Article Text

Abstract
Background Cancer of unknown primary (CUP) is a heterogeneous group of metastatic cancers where a primary tissue of origin (TOO) is uncertain. Most patients with CUP have limited treatment options and poor survival outcomes. Immune checkpoint inhibitors (ICIs) can be efficacious in some patients with CUP, but the optimal predictive biomarkers are unknown. We therefore assessed immune and genomic biomarkers as well as predicted TOO in patients with CUP, including a subset treated with ICIs.
Methods Patients with CUP were subject to gene-expression profiling (GEP) and DNA panel sequencing. Immune and stromal-related gene expression was explored by NanoString, including genes associated with immunotherapy response (IR) in other solid malignancies. ICI responsive cancer types were assigned based on Food and Drug Administration-approved indications, and either detection of a latent primary tumor or the TOO was suspected based on genomics informed pathology review. Tumor mutation burden (TMB) and gene mutations were also assessed.
Results A total of 219 patients with CUP were included, 215 assessed for TOO in a previous study, with the majority (163) receiving both RNA and DNA tests. Of GEP profiled cases, 33% (59/175) had a high IR gene-expression score. Of the DNA sequenced cases, 16% (32/203) had high TMB (>10 mutations/Mb), including two with mismatch repair deficiency. Low correlation was observed between TMB and an IR score (R=0.26, p<0.001). Among 110 CUPs with a latent primary or suspected TOO, 47% (52/110) belonged to ICI-responsive cancer types. More than half of the CUPs had at least one feature that may predict ICI response (high IR score, high TMB, ICI-responsive cancer type). Among patients with CUP treated with ICIs, 8/28 (29%) responded (2 complete responses and 6 partial responses). Among non-responders, 9 had stable and 11 had progressive disease. All responders had a high IR score (7/8) and/or high TMB (3/8), while most (5/8) belonged to ICI-responsive cancer types. These features were detected at a lower frequency in non-responders and mostly in patients with stable disease.
Conclusions A significant fraction of CUP tumors had genomic features previously associated with ICI response. High IR score was the most sensitive predictive feature of ICI response, warranting evaluation in a larger patient series.
- Immunotherapy
- Biomarkers, Tumor
- Gene Expression Profiling
- Genetic Markers
- Translational Medical Research
Data availability statement
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- Immunotherapy
- Biomarkers, Tumor
- Gene Expression Profiling
- Genetic Markers
- Translational Medical Research
WHAT IS ALREADY KNOWN ON THIS TOPIC
Immune checkpoint inhibitors (ICIs) can be effective for the treatment of cancer of unknown primary (CUP). However, given the heterogeneity of CUP, only a subset responds to ICIs, highlighting the need for predictive biomarkers. Molecular profiling has been used to predict ICI response in other cancer types, specifically by estimating the tumor mutation burden (TMB) and detection of immune or inflammatory biomarkers in the tumors. Therefore, these molecular features are rational biomarkers of treatment response to assess in patients with ICI-treated CUP.
WHAT THIS STUDY ADDS
Our study shows that genomic profiling may be useful to select ICI-responsive patients with CUP. Immune gene-expression profiling, in particular, was the most sensitive predictive biomarker, whereas high TMB and the classified cancer type had lower predictive value when used as single biomarkers.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study provides important evidence for the use of genomic biomarkers to select patients with CUP for ICI treatment and particularly supports incorporating immune gene expression as a predictive biomarker in future ICI clinical trials.
Introduction
Cancer of unknown primary (CUP) is a heterogeneous group of metastatic cancers where a primary tissue of origin (TOO) cannot be identified after a standardized clinical and pathological workup.1 Historically, patients with CUP have been treated with empirical regimens involving platinum and taxane chemotherapies that do not provide durable responses in most cases, with an average survival of less than 1 year.2 As cancer treatment is predominantly based on TOO, high value is still placed on resolving the primary cancer diagnosis. In this context, molecular profiling has been used to help resolve TOO, although whether any survival benefit is gained from directing site-specific treatment guided by molecular profiling remains controversial.3–6 A significant improvement in CUP patient survival outcome will therefore likely require access to more effective targeted treatments, including immunotherapies, that are now standard of care for a growing number of cancer types.
It can be argued that resolving the primary TOO of CUP may become less important with the use of targeted treatments or immune checkpoint inhibitors (ICIs). Among CUPs with an unfavorable outcome, ~30% have been found to harbor genomic features that could direct the use of a targeted therapy.7 Furthermore, companion biomarkers predictive of ICI response are frequently detected in CUPs, including 12% that have high tumor mutation burden (TMB) and ~2% that have microsatellite instability.8 9 In favor of the importance of identifying a TOO, a retrospective study using a gene-expression profiling (GEP) TOO test (CancerTYPE ID) found that a significant fraction (~40%) of CUPs were classified as cancer types approved for access to ICI-based therapies by the US Food and Drug Administration (FDA).10 Additionally, many CUP tumors have been found to have heavy T-cell infiltrates or express T-cell inflammatory markers, similar to other ICI-responsive cancer types.5 While individual case reports of exceptional response to ICI treatment in CUP have been described,11–13 two clinical trials have also recently reported objective response to ICI treatment in approximately 20% of CUP patients.14 15
The current study involved a retrospective evaluation of immune and genomic biomarkers in CUP tumors, including a subset treated with ICIs. Previously, we used GEP and DNA sequencing combined with clinicopathology review to predict TOO.16 We have now complemented this data with immune GEP and the interpretation of somatic DNA features in the same patients. We specifically investigated the biomarkers of TMB and inflammatory gene-expression signatures, as well as the predicted TOO. We hypothesized that one or more of these biomarkers would correlate with treatment response in the subset of patients treated with ICIs in our patient cohort.
