Article Text

Abstract
Background Immunological contexture differs across malignancies, and understanding it in the tumor microenvironment (TME) is essential for development of new anticancer agents in order to achieve synergistic effects with anti-programmed cell death protein-1 (PD-1) therapy. TYRO3, AXL, and MERTK receptors are bi-expressed in both cancer and immune cells, and thus emerge as promising targets for therapeutic intervention. Whereas AXL and MERTK have been extensively studied, the role of TYRO3, in the TME, is still undetermined.
Methods Here, we screened the TYRO3-focused chemical library consisting of 208 compounds and presented a potent and highly selective TYRO3 inhibitor, KRCT87. We explored the role of TYRO3 using mouse engrafting MC38 or 4T1 tumors. We validated the results using flow cytometry, RNA sequencing analysis, gene knockdown or overexpression, ex vivo immune cells isolation from mouse models, immunoblotting and quantitative PCR. Flow cytometry was used for the quantification of cell populations and immunophenotyping of macrophages and T cells. Co-cultures of macrophages and T cells were performed to verify the role of CCN1 in the tumors.
Results TYRO3 blockade boosts antitumor immune responses in both the tumor-draining lymph nodes and tumors in MC38-syngeneic mice models. Moreover, the combination of KRCT87 and anti-PD-1 therapy exerts significant synergistic antitumor effects in anti-PD-1-non-responsive 4T1-syngeneic model. Mechanistically, we demonstrated that inhibition of TYRO3-driven CCN1 secretion fosters macrophages into M1-skewing phenotypes, thereby triggering antitumor T-cell responses. CCN1 overexpression in MC38 tumors diminishes responsiveness to anti-PD-1 therapy.
Conclusions The activated TYRO3-CCN1 axis in cancer could dampen anti-PD-1 therapy responses. These findings highlight the potential of TYRO3 blockade to improve the clinical outcomes of anti-PD-1 therapy.
- tumor microenvironment
- macrophages
- CD4-CD8 ratio
- drug evaluation, preclinical
- immunotherapy
Data availability statement
Data are available in a public, open access repository. Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
TYRO3 receptor, which is bi-expressed in both cancer cells and immune cells, is a promising target in cancer. Despite this emerging role of TYRO3 in cancer, the understanding of TYRO3 in the tumor microenvironment (TME) has not been elucidated yet.
CCN1, a matricellular protein, is closely related to cancer progression including cancer cell proliferation, metastasis, and poor survival outcomes.
WHAT THIS STUDY ADDS
This study first presents the potent selective TYRO3 inhibitor, KRCT87, and its clinical potential in combination use with anti-programmed cell death protein-1 (PD-1) therapy.
The crucial relationship between the TYRO3-CCN1 axis and tumor-associated macrophages is identified. Blocking this axis reshapes the TME, thereby increasing the responsiveness of anti-PD-1 therapy.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The role of CCN1, which has been mainly studied in cancer cells, could be extended to the regulation of the TME.
Considering the abundance of TYRO3 ligands and CCN1 in the TME, new therapeutic agents targeting the TYRO3-CCN1 axis could be applicable for immune checkpoint blockade-refractory patients as a potentially successful anticancer strategy.
Introduction
Immune checkpoint blockade (ICB) effectively suppresses cancer malignancy and promotes tumor remission, showing promising therapeutic outcomes for diverse cancer types. Nonetheless, the response rate to ICB across cancer types is only around 30%, and so the clinical key challenge is to enable ICB’s full potential to be reached.1 Tumor microenvironment (TME) differs across malignancies and various cell types consisting of the TME could determine ICB responsiveness. Considering that communication between immune cells and cancer cells shape the immunological contexture, understanding of this interaction is pivotal in anticancer research.2 The well-studied innate immune cell subsets in the TME include myeloid-derived suppressor cells (MDSC), tumor-associated macrophages (TAMs), dendritic cells (DC), natural killer (NK) cells, and neutrophils.3 Sensing of tumor-derived factors by these innate immune cells modulates T-cell infiltration and their cytotoxic function, which are important prognostic markers for ICB responsiveness.2 4 In particular, TAMs are the most abundant population in immune cells across multiple human cancer types.5 TAMs usually exist in the TME as an M2-skewing type that fuels cancer progression and hampers tumor immunity. High infiltration of TAMs in the TME is associated with resistance to anti-programmed cell death protein-1 (PD-1) therapy and bad prognoses.6
Overexpression of the TYRO3, AXL, and MERTK family of receptor tyrosine kinases (TAM RTKs) has been detected in a variety of cancer types.7 In recent years, there has been increasing interest in TAM RTKs due to their bi-expression in both immune and cancer cells. TAM RTKs possess dual critical roles in tumors. The first is facilitating chemoresistance and metastasis in cancer cells, and the second is stimulating immune tolerance via efferocytosis in innate immune cells.7 8 The roles of both AXL (frequently overexpressed in cancer cells)9 and MERTK (expressed in various immune cells)10 11 have been extensively studied among the three TAM RTKs. However, much less is known about the function of TYRO3 in both cancer and immune cells. We have recently demonstrated that circulating small extracellular vesicle (csEV)-activated TYRO3 in cancer cells drives cancer cell survival and metastasis under the growth factor-restricted condition.12 TYRO3 has been posited as a key receptor for acquisition of resistance to anti-PD-1 therapy, specifically by limiting ferroptosis of cancer cells.13 Nonetheless, comprehensive understanding of the expression pattern and function of TYRO3 in the TME is still lacking.
We and others have demonstrated that TAM RTKs are coupled with Hippo pathway-dependent yes-associated protein (YAP) activation.12 14 YAP is a transcriptional activator and a major contributor to cancer progression in terms of proliferation, metastasis, and chemoresistance within the TME.15 Moreover, YAP activity is critical to the regulation of the tumor immune surveillance system, as it involves recruitment of MDSC,16 regulatory T cells (Treg)-dependent suppression of tumor immunity,17 and downregulation of CD8+ T cell-mediated anticancer effects.18 However, the mechanism underlying YAP’s or YAP-related genes’ regulation of TAMs characteristics has not been elucidated. Specifically, the CCN family (CCN1 (CYR61), CCN2 (CTGF)) is the representative transcriptional target genes of YAP. YAP-dependent production of CCN1, a secreted extracellular matrix protein, contributes to tumor remodeling.19 20 CCN1 not only regulates the adhesion or migration process of immune cells but also mediates the production of various chemokines and cytokines.21 Although these complex functions of CCN1 are expected to play pivotal roles in the control of immune surveillance, few studies have explored the role of CCN1 in the regulation of immune cell function in the TME.
