Article Text

Abstract
Background Anti-PD-1 immune checkpoint blockade is approved for first-line treatment of recurrent/metastatic head and neck squamous cell carcinoma (HNSCC), but few patients respond. Statin drugs (HMG-CoA reductase inhibitors) are associated with superior survival in several cancer types, including HNSCC. Emerging data suggest that manipulation of cholesterol may enhance some aspects of antitumor immunity.
Methods We used syngeneic murine models (mouse oral cancer, MOC1 and TC-1) to investigate our hypothesis that a subset of statin drugs would enhance antitumor immunity and delay tumor growth.
Results Using an ex vivo coculture assay of murine cancer cells and tumor infiltrating lymphocytes, we discovered that all seven statin drugs inhibited tumor cell proliferation. Simvastatin and lovastatin also enhanced T-cell killing of tumor cells. In mice, daily oral simvastatin or lovastatin enhanced tumor control and extended survival when combined with PD-1 blockade, with rejection of MOC1 tumors in 30% of mice treated with lovastatin plus anti-PD-1. Results from flow cytometry of tumors and tumor-draining lymph nodes suggested T cell activation and shifts from M2 to M1 macrophage predominance as potential mechanisms of combination therapy.
Conclusions These results suggest that statins deserve further study as well-tolerated, inexpensive drugs that may enhance responses to PD-1 checkpoint blockade and other immunotherapies for HNSCC.
- Head and Neck Neoplasms
- Macrophages
- Programmed Cell Death 1 Receptor
Data availability statement
Data are available on reasonable request. Raw data are available on reasonable request with data transfer agreement.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Cholesterol may impact antitumor immune responses, and a few prior studies suggest that statin drugs may increase responses to immune checkpoint blockade.
WHAT THIS STUDY ADDS
This study is the first to investigate activity of all statin drugs and clinically relevant, oral doses of two statins in combination with PD-1 immune checkpoint blockade.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
These results suggest that daily oral statin therapy may be an inexpensive, safe strategy for enhancing responses to immunotherapy.
Background
Approximately half of patients with head and neck squamous cell carcinoma (HNSCC) will develop recurrent/metastatic disease. Traditionally, cytotoxic chemotherapy was used in this setting, with substantial toxicity.1 In 2016, anti-PD-1 immune checkpoint blockade (ICB) was approved by the Food and Drug Administration (FDA) for chemotherapy-refractory HNSCC based on large trials showing durable responses and improved survival with pembrolizumab or nivolumab versus second-line chemotherapy.2 3 In 2020, pembrolizumab was FDA approved as a single agent or in combination with chemotherapy in the first-line recurrent/metastatic setting based on results from the KEYNOTE-048 trial.4 Although pembrolizumab is relatively well tolerated, only a minority of patients with recurrent/metastatic disease will respond.4 There is an unmet clinical need for agents that enhance responses to anti-PD-1 ICB without excessive toxicity. The ideal agent would also be inexpensive, since pembrolizumab itself is costly.
HMG-CoA reductase inhibitors, also known as statin drugs, are commonly used to treat hyperlipidemia. It is well established that statins may also have anticancer, anti-inflammatory, and other effects.5 6 Numerous epidemiological studies in different cancer types have shown improved survival outcomes in patients taking statins, versus patients not taking these drugs.7–12 In head and neck cancer patients, the use of statins is associated with improved survival outcomes,11 12 lower incidence of cisplatin-induced hearing loss,13 and reversal of radiation-induced fibrosis,14 by mechanisms that are not currently understood. The potential benefit of statins in patients with recurrent/metastatic head and neck cancer treated with anti-PD-1 ICB have not yet been explored. Studies in several other cancer types suggest that statins are associated with increased rates of response to anti-PD-1 ICB.15 16 However, the off-target effects of different statins can vary dramatically,13 15 and studies comparing the potential of all seven commercially available statin drugs to enhance responses to ICB are lacking.
Emerging studies suggest that manipulation of cholesterol in the serum, tumor microenvironment, or peripheral lymphoid tissue may enhance some aspects of antitumor immunity.17–25 Proposed mechanisms based on preclinical studies are numerous. These include enhanced activation of T cells or dendritic cells, decreased T cell exhaustion, improving antigen presentation, ER stress, elevating immunogenic cell death, and transient inhibition of type I interferon.17–25 However, conclusions from many of these studies were drawn from use of supraphysiological (micromolar)26 27 doses and non-oral routes of administration. In the present study, we used a high-throughput, ex vivo T cell killing platform to compare the activity of all seven available statin drugs at a range of clinically relevant (nanomolar)26 27 concentrations. We then used human papillomavirus (HPV)-negative and HPV-positive syngeneic mouse models to determine whether daily oral administration of statins, such as would occur in a head and neck cancer patient taking a statin, could enhance the tumor growth delay afforded by PD-1 ICB by immune or non-immune mechanisms.
Methods
Cell lines
Mouse oral cancer (MOC1) cells were obtained from Kerafast and maintained as previously described.28 29 Mouse TC-1 tumor cells were obtained as a kind gift from Dr. T.C. Wu of Johns Hopkins University and maintained as previously described.30 All cell lines were regularly tested for Mycoplasma contamination and cultured for no longer than 3 months or 20 passages before use.