Methods
Study cohort and clinical data
Two hundred and nineteen patients with CUP were recruited with informed consent by treating clinicians at participating sites for the Australian CUP study Solving Unknown Primary Cancer (SUPER) between 2014 and 2020.
Eligibility criteria for the study has been previously described.16 Briefly, inclusion criteria included: patients presenting with carcinoma of no identifiable primary site following a preliminary diagnostic workup as outlined in the European Society of Medical Oncology guidelines2; patient had not yet commenced treatment or commenced treatment less than 6 months prior to recruitment; could read and write in English and could provide written informed consent. Exclusion criteria: patients under 18 years of age; poor Eastern Cooperative Oncology Group (ECOG) performance status (ECOG ≥3); uncontrolled medical or psychological conditions that may have prevented completion of study requirements.
Molecular data for 219 CUP cases were analyzed retrospectively, as outlined in figure 1. Clinical, histopathology, and treatment information were collected at recruitment, 6 months, and 12 months after or until the time of death. Survival was used as an endpoint and followed-up 12 months post-recruitment. The patient’s treating clinician made treatment decisions, and the information was recorded. Treatment was either systemic (eg, chemotherapy, targeted therapy, ICI) or non-systemic (eg, radiotherapy, surgery) unless no treatment was recorded. Treatment information before recruitment was not recorded; therefore, every treatment used during the study was counted as a line starting from the time of recruitment. Response to ICI treatment was assessed by the treating medical oncologist using the Response Evaluation Criteria in Solid Tumors V.1.1 (RECIST) scoring system for solid tumors: complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD).17
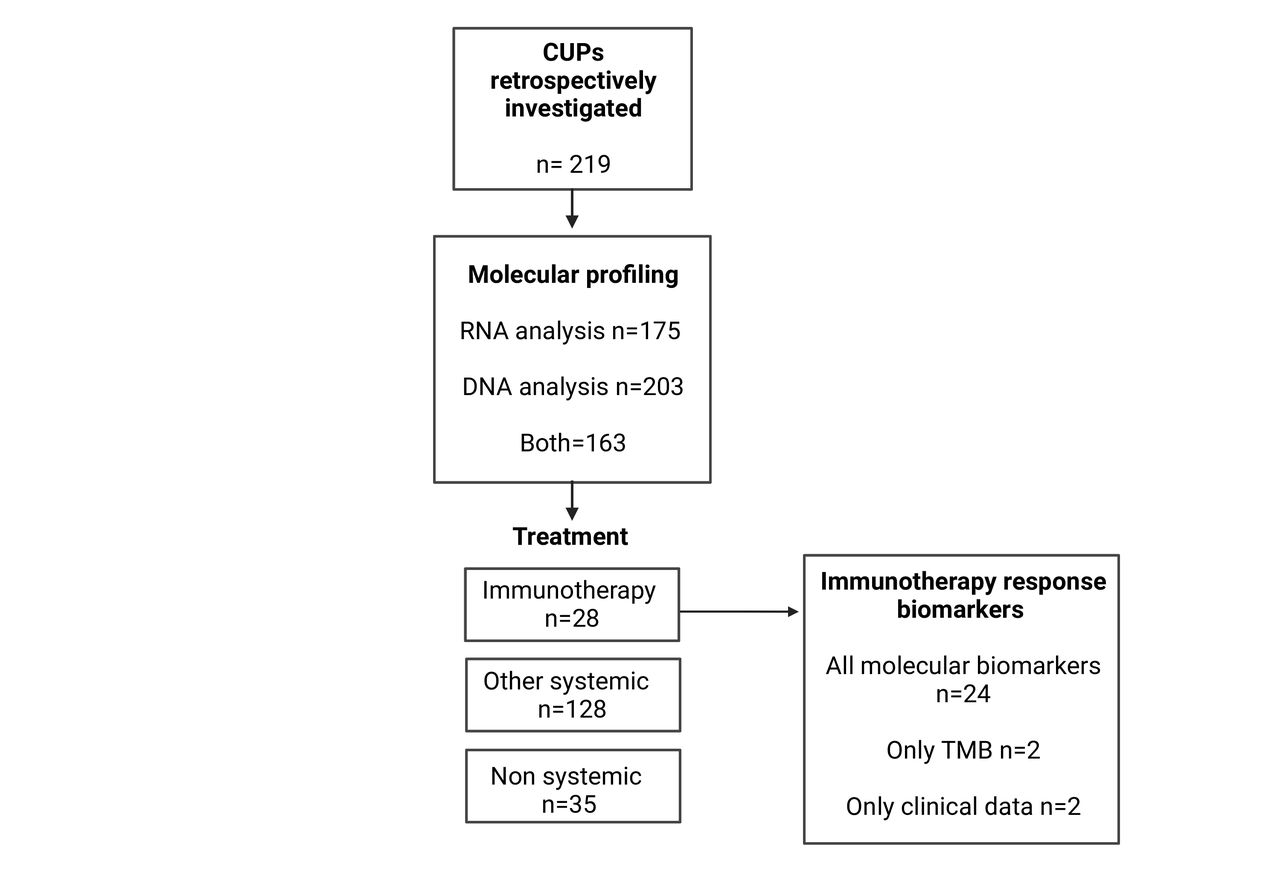
Patients with CUP included in the study subject to retrospective molecular, pathological, and clinical analysis. The 219 patients had either DNA sequencing, NanoString RNA profiling, or both assays performed. Treatment information was recorded for 191 patients. Twenty-eight patients with CUP were treated with immunotherapy and used for ICI-biomarker analysis. CUP, cancer of unknown primary; TMB, tumor mutation burden.