Here, we explored the role of TYRO3 in the TME using syngeneic tumor models. We documented the characterization of KRCT87, a highly potent and selective TYRO3 inhibitor. Administration of KRCT87 significantly elevated the overall antitumor immune responses and enhanced the responsiveness to anti-PD-1 therapy in syngeneic mouse models. Mechanistically, we demonstrated that TYRO3-dependent CCN1 secretion evokes tumor-immunosuppressive reactions via TAMs. CCN1-overexpressing MC38 tumors showed less responsiveness to anti-PD-1 therapy as compared with control tumors. The findings of this study highlight the fact that TYRO3-driven changes in the tumor immune structure mediate anti-PD-1 therapy responsiveness.
Methods
Detailed methods are listed in online supplemental information.
Supplemental material
Mouse experiments
All animal procedures were approved by the Institutional Animal Care and Use Committee of Seoul National University (SNU IACUC) (Approval #: SNU-200218-3-1, SNU-200827-2-3, SNU-210513-6-2, SNU-210524–1, SNU-211201–4, SNU-211210–4).
Results
Expression of TYRO3 in TME
We have previously reported that TYRO3 is upregulated in metastatic or chemo-resistant cancer cells, which sustains cancer cell survival and promotes aggressiveness as well.12 To assess the expression pattern of TYRO3 in the TME, we compared the expression of TYRO3 in various cell types isolated from MC38-derived tumor tissues (figure 1A), adhering to the gating strategy as shown in online supplemental figure S1A. Of note, the frequency of TYRO3 was remarkably higher in the cells isolated from the MC38 tumor tissues than in the corresponding mice spleen cells (figure 1B). The TYRO3high population was predominantly found in CD45−CD90− double-negative cells (enriched for cancer cells), and the second subpopulation was CD45+ cells (enriched for immune cells) averaging approximately 10% (figure 1C). Next, we analyzed immune cell subsets including macrophages (MQ), DC, neutrophils, monocytes, B cells, and T cells. The TYRO3highCD45high population was mainly occupied by MQ in tumors (33.2%) (figure 1D). We analyzed the frequency of TYRO3 positivity in each cell subset isolated from the MC38 tumors and corresponding spleens. The frequency of TYRO3 in each cell is higher in MQ (22%) and CD45−CD90− population (43%) relative to other cell types in the tumor, which illustrates the potential importance of TYRO3 expression in TAMs. On the other hand, DC has the highest proportion of TYRO3 in the spleen (figure 1E). These overall results indicate that TYRO3 may be more actively involved in the TME than under normal physiological conditions and that cancer cell-rich population and TAMs exhibit highly upregulated TYRO3 expression.

Expression of TYRO3 in MC38 tumor microenvironment. (A) Schematic illustration of in vivo sample collection in MC38 tumor-bearing mouse. On day 0, C57BL/6 mice were challenged subcutaneously with 1×105 MC38 cancer cells, and tumors were analyzed on day 17 (n=4 mice). (B) Frequencies of TYRO3+ cells in the MC38 tumors and the corresponding spleens (left) and representative FACS plots of them (right). (C) The percentage of CD45+ and CD45−CD90− cells in TYRO3+ population in MC38 tumors (left) and representative FACS plots of them (right). (D) Flow-cytometric analysis showing percentage of B cells, CD4+ T cells, CD8+ T cells, DC (Ly6C−CD11c+MHC-II+), MQ (Ly6C−CD11b+F4/80+), monocytes (CD11b+Ly6Chigh) and neutrophils (CD11b+Ly6Cmed) among CD45+TYRO3+ cells isolated from MC38 tumor. (E) Frequencies of TYRO3+ cells in each cell subset in MC38 tumors (up) and representative FACS plots of them (down). All data represent the mean±SD. The statistical significance of the differences in E was determined by one-way analysis of variance followed by Tukey post hoc test and that in B and C was determined by an unpaired two-tailed Student’s t-test. **, p<0.01, ***, p<0.005, ****, p<0.001 significant as compared with the indicated group. CD4+ T, CD4+ T cells; CD8+ T, CD8+ T cells; DC, dendritic cells; MQ, macrophages.
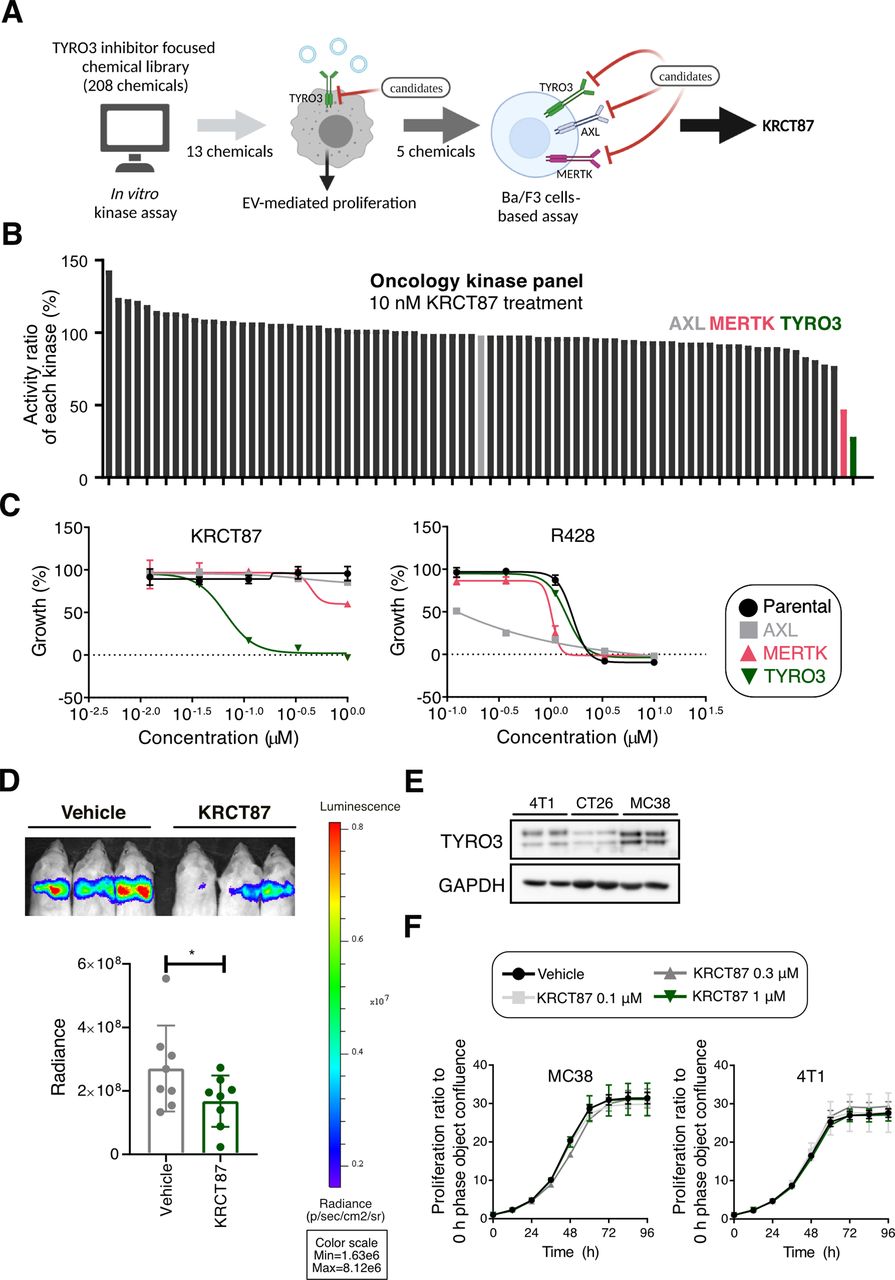
Identification of KRCT87, a potent and selective TYRO3 inhibitor
Even though the function of TYRO3 in cancer progression has been elucidated in several types of malignancies, there are still no highly selective TYRO3 small molecule inhibitors (SMI) for targeting of cancers. Most TYRO3 SMI developed so far only non-selectively inhibit the activity of other TAM family kinases in the effective concentration ranges.22 To develop TYRO3 SMI, a TYRO3-focused chemical library consisting of 208 compounds23 24 was initially screened to find highly selective TYRO3 inhibitors. Through a stepwise selection procedure, we determined that KRCT87 is a potent and highly selective TYRO3 inhibitor (figure 2A). We first selected 13 chemicals displaying at least 10-fold selectivity for TYRO3 over both MERTK and AXL with IC50 values from an in vitro kinase assay (online supplemental table S1). Since csEV promotes LNCaP-SL cell proliferation via