Antibodies and reagents
Antibodies for in vivo mouse treatments specific for PD-1 (clone RMP1-14) and CD8 (clone YTS 169.4) were from BioXCell. Fluorescent-conjugated flow cytometry antibodies for mouse tumor experiments were obtained from BD Biosciences, Biolegend, Miltenyi or Abcam (see online supplemental methods table S1). Statin drugs (simvastatin, lovastatin, fluvastatin, pitavastatin, atorvastatin, rosuvastatin, pravastatin, table 1), were obtained from VWR International. For in vitro experiments, stock solutions were made with sterile DMSO and frozen at −20°C until use. For in vivo administration, statins were prepared as an oral suspension with 6% polyethylene glycol 400, 1% propylene glycol, and 0.1% Tween (all from Sigma) in water, as previously described.31 Aliquots of statin suspension were frozen at −20°C for up to 2 weeks prior to administration to mice by oral gavage.
Supplemental material
Comparison of the degrees of tumor-cell killing, enhancement of immune response (in vitro and/or in vivo), toxicity in vivo, and suppression of T cell proliferation among seven commercially available statin drugs
In vivo murine studies
Wild-type, female C57BL/6 mice at 6–8 weeks were obtained from Taconic. Mice were injected in the right flank with MOC1 cells (5×106, in Matrigel) or TC-1 cells (1×105) cells and allowed to grow for 7–14 days, then randomized into treatment groups. Mice were then treated with statins (60 mg/kg for lovastatin, simvastatin and fluvastatin and 3 mg/kg for pitavastatin) or vehicle (0.2 mL) by oral gavage once daily for up to 3 weeks. A subset of mice also received anti-PD-1 (200 µg, two times per week) by intraperitoneal injection for up to five doses. CD8+cells were depleted in a subset of mice as previously described and validated.32 Because previous experiments have shown no difference in MOC1 tumor growth whether isotype controls for anti-PD-1 or CD8 antibodies (rat IgG2a and IgG2b, respectively) were included or omitted,29 33–38 control animals were not treated with isotype control antibodies in these experiments. In a cohort of mice from each experimental group (n=5), tumors and spleens were harvested prior to completion of treatment, processed into single cell suspensions as previously described,32 and analyzed by flow cytometry. Tumor draining lymph nodes (TDLN) were also collected.
Flow cytometry
Single-cell suspensions from tumors, spleens and lymph nodes were rinsed in FACS buffer, then stained with surface antibodies for 30 min, followed by additional rinsing, fixation, and permeabilization with the eBioscience kit where needed for intracellular staining, which was performed in a similar manner. Samples were analyzed on a BD Symphony A3 cytometer, then further analyzed using FlowJo (V.10.8.1) software. ‘Fluorescence minus one’ controls were tested for each multicolor flow panel.
T cell assays
For ex vivo T cell killing assays, MOC1 and TC-1 tumors were harvested from mice 7–14 days after tumor cell inoculation, cultured with murine IL-2 (100 U/mL, Biolegend, replenished every 2 days) to expand tumor infiltrating lymphocytes (TILs), and magnetically sorted using a CD8 negative selection kit (Miltenyi) to generate effector CD8+TIL (figure 1A), as previously described.38 Briefly, 10,000–12,000 target cells (MOC1 or TC-1) were plated in a 96-well E-Plate (ACEA Biosciences) and allowed to adhere and grow for 24 hours prior to adding statins or effector cells. Respective TILs were subsequently added with or without statin drugs at a range of effector-to-target cell ratios. Each statin-containing well was replenished with additional drug at 72 hours, based on preliminary experiments that showed cell recovery at that time point when additional statin was not added. Alteration of impedance was acquired using the xCELLigence Real-Time Cell Analysis (RTCA) platform per manufacturer instructions. Triton X-100 (0.2%) was added to some wells to verify complete loss of cell index with total cell lysis. Controls with maximum concentrations of DMSO were also included to rule out effects of the statin vehicle. Changes in impedance were recorded using the xCELLigence RTCA platform as previously described.37 39 40