As previously described,16 the diagnostically unresolved CUPs were classified into the Memorial Sloan Kettering OncoTree classes for CUP,18 modified to incorporate immunohistochemistry (IHC) stains for cytokeratin (CK) 7 and 20 and p16INK4A. Four patients included in the current study were not a part of the previous cohort and not diagnostically evaluated.
Known metastatic tumors analyzed for comparative immune GEP were sourced under separate HREC approved protocol (HREC protocol 11/117). All research was conducted according to the ethical standards of the Australian National Health and Medical Research Council.
GEP
DNA and RNA were extracted from formalin-fixed paraffin-embedded (FFPE) tissues using the AllPrep DNA/RNA FFPE Kit (Qiagen, Germany) according to the manufacturer’s instructions, as previously detailed.16 GEP was done proximal to the time of patient recruitment and blinded to treatment and outcome data. Gene-expression analysis involved a custom NanoString nCounter assay performed according to the manufacturer’s instructions (NanoString Technologies, Seattle, Washington, USA). Genes in the immune panel included markers of T cells (CD8A, CD8B) and their associated transcription factors (TBX21, EOMES, and PRDM1), natural killer (NK) cell markers (CD160, CD244), T-regulatory cells (FOXP3), macrophages (CD68), B cells (CD19, CD79B, and MS4A1), endothelial cells (ESM1 and PECAM1/CD31), fibroblasts (COL1A1, CTGF, FAP, and THBS2), inflammatory molecules (CXCL9, CXCL8, CXCL10, IL6, PTGS2, IFNG), cytotoxic molecules (GZMA, GZMB, and PRF1) and immune checkpoint molecules (LAG3, IDO1, CD80, CTLA4, HAVCR2 (TIM3), CD274 (PD-L1), PDCD1LG2 (PD-L2), PDCD1 (PD-1)). The immunotherapy response (IR) score was calculated using the genes CD8A, CD8B, PCDC1, TBX2, GZMA, GZMB, PRF1, IFNG, CXCL9, CXCL10, CD274, EOMES, LAG3, PCDC1LG2, IDO1 that had been previously described in signatures of ICI response in other cancer types.19–22
Viral transcripts encoding capsid proteins for human papillomavirus (HPV)-16 L1, HPV18 L1, and Merkel cell polyomavirus (VP2) were included in the panel. Detection of viral RNA transcripts was complemented by detection of viral DNA sequences from panel DNA sequencing data as previously described.16 Epstein-Barr virus (EBV) was detected from DNA sequencing data only.16 All raw NanoString count data for immune and housekeeping genes are available in online supplemental table 1. Raw count data was normalized using the geometric mean of housekeeping genes from the reference set of known metastatic tumors. An RNA reference pool consisting of RNA from 18 metastatic known origin tumors was used to monitor the reproducibility and performance of each NanoString experimental batch. Gene expression values were transformed by z-score normalization across tumors. For immune/stromal and IR-score gene sets, the mean z-score transformed values were used. Unsupervised hierarchal clustering was performed on the z-score normalized data using the R package ComplexHeatmap with the default settings.23 The known metastatic tumors were used as a reference for calculating the percentile of expression. The top ≥75th percentile expression of a particular gene or gene set was considered high, the bottom ≤25th, low, and the 26–74th intermediate expression.
Supplemental material
DNA sequencing
Targeted DNA sequencing was performed proximal to time of patient recruitment on DNA extracted from tumors and matched whole blood. Germline DNA Extraction was performed using the DNeasy Blood & Tissue Kit (Qiagen, Germany). KAPA Library Preparation Kit (Roche Holdings, South San Francisco, California, USA) was used to prepare DNA libraries and SureSelectXT Target Enrichment assay (Agilent Technologies, Santa Clara, California, USA) for targeted enrichment. Data and all procedures used for nucleic acid extraction, sequencing methods, DNA tumor and matched germline alignment, variant calling, dominant single-nucleotide variant (SNV)-96 trinucleotide mutational signatures (Catalogue Of Somatic Mutations In Cancer (COSMIC) V.2),24 and detection of viral DNA in off-target reads (not targeted with panel capture bait design) have been previously described.16 TMB was calculated based on non-synonymous, coding somatic mutations (base pair substitutions, small insertions and deletions) divided by exon-level DNA sequence capture size.25 The R package maftools was used to visualize the mutation data.26
IHC validation
IHC for programmed death ligand-1 (PD-L1) staining was performed using the Ventana Discovery ULTRA autostainer (Ventana Medical Systems, Arizona, USA). PD-L1 staining and scoring were done blinded to RNA gene expression, patient treatment, and survival outcome data.