TYRO3 receptor under the serum-deprivation condition,12 we further evaluated the inhibitory effect of candidate compounds on csEV-induced proliferation. Among them, five chemicals including KRCT31, KRCT62, KRCT77, KRCT87, and KRCT203 showed superior inhibition effects on LNCaP-SL cells (online supplemental figure S2A). Moreover, in oncology-focused protein kinase panel assays composed of 79 kinases, 10 nM KRCT87 reduced the activities of TYRO3 to 28% and MERTK to 47% (figure 2B and online supplemental table S2). We then tested the effects of five candidate chemicals and R428 (a representative AXL selective inhibitor as a reference compound25 using Ba/F3 cell-based tyrosine kinase inhibition assays (online supplemental table S3). Of note, KRCT87 was the most potent and selective TYRO3 inhibitor, showing a TYRO3 growth inhibition 50 (GI50) value of 64.1 nM (an estimated MERTK GI50 value=9.05 µM, AXL GI50 value >1 µM) (figure 2C). In addition, it was determined that KRCT203 could be a dual inhibitor concomitantly acting on TYRO3 and MERTK (TYRO3 GI50=137.1 nM, MERTK GI50=152.4 nM) (online supplemental figure S2B). As next step, we investigated the inhibitory function of TYRO3 in apoptotic cell uptake by MQ because TYRO3 is also involved in apoptotic cell clearance although MERTK could play a more prominent role in efferocytosis.26 27 To investigate the inhibitory function of KRCT87 in apoptotic cell uptake by MQ, we employed pHrodo-labeled apoptotic thymocytes as the target cells and mouse bone-marrow-derived macrophages (BMDM) as the phagocytes. A pH-sensitive dye called pHrodo can be used to detect phagocytic processes due to the increasing fluorescence intensity under acidic conditions. Treatment with KRCT87 or UNC2250 (a selective MERTK inhibitor28) significantly decreased the ability of phagocytosis in BMDM (online supplemental figure S2C).

Identification of KRCT87, a potent and selective TYRO3 inhibitor. (A) Schematic representation of stepwise TYRO3 inhibitor selection procedure following three rounds of screening TYRO3-focused library consisting of 208 compounds. (B) Effect of KRCT87 on the activity ratio of oncology kinase panels composed of 78 kinases (tested at 10 nM). (C) Growth inhibition by R428 and KRCT87 in parental, MERTK, AXL, or TYRO3-overexpressing Ba/F3 cells. (D) Representative bioluminescent images showing the effect of KRCT87 in 4T1-luc engrafting Balb/c mice (up) and quantitative evaluation of metastasis (down). Region of interest (ROI) was defined and the total luciferin signal in ROIs was calculated as photons/second/cm2 (total flux/area) (n=8 mice). (E) Expression of TYRO3 in 4T1, CT26 and MC38 cells. (F) Effect of KRCT87 on the cell proliferation of MC38 and 4T1 cells under 10% FBS containing medium (10% FBS) (n=3). All data represent the mean±SD. The statistical significance of the differences in D was determined by an unpaired one-tailed Student’s t-test. *, p<0.05; significant as compared with the vehicle-treated group. FBS, fetal bovine serum.
We have previously demonstrated that TYOR3 marginally affects the proliferation of human cancer cells, but is critical to metastasis and cancer cell survival.12 As expected, KRCT87 significantly inhibited lung metastasis in Balb/c mice injected with 4T1-luciferase (luc) cells into the tail vein (figure 2D). Since TYRO3 expression is higher in both 4T1 (mouse mammary cancer) and MC38 (mouse colon cancer) cells relative to CT26 (mouse colon carcinoma) cells (figure 2E), we further evaluated proliferation of MC38 cells and 4T1 cells in the presence of KRCT87. KRCT87 did not affect the cell proliferation under the 10% fetal bovine serum condition, as is consistent with our previous study12 (figure 2F).
TYRO3 blockade transforms TAMs into efficient antigen-presenting cells
As a first step in investigating the effect of KRCT87 in MQ in vitro, we compared the expression of TAM RTKs in BMDM and bone marrow-derived dendritic cells (BMDC). The messenger RNA (mRNA) level of Mertk was higher in BMDM than in BMDC, whereas that of Tyro3 was relatively higher in BMDC than in BMDM (online supplemental figure S3A), as is consistent with a previous report.29 An immunoblot result showed an additional ~110 kDa TYRO3 band in the cancer cell lines, presumably due to the formation of various glycosylation forms (theoretical molecular weight: ~ 96 kDa) (online supplemental figure S3B).30 Since M1/M2 polarization status in MQ generally reflects the antitumor versus protumor activity of MQ,31 we treated KRCT87 to BMDM under the M2-skewing condition (20 ng/mL interleukin-4) in order to estimate the polarization of MQ. Treatment with KRCT87 significantly downregulated the mRNA levels of Arg1 and Ciita, representative M2 markers (online supplemental figure S3C). Moreover, Arg1 expression upregulated by MC38 cancer cell-conditioned medium (CCM) in BMDM was suppressed by KRCT87 in turn, indicating that TYRO3 inhibition suppresses cancer-cell-mediated M2 skewing in MQ (online supplemental figure S3D).
