Article Text

Abstract
Background As a major driver of lymphocyte proliferation and activation interleukin 2 (IL-2) is a crucial mediator for antitumor responses. Despite promising activity in a subset of patients, wider therapeutic utility of IL-2 (aldesleukin) has been hampered by severe dose-limiting toxicities, the expansion of immunosuppressive regulatory T cells and a poor pharmacokinetic (PK) profile. Recent engineering efforts, including non-α IL-2 variants, have lowered the toxicity profile, but have yet to induce meaningful antitumor activity in a wider patient population.
Methods We engineered INBRX-120, a CD8α-targeted Cisleukin™ molecule consisting of an affinity tuned IL-2 (IL2-x) connected to two high affinity CD8α-specific single domain antibodies via an effector-silenced Fc domain. To show that this large affinity differential enables directed IL-2 cis-signaling exclusively on CD8α-expressing tumoricidal effector cell populations, INBRX-120 effects on target cell expansion, activation and antitumor activity were tested in vitro. In vivo antitumor efficacy was evaluated in syngeneic mouse models alone or in combination with programmed cell death protein-1 (PD-1) blockade. Preclinical safety, as well as pharmacodynamic (PD) and PK profiling was carried out in non-human primates.
Results INBRX-120 effectively expanded and enhanced the cytotoxic capacity of CD8 T cells and natural killer cells towards tumor cells without affecting regulatory T cells in vitro and in vivo. In syngeneic mouse models, INBRX-120 surrogate showed safe, potent, and durable antitumor efficacy alone and in combination with PD-1 blockade. In non-human primates, INBRX-120 expanded and activated CD8α-expressing effector cells, showed a favorable PK profile, and was well tolerated up to a dose of 1 mg/kg.
Conclusions Through its unique cis-signaling activity on CD8α-expressing effector cells, INBRX-120 overcomes the major limitations of IL-2-based therapy and effectively harnesses IL-2’s potent intrinsic antitumor activity. This novel therapeutic strategy promises safer clinical activity that could induce meaningful antitumor efficacy in a wider set of patients with various cancer indications.
- Immunotherapy
- Cytokines
- Cytotoxicity, Immunologic
- Lymphocyte Activation
- T-Lymphocytes
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Interleukin 2 (IL-2)-based cancer therapies have proven to be incredibly efficacious, however safe antitumor responses remain limited to a small subset of patients due to the pleiotropic activity of IL-2 on both immunostimulatory and immunosuppressive cell populations and severe toxicities induced by high-dose IL-2 therapy.
WHAT THIS STUDY ADDS
INBRX-120 provides a novel IL-2-based therapeutic strategy, wherein potent IL-2 signaling is directed specifically to CD8α-expressing tumoricidal effector cell populations, limiting toxicity and immunosuppression induced by signaling on other cell populations. INBRX-120 effectively induces the expansion of tumoricidal effector cells, enhances their cytotoxicity in vitro and in vivo, and has a favorable safety and pharmacokinetic profile in non-human primates.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The selective activity of INBRX-120 provides a safer and more effective IL-2-based therapy and the potential for improved response rates in larger patient populations. INBRX-120 could deliver benefit to a broad array of cancer indications and be used in combination with strategies ranging from checkpoint blockade to adoptive T or natural killer cell therapy.
Background
Interleukin-2 (IL-2) is a key modulator of lymphocyte proliferation and activation.1 2 Starting in the 1980s, this activity was translated into the first effective cancer immunotherapy (aldesleukin) producing durable and complete responses in metastatic melanoma and renal cell carcinoma.1 3–6 Importantly, this demonstrated that driving T and natural killer (NK) cell expansion can cause potent antitumor responses, particularly in patients with high T cell and NK cell tumor infiltration.3 7 Unfortunately, clinical application of IL-2 has been limited by severe toxicities induced by the inherent biological activity of the cytokine, hence reducing the treatment eligible patient population.