Four µm thick sections of paraffin-embedded tissue mounted on SuperFrost plus slides were heated at 72°C and deparaffinized using the discovery wash solution (REF 950–510 Ventana Medical Systems). Heat-induced antigen retrieval was carried out using Cell Conditioning 1 (REF 950–500; Ventana Medical Systems) for 64 min at 98°C. A primary antibody targeting PD-L1 (SP263 REF 790–4905, Ventana Medical Systems) was applied to the tissue section for 48 min at 36°C. The Discovery ChromoMap DAB Detection kit (REF 760–159; Roche) was used to visualize PD-L1. The slides were counterstained with hematoxylin II (REF 790–2208 Ventana Medical Systems) and Bluing Reagent (REF 760–2037 Ventana Medical Systems). The slides underwent rapid dehydration and clearing in ethanol and xylene and were cover slipped using dibutylphthalate polystyrene xylene mounting medium. PD-L1 expression scoring was performed by a pathologist using the tumor proportion score method of scoring.27
Scoring of tumor-infiltrating mononuclear leukocytes was performed by a single pathologist (OWJP) blinded to clinical and molecular data using tumor hematoxylin and eosin stained sections according to a previously described breast cancer scoring system.28
Statistics
The distance measure used for unsupervised hierarchal clustering of genes and samples was Pearson’s correlation (ComplexHeatmap). A paired sample t-test was used to compareICI response groups using the R package rstatix (V.0.6.0). The R package ggpubr (V.0.3.0) calculated Pearson’s correlation in scatter plots. Overall survival (OS) was measured from the date of CUP diagnosis (histologically confirmed) to the date of death from any cause up to 12 months. Patients without a recorded death were censored at 12 months. Survival rate was calculated as a fraction of the number of patients that reached 12 months survival over the total number of patients in a treatment group. Survival analysis was performed using the R package survival (V.3.1–12), and plots were made using the survminer package (V.0.4.8.999). Kaplan-Meier estimates of OS were presented along with log-rank tests for comparison between ICI treated CUPs, other systemic therapy treated CUPs and no systemic treatment groups.
Results
Immune and stromal gene expression patterns in CUP and known cancers
To interrogate immune and stromal cell gene expression in CUP tumors, we analyzed 35 genes across 175 CUP tumors using a custom NanoString panel (figure 2A, online supplemental table 2).19–22 We also profiled 188 metastatic cancers of known primary origin for direct comparison (online supplemental table 3). Two-dimensional unsupervised hierarchical clustering of all samples using all immune/stromal genes showed that CUP tumors and the known metastatic cancers co-clustered and were interspersed across four major sample clusters, exhibiting a gradient of gene expression for the immune and stromal gene sets (figure 2A).
Supplemental material
Supplemental material

Immune gene-expression profiling in CUP (A) Z-score normalized expression of 35 immune and stromal cell gene markers across 175 CUPs and 188 metastatic tumors of known origin clustered using unsupervised hierarchical clustering (distance of dendrogram branches for samples and genes represents Pearson’s correlation coefficient). Genes are annotated alongside cell type or immune cell function and their inclusion within the immunotherapy response score (IR score). (B) Normalized expression of gene sets for cell types and IR score in the known metastatic cancers and CUP tumors. Lymphocyte class represent combined gene markers for B cells, T cells and regulatory T cells (Treg), and natural killer (NK) cells. CUP, cancer of unknown primary.
To validate that bulk-RNA gene expression reflected the immune-cell infiltration pattern in tumors, a random subset of CUP tumors (n=40) was scored for the presence of mononuclear leukocytes by light microscopy on hematoxylin and eosin stained sections. A positive correlation was observed between the number of immune cells seen in tumors and the normalized expression of mononuclear-cell marker genes (all T, B, NK cell, and macrophage messenger RNA (mRNA) markers combined to mononuclearcell score, R=0.65, p<0.001) (online supplemental figure 1).28
Supplemental material
Among known metastatic cancers, immune and stromal gene expression was also consistent with the known molecular and histopathological features of the cancer types (figure 2B). For instance, high expression of endothelial cell markers was observed among kidney cancers (average z score=1.85), consistent with their known vascularized features.29 Elevated expression of fibroblast-associated genes was also observed in pancreatic adenocarcinomas (average z score=0.4) and cholangiocarcinomas (average z score=0.65), reflecting the desmoplastic stroma observed in these cancers.30 31 Higher expression of lymphocyte markers (average z score=0.9) was detected in lung cancers, consistent with the immunogenic phenotype of these tumors.32 Meanwhile, CUP tumors had a near-average expression for immune and stromal gene sets (average z score=0.05), consistent with CUP representing a broad spectrum of solid cancer types (figure 2B).
In addition to measuring immune cell type gene expression, we calculated an IR score based on the combined expression of genes known to be predictive of ICI response in other cancer types (see online supplemental table 2).19–22 These genes included markers of cytolytic T/NK cells (eg, CD8A, GZMA, PRF1), interferon-γ (IFN-γ), and IFN inducible genes (eg, CXCL9, CXCL10, CD274) as well as T-cell exhaustion markers (eg, EOMES) (figure 2A,B). Similar to lymphocyte markers, the mean IR score among CUP tumors was near the mean IR score observed across known metastatic cancers (mean z score=0.01). Among CUPs, 33% had a high IR score (≥75th percentile, z score >0.42), while 27% had low IR scores (≤25th percentile, z score <−0.46).
Integrated biomarker analysis identifies potentially immunogenic and ICI-responsive CUP subgroups
We next assessed the association between predicted cancer type and the immune profile of CUPs. We previously found that 51% of CUPs (110/215) in the SUPER cohort had either a latent primary tumor discovered during the patient’s treatment (22/215, 10%) or were assigned a likely TOO based on a centralized review of clinical and histopathological data alone (27/215, 12.5%) or in combination with DNA sequencing and gene expression data (61/215, 28%).16 The remaining 49% of CUPs (105/215) were diagnostically unresolved. Among CUPs assigned a single TOO (termed resolved CUP), 47% (52/110) were classified to cancer types with FDA approval for an ICI drug as of June 2020; subsequently referred to as ICI-responsive cancer types (figure 3A, online supplemental table 4). CUPs that were assigned to ICI-responsive cancer types, such as melanoma, kidney, HPV-associated squamous cell carcinomas (SCCs), gastric, and lung cancers, had higher IR scores. Conversely, CUPs assigned to non-immunogenic cancers, including some sarcomas and pancreatobiliary cancers, had low IR scores (figure 3A). Similarly, among resolved CUPs, the stromal gene-expression patterns were consistent with the expected cancer type. For instance, kidney-CUPs had elevated endothelial markers, while cholangiocarcinoma-CUPs had high fibroblast marker expression (online supplemental figure 2).