We determined the plasma concentration of KRCT87 in mice intraperitoneally injected with 5 mg/kg KRCT87 in a time-dependent manner (online supplemental figure S3E). The peak Cmax is 4.8 µM and the plasma concentration of KRCT87 rapidly dropped to 0.86 µM within 4 hours with a half-life 189 min, confirming that 5 mg/kg dosage is in the range of selectively inhibiting TYRO3. We next performed an immune-phenotyping analysis on MC38 tumor-infiltrating leukocytes in vivo. Syngeneic mice with established MC38 tumors were intraperitoneally injected with vehicle (5% isotonic glucose) or KRCT87 (5 mg/kg/day) for 9 days (figure 3A). The tumor volume was slightly but not significantly diminished by the administration of KRCT87 in the MC38-syngeneic mice (figure 3B). However, the absolute number of CD45high-infiltrating cells was significantly increased in the KRCT87-treated group relative to the vehicle-treated group (figure 3C). In addition, a flow-cytometric analysis confirmed that KRCT87 elevated the intensity of major histocompatibility complex class II (MHC-II, an M1 marker) in TAMs (gated as F4/80+CD11b+Ly6G−Ly6C−) (figure 3D). Notably, the ratio of CD8+ T cells to Foxp3+ Treg cells was higher in KRCT87-treated tumors than in control tumors (figure 3E). We further assessed the possible DC involvement in TYRO3-mediated immune suppression but no increase in MHC-II mean fluorescence intensity was detected in both CD11b+ DC and CD103+ DC (online supplemental figure S3F).

Effect of KRCT87 on tumor infiltrating leukocytes in MC38 tumors. (A) Schematic illustration of the treatment schedule and the leukocytes collection in MC38 tumor engrafting mice. Data are representative of two independent experiments (n=7 mice). (B) Assessing the growth of MC38 tumors in mice treated with KRCT87 or vehicle. (C) Absolute counts of CD45+ cells normalized by tumor weight in MC38 tumor-bearing mice treated with KRCT87 or vehicle. (D) Flow-cytometric analysis of MHC-II expression in TAMs in MC38 tumors (left) and the gating strategy of TAMs (right). (E) The ratio of CD8+ T cells to Foxp3+ Treg cells in MC38 tumors after treatment with vehicle or KRCT87. (F) Schematic illustration of transcriptomic analysis of F4/80+CD11b+ cells collected from MC38 tumors. For each group, tumors from three mice were pooled (n=9 mice). (G) Heat map of the one-way hierarchical clustering using z-score for normalized value (log2 based). (H) The top four upregulated Hallmark GSEA enriched in KRCT87-treated group versus vehicle-treated group using data from G. (I) GSEA of TNF-α signaling (left) and Inflammatory response (right) using data from G. All data represent the mean±SD. All statistical significance of the differences was determined by an unpaired two-tailed Student’s t-test. *, p<0.05; significant as compared with the vehicle-treated group. GSEA, gene set enrichment analysis; IL, interleukin; MFI, mean fluorescence intensity; MHC-II, major histocompatibility complex class II; TAMs, tumor-associated macrophages; TNF, tumor necrosis factor; Treg, regulatory T cells.
Since TYRO3 is highly expressed in MQ in the TME compared with other immune cells (figure 1E), we sought to determine whether KRCT87 changes the characteristics of TAMs. We first pooled tumors isolated from three mice each (total: nine mice per group) after 8 days of treatment of KRCT87, and isolated F4/80+CD11b+ MQ by flow-sorting (figure 3F). After profiling the transcriptomes of TAMs, a one-way hierarchical clustering analysis of significant differentially expressed genes (DEG) showed that the KRCT87-treated group was classified as a similar hierarchy and displayed a prominent change in DEG relative to the vehicle group (figure 3G). To identify distinctive transcriptional programs activated by KRCT87, a gene set enrichment analysis (GSEA) was performed using 50 hallmark gene sets. Noticeably, the top four sets enriched in the KRCT87-treated group were linked to M1-skewing MQ phenotypes (figure 3H). The GSEA revealed prominent enrichment of tumor necrosis factor α (TNF-α) signaling (normalized enrichment score (NES)=3.04) and inflammatory response (NES=2.58) in the transcriptomes from mice administered with KRCT87 (figure 3I). These results together demonstrate that KRCT87 treatment resulted in an M1-rich tumor environment in vivo.
TYRO3 blockade enhances antitumor T-cell responses
Blocking TAM RTK can also boost T-cell-dependent antitumor responses.11 32 We performed immune-phenotyping of tumor-infiltrating lymphocytes (TILs) from days 3 and 9 after treatment of KRCT87 in MC38-syngeneic mice (figure 4A). T-cell priming begins in the tumor-draining lymph nodes (TdLNs) for the progress of antitumoral immunity.33 From 3 days after KRCT87 administration, the weight of TdLN and the absolute numbers of CD45+ cells, CD4+ and CD8+ T cells accumulated in TdLN were significantly enhanced (figure 4B). These results show that initial exposure to KRCT87 makes for a TdLN antitumorigenic environment in which immune cells are abundant.

TYRO3 blockade enhances antitumor T-cell responses. (A) Schematic overview of immune profiling during and after KRCT87 treatment. On day-11, 24 C57BL/6N mice were challenged subcutaneously with 1×105 MC38 cells (n=6 mice) and administered intraperitoneally with KRCT87 (5 mg/kg) or vehicle daily. Tumors were analyzed on days 3 and 9 after the first injection of KRCT87 (black arrows). (B) Analysis of TdLN weight, and absolute counts of CD45+ cells, CD8+ cells and CD4+ cells in TdLNs on day 3. (C) Frequencies of IFN-γ or TNF-α in CD4+ cells and CD8+ cells in MC38 tumors on days 3 or 9 after first injection. (D) Representative FACS plots (left) and bar graphs (right) showing the average percentage of CD4+ and CD8+ T cells that express both IFN-γ or TNF-α in MC38 tumors on day 9. All data represent the mean±SD. All statistical significance of the differences was determined by an unpaired two-tailed Student’s t-test. *, p<0.05; **, p<0.01; ****, p<0.001 significant as compared with the vehicle-treated group. IFN, interferon; TdLNs, tumor-draining lymph nodes; TNF, tumor necrosis factor.
Since TIL-producing interferon (IFN)-γ and TNF-α play a pivotal role in tumor regression,34 we next investigated the effect of KRCT87 on the production of IFN-γ and TNF-α in TILs. Production of these cytokines in CD4+ and CD8+ T cells was comparable between the vehicle-treated and KRCT87-treated group on day 3 after administration of KRCT87 (figure 4C). However, KRCT87 treatment led to a remarkable intratumoral accumulation of IFN-γ+ and/or TNF-α+ in CD4+ and CD8+ T cells at day 9 (figure 4C–D). In addition, we observed that KRCT87 treatment polarized MQ toward an M1-like phenotype, as characterized by increased frequency of CD86 and decreased frequency of CD206 in TAMs (online supplemental figure S4A). Together, these results suggest that blocking of TYRO3 in tumors improves antitumor immune responses.