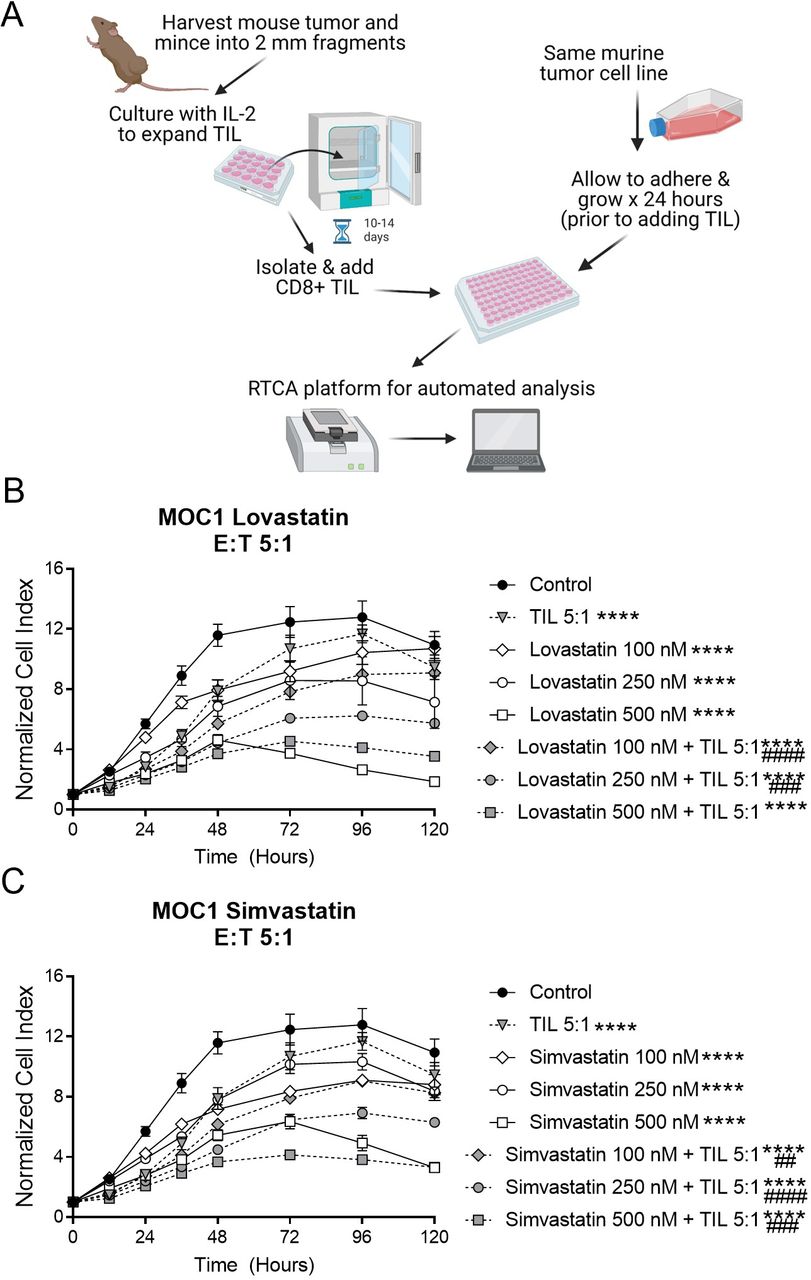
MOC1 tumor-cell proliferation was reduced by simvastatin and lovastatin, with further reduction on addition of T cells. (A) Schema of tumor cell and TIL coculture experiments. Created with Biorender.com, with license. (B, C), MOC1 cells were plated in 96-well plates and allowed to adhere overnight, then some wells treated with simvastatin (B) or lovastatin (C) and/or TIL at a 5:1 effector:target (E:T) ratio for 120 hours. Data represent mean±SEM of 4 replicates, normalized to a cell index of 1.0 when statin and/or TIL were added (time 0 on graph). Graphs are representative of at least two independent experiments done in quadruplicate. MOC1, mouse oral cancer; RTCA, real-time cell analysis; TIL, tumor infiltrating lymphocytes. ****p<0.0001 versus control, ##p<0.01, ###p<0.001, ####p<0.0001 versus statin or TIL alone.
For T cell proliferation/activation assays, peripheral blood mononuclear cells from one healthy donor and two previously untreated patients with head and neck cancer were used. Cells were labeled with CellTrace Violet (ThermoFisher) per manufacturer instructions, then stimulated with CD2/CD3/CD28 beads (Miltenyi) per manufacturer instructions and human IL-2 (50 IU/mL, Biolegend) for 5 days in the presence or absence of statin drugs (statin and IL-2 replenished on day 2), then stained for surface CD107a and analyzed by flow cytometry. For murine T cell proliferation assays, splenocytes from an untreated C57BL6 mouse were stained with CellTrace Violet and cultured with murine IL-2 for 5 days, then analyzed by flow cytometry.
For ex vivo assays on T cells taken from TDLN, T cells were rested overnight, then treated with eBioscience Cell Stimulation Cocktail (PMA/ionomycin) for 4 hours or CD3/CD28 Dynabeads (ThermoFisher) for 24 hours, at concentrations recommended by the manufacturers. Cells were then labeled for intracellular interferon-γ and analyzed by flow cytometry.
Statistical analyses
Data were analyzed by Student’s t-test, and one-way or two-way analysis of variance with post hoc Tukey analyses where appropriate. Animal survival curves were made using the Kaplan-Meier method with comparison by using log-rank (Mantel-Cox) testing. GraphPad Prism software was used for statistical testing, and a p<0.05 was considered statistically significant.
Results
Statins induce direct killing and enhance T-cell-mediated killing of cancer cells
We hypothesized that any statin drug could bolster T cell-mediated tumor destruction, with variable efficacy. To test this idea, we first performed a series of in vitro experiments to determine whether any of the seven commercially available statin drugs, when used at physiological concentrations, could affect the growth of HNSCC cell lines in the presence or absence of T cells. We used an HPV-negative model (MOC1) and an HPV-positive model (TC-1). The serum concentration achieved in humans after oral administration of statin drugs is in the nanomolar range, typically 500 nM or less,26 27 41 and lower for rosuvastatin versus other statins.41 To mimic these conditions, MOC1 cells were seeded into 96-well Agilent RTCA plates and allowed to adhere overnight prior to addition of statins or T cells. TIL were obtained from murine tumors and cultured ex vivo for 10–14 days with IL-2 prior to adding them to the wells containing tumor cells (figure 1A). All seven statin drugs inhibited MOC1 proliferation in a dose-dependent fashion, and the most pronounced effects were seen with simvastatin, lovastatin, fluvastatin and pitavastatin (figure 1, online supplemental figure S1, table 1), which are all lipophilic statins. The inhibition of cell growth seen when TIL were added was modestly enhanced by a subset of statins, with most pronounced effects mediated at moderate doses of simvastatin and lovastatin (figure 1B,C and table 1). The combined antitumor activity of statins and TIL appeared to be additive, not synergistic.