Supplemental material
Supplemental material

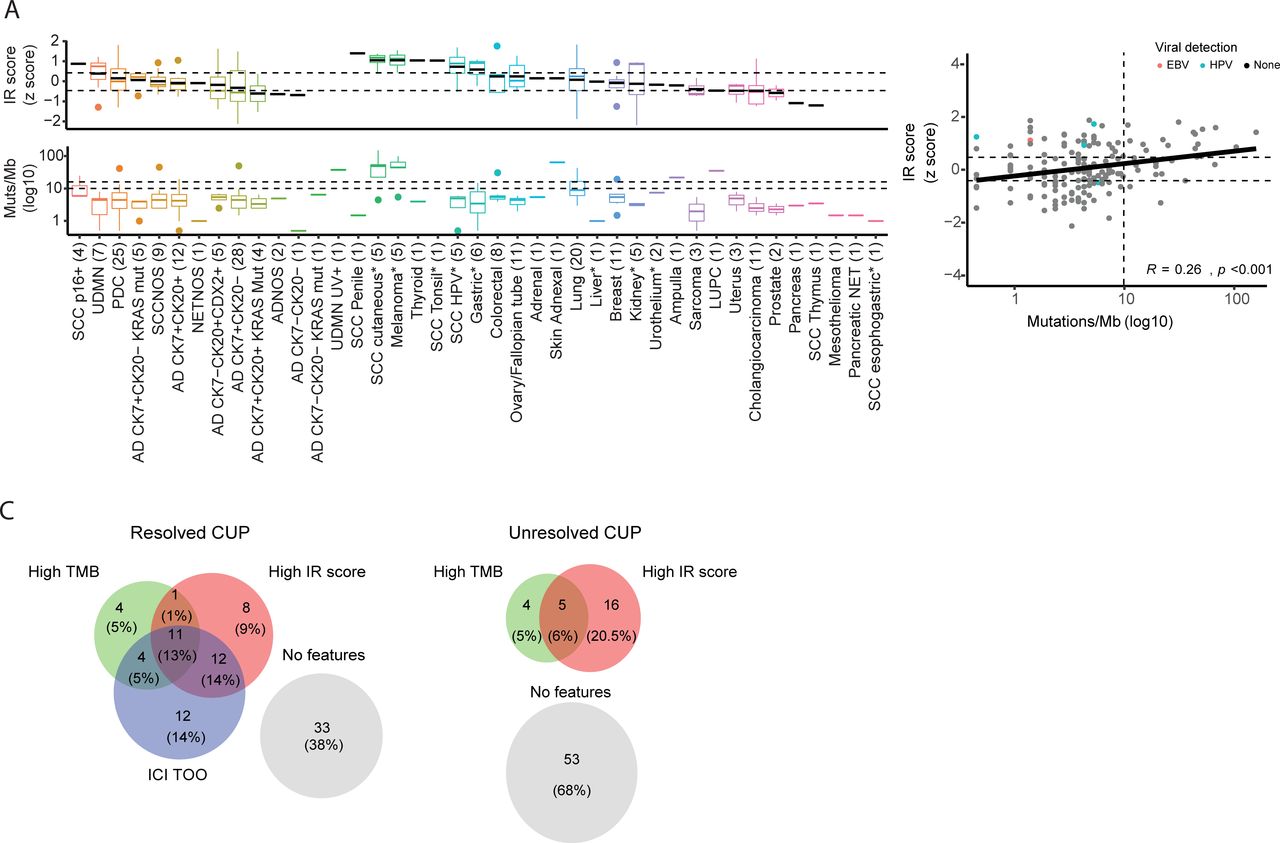
Latent primary or predicted tissue of origin and associated immune and genomic features (A) Immunotherapy response (IR) score and tumor mutation burden (TMB) in tumors grouped by their latent primary diagnosis and predicted tissue of origin. Unresolved CUPs are arranged based on their modified OncoTree classification. CUP groups were ordered by decreasing mean IR score. Group mean values are represented as a black line in a boxplot. Tumor classes with an asterisk (*) indicate cancer types eligible for immune checkpoint inhibitor treatment based on Food and Drug Administration approval as of June 2020. Dotted horizontal lines indicate the top 75th and bottom 25th IR score, and 10 and 16 mutations/Mb in the TMB plot. (B) Correlation between IR score and TMB in CUP tumors, using a Pearson’s correlation coefficient (R). Cases with oncoviral DNA or RNA detected are annotated. (C) Biomarkers observed in unresolved CUP (n=75) and resolved CUP (n=87) groups where both molecular tests were performed. CUP, cancer of unknown primary; AD, adenocarcinoma; ADNOS, adenocarcinoma not otherwise specified; SCC, squamous cell carcinoma; PDC, poorly differentiated carcinoma; UDMN, undifferentiated malignant neoplasms; NETNOS, neuroendocrine tumor not otherwise specified; LUPC, pleomorphic carcinoma of the lung; p16, p16INK4A.