IL-2 has pleiotropic activity, regulating both immune inflammatory responses through effector T cells, NK cells and B cells, while limiting autoimmune responses through regulatory T cells (Tregs). These opposing functions are facilitated through differential expression of three IL-2 receptor (IL-2R) subunits (CD25 (IL-2Rα), CD122 (IL-2Rβ), and CD132 (IL-2Rγ)) on effector cells along with variable affinity of IL-2 to specific receptor configurations.5 Resting NK and CD8 T cells, for example, express only the intermediate affinity IL-2R (CD122/CD132), while Tregs express the high-affinity IL-2R (CD25/CD122/CD132). Thus, high local doses of IL-2 are necessary to activate antitumor effector cells whereas suppressive Tregs can be activated at much lower concentrations. Furthermore, the short half-life of IL-2 necessitates frequent dosing of IL-2 to achieve therapeutic benefit.2 High IL-2 doses in turn lead to severe toxicities such as vascular leak syndrome attributed to IL-2R expression on pulmonary endothelial cells (PEC).8 9 Along with the toxicity caused by IL-2’s powerful stimulation of pro-inflammatory cytokine secretion this makes high-dose IL-2 therapy extremely difficult to administer in a safe and efficacious manner.10–15
To improve the drug-like properties of IL-2 and tip the balance towards effector responses, numerous strategies have been deployed to bias IL-2 binding toward the intermediate affinity receptor complex, resulting in non-α IL-2 variants, and improve pharmacokinetic (PK) properties. Engineering approaches have included polyethylene glycol modifications on IL-2,16 design of de novo mimetics and IL-2 muteins,17 18 IL-2 fusion proteins,19 and antibody/IL-2 complexes.20 However, these strategies can result in reduced activity towards effector cells, reduced stability of the IL-2-based drug, and most do not fully eliminate activity on Tregs. Thus, alternative approaches to effectively target IL-2 to antitumor effector cells are necessary to realize the cytokine’s full therapeutic potential.
Here we describe the development of INBRX-120, a CD8α-targeted Cisleukin™ molecule. INBRX-120 consists of a detuned, low-affinity IL-2 (IL2-x) connected to two high affinity CD8α-targeted single domain antibodies (sdAbs) via an effector-silenced Fc domain. CD8α is part of the heterodimeric CD8αβ co-receptor expressed on cytotoxic CD8 T cells but can also be expressed as a homodimeric CD8αα complex on other cytotoxic cell types including NK cells and a small subset of γ/δ T cells.21 22 INBRX-120 selectively targeted these cell populations and enhanced the proliferation and cytotoxic capacity of CD8 T cells and NK cells. Uniquely, our strategy enabled the activation of these effector cells with potencies comparable to wild type IL-2 while sparing activation of undesired cell populations like Tregs and PEC. Using a surrogate of INBRX-120 we demonstrated robust in vivo antitumor efficacy. Furthermore, INBRX-120 was found to have a favorable PK and safety profile in non-human primates (NHP). Together, the data highlight the potential of this novel therapeutic concept to provide a safe and efficacious IL-2-based therapy that could be applied to various cancer indications.
Methods
Animal studies were conducted in accordance with Association for Assessment and Accreditation of Laboratory Animal Care International guidelines and were approved by the Institutional Animal Care and Use Committee for Explora BioLabs (EB17-004-019), Champions Oncology (2020-TOS-001-Am15) and BTS Research (17–047 Enrollment 39). Biopsies from consented patients were collected by the Biorepository Tissue Technology Shared Resource of the University of California, San Diego (IRB# 090401).
IL-2 reporter cell activity
CD8α-expressing IL-2 reporter cells were generated as described in online supplemental figure 3. To measure IL-2R signaling, reporter cells were co-incubated with test article dilutions for 20 hours. As a surrogate for IL-2R signaling, levels of secreted embryonic alkaline phosphatase in the cell supernatants were quantified using QUANTI-Blue solution.
Supplemental material
STAT5 signaling assay
Human peripheral blood mononuclear cells (PBMC) were stimulated with test articles for 15 min at 37°C and immediately fixed and permeabilized. Changes in the levels of phosphorylated signal transducer and activator of transcription 5 (STAT5) on specific cell populations were measured by flow cytometry (see Online supplemental file 1). Half maximal effective concentrations (EC50) were calculated using the median fluorescence intensities of the antibody detecting phosphorylated STAT5.