We then repeated these experiments with a cell line expressing HPV oncoproteins (TC-1). Once again, simvastatin, lovastatin, fluvastatin and pitavastatin induced the most pronounced antitumor activity (figure 2, online supplemental figure S2). Both simvastatin and lovastatin enhanced tumor-cell killing by TIL, which was more robust in this model (figure 2A,B). To explore whether statins have any direct effect on the T cells in addition to the tumor cells, we pretreated TIL from TC-1 with simvastatin or lovastatin for 24 hours prior to adding them to the 96-well plates with tumor cells (without any statin drugs in the tumor cell culture). This pretreatment of TIL enhanced tumor cell killing versus untreated TIL (figure 2C), suggesting that statins do have some direct, beneficial effects on T cell-induced killing of tumor cells. Taken together, these results suggest that statins, particularly simvastatin and lovastatin, can directly inhibit proliferation of tumor cells and enhance tumor-cell killing by T cells.

TC-1 tumor-cell proliferation was reduced by simvastatin and lovastatin, with further reduction on addition of T cells. In (A, B), TC-1 cells were plated in 96-well plates and allowed to adhere overnight, then some wells treated statin drug and/or TIL at a 0.5:1 or 1:1 effector:target (E:T) ratio for 48 hours. In (C), TIL were pretreated with statin drugs for 24 hours prior to adding to the tumor cell cultures (without any statin drug). Data represent mean±SEM of 4 replicates, normalized to a cell index of 1.0 when statin and/or TIL were added (time 0 on graph). Graphs are representative of at least two independent experiments done in quadruplicate. TIL, tumor infiltrating lymphocytes. ****p<0.0001 versus control, #### p<0.0001 versus statin or TIL alone.
Statins enhance responses to PD-1 ICB
We next wondered if oral statins could enhance the response to PD-1 ICB in vivo. Once again, we used both HPV-negative (MOC1) and HPV-positive (TC-1) mouse models. Given the apparent direct killing and enhancement of T-cell-induced killing of tumor cells by statins, we next treated MOC1 tumor-bearing mice with statins and anti-PD-1 ICB, alone or in combination. We began with pilot experiments including the four drugs with best performance in our in vitro experiments (lovastatin, simvastatin, fluvastatin, pitavastatin). To simulate the typical clinical/oral dosing of statin drugs, mice were treated with statins by oral gavage every day for 3 weeks. Anti-PD-1 was given IP two times weekly for five doses (figure 3A). Although fluvastatin and pitavastatin delayed tumor growth in MOC1 bearing mice when used alone or in combination with anti-PD-1 antibody (online supplemental figure S3), these drugs also had notable toxicity, with 20%–30% of the mice in these groups showing weight loss and other signs of severe illness prior to reaching tumor endpoints. Simvastatin and lovastatin did not have any appreciable effect on tumor growth when used alone, but these drugs both enhanced tumor growth delay and survival when used in combination with anti-PD-1 (vs anti-PD-1 alone; figure 3B–E). Lovastatin was particularly effective in combination with anti-PD-1, resulting in rejection of tumors in 30% of animals and significantly increased survival (figure 3D,E). Two of the three cured mice were rechallenged at 90 days with MOC1 in the left flank and failed to grow new tumors within 60 days, suggesting development of immunological memory. As expected, when CD8+T cells were antibody depleted, the effects of lovastatin/simvastatin plus anti-PD-1 were lost (figure 3B–D), suggesting a major role of CD8+T cells in mediating tumor regression. We repeated the experiment with simvastatin in the TC-1 model, which is more aggressive versus MOC1 in vivo. The effects of simvastatin+anti-PD-1 were minimal in TC-1 in vivo, with apparent responses in only one mouse in the simvastatin group and one mouse in the combination group (figure 4). Based on the overall effects of different statins on tumor-cell growth, enhancement of immune responses, and in vivo toxicity (table 1), we chose simvastatin and lovastatin for more in-depth studies.

Simvastatin and lovastatin enhance responses to PD-1 ICB in vivo. MOC1 cells were injected into the right flank, and animals were randomized on days 10–12 to treatment with simvastatin or lovastatin (60 mg/kg/day by oral gavage), anti-PD-1 (200 µg IP two times per week), statin+anti-PD-1, or statin+anti-PD-1 + anti-CD8 (200 µg IP two times per week) for 3 weeks. (A), Schema of experiment. (B, D) Tumor growth curves showing individual animals, compared with control. (C, E) Kaplan-Meier survival curves. Data are combined from two independent experiments, total n=5–10 per group as noted. *p<0.05, **p<0.01, ***p<0.001 versus control. ICB, immune checkpoint blockade; MOC1, mouse oral cancer;

Simvastatin minimally enhances response to PD-1 ICB in a small subset of TC-1-bearing mice in vivo. TC-1 cells were injected into the right flank, and animals were randomized on day 7 to treatment with simvastatin (60 mg/kg/day by oral gavage), anti-PD-1 (200 µg IP two times per week), statin+anti-PD-1, or statin+anti-PD-1 + anti-CD8 (200 µg IP two times per week) for 2 weeks. (A) Schema of experiment. (B) Tumor growth curves showing mean±SEM. (C) Tumor growth curves showing individual animals, compared with control. (D) Kaplan-Meier survival curves. n=5 animals per group. ICB, immune checkpoint blockade.