High TMB has been previously associated with a greater frequency of intratumoral T cells in solid cancers.33–37 The mean TMB across the CUP cohort was 9.2 mutations/Mb, and the median TMB was 4.4 mutations/Mb. Of the 219 patients investigated, 203 had DNA sequencing results. Sixteen per cent of CUPs (32/203) had a TMB >10 mutations/Mb; the current FDA-approved threshold for treatment with pembrolizumab.38 Twelve per cent (24/203) of CUPs had a TMB above 16 mutations/Mb; the threshold for single agent atezolizumab treatment in the CUPISCO phase II randomized trial (figure 3A).39 Two patients with high TMB had a DNA mismatch repair deficiency. We observed a significant but weak correlation between TMB and the IR score (R=0.26, p<0.001) (figure 3B). Importantly, marked discordancy was observed between TMB and IR score in some cancer types, including viral-associated CUPs (figure 3B) and kidney-CUPs, with both groups having low TMB but high IR scores (figure 3A).
Where data was available for all three features tested (ICI-responsive cancer type prediction, TMB, and IR score), 62% (53/85) of resolved CUPs (latent primary and predicted cancer type) had at least one positive biomarker, and 13% had all three features (figure 3C). Thirty-two per cent (25/78) of the unresolved CUPs had either a high IR score or high TMB, with a high IR score in 20.5% (16/78) cases and 6% (5/78) having both a high IR score and high TMB (figure 3C). Notably, among the unresolved CUPs, undifferentiated malignant neoplasms (UDMN) had higher IR scores (mean z score=0.38) but low TMB (mean 3.7 mutations/Mb) (figure 3A,C).
High IR score was the most common feature among ICI-responsive CUPs
Immune and genomic biomarkers thought to be predictive of ICI-treatment response were retrospectively evaluated in patients with CUP treated with these drugs. Twenty-eight patients with CUP were treated with an ICI as any line of treatment. Patients were treated with a single agent or combined with another targeted therapy. Treatment information is summarized in table 1 and online supplemental table 5.
Supplemental material
Summary of immunotherapy treatment cancer of unknown primary cohort
Response to treatment was retrospectively assigned using the RECIST solid tumor response scoring system.17 The overall response rate was 29%. Eight patients responded—two with a CR and six with a PR. Nine patients had SD (32%), while 11 (39%) had PD.
The survival outcomes of patients with CUP treated with ICIs were compared with those treated with other systemic therapies (n=118) and those that did not receive systemic therapy (n=34), in cases where treatment and survival data was recorded. Log-rank testing showed longer OS in ICI-treated CUPs (p<0.05) and 77% of ICI-treated CUPs achieved 12-month survival compared with 53% for non-ICI treated CUPs and 52.5% for non-systemic therapy treated CUPs (figure 4A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immune and genomic tumor features of immunotherapy responsive and non-responsive patients with CUP patients (A) Kaplan-Meier analysis with log-rank test for survival of CUPs treated with immune checkpoint inhibitor (ICI) compared with those treated with other systemic or no systemic therapy. (B) Immune and genomic features of CUP tumors for 28 patients treated with ICIs. (B) Gene set expression for cell types and immunotherapy response (IR) score shown as ordered heat map. CD274 messenger RNA gene expression, with a black dot indicating cases where PD-L1 IHC staining was done and >1% of cells stained positive. ‘H’ indicates a high IR score (>75th percentile). The top 10 mutated genes and genes reported to be associated with resistance to ICI treatment were included (PTEN, STK11). Other mutation features include the presence of dominant COSMIC 96-SNV mutational signatures (UV and tobacco smoking) and TMB. The assigned tissue of origin (TOO) and OncoTree classes were annotated with assignment for Food and Drug Administration-approved ICI eligible cancer types. Cases that did not have a retrospective histopathology review performed were labeled with an asterisk. (C) Boxplot of IR score and TMB (mutations/Mb) grouping CUP tumors by treatment response. Significance tested using a paired sample t-test. An (*) indicates p<0.05. The dashed line showing TMB high (10 mutations /Mb) threshold. CR, complete response; CUP, cancer of unknown primary; IHC, immunohistochemistry; PD, progressive disease; PD-L1, programmed death ligand-1; PR, partial response; AD, adenocarcinoma; SCC, squamous cell carcinoma; SD, stable disease; TMB, tumor mutation burden; TOO, tissue of origin; UDMN, undifferentiated malignant neoplasms; UV, ultraviolet; CK7, cytokeratin 7; CK20,cytokeratin 20.
Of the 28 ICI-treated patients, 24 had histopathology, clinical and genomic data reviewed for TOO prediction (figure 4B), while the remaining four patients were not reviewed due to insufficient data. Seventeen of 24 CUPs had a primary TOO assigned, 12 of which corresponded to an ICI-responsive cancer type (table 1, figure 4B). These ICI-responsive cancers included lung cancer (n=8), melanoma (n=2), and cutaneous SCC (n=2).
CUP tumors in seven of eight ICI-responsive patients (CR/PR) had a high IR score, and the remaining responding patient had a high TMB. Two of eight tumors had a high IR score but had PD. Overall, responding patients had significantly higher IR scores than tumors from patients with PD (CR/PR average z score=0.97, PD average z score=−0.23, p=0.016). No significant differences in IR score were observed comparing CR/PR and SD groups (figure 4B,C). Aggregate gene-expression scores for lymphocytes, macrophages, fibroblasts, and endothelial cells gene sets were not significantly different between any of the clinical response groups (online supplemental figure 3A). These results indicate that IR score may be more predictive of ICI response than a measure of total immune cell infiltration.