In vitro cell proliferation
Human PBMC or dissociated tumor cells (DTC) were labeled with the CellTrace Violet (CTV) Cell Proliferation Kit for flow cytometry (Invitrogen, #C34557). Cells were incubated with test article dilutions and cell proliferation was measured by flow cytometry after 6 days (PBMC) or on day 7 (DTC). Cells with CTV fluorescence intensities below the undivided parental peak were considered proliferating.
Tumor target cell killing assays
Killing of A431 epidermoid carcinoma cells by CD8 T cells or NK cells was measured using an Incucyte Live-Cell Analysis System (Sartorius). Target cells were labeled with the CYTO-ID Red Long-Term Cell Tracer Kit (Enzo Life Sciences, #ENZ-51037-K025) and co-incubated with prestimulated or resting PBMC (for NK cell killing assays) or prestimulated enriched CD8 T cells to a defined effector-to-target ratio (optimal 10:1). Incucyte Caspase-3/7 dye (Sartorius, #4440) was added to the co-cultures to determine target cell apoptosis induced by NK cells or CD8 T cells. Live cell images for time-course experiments were taken every 2 hours. Target cell death was quantified via the overlap area between the Caspase-3/7 dye and the target cell counterstain (total object area in µm/image).
Antigen-specific CD8 T cell stimulation
CD8 T cell response to antigen-specific stimulation was measured using a human interferon (IFN)-γ/tumor necrosis factor (TNF)-α/granzyme B Three-Color FluoroSpot assay (Cellular Technology Limited). MART antigen-specific enriched CD8 T cells were co-cultured on FluoroSpot plates with test articles and an equal number of mitomycin C-treated T2 cells that had been preincubated with 1 fg/mL MART peptide (ELAGIGILTV) for 4 hours. After 19 hours, plates were developed according to the manufacturer’s manual and imaged with an S6 Universal M2 analyzer. Spot counts for cells secreting IFN-γ, TNF-α or granzyme B were quantified using the integrated software.
In vivo cell expansion
Female mice (C57BL/6 or BALB/c), 6–8 weeks old, were dosed with a single intravenous injection of INBRX-120 surrogate into the tail vein. For 1 week after dosing blood was collected daily and analyzed by flow cytometry.
Syngeneic tumor mouse models
Female C57BL/6 or BALB/c mice, 6–8 weeks-old, were implanted with 500,000 MC-38 or 300,000 CT-26 cells subcutaneously in their left flanks as previously described.23 24 Mice were randomized into groups of 10 animals when tumors reached an average tumor volume (TV) of 100 mm3. Mice were dosed with a single intravenous dose of INBRX-120 surrogate at 1 mg/kg, intraperitoneal doses of anti-programmed cell death protein-1 (PD-1) (clone RMP1-14) at 10 mg/kg two times a week, or a combination of both. Phosphate-buffered saline was injected two times a week intraperitoneally in the control group. TV were measured using the formula TV=width×length×0.5 until the control groups reached the maximal allowable TV. Mice with completely cleared CT-26 tumors were rechallenged with CT-26 in the contra-lateral flank. Matched naïve mice were inoculated with CT-26 in parallel and TV were followed for 4 weeks.
Activity in NHP
Male cynomolgus monkeys, 4–8 years-old, were dosed with a single intravenous bolus of INBRX-120 at different dosages. Cell counts in peripheral blood were quantified by flow cytometry before dosing and after 24 hours, 72 hours, 168 hours and 336 hours. Serum samples for PK analysis were collected after 30 min, 24 hours, 48 hours, 72 hours and 96 hours. Soluble CD25 in the serum was quantified using the Human CD25/IL-2R alpha Quantikine ELISA Kit (R&D, #SR2A00). Levels of free INBRX-120 in the serum were captured on plates coated with 1 µg/mL recombinant human CD8α (Sino Biologics, #10980-H08H). Bound INBRX-120 was then detected with a horseradish peroxidase (HRP)-conjugated mouse anti-human IgG secondary antibody (SouthernBio, #9040–05) and antibody binding was quantified using a 3,3',5,5'-Tetramethylbenzidine (TMB) substrate solution (Seracare, #5120–0077).