Statins enhance T cell activation but inhibit T cell proliferation at high doses
To explore possible mechanisms by which statins may enhance responses to ICB, a subset of MOC1 tumor-bearing mice were sacrificed during the second week of treatment, followed by harvest of tumors, spleens, and TDLN. As expected, PD-1 ICB increased the number of intratumoral CD8+T cells (figure 5A). There was also a trend toward increases in overall number of CD45+cells and CD4+T cells in mice receiving PD-1 ICB (online supplemental figure S4A and B). Importantly, the infiltration of CD8+T cells was much lower in the control group of the lovastatin experiment versus the control group of the simvastatin experiment (figure 5A), whereas the number of intratumoral myeloid derived suppressor cells (MDSCs) was much higher (online supplemental figure S4C–F). In general, MOC1 tumors tend to lose CD8+T cells and rapidly gain MDSCs as they grow, with variations in the speed of these changes from one experiment to another.33 Interestingly, it appeared that the combination of lovastatin with PD-1 ICB was able to reverse this pattern (online supplemental figure S4E,F). Cells expressing Tim-3 and NK1.1 were not altered by statins (online supplemental figure S4G,H), suggesting that statins do not have a major impact on T cell exhaustion or NK cell infiltration.

Simvastatin and lovastatin enhanced T cell activation within the tumor draining lymph node (TDLN). MOC1 tumor-bearing mice were treated with simvastatin/lovastatin and anti-PD-1 as in figure 3. Mice were sacrificed during the second week of treatment, then tumors and TDLN were harvested. (A) Density of intratumoral CD8+T cells was analyzed by flow cytometry. (B, C) TDLN from individual animals in the four treatment groups were mechanically digested and cultured overnight, then stimulated with PMA/ionomycin for 4 hours or CD3/CD28 Dynabeads for 24 hours without any inhibitors of secretion. (B) PMA/ionomycin-stimulated CD8+T cells were compared with their unstimulated counterparts for expression of surface CD107a. (C) Stimulated CD8+T cells were compared with their unstimulated counterparts for expression intracellular IFN-γ by flow cytometry. Data are mean±SEM, n=5, *p<0.05, **p<0.01, ***p<0.001, or indicated p value versus control. MOC1, mouse oral cancer.