Supplemental material
Due to limited tissue availability, we could not perform PD-L1 IHC on ICI treated cases using a single standardized method, although PD-L1 IHC staining results were available from pathology reports in some cases. Alternatively, we used CD274 mRNA expression in place of PD-L1 staining, given that we found standardized PD-L1 IHC scoring in an independent tumor series was positively correlated with CD274 expression measured by NanoString (R=0.59, n=39) (online supplemental figure 3B). CD274 expression was not significantly different between the CR/PR and PD groups and the clinically reported PD-L1 IHC results did not appear predictive of response given that five of nine patients reported to have high PD-L1 staining were unresponsive (SD/PD) (figure 4B and online supplemental table 5).
TMB was higher in the CR/PR group (median=8.4 mutations/Mb, range 4–149 mutations/Mb) compared with the SD group (median=6.7 mutations/Mb, 1–99 mutations/Mb) and the non-responsive PD group (median=3.9 mutations/Mb, 1–7 mutations/Mb); however, did not reach significance as a continuous variable contrasting any groups (figure 4B,C). A high TMB was frequently associated with a dominant SNV-96 trinucleotide mutational signature, including Signature 7 (ultraviolet (UV) light) and Signature 4 (tobacco smoking) that supported TOO diagnoses of skin and lung cancer, respectively (figure 4B).
We investigated whether the lack of ICI-treatment response may be linked to mutated cancer genes. Mutations in genes previously associated with ICI resistance were detected in both responders and non-responders. For instance, STK11 mutations, previously associated with poor response to ICI treatment in lung cancer,40 41 were detected in lung-CUPs and one unresolved CUP (adenocarcinoma, CK7+, CK20–), and these patients did not respond to ICI treatment. A splice-site PTEN mutation was found in a lung-CUP (7031) with a CR (figure 4B), contrary to reports of deleterious PTEN mutations associated with ICI resistance in other cancers.42
In summary, 58% (7/12) of ICI-treated CUPs with a high IR score responded to treatment, while 50% (3/6) with a high TMB responded. All but one ICI responsive (CR/PR) CUP had a high IR score, while high TMB was detected in less than half of these cases (3/8). Conversely, only two patients with PD had a high IR score, and none had a high TMB. Unresponsive patients with SD had mixed results. Additionally, most of the CR/PR patients were predicted to belong to ICI-responsive cancer types (5/8), which was a similar frequency among patients with SD (5/9), while only a minority of the PD group (3/11) were assigned to ICI-responsive cancer types (figure 4B and online supplemental table 5).
Discussion
Without effective treatments for many CUP patients, there is a strong rationale to use ICIs given their pan-cancer efficacy. This study explored three features that may be predictive of ICI response in CUP: the likely cancer type based on genomic and clinicopathological data, TMB, and tumor immune profile measured by GEP. Based on genomics informed pathology review, approximately one-quarter of CUPs could be classified as an ICI-responsive cancer type, but this classification did not have a high specificity to predict response among the ICI treated patients. Consistent with prior studies, a significant minority of CUPs had high TMB; between 12–16% of cases depending on TMB threshold applied.33 34 43 Conversely, immune GEP, including the IR score described in the current study, showed one-third of CUPs had high scores that likely reflected the higher inflammatory status of these cancers. Moreover, among ICI-treated patients, those who responded to treatment were more likely to have a high IR score than a high TMB, suggesting that assessment of the tumor immune status is likely to be more sensitive or at least complementary to TMB as a predictive biomarker.
Our observations in CUP are supported by similar findings in other cancer studies that showed immune GEP profiling or scoring immune cells in tumors can be a predictive or prognostic biomarker.35 43 44 Our data also validates prior studies investigating the immune profile of CUP tumors using IHC staining or mRNA expression analysis.5 8 In our study, one-third of CUP tumors had elevated immune markers or IR score, indicating the tumors are likely to be highly immunogenic. In contrast, we found that less than half of immune high cases were explained by high TMB. Indeed, it is known that the link between TMB and cancer immunogenicity (and ICI response) is only valid for certain cancer types. For instance, TMB has limited predictive value in kidney cancers and viral-associated tumors.44 45 We also observed several examples of viral-associated cancers and kidney-CUPs in our cohort, most with a low TMB but elevated immune scores. In the absence of high TMB or a confirmed ICI-responsive cancer type diagnosis, an assessment of the tumor immune status may therefore identify patients not otherwise considered suitable for ICI treatment.
Not surprisingly, among CUPs classified as ICI-responsive cancer types, these tumors often had high TMB and/or high IR scores. Diagnostically unresolved CUPs infrequently had high TMB, but this can be explained as hypermutation and mutational signatures caused by environmental exposures such as tobacco smoke and UV sunlight have high diagnostic value and, therefore, more likely undetected among the unresolved cases. Aside from HPV-associated SCCs, the UDMN cohort had an above-average IR score but low TMB, suggesting they may benefit from ICI treatment. Two UDMN CUP cases received ICI treatment—one had PR and another SD. Notably, the UDMN patient who responded had a high IR score, while the non-responder had a low IR score. While no common mutational or histopathological features were observed within the UDMN CUPs,16 an immunogenic phenotype has been reported among some undifferentiated tumors.46 47 Further analysis of additional UDMN CUPs is required to validate the association of elevated immune infiltration and ICI response in this group.