Data analysis and statistics
All data were analyzed and graphed in GraphPad Prism V.9. Apparent affinities from binding curves were calculated using a one site – total binding curve fit (Y=Bmax×X/(Kd+X) + NS×X+Background). EC50 values were calculated using the equation (agonist) versus response – variable slope (Y=Bottom + (X∧Hillslope)×(Top-Bottom)/(X∧HillSlope+EC50∧HillSlope). INBRX-120 serum concentrations were calculated based on an 8-point standard curve using a 4-Parameter Logistic curve fit in SoftMax Pro (Molecular Devices). Statistical analysis was performed in Prism V.9 and the statistical tests used are indicated in each relevant figure legend. No statistical analyses were performed on the cynomolgus monkey data due to the small number of animals per group.
Results
INBRX-120 delivers a detuned IL-2 specifically to cells expressing CD8α
Therapeutic benefits of IL-2 are limited by the cytokine’s pleiotropic activity on diverse cell populations expressing high-affinity and intermediate-affinity IL-2R, including both tumoricidal effector cells as well as immunosuppressive Tregs and PEC responsible for IL-2 mediated toxicity.2 8
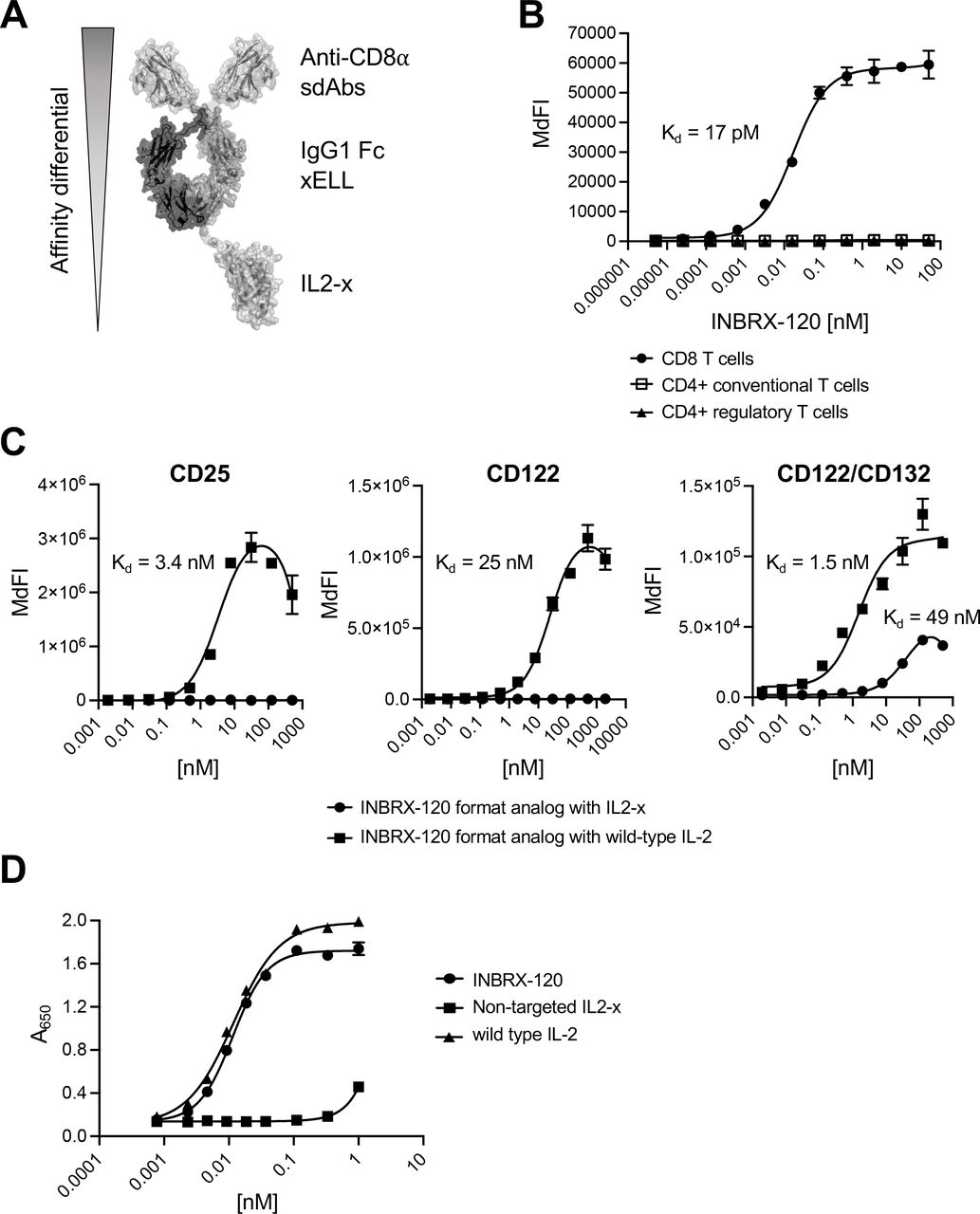
To focus IL-2 activity exclusively on antitumor effector cells, we engineered IL2-x with significantly detuned IL-2R binding affinity that requires high-affinity binding to a secondary surface receptor on the target cell of interest to recover IL-2 signaling. INBRX-120 (figure 1A) consists of a heterodimeric Fc, with N-terminal humanized sdAbs targeting CD8α and IL2-x fused to the C-terminal end of one chain of the Fc. The IgG1 Fc contains deletions (ΔGlu233, ΔLeu234 and ΔLeu235) that disable effector functions mediated by Fc gamma receptor or complement interactions but leave FcRn-mediated recycling intact (referred to as xELL; online supplemental figure 1).25 CD8α is expressed on cytotoxic CD8 T cells and NK cells, as well as subsets of natural killer T (NK-T) cell-like populations and γ/δ T cells, enabling efficient targeting of these effector cell types.21 22 26 27 INBRX-120’s two N-terminal humanized sdAb units were highly specific and bound with sub-nanomolar affinity to CD8 T cells (Kd=17 pM), NK cells (Kd=10 pM) and CD3+CD56+ NK-T cells (Kd=19 pM), but not to regulatory or conventional CD4 T cells (figure 1B and online supplemental figure 2A and 2B). Numbers of CD8 expressing γ/δ T cells in the peripheral blood were too low to see specific