KRCT87 restores anti-PD-1 responsiveness in 4T1 tumor
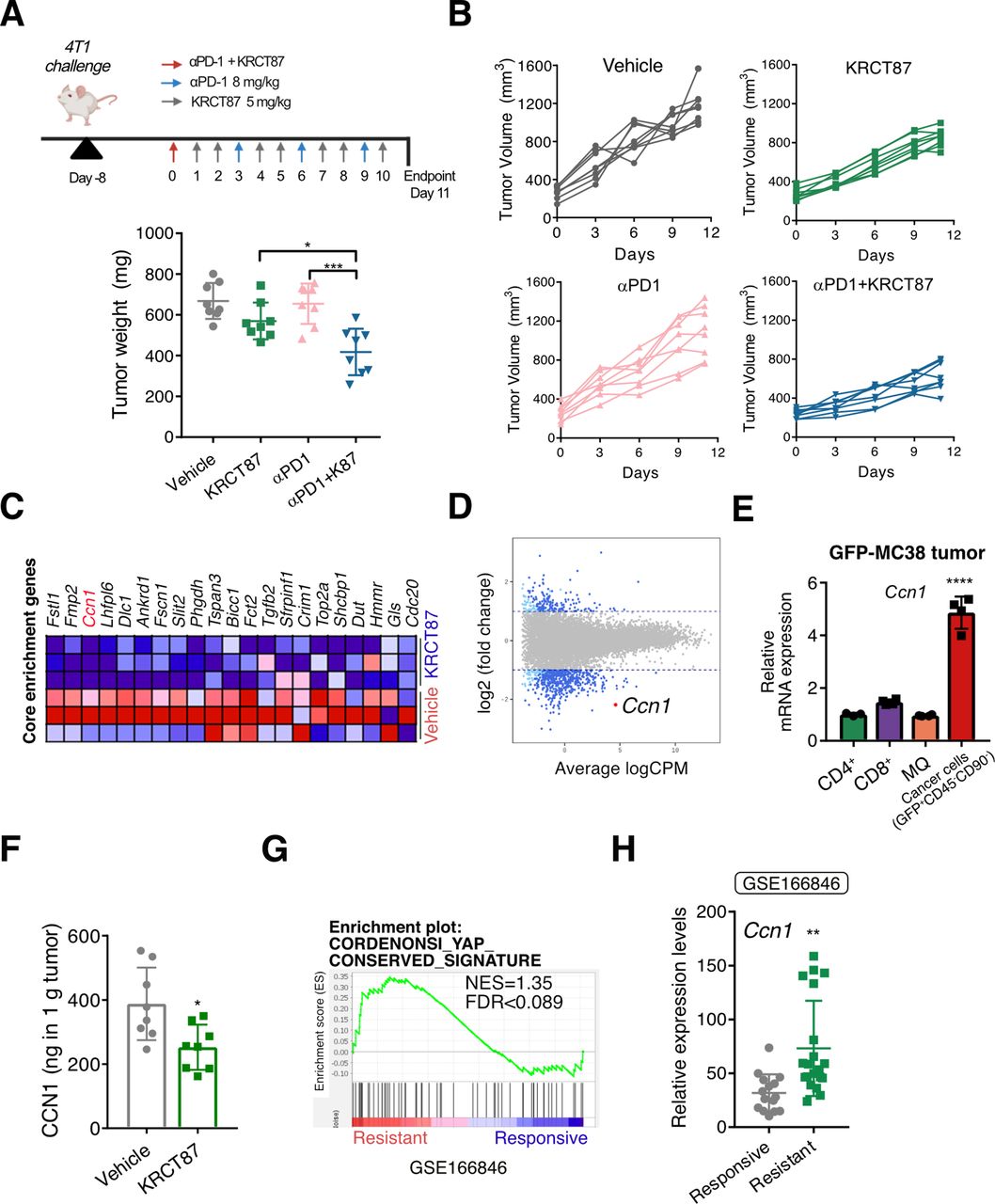
We then explored if KRCT87 improves the efficacy of anti-PD-1 therapy in a syngeneic tumor model. We selected 4T1 breast cancer cells as a model cell line, because 4T1-syngeneic tumors exhibit intrinsic resistance to anti-PD-1 therapy.35 To accurately reflect clinical circumstances, drug administration was initiated when the average tumor volume had reached 260 mm3. Notably, co-administration of KRCT87 and anti-PD-1 showed a significant and synergistic antitumor effect compared with each monotherapy in the 4T1-syngeneic tumor model (figure 5A). We pooled tumors isolated from two mice each (total: eight mice per group) and analyzed the tumor-infiltrating cells (online supplemental figure S5A–E). Treatment with KRCT87 increased the frequency of CD45+ cells, the ratio of CD8+ T cells to Foxp3+ Treg cells in CD45+ cells, frequencies of PD-1+CD4+ T cells and PD-1+CD8+ T cells (online supplemental figure S5A–D). Additionally, significantly fewer TAMs (F4/80+CD11b+Ly6G−Ly6C−) were detected in 4T1 tumor tissues from mice co-administered with anti-PD-1 and KRCT87 (online supplemental figure S5E). Since TAMs-infiltration increment is generally linked to poor prognosis in cancer, these data raise the possibilities that KRCT87 may enhance the effectiveness of anti-PD-1 therapy by modulating TAMs.36

TYRO3 blockade restores anti-PD-1 responsiveness in 4T1 tumor. (A) Assessing the growth of 4T1 tumors in mice treated with the indicated agents. On day-8, 32 Balb/c mice were challenged subcutaneously with 1×106 4T1 cells (n=8 mice) and administered intraperitoneally with KRCT87 (5 mg/kg) and/or anti-PD-1 ab. (B) The growth curves of 4T1 tumors. (C) Heatmap of YAP-related genes in vehicle- and KRCT87-treated groups from the data in figure 3G. (D) The smear plot visualization showing differential expression genes of KRCT87-treated versus vehicle-treated MC38 tumor infiltrating CD11b+F4/80+ cells. This plot was produced by the data in figure 3G. (E) The mRNA levels of Ccn1 in CD4+, CD8+, F4/80+ and cancer cells isolated from the GFP-MC38 tumor. (F) The secretion level of CCN1 in 4T1 tumors in mice treated with vehicle or KRCT87 (n=8 mice). (G) Gene set enrichment analysis (GSE168846) showing the enrichment of YAP-conserved signature in anti-PD-1 ab resistant tumors. (H) Relative expression levels of Ccn1 in indicated groups (GSE168846) (n=15, responsive; n=20, resistant). All data represent the mean±SD. All statistical significance of the differences was determined by one-way analysis of variance followed by Tukey post hoc test except F and H (an unpaired two-tailed Student’s t-test). *, p<0.05; **, p<0.01; ***, p<0.005; ****, p<0.001 significant as compared with the vehicle-treated group or the indicated group. MQ, macrophages; NES, normalized enrichment score; PD-1 ab, programmed cell death protein-1 antibody; YAP, yes-associated protein; mRNA, messenger RNA.
