five which gave similar results. Quantitative data represent mean values±SEM of 5 independent experiments. *p<0.05 vs control or indicated events.
Blood plasma of LN-18-injected mice contained clearly detectable amounts of human galectin-9 (undetectable in the control group). Lysates of T cells of these mice also showed high levels of human galectin-9, which was undetectable in T lymphocytes obtained from control animals (figure 7B). Additionally, blood plasma TGF-β levels were highly upregulated (figure 7B).
We also assessed whether human galectin-9 affected the anticancer cytotoxic activity of mouse T lymphocytes. For this purpose, mouse T cells were cocultured with equal amounts of K562 cells pretreated with 100 nM PMA for 24 hours at a ratio of 1:1 with primary mouse T cells isolated from control and LN-18-injected mice. Cells were coincubated for 24 hours followed by viability measurements of K562 cells. We found that viability of K562 cells cocultured with T cells from control group mice was significantly lower compared with cells which were not cocultured with mouse T lymphocytes and those which were cocultured with T cells obtained from mice injected with LN-18 cells (figure 7C). We can, therefore, conclude that human galectin-9 downregulates the cytotoxic activities of mouse T lymphocytes.
Discussion
In this work, we aimed to investigate the mechanism of galectin-9 involvement in the suppression of T cell activities during anticancer immune evasion. Our previous work demonstrated that solid tumor cells rarely secrete galectin-9 at rest.10 However, we also found that cytotoxic T cell suppression can be achieved through ‘opsonization’ of T cells.12 As a result, T cells display a so-called ‘eat me’ signal by externalizing phosphatidyl serine.12 As such, we investigated whether the interaction with T cells triggers galectin-9 secretion in solid tumor cells. We found that T helpers and cytotoxic cell lines (Jurkat T and TALL-104, respectively) as well as primary human CD3-positive T cells induce the secretion of galectin-9 from various solid tumor cell types including breast and kidney cancers as well as high grade glioblastoma cells. Interestingly, non-malignant HaCaT keratinocytes also secreted galectin-9 when they were exposed to T cells (figure 1), although they secreted less galectin-9 than cancer cells. Primary human embryonic cells (13–14 weeks), which express high levels of galectin-99 12 and translocate it onto the surface,12 were almost unable to secrete it on exposure to T cells (figure 1). Importantly, in each case a protein carrier was required for galectin-9 secretion as reported before only for blood cancer (AML) cells. When Tim-3 was available, this protein serves as a carrier (irrespective of other proteins with affinity to galectin-9) since it probably has the highest affinity to galectin-9 compared with other proteins (eg, VISTA).7 14 However, in one case (WT3ab Wilms tumor cells), Tim-3 was not available in these cells but they expressed high levels of VISTA. In this case, VISTA was used to carry galectin-9 during the process of secretion.
Galectin-9 secretion appears to be employed for immune evasion not only by cancer cells but also by non-malignant cells (HaCaT, figure 1). This could mean that galectin-9 could be involved in the development of disorders such as psoriasis, where keratinocytes are attacked by T cells, though in this case keratinocytes manage to suppress T cells and proliferate.21 Our results suggest that galectin-9 could be used by keratinocytes to suppress the activity of T cells during autoimmune attack. Embryonic cells clearly do not aim to suppress the activities of T cells in general, since they participate in protection of mother’s body and, respectively, the embryo, from infections. However, they do translocate galectin-9 onto the cell surface, which means that embryonic cells use galectin-9 to suppress T cells which attack the embryo directly. In contrast, cancer cells can suppress both attacking T cells in the tumor microenvironment as well as T cells in general if high levels of galectin-9 are secreted into the circulation. Importantly, translating our findings into a real-life scenario in solid tumors where T cells are often sparse or located peripherally, galectin-9 secretion is most likely triggered on the interaction of T cells with malignant cells (in a CD4/CD8-independent process) protects the tumor microenvironment from T cell attack. The localized involvement of galectin-9 in this process may also explain why galectin-9 levels are not always elevated in the blood plasma of patients with cancer10, where tumor cells express it.
Using pharmacological inhibitors, we found that human cancer cells derived from solid malignant tumors secrete galectin-9 by proteolytic shedding of it by matrix metalloproteinases from the cell surface, where it was translocated to together with a protein carrier (Tim-3 or VISTA). This occurs (in the systems we studied) in a PKC-dependent but PLC-independent manner. Given the increased intracellular calcium levels in cancer cells in the presence of T cells and the inhibiting effect of Gö6983 on the process, one could suggest a role for Ca2+-dependent and DAG-independent PKC isoforms. Intracellular Ca2+ levels were most likely upregulated in PLC-independent manner, since PLC inhibitor U73122 had no impact on intracellular calcium levels (online supplemental figure 5) and galectin-9 secretion (figure 4B).
An alternative mechanism for galectin-9 secretion involves autophagy, including formation of autophagosome with galectin-9 and Tim-3/VISTA most likely located on the cell surface. An autophagosome is then merged with the lysosome for secretion (lysosomal exocytosis).22 Lysosomal proteases shed off galectin-9 inside the secretion lysosome and galectin-9 is then released by exocytosis from the cell.