binding, but we were able to detect specific binding to in vitro expanded γ/δ T cells indicating that at least a subset of these cell types expresses CD8α (online supplemental figure 2C). IL2-x did not meaningfully contribute to INBRX-120 binding due to its highly detuned IL-2R affinity. This was achieved through mutations within the CD25 and CD122 binding interfaces of IL-2 which abrogated any appreciable affinity of IL2-x for CD25 or CD122 overexpressed on 293 cells (figure 1C). However, IL2-x bound the CD122/CD132 complex, albeit with considerably reduced affinity (Kd=49 nM) compared with wild type IL-2 (Kd=1.5 nM). As anticipated, the lack of CD25 binding and reduced affinity for CD122 also prevented effective binding of INBRX-120 to CD8α negative endothelial cells. While wild type IL-2 bound to HULEC-5a, a microvascular endothelial cell line, INBRX-120 showed little to no appreciable interaction with these cells (online supplemental figure 2D).

INBRX-120 delivers a detuned IL-2 specifically to cells expressing CD8α. (A) Schematic representing the design and functional units of INBRX-120. A detuned IL-2 (IL2-x) is fused to the C-terminal end of one chain of a heterodimeric sdAb IgG1 Fc polypeptide targeting CD8α. The IgG1 Fc contains a deletion that removes Fc gamma receptor binding and disables Fc effector function (xELL). (B) Binding curves for INBRX-120 on T cell populations within the peripheral blood of a representative human donor as determined by flow cytometry. (C) Representative binding curves on 293 cells overexpressing the individual IL-2 receptor subunits CD25 or CD122, or the heterodimeric intermediate affinity IL-2 receptor consisting of CD122 and CD132 for an INBRX-120 format analog containing either wild type IL-2 or IL2-x fused to the C-terminal end of one chain of a heterodimeric sdAb IgG1 Fc polypeptide targeting a protein not expressed in 293 cells. Binding was measured via flow cytometry and shown is the median fluorescence intensity for the Alexa Fluor 647-conjugated detection antibody specific to human IgG Fc. (D) Signaling on a HEK-Blue IL-2 reporter cell line overexpressing CD8α in addition to the IL-2 receptor subunits CD25, CD122 and CD132 induced by INBRX-120 or wild type IL-2 compared with IL2-x not targeted to a surface receptor expressed on the reporter cell line. IL-2, interleukin 2; IL2-x, low-affinity IL-2; MdFI, median fluorescence intensity; sdAb, single domain antibody.