In the next step, we investigated the mechanisms underlying the antitumor effect of TYRO3 blockade. We previously demonstrated that csEV-stimulated TYRO3 causes YAP activation in several cancer cell lines.12 In tumor immunity, YAP highly relates to immune suppression via Treg17 and CD8+ T cells18 in the TME. Seeking clues, we reviewed transcriptomic data derived from TAMs in KRCT87-treated MC38 tumors (figure 3G). A GSEA in TAMs from MC38 tumors revealed that the mRNA expression levels of YAP-target genes and the YAP-conserved signature were both diminished in TAMs from the KRCT87-treated group relative to those from the vehicle-treated group (figure 5C and online supplemental figure S6A). The DEGs were visualized using a smear plot illustrating average signal intensity (X-axis) and log2 fold change (Y-axis) (figure 5D). We especially focused on a secreted angiogenic inducer, cysteine-rich 61 (Ccn1) among the changed DEGs, since the secreted protein is (i) a representative transcriptional target of YAP, (ii) relatively abundant in TAMs, and (iii) significantly downregulated by KRCT87 treatment. To estimate the possibility that CCN1 regulates the immune responses in MQ, we treated recombinant CCN1 to BMDM. This increased the frequency of CD206+ BMDM both in a concentration-dependent manner (online supplemental figure S6B) and in the presence of MC38 CCM (online supplemental figure S6C). Given that CCN1 is a secretory protein and plays a role in tumor remodeling, the major source of CCN1 in the TME needs to be elucidated. We postulated that CCN1 may be mainly secreted by cancer cells in the TME, because YAP is highly overactivated in cancer cells. The amount of secreted CCN1 is significantly lower in BMDM than in 4T1 or MC38 cancer cells (online supplemental figure S6D). We further confirmed its expression in diverse cell types isolated from green fluorescent protein (GFP)-overexpressing MC38 (GFP-MC38) tumors in order to find the major source of CCN1 in the TME. As expected, the mRNA level of Ccn1 was highest in cancer cells (GFP+CD45−CD90−) compared with F4/80+ MQ, CD4+, and CD8+ T cells isolated by MACS microbeads (figure 5E). Therefore, we postulated that cancer-cell-secreted CCN1 could shape the phenotypes of MQ.
We further investigated whether TYRO3 inhibition downregulates the expression of CCN1 in tumor tissues and MQ. In the results, CCN1 secretion was significantly decreased in KRCT87-treated 4T1 tumor tissues relative to the vehicle-treated group (figure 5F). In addition, KRCT87 reduced the mRNA levels of Ccn1 in BMDM primed with MC38 culture media, MC38, and 4T1 cells, whereas UNC2250 could not decrease the mRNA expression of Ccn1 (online supplemental figure S6E,F). Ccn1 expression was also suppressed by TYRO3 siRNAs (online supplemental figure S6G), and we further confirmed that secretion of CCN1 was downregulated in stable TYRO3 knockdown 4T1 cells (online supplemental figure S6H).
To assess the potential role of CCN1 in responsiveness to anti-PD-1 therapy, a GSEA was performed using transcriptome data from various tumors in mice injected with anti-PD-1 antibody (ab) (GSE168846). We found that an YAP-conserved signature was enriched in anti-PD-1 ab-resistant tumors compared with anti-PD-1 ab-responsive tumors (figure 5G). When we compared the expression levels of Ccn1 between anti-PD-1 ab-resistant tumors (4T1, TC1, B16-F10, and LL/2) and anti-PD-1 ab-responsive tumors (MC38, MBT-2, and CM3),35 Ccn1 is significantly upregulated in anti-PD-1 ab-resistant tumors (figure 5H). The data show that CCN1 could be a key player in deciding responsiveness to anti-PD-1 therapy.
CCN1 modulates immune contexture of TME
Since cancer cells are a major source of CCN1 in the TME (figure 5E), MC38 cells were treated with KRCT87 for 72 hours, and a cancer CCM was used for co-culture models with MQ and CD4+ T cells (KRCT87-I group). In another group (the KRCT87-D group), KRCT87 was directly applied to MQ and CD4+ T cells when starting the co-culture as shown in online supplemental figure S7A. Even though the frequency of Treg cells was slightly decreased in the KRCT87-D group, it was more potently decreased in the KRCT87-I group (online supplemental figure S7B). Moreover, the reduced frequency of IFN-γ+ in CD4+ T cells induced by CCM treatment was restored in the KRCT87-I group (online supplemental figure S7C), indicating that TYRO3 inhibition in cancer cells is essential for phenotypic changes of CD4+ T cells toward immune activation. Intriguingly, KRCT87 treatment did not change in vitro differentiation of CD4+ T cells solely (online supplemental figure S7D), suggesting that TYRO3-mediated immune effects require the presence of MQ in the TME.
To investigate the role of CCN1 in responsiveness to anti-PD-1 therapy, we established CCN1-overexpressing MC38 cells (online supplemental figure S8A). Notably, CCN1-MC38 engrafted mice showed lower responsiveness to anti-PD-1 ab (figure 6A,B). Anti-PD-1 ab treatment increased the frequency of CD86+ in TAMs in the control tumor group, whereas it was not altered by anti-PD-1 ab treatment in the CCN1-overexpressing tumor group (figure 6C). Moreover, the frequency of grazyme B+CD8+ T cells was increased significantly in the control MC38 tumor group relative to the CCN1-MC38 tumor group (figure 6D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CCN1 modulates the immune contexture of the tumor microenvironment. (A) Assessing the tumor volumes treated with anti-PD-1 ab in CTRL-MC38 and CCN1-MC38 bearing tumors (n=6 mice). (B) The growth curves of MC38 tumors treated with indicated agents. (C) Frequencies of CD86+ in tumor associated macrophages in indicated groups. (D) Frequencies of granzyme B+ in CD8+ T cells in indicated groups. (E) Frequencies of CD86+ in BMDM after incubation with CTRL-MC38 CCM or CCN-MC38 CCM for 18 hours (n=7). (F) The experimental scheme of co-culture of either CD4+ or CD8+ T cells with MQ. (G) Frequencies of FOXP3+ in CD4+ T cells co-cultured with indicated MQ under regulatory T cells-skewing condition (n=4). (H) Granzyme B production in CD8+ T cells co-cultured with indicated MQ under LPS 100 ng/mL condition (n=4). All data represent the mean±SD. All statistical significance of the differences was determined by one-way analysis of variance followed by Tukey post hoc test. *, p<0.05; **, p<0.01; ***, p<0.005; ****, p<0.001 significant as compared with the indicated group. CCM, cell-conditioned medium; CTRL, a control vector; BMDM, bone-marrow-derived macrophages; IFN, interferon; MQ, macrophages; αPD1, anti-programmed cell death protein-1 antibody; TGF, transforming growth factor.