Our studies further demonstrated that galectin-9, in cooperation with other immune checkpoint proteins, can suppress both T helper and cytotoxic T cell activities. On one hand, it is known that the interaction of galectin-9 with Tim-3 on the cell surface activates the PI-3K pathway.14 We confirmed that this is the case for helper T cells (figure 5A), where galectin-9 was derived from solid tumor MCF-7 human breast cancer cells. In addition, the PI-3K pathway leads to the activation of IL-2 expression and production.20 Our studies (figure 5A) confirmed that galectin-9 derived from MCF-7 cells also upregulates IL-2 production in Jurkat T cells. This suggests that, in order to block IL-2 production, another checkpoint protein or suppressor may be required. It is known that PD-L1 can upregulate SHP2 phosphatase and thus block PI-3K and MAP kinase pathways thus attenuating IL-2 production.23 This mechanism also can prevent the activation of the anti-apoptotic protein BCL-XL23 and so possibly lead to the death of cytotoxic T cells caused by galectin-9-induced granzyme B leakage. Importantly, MCF-7 cells used for this work do not produce PD-L1.24 Interestingly, they also do not produce VISTA, which similarly belongs to the B7 family of proteins (as PD-L1).16 We hypothesized that VISTA, which was reported earlier to enhance cytotoxic cell killing effects of galectin-9, can also trigger downregulation of the PI-3K pathway/IL-2 production and thus ‘help’ galectin-9 to suppress both T helper and cytotoxic T cell activities. We found that this was indeed the case by coculturing LN18 high grade glioblastoma cells and Jurkat T cells as well as LN18 and primary human or mouse CD3-positive T cells. Neutralization of VISTA led to upregulation of PI-3K activity and IL-2 secretion in Jurkat T cells, which suggests that VISTA can block the pathway. LN18 cells express barely detectable amounts of PD-L1.25 A similar effect was observed with primary mouse T cells, when neutralization of either galectin-9 or VISTA led to decreased viability of LN18 cells as well as increased granzyme B release (figure 6B). Increased granzyme B release on neutralization of VISTA is in line with our previous reports regarding the interaction of galectin-9 as a ligand with VISTA (acting as a receptor) on the surface of T cells7 and the downregulatory effects of VISTA on PI-3K activity and IL-2 production in T helpers as well as the activities of cytotoxic T cells.26
We also confirmed the observed effect of human galectin-9 secretion by human cancer cells derived from solid malignant tumors in in vivo studies. We found that LN18 cells, which were subcutaneously injected to mice interacted with T cells (figure 7), resulted in the release of galectin-9 into the microenvironment. We also found that human galectin-9 was present in the blood plasma of LN18-injected mice. Finally, isolated mouse T cells were “opsonised” with human galectin-9. Interestingly, TGF-β levels in the blood plasma of LN18-injected mice was highly upregulated compared with mice from the control group. In this case, ELISA detected both human and mouse TGF-β (unlike galectin-9, where we detected only human protein). Most likely, given the autocrine and paracrine rather than endocrine activities of TGF-β, this TGF-β was produced by T cells affected by human galectin-9.9 As we confirmed earlier (figure 6B), human galectin-9 affected mouse T cell functions. To confirm this effect, we exposed K562 cells, which do not operate studied immune evasion mechanisms,6 to primary T cells isolated from wild type and LN-18-injected mice. We found that K562 cell viability was reduced when they were cocultured with T cells from wild type mice but not LN18-injected mice. This suggests that the injection of LN18 cells leads to suppression of mouse T cells, which is in line with the fact that they were opsonised with human galectin-9 (figure 7).
Taken together, our results show for the first time that human cancer cells derived from solid malignant tumors are highly capable of secreting galectin-9 in response to their interaction with T cells. Galectin-9 is secreted by translocation onto the cell surface followed by proteolytic shedding as well as by lysosomal secretion associated with autophagy and proteolytic shedding. This strategy also applies when non-malignant cells (like keratinocytes) are exposed to T cells and thus explains how keratinocytes may suppress T cells during psoriasis development associated with the suppression of T cells attacking keratinocytes followed by keratinocyte proliferation. Foetal cells do not secrete galectin-9 when exposed to T cells, suggesting that embryonic cells aim to suppress only T cells attacking the embryo rather than suppressing T cell immunity per se. VISTA potentiates the immunosuppressive effects of galectin-9 also by suppressing IL-2 production by helper T cells and possibly further preventing cytotoxic T cell function. This involves a negative impact on signaling pathways like PI-3K, affecting the activity of BCL-XL anti-apoptotic proteins, as well as by altering their membrane potential as previously reported.7 Therefore galectin-9 itself and the expression systems associated with its production (like the TGF-β/Smad3 signaling pathway) could be considered as specific and highly efficient targets for immunotherapy in a wide range of cancers.
Data availability statement
Data are available on reasonable request. The datasets used and/or analysed during the current study are available from the corresponding author.
Ethics statements
Patient consent for publication
Acknowledgments
We thank Diamond Light Source for access to B23 beamline and funding the projects number SM24509, SM20755, and SM21202).
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors SS conducted most of the experiments presented in the paper together with VVS. GL, IY, EF-K and BFG helped with in vivo experiments and studies including primary human T cells. SR, DC, NA and SMB helped with experiments on primary human embryonic cells, keratinocytes, glioblastoma and pediatric tumor cells. VVS is acting as a guarantor.
Funding This work was also supported by the Swiss Batzebär grant (to EF-K and SMB).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.