The large affinity differential between the two ends of the molecule enabled specific targeting to and signaling on defined cells of interest, an effect termed cis-signaling. The low affinity of IL2-x alone was insufficient to induce functional IL-2R engagement, but an avidity increase on cells expressing CD8α, driven by the high-affinity sdAb, rescued functional IL-2 signaling on these cells. INBRX-120 induced signaling on IL-2 reporter cells expressing CD8α, while an untargeted IL2-x was not able to activate these cells (figure 1D). The EC50 for this targeted signaling was comparable to wild type IL-2. Similarly, INBRX-120 displayed reduced activity on CD8α negative IL-2 reporter cells (online supplemental figure 3).
These data show that through its differential affinity tuning, INBRX-120 can deliver IL2-x specifically to cells expressing CD8α, including important antitumor effectors like CD8 T cells and NK cells, but does not affect undesired cell populations like Tregs or lung endothelial cells.
CD8α-targeted IL2-x induces functional IL-2R signaling on CD8α-expressing cells and expands CD8 T cells in vitro and in vivo
Functional IL-2R engagement induces a chain of signaling events that starts with phosphorylation of the STAT family by Janus kinases constitutively associated with the intracellular domains of IL-2R. Phosphorylated STAT5 dimers then translocate to the nucleus and induce transcription of genes required for outcomes like cell proliferation.5
INBRX-120 enabled induction of STAT5 phosphorylation specifically in CD8 T cells and both NK cell populations (CD56dim cytotoxic NK cells and CD56bright cytokine-producing NK cells), while not affecting regulatory or conventional CD4 T cells (figure 2A and online supplemental figure 4A). In line with the ability of INBRX-120 to bind to CD8α-expressing expanded γ/δ T cells, these cells also respond with an increase in STAT5 phosphorylation in response to INBRX-120 stimulation (online supplemental figure 4B). HULEC-5a endothelial cells do not respond with appreciable STAT5 signaling to INBRX-120 stimulation, even at high concentrations of 1 µM. Wild type IL-2, on the other hand, induces noticeable increases in the levels of phosphorylated STAT5 indicating that IL-2R on these cells, while low in expression as described previously for primary human pulmonary microvascular endothelial cells, are still capable of functional signal transduction (online supplemental figure 2E and 4C).9

INBRX-120 induces functional IL-2 receptor signaling on CD8α-expressing cells and expands CD8 T cells in vitro and in vivo. (A and B) Dose-response curves of STAT5 signaling in resting CD8 T cells, as well as regulatory and conventional CD4 T cells (A) and proliferation of resting CD8 T cells within PBMC of representative human donors (B) induced in vitro by INBRX-120, a bivalent CD8α-targeting sdAb-Fc fusion without IL2-x, a heterodimeric sdAb IgG1 Fc polypeptide targeting a non-mammalian protein fused to IL2-x (non-targeted IL2-x) and wild type IL-2. (C) Fold-increase in cell counts after in vitro co-incubation of dissociated tumor cells from patients with head and neck (H&N, n=2), kidney (n=2) or colon (n=1) cancer with 10 nM of INBRX-120, non-targeted IL2-x or wild type IL-2. n.s. denotes p>0.05 and ** indicates p<0.005 by an ordinary one-way analysis of variance with Šidák’s multiple comparison correction. (D) Expansion of T cell subpopulations within the peripheral blood of healthy C57BL/6 mice (n=6) dosed with a single 1 mg/kg dose of an INBRX-120 surrogate consisting of IL2-x fused to the C-terminal end of one chain of a heterodimeric sdAb IgG1 Fc polypeptide targeting mouse CD8α. IL-2, interleukin 2; IL2-x, low-affinity IL-2; MdFI, median fluorescence intensity; PBMC, peripheral blood mononuclear cells; pSTAT5, phosphorylated STAT5; sdAb, single domain antibody; STAT5, signal transducer and activator of transcription 5.