To further explore the role of CCN1 in MQ, BMDM were incubated with either CCN1-MC38 or control (CTRL)-MC38 CCM for 18 hours. The CCN1-MC38 CCM polarized MQ toward a less M1-like phenotype characterized by decreased CD86+ population compared with the CTRL-MC38 CCM treated group, indicating the inhibitory effect of CCN1 in M1-skewing of MQ (figure 6E). The CD86+ population was not affected by CCN1-MC38 CCM in BMDC (online supplemental figure S8B). We next determined the effect of CCM-educated MQ on differentiation of CD4+ or CD8+ T cells by co-culturing with naive CD4+ T cells and CCM-pretreated MQ for 72 hours (figure 6F). Interestingly, the enhanced production of IFN-γ in CD4+CD44+ T cells was significantly decreased by CCN1-MC38 CCM exposure in MQ (online supplemental figure S8C). Conversely, the frequency of Foxp3+ Treg was significantly higher in CCN1-MC38-CCM-treated MQ than in CTRL-MC38-CCM-treated MQ under the Treg-skewed condition (figure 6G). The frequencies of Foxp3+ Tregs were not altered by CCN1-MC38-CCM-educated CD11c+ DC relative to CTRL-MC38-CCM-educated CD11c+ DC (online supplemental figure S8D). Similarly, to the in vivo data (figure 6D), the absolute cell counts of CD8+ T cells and the production of granzyme B in CD8+ T cells were decreased by co-culturing with CCN1-MC38-CCM-educated MQ (online supplemental figure S8E,F). When CD4+ T cells were incubated with MC38 CCM under the Th1 or Treg-skewing condition, expression of IFN-γ and FOXP3 was comparable between the groups, excluding the possibility that CCN1 directly affects the production of cytokines in T cells (online supplemental figure S8G–H).
We also obtained CCM from stable TYRO3 knockdown 4T1 cells (online supplemental figure S9A and S6H). This CCM-educated MQ accelerated granzyme B and IFN-γ production in CD8+ T cells (online supplemental figure S9B,C). We finally evaluated whether KRCT87 mediated-immune boosting effects are blocked by CCN1 addition. MQ, educated by MC38-CCM were prepared as shown in online supplemental figure S9D, adhering to the method displayed in figure 6F. CD8+ T cells, co-cultured with KRCT87 pretreated MC38 CCM-educated MQ (MC38-MQ (K87-I)) have remarkably higher granzyme B, IFN-γ, and TNF-α production relative to control group (MC38-MQ). These elevated changes were completely reversed by CCN1 addition, confirming that TYRO3-mediated immune suppression was induced by CCN1 (figure 6H, online supplemental figure S9F–G). On the other hand, CCN1 addition to DC could not nullify KRCT87-mediated immune-boosting effects (online supplemental figure S9E–G). Taken together, these results indicate that abundant CCN1 secretion in the TME blunts antitumor immune responses by anti-PD-1 therapy.
Discussion
Since immune function within the TME varies with malignancies, a pan-cancer immunotherapeutic approach is not applicable to all types of patients with cancer. In recent clinical trials, investigational new drugs and diverse approved agents have been actively applied to assess their synergistic or additive anticancer effects in combination with ICB.37 TAM RTKs are expressed in both cancer and innate immune cells (i.e. MQ and DC) and thus they have been noted as emerging targets in cancer therapies. The pan-TAM SMIs (e.g., RXDX-10632,BMS-77760729 and anti-MERTK antibodies11) have exhibited excellent tumor-growth-inhibitory effects when used as a single agent or in combination with anti-PD-1 ab in mouse models, thereby suggesting the potential for targeting of TAM RTKs in immune-oncology fields. In particular, various SMIs targeting AXL and MERTK have been and are being tested in clinical and preclinical trials.22 However, little research on the blockade of TYRO3 receptor has been conducted, even though TYRO3 has emerged as a promising therapeutic target in the oncology field.38
The specialized functions of TAM receptors in antigen-presenting cells (APC) have been diversified by several studies. As the first detailed study of AXL function in APC, Rothlin et al reported that AXL activation negatively regulates both toll-like receptor (TLR) activation and cytokine production in DC.39 Additionally, the regulation of MERTK and AXL expression is clearly different. Immunosuppressive stimuli that upregulate MERTK tend to inhibit AXL expression, whereas TLR agonists that elevate AXL expression tend to downregulate MERTK.40 These studies suggest that the regulation of gene expression and signaling of TAM receptors can be controlled by various pathways even though all TAM receptors are frequently activated in cancer.
There is a growing body of literature about TAM receptor expression in T cells, which have long been regarded as lacking TAM receptors. MERTK is detected in activated human CD4+ T cells,41 CD8+ T cells,42 and B cells.43 PROS1-mediated MERTK signaling, functioning as a late costimulatory signal, increased the secretion of effector-associated and memory-associated cytokines in human CD8+ T cells.42 NK cells also express TAM receptors, which seems to be related to the development of effective and functional NK cells. GAS6-stimulated activation of TAM receptors suppressed IFN-γ production in mouse NK cells, thus regulating lung metastasis.44 TYRO3 expressed in tumor cells could transfer to human NK cells via trogocytosis, which enhances effector functions and proliferation.45 Despite these influential roles of TAM receptors in T cells and NK cells, further studies are required to elucidate the comprehensive roles of TAM receptors in both the cell types.
In this study, we screened the TYRO3-focused chemical library and identified KRCT87 as a selective TYRO3 inhibitor. KRCT87 selectively inhibited TYRO3 among 79 oncology-focused protein kinases (figure 2B). Moreover, it displayed at least a 100-fold more selective inhibitory effect towards TYRO3 than did MERTK or AXL in each TAM-receptor-overexpressing Ba/F3 assay (figure 2C), indicating its superior efficacy as a TYRO3 SMI. Administration with KRCT87 increased M1-polarized TAMs and overall antitumor T-cell responses in the mouse model (figures 3–4).