There appeared to be a trend toward decreased T cell infiltration in tumors when ICB was combined with simvastatin, but not lovastatin (figure 5A), suggesting a possible detrimental effect of simvastatin on T cell infiltration and/or proliferation. It was unclear whether this difference was due to the specific drug, chosen dose, or starting number of T cells; however, decreased T cell proliferation in the presence of statin drugs has been reported.42 43 To explore this further, we performed T cell proliferation assays on human T cells from peripheral blood and splenocytes from untreated mice, treating the cells with T cell media in the presence or absence of simvastatin/lovastatin at a range of concentrations. We found that both simvastatin and lovastatin inhibited T cell proliferation at higher concentrations (online supplemental figure S5A,B). Although additional experiments are needed to determine how in vivo dosing impacts intratumoral T cell function, these ex vivo experiments do suggest that higher doses of statins may have some detrimental effects.
T cells obtained from draining lymph nodes were stimulated for 4 hours with PMA/ionomycin or for 24 hours with CD3/CD28 Dynabeads, followed by flow cytometry for surface CD107a and intracellular interferon-gamma (IFN-γ). CD107a expression was inconsistent after PD-1 ICB (possibly due to the very different CD8/MDSC dynamics between the two experiments as noted above) but consistently elevated in T cells from TDLN of animals treated with simvastatin/lovastatin alone (figure 5B). We also noted increased CD107a in human T cells treated with statins (online supplemental figure S5C). The CD8+T cells from TDLN of animals receiving combination therapy released more IFN-γ, as evidenced by decreased intracellular IFN-γ within the T cells on stimulation versus their unstimulated counterparts (figure 5C), suggesting that the T cells are more activated in the TDLN with combination therapy. Effects of statin or combination therapy on T cells in the spleen were inconsistent (online supplemental figure S6A,B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
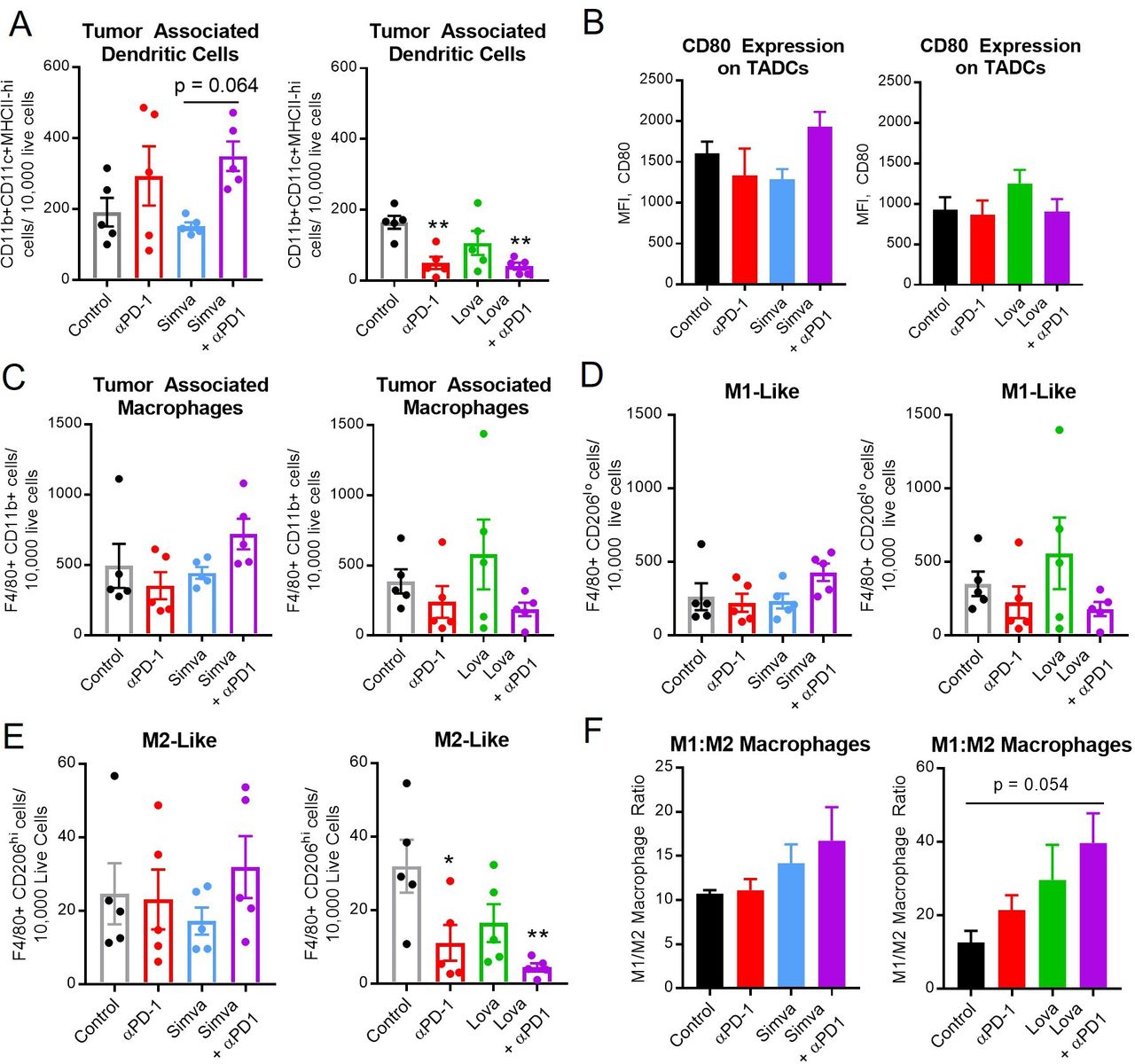
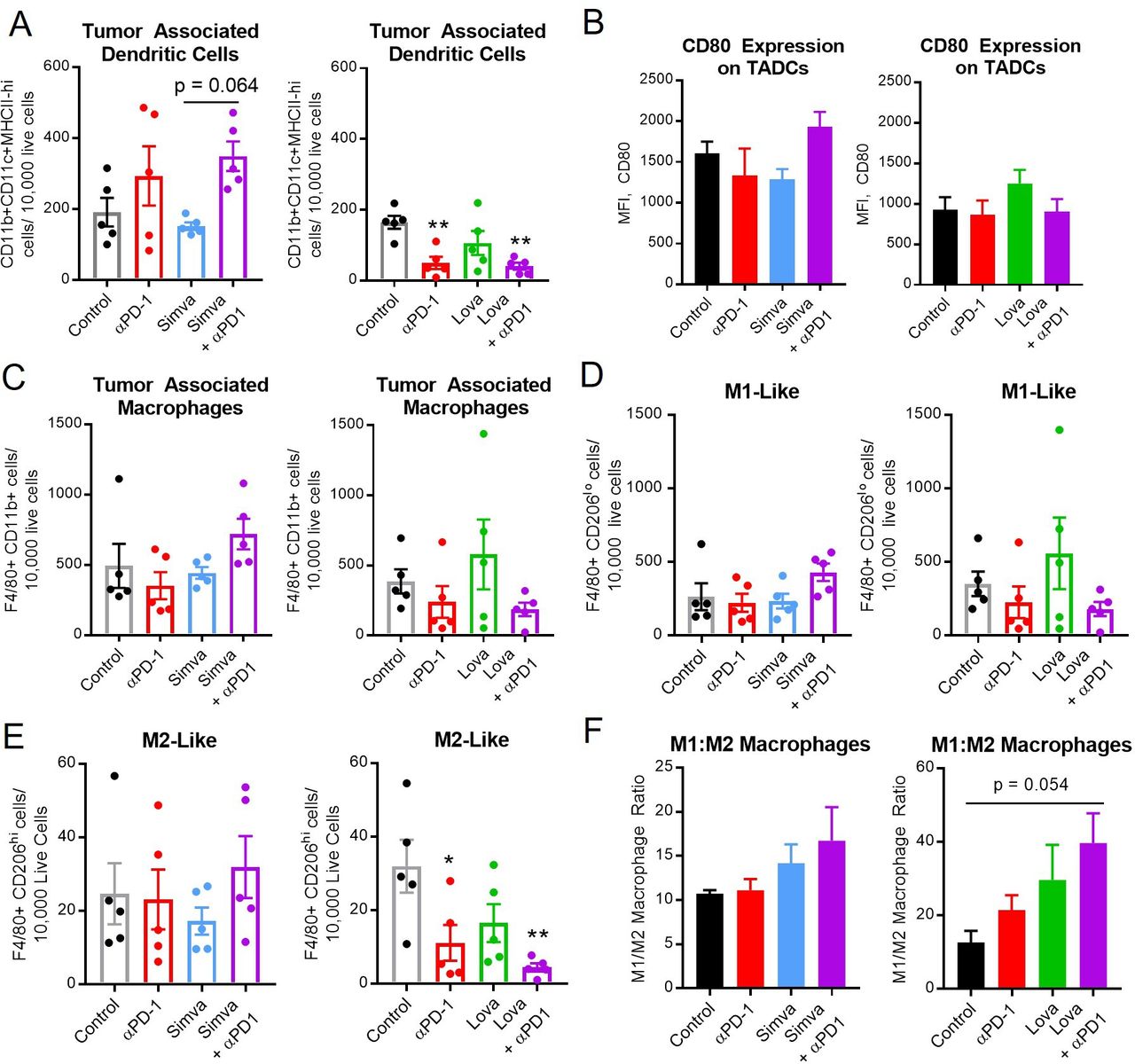
Simvastatin and lovastatin may alter the ratio of M1:M2 macrophages. MOC1 tumor-bearing mice were treated with simvastatin/lovastatin and anti-PD-1 as in figure 3. Mice were sacrificed during the second week of treatment, then tumors were harvested and analyzed by flow cytometry for myeloid cells, including dendritic cells (A, B, CD11b+CD11c+F4/80-), and macrophages (C–F, CD11b+F4/80+), including M1-like (D, F, CD206 low) and M2-like (E, F, CD206 high) macrophages. Data are mean±SEM, n=5, *p<0.05, **p<0.01, versus control or as indicated. MOC1, mouse oral cancer; TADCs, tumor-associated dendritic cells.
Impact of statins on the ratio of M1 to M2 macrophages
Given the apparent enhancement of T-cell activation by statins, we hypothesized that upstream antigen-presenting myeloid cells might also be activated. We next explored the density and phenotypes of dendritic cells and macrophages in the tumor and spleen. The effects of PD-1 ICB on overall numbers of dendritic cells (CD11b+CD11c+F4/80-) and macrophages (CD11b+F4/80+) in the tumor and spleen was inconsistent (figure 6, online supplemental figure S6C and D), likely related to the very different milieu noted among the control animals in the simvastatin versus lovastatin experiments. There was no obvious difference in the overall number of macrophages or M1-like macrophages. However, lovastatin+PD-1 ICB significantly decreased the number of intratumoral M2 macrophages (figure 6E). In both experiments we noted a potential shift in the ratio of ‘M1-like’ (CD206 low) to ‘M2-like’ (CD206 high) macrophages (figure 6F), although this did not reach statistical significance. In summary, no strong or consistent patterns were seen in the myeloid compartment, suggesting that myeloid cells play a less significant role versus T cells in the antitumor immune response to PD-1 ICB combined with statins.
Discussion
In this study, we show that all seven commercially available statins mediate antitumor activity against murine cell lines, but a subset of statins also enhance cytotoxic T cell responses to PD-1 checkpoint blockade. We divulge concentration-dependent effects on T cells, with enhanced activation at moderate concentrations, but reduced in vitro proliferation at higher concentrations. Importantly, our in vivo studies are the first to be designed to mimic the clinical situation wherein a cancer patient is taking a statin for hyperlipidemia, with daily oral dosing of high but tolerable doses of statin drugs. Statins block the breakdown of HMGCoA into mevalonate and downstream metabolites, including cholesterol and other metabolites important for protein prenylation.24 Rapidly dividing cancer cells are dependent on these metabolites and may be effectively ‘starved’ in the presence of statin drugs.44 Not surprisingly, statins have activity against a variety of cancer types.5 In addition, a growing body of studies suggests that manipulation of cholesterol, either with statins, PCSK9 inhibitors, or other means, has important implications for antitumor immunity.6 15–17 21–24
Several recent studies have focused on immune effects of statin drugs and/or shown enhanced responses to immunotherapy with statins, including studies in preclinical models of HNSCC. Kwon et al recently showed that simvastatin enhanced responses to PD-1 ICB combined with cisplatin chemotherapy in another murine model of HNSCC.17 The authors noted increased density and proliferation of CD8+T cells in the tumor microenvironment after treatment with simvastatin alone, in contrast to our study; however, simvastatin was given by IP injection at a sixfold lower dose. Whether the divergent data from that study and the present work are related to differences in mouse models, dosing, or route of administration are unclear. We suspect that our in vivo dose of simvastatin may have been too high for optimal T cell proliferation, though we did not observe this effect with the same dose of lovastatin. The statin doses in our in vitro experiments were carefully chosen to mimic serum concentrations achieved after oral dosing of statins in humans. It is unclear whether the doses we selected for in vivo murine studies accurately reflect oral human doses, since few pharmacokinetic studies have been published in mice. Further, it has been shown that higher serum concentrations are needed in mice to achieve lowering of serum cholesterol.26 45 46 However, oral simvastatin/lovastatin doses of 60–100 mg/kg per day are considered relatively high doses in mice and have been shown to produce pleiotropic effects on tissues outside of the liver and serum.31 45 46 Further in vivo studies with a range of statin doses are needed to determine whether dose has a major impact on T-cell infiltration into the tumor microenvironment.
Preclinical studies have shown enhanced immune responses against several different tumor types by statins. In another study with the same HPV-positive (TC-1) model used in our study, intramuscular (IM) injection of simvastatin along with E7 vaccination dramatically improved tumor control, particularly when PD-1 ICB was also added.24 In colorectal cancer, in vivo administration of statins elicited antitumor immune activity specifically in KRAS-mutant models.21 Immune activity has also been demonstrated in models of melanoma and lung cancer.20 23 24 Most of these studies have shown positive results with simvastatin, lovastatin, and atorvastatin,17 20–23 suggesting that lipophilic statins have the most activity, consistent with our results. Routes of administration in these preclinical studies have included IP injection, IM injection, and intratumoral injection,17 20 23 and doses used for in vitro studies are often in the supraphysiologic, micromolar range. As a result, the clinical implications of such studies are unclear.