Targeting of CD8α-expressing cells over other IL-2R positive cell populations was found to lower the effective concentration required for optimal stimulation. The EC50 for STAT5 phosphorylation in human CD8 T cells in vitro was 23 pM for INBRX-120, 70-fold lower than for wild type IL-2 (EC50=1.6 nM). Subsequently, INBRX-120 signaling caused targeted cells within preparations of human donor PBMC to proliferate (EC50=69 pM), and maximal proliferation was comparable to or better than the effect caused by wild type IL-2 (figure 2B and online supplemental figure 4D). Neither the CD8α-targeting sdAb portion of INBRX-120 alone, nor non-targeted IL2-x induced STAT5 signaling or proliferation, highlighting the requirement for both components to recover the detuned activity of IL2-x on targeted cells. In line with the therapeutic design of the molecule, functional outcomes depended on cell surface expression of CD8α, and the lower CD8α levels on NK cells compared with CD8 T cells could explain the lower maximally induced cell proliferation (online supplemental figure 4E). INBRX-120 activity was not limited to cells from the peripheral blood of healthy donors. In DTC from patients with head and neck, kidney, or colon tumors INBRX-120 exerted similar specific activity and increased the numbers of CD8 T cells, but not the numbers of CD4 T cells present in the samples, an average of eightfold after 7 days in culture (figure 2C). CD8 T cell expansion by wild type IL-2 in this experimental set-up was comparable, but not specific, as CD4 T cells were also significantly expanded.
Due to sequence differences between CD8α from human and mouse origin (<50% sequence identity between extracellular domains, online supplemental figure 5) the CD8α sdAb in INBRX-120 does not bind to mouse CD8α (data not shown). IL2-x, however, is cross-reactive to mouse. We therefore generated an INBRX-120 surrogate molecule consisting of a heterodimeric Fc, with N-terminal sdAbs targeting mouse CD8α and IL2-x fused to the C-terminal end of one chain of the Fc. The INBRX-120 surrogate was selected to match the functional activity of INBRX-120 on human cells as defined by its ability to specifically activate STAT5 signaling with a comparable EC50 (EC50=52 pM, online supplemental figure 6A). The CD8α affinity of the INBRX-120 surrogate was equally specific but lower (Kd=2.4 nM) than for INBRX-120 on human CD8 T cells (online supplemental figure 6B). With this INBRX-120 surrogate, specific expansion of CD8 T cells was also achievable in healthy C57BL/6 mice (figure 2D). A single 1 mg/kg dose of INBRX-120 surrogate induced a 13-fold expansion of CD8 T cells on day 3 compared with pre-dose levels, while CD4 T cell numbers did not change.
These findings highlight that INBRX-120 specifically and potently expands CD8α-expressing effector cells in vitro and in vivo while leaving CD8α-negative cells unaffected. Due to the mixed nature of the cell samples used in these experiments (consisting of CD8α positive and CD8α negative cell types) we can further conclude that INBRX-120 only induced IL-2R signaling in cis on cells expressing CD8α and did not cause trans signaling on CD8α negative cells even when arrayed on other cells.
INBRX-120 enhances the cytotoxicity of CD8 T cells and NK cells
IL-2 is a key promoter of lymphocyte proliferation, but also modulates the activation state of cytotoxic cell populations to prime their effector functions. IL-2 stimulation increases expression levels of activation markers and cytotoxic mediators like granzyme B, thereby facilitating effective target cell killing.28 29
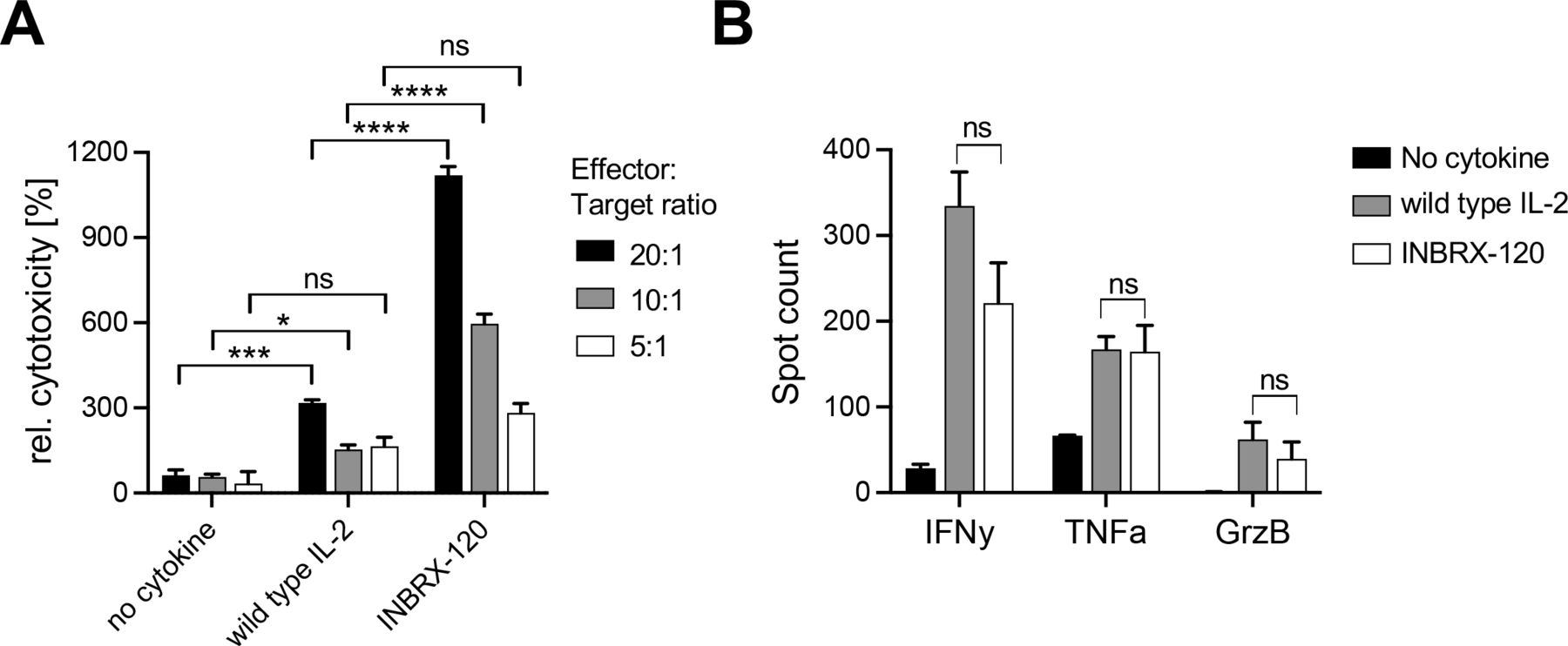
The ability to enhance the cytotoxic capacity of effector cells was retained in INBRX-120. Treatment of enriched human CD8 T cells with INBRX-120 during prestimulation with the CD3-specific antibody clone OKT3 improved the target cell killing of A431, an epidermoid carcinoma cell line (figure 3A). At two out of the three different tested effector-to-target cell ratios, INBRX-120 pretreated cells significantly outperformed those stimulated with anti-CD3 antibodies alone. Furthermore, the relative toxicity after INBRX-120 co-treatment was almost fourfold higher than after treatment with an equimolar concentration of wild type IL-2, exemplifying the unique potency of INBRX-120. Improved target cell killing may have been at least in part due to changes in the cytokine response or an improved secretion of cytotoxic factors. During antigen-specific stimulation of CD8 T cells using MART as a model antigen, the presence of INBRX-120 or wild type IL-2 equally increased such factors. MART-reactive CD8 T cells stimulated with antigen presenting cells loaded with suboptimal concentrations of MART peptide secreted more IFN-γ, TNF-α and granzyme B (figure 3B and online supplemental figure 7).

INBRX-120 enhances the cytotoxicity of CD8 T cells. (A) Target cell killing of A431 epidermoid carcinoma cells by CD8 T cells prestimulated with a monoclonal antibody specific to CD3 (OKT3) in the presence of 1 nM INBRX-120 or wild type IL-2. Target cell death was measured at different effector:target ratios and was visualized using live-cell imaging. Shown is the relative cytotoxicity after 20 hours compared with the maximal target cell death observed at each effector:target ratio without cytokine stimulation. (B) Protein secretion by MART-specific CD8 T cells stimulated with MART peptide loaded T2 cells in the presence of 1 nM INBRX-120 or wild type IL-2. Spot counts represent the numbers of responding cells as measured by FluoroSpot. GrzB, granzyme B; IFN-γ, interferon-γ; IL, interleukin; TNF-α, tumor necrosis factor-