Of 28 CUPs that received programmed cell death protein-1 or PD-L1 inhibitors, 28% responded to treatment, similar to the overall response rates reported in two CUP ICI clinical trials.14 15 The NivoCUP trial (UMIN-CTR ID UMIN000030649) reported a 21% (12/56) overall response (CR, PR) to nivolumab in unfavorable CUPs, while a Phase 2 basket trial involving rare tumors treated with pembrolizumab (NCT02721732) reported that 20% (5/25) of patients with CUP had a PR.14 15 48 Interestingly, with respect to the biomarker analysis in these clinical trials, similar observations were made to our own study. In both clinical trials, better efficacy was observed if a patient’s tumor had a high number of infiltrating lymphocytes. The NivoCUP trial also reported higher TMB in the nivolumab responsive patients when compared with unresponsive patients, whereas molecular profiling to classify CUPs to ICI responsive cancer types was not predictive of ICI efficacy.
While immune gene-expression signatures and TMB can have predictive value, there may be compounding mechanisms of ICI resistance explained by the underlying genetic background of the tumor. Two of the 11 patients in our cohort with PD had high IR scores. One unresponsive case with a high IR score (ID B015) was treated with five cycles of single-agent nivolumab (accessed through high PD-L1 staining (>90%) and presumed to be of lung origin). Lack of treatment response may be explained by a somatic mutation in STK11, given that STK11 mutations have been linked to poor survival outcomes in non-small cell lung cancer,49 50 although conflicting evidence involving STK11 mutations and ICI resistance has also been reported.51 We also identified a case where a putative ICI resistance gene mutation appeared to have no impact on treatment response. A deleterious PTEN mutation was detected in a lung-CUP where the patient had a CR (7031), despite PTEN loss being linked to resistance in several cancer types, including non-small cell lung cancer.42 In reality, the detection of putative ICI resistance mutations may have limited predictive value in individual patients with CUP, given the heterogeneity of these tumors.
It is also important to consider that while some tissue-based biomarkers may have use, there are still inherent challenges to applying these in practice. The reliability of immune biomarkers can be adversely affected by biopsy tissue sampling compounded by tumor heterogeneity within and between tumor lesions. Alternative approaches may therefore involve the use of positron emission tomography (PET) imaging, employing radionuclide conjugates to detect tumor infiltrating CD8 T cells.52 PET imaging can also provide quantitative data on tumor volume and metabolic activity, which have also been shown to have value in predicting ICI-treatment response in other cancers.53
A clear limitation of our study was the small number of patients treated with ICIs, and that patient selection for treatment was not randomized. Although our observations are in broad agreement with observations from clinical trials involving CUP, further validation is required in a larger patient series. Current prospective trials investigating ICIs in CUP (NCT03752333), poor prognosis CUPs (NCT03391973, NCT04131621), ICIs concurrent with radiation (NCT03396471), and guided by comprehensive genomic profiling (CUPISCO, NCT03498521) are underway, and biomarker analysis employed as part of these trials may help to further validate some of the findings of our study.
Data availability statement
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
All participating patients provided informed consent to partake in this study. This study was approved by the Peter MacCallum Cancer Centre (PMCC) human research ethics committee (HREC protocol: 13/62).
Acknowledgments
We acknowledge Cameron Patrick of the Melbourne Statistical Consulting Platform for providing statistical support. We wish to thank Jillian Hung and Niklyn Nevins, SUPER study coordinators at Westmead and Blacktown, Lisa Kay at Nepean, Karin Lyon (ethics and governance), and acknowledge the contributions of the Nepean Cancer Biobank and the Westmead GynBiobank (funded by the Cancer Institute NSW, NHMRC and the Department of Gynaecological Oncology, Westmead Hospital) for facilitating the study. We wish to acknowledge the patients who have contributed to this study and the CUP consumer steering committee, Cindy Bryant (chair), Kym Sheehan, Christine Bradford, Clare Brophy, Dale Witton, and Frank Stoss.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @UMCCR_RADIO_Lab
RWT and LM contributed equally.
Contributors RWT and LM conceived the study. AP performed the analysis, drafted the figures and tables. OWJP undertook the histopathology review and immunohistochemistry sample scoring. TS and LM reviewed the clinical data. CW, KF, and SW collected the data for the study. ADeF, NW, CSK, BG, NW, CS, MS, IMMC, GR, MW, and NK screened patients for eligibility. DE and NT developed the NanoString classifier. DE and AP performed the gene-expression profiling. AF and SBF performed the mutation profiling. AP and RWT co-wrote the manuscript. RJH provided clinical expertise. All authors critically reviewed the manuscript. PS, DB, LM and RWT are the principal investigators and obtained research funding to support the study. LM is the guarantor of the study.
Funding This study was supported by funding from Cancer Australia (APP1048193, APP1082604) and the Victorian Cancer Agency (TRP13062). RWT was supported by funding from the Victorian Cancer Agency (TP828750). SBF received funding from National Health and Medical Research Council (APP1193630). ADeF and the Westmead, Blacktown and Nepean study sites were supported by the Cancer Australia, Cancer Institute NSW, Medical Research Future Fund, New South Wales Ministry of Health, National Health and Medical Research Council, Rivkin Center for Ovarian Cancer (USA), University of Sydney, U.S. Army Medical Research and Materiel Command (USA), AstraZeneca, and Sydney Health Partners.
Competing interests All authors have completed the Unified Competing Interest form (available on request from the corresponding author) and declare: RJH is a shareholder in Telix Pharmaceuticals and a Founder and Director of PreMIT. CSK is on the advisory board of AstraZeneca, BMS and Roche. CS is on the advisory board of MSD, Sanofi, Janssen, and GSK. ADeF has received grant funding from AstraZeneca, unrelated to this study; no other relationships or activities that could appear to have influenced the submitted work.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.