Putative mechanisms of statin-induced immune enhancement suggested by prior studies include elevated MHC class I on tumor cells, decreased T cell exhaustion markers, immunogenic cell death, and increased intratumoral CD8+T cells.17 21 22 We did not see any obvious evidence of these putative mechanisms in the present study (online supplemental figure S4, S7) with daily oral administration of simvastatin or lovastatin in preclinical models of HNSCC.
Statins may enhance antitumor immunity in much the same way as chemotherapy, radiation, and other cytotoxic modalities: mevalonate starvation results in dying cells, releasing antigens to dendritic cells, thereby using the tumor as an in situ vaccine and activating T cells in the TDLN.47 Statins may also increase tumor uptake of anti-PD-1 antibodies, since these drugs can increase uptake of other antibodies such as cetuximab and panitumumab.48 However, our experiments suggest that simvastatin and lovastatin directly bolster T cell functions. We speculate that optimally dosed, lipophilic statins enhance activation of T cells by mechanisms that are not yet entirely understood. Above the optimal dose of statins, T cells, which also require mevalonate and its metabolites, may struggle to proliferate. We also noted a shift in tumor-associated macrophages toward a more M1-like phenotype, suggesting that myeloid cells may also play a role; however, those results were less consistent and require further study.
Our results have important clinical implications. The responses to PD-1 ICB alone in HNSCC are infrequent, and there is an unmet need for agents that can be given in combination with these expensive drugs to increase the proportion of patients who will benefit. Statins are well tolerated, are inexpensive, and may have other benefits for head and neck cancer patients including prevention of cisplatin-induced hearing loss13 and radiation fibrosis.14 A retrospective study including patients with non-small cell lung cancer (NSCLC), renal cell carcinoma, and melanoma showed a 1.6 odds ratio f response to checkpoint blockade in patients taking statins, though the effects of specific statins were not detailed.16 The optimal drug and dose of that drug to use in the clinical setting is unclear, as responses to statins in humans may differ from those in mice. Although simvastatin was the drug we found most in preclinical studies of statin-induced immunity, a retrospective study showed increased responses to checkpoint blockade in NSCLC and mesothelioma patients taking rosuvastatin and atorvastatin, but not simvastatin.15 Atorvastatin and rosuvastatin are high-intensity statins, suggesting that lipid lowering may be critically important. Additional retrospective studies and prospective, randomized clinical trials would help to address these questions.
Our study has several limitations, including the small number of animals in flow cytometry experiments, limiting our ability to detect subtle effects on the tumor immune microenvironment. The decrease of intratumoral T cells seen in mice treated with simvastatin might suggest that our in vivo dose was too high for optimal T cell function, and the in vivo dose–response relationship of simvastatin and lovastatin combined with checkpoint blockade deserves further study.
In conclusion, daily oral administration of statin drugs enhances responses to PD-1 ICB in preclinical models of HNSCC, with apparent direct effects on tumor cells as well as direct and indirect effects on T cell function. These data suggest that further preclinical and clinical study is warranted to determine whether oral statins can be quickly adopted as a well-tolerated, inexpensive way of expanding the proportion of patients with recurrent or metastatic head and neck cancer who may benefit from currently available forms of immunotherapy.
Data availability statement
Data are available on reasonable request. Raw data are available on reasonable request with data transfer agreement.
Ethics statements
Patient consent for publication
Ethics approval
All animal procedures were approved by the Institutional Animal Care and Use Committee at Emory University (Protocol #202100008). Blood from human head and neck cancer patients was obtained under a protocol approved by the Institutional Review Board at Emory University (Study #00002286) with written informed consent from each patient prior to participation and blood collection.
Acknowledgments
This study was supported by Winship Cancer Institute, the Department of Otolaryngology at Emory University School of Medicine, and the Morningside Center for Innovative and Affordable Medicine (through the generous support of donors and Emory University). This work was supported in part by the Pediatrics/Winship Flow Cytometry Core of the Winship Cancer Institute of Emory University, Children’s Healthcare of Atlanta, and NIH/NCI under award number P30CA138292. The content is solely the responsibility of the authors.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @Kansal_Vikash, @DrNabilSaba, @PaulosLab, @LesinskiLab, @NikkiSchmittMD
Correction notice This article has been corrected since it was first published. The middle initial has been added to author Nabil F Saba.
Contributors VK, AJB, BLCK and NFS performed the experiments, collected and analyzed the data. VK and NCS drafted and revised the manuscript. NFS, CP, GBL and ZSB provided feedback on preliminary data, suggested some of the experiments, and revised the manuscript. NCS conceived and supervised the study. NCS accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Funding This study was supported by Winship Cancer Institute, the Department of Otolaryngology at Emory University School of Medicine, and the Morningside Center for Innovative and Affordable Medicine (through the generous support of donors and Emory University).
Competing interests NCS: Consulting: Checkpoint Surgical, Sensorion Book Royalties: Plural Publishing Clinical Trial Funding: Astex Pharmaceuticals CP—Research funding through a sponsored research agreement between the Medical University of South Carolina and Obsidian, Lycera, ThermoFisher and is the Co-Founder of Ares Immunotherapy GBL—Grant/Research Support through sponsored research agreements between Emory University and from Merck and Co., Bristol-Myers Squibb, Boerhinger-Ingelheim and Vaccinex and a consulting or advisory role for ProDa Biotech.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.