We explored the expression of TYRO3 in the TME. Although the frequency of TYRO3-positive cells was highest in TAMs in immune cell subsets, the absolute number of TYRO3-positive cells in tumors was highest in cancer cells, presumably due to the high proportion of cancer cells in the overall cell population in the TME. In addition, the expression of TAM RTKs may appear differentially depending on the culture condition or pathological conditions.26 29 46 Membrane TYRO3 was more abundant in DC than in MQ in the mouse spleens (figure 1E), as is consistent with the notion that the function of TYRO3 is more prominent in DC than in MQ under physiological conditions.26 On the other hand, this tendency was completely reversed in immune cells isolated from MC38 tumors; the frequency of TYRO3-positive cells was markedly increased in MQ compared with DC (figure 1D,E), suggesting a potential role for TYRO3 in MQ in TME.
The characteristics of cancer cells determine the immune structure in tumors, and understanding of immune cell–cancer cell interaction may be crucial to the development of promising anticancer agents.2 In this study, we revealed that there are two mechanisms underlying TYRO3-dependent MQ-phenotypic changes: TYRO3 expressed in not only MQ but also in cancer cells could change the immune structure in the TME. First, the main physiological role of TAM RTKs in innate immune cells is efferocytosis, which evokes immunosuppressive effects via the secretion of anti-inflammatory cytokines. As expected, we found that TYRO3 inhibition in MQ reduced the expression of representative M2-skewing markers (Arg1 and Ciita) (online supplemental figure S3C,D). Second, we found that TYRO3 expressed in cancer cells also mediates upregulation of M2 phenotypes in tumors, which would be the most interesting finding of this study. TYRO3 blockade in both MQ and MC38 cells dramatically enhances antitumor T-cell response compared with TYRO3 blockade in MQ (online supplemental figure S7B). As a submechanism to this, we present the TYRO3-CCN1 axis in cancer cells. CCN1 is primarily produced by cancer cells rather than other cell types in the TME, and TYRO3 inhibition in cancer cells reduced its expression in the tumors and cells (figure 5E and online supplemental figure S6E–H). TYRO3-CCN1 axis in cancer cells could mediate phenotypic changes in TAMs, leading to production of antitumor T-cell cytokines such as IFN-γ, TNF-α, and granzyme B in T cells (figure 6 and online supplemental figure S8 and S9).
Even though YAP recently has been posited as a key player in the tumor immune surveillance system, immunosuppressive effects of its downstream targets in the TME are not yet fully understood. CCN1 is a representative downstream target of YAP, as is Ras homolog family member A (RhoA), activation of which is linked to the TAM RTKs.12 22 This study shed light on the role of CCN1 in the immune-oncological view, which has been studied in cancer cells solely. In order to precisely reflect the effect of CCN1 secreted from cancer cells on TAMs, BMDM were exposed to CCN1-MC38 CCM. Treatment with this CCM reduced the M1-skewing phenotype of MQ (figure 6E). Moreover, MQ educated with CCN1-abundant CCM possesses the ability to make CD4+ and CD8+ T cells tumor-friendly (figure 6G and online supplemental figure S8E,F,C). These results coordinately suggest that the TYRO3-CCN1 axis in cancer cells reflects the immune texture of the TME. Indeed, the syngeneic tumor model of CCN1-overexpressing MC38 cells showed a marked reduction of anti-PD-1 therapy responsiveness (figure 6A–D).
Although we have suggested that TYRO3-driven CCN1 secretion mediates immune suppression in the TME, there have been controversial reports on the function of CCN1 in MQ. Direct exposure of higher amounts of CCN1 to mouse peritoneal MQ promotes differentiation into M1-skewing MQ producing pro-inflammatory cytokines.47 However, 100 µg/mL CCN1, used in this study, may not reflect the physiological concentration of CCN1. Herein, we depict that 10–500 ng/mL CCN1 increases the frequency of M2-skewed MQ (online supplemental figure S6B). Similarly to our finding, earlier studies have found that CCN1 secreted from cancer cells increased the migration and invasion of M2-phenotypic MQ in mouse brain tumors48 and in esophageal squamous cell carcinoma cell lines,49 showing that CCN1 may differentially affect MQ phenotypes according to the specific pathological environment (e.g., cancer).
A limitation of this study is its lack of direct clinical evidence, most of the experiments having been performed in mouse tumor models. Thus, our findings invite further clinical studies that will confirm whether TYRO3-dependent CCN1 secretion dictates anti-PD-1 responsiveness. In addition, it could be pointed out that CCN1 is not solely regulated by TYRO3, since its upstream activators, YAP and RhoA, have been known to be regulated by diverse signaling pathways including the multiple tyrosine kinase families. However, we assume that TYRO3 is highly activated in the TME, owing to the abundance of its phosphatidylserine-containing endogenous ligands (e.g., apoptotic cells, tumor-derived exosomes, and csEV), and that as such, it could be a critical receptor for regulation of YAP-dependent CCN1 expression and subsequent secretion to TME. We surprisingly found that MERTK inhibition did not affect CCN1 expression in MQ primed with CCM (online supplemental figure S6E); regardless, further studies using other cell lines and animal models will be needed to clarify the interaction between TAM RTKs and CCN1.
Conclusion
These findings demonstrate the comprehensive role of TYRO3 in TME. Intratumoral TYRO3 inhibition not only increases the M1 phenotype via acting on MQ TYRO3 but also suppresses M2 induction of MQ by blocking cancer-cell-derived CCN1 secretion, thereby triggering antitumor immune responses. In particular, we present the proof-of-concept that a selective TYRO3 inhibitor, KRCT87, boosts the effect of anti-PD-1 therapy. The clinical success of future therapeutics targeting cancers depends on individual customization, which is based on the understanding of the complexity of the TME. Considering the abundance of TYRO3 ligands and CCN1 in the TME, new therapeutic agents targeting the TYRO3-CCN1 axis could be applicable for ICB-refractory patients as a potentially successful anticancer strategy.
Data availability statement
Data are available in a public, open access repository. Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
The authors appreciate members of our laboratories for their kind suggestions. We would like to thank Dr. Chang-Yuil Kang and Dr. Youngro Byun for their kind donation of cell lines. We also thank Dr. Sung Baek Jeong in Daegu-Gyeongbuk Medical Innovation Foundation (DGMIF), for providing the data for Ba/F3 cell-based tyrosine kinase inhibition assays.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MP: conceptualization, methodology, writing—original draft, data curation, formal analysis, validation, project administration, investigation, writing—review and editing. D-SK: methodology, investigation. JP: investigation. MC: investigation. YK: investigation. ECR: methodology, investigation. YJC: investigation. YGK: resources, supervision. YC: resources, supervision. SYC: resources, supervision. KWK: conceptualization, resources, supervision, writing—review and editing, funding acquisition. MP and KWK act as the guarantor of the manuscript.
Funding This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government ((2021R1A2C2093196 (KWK) and 2022R1I1A1A01054388 (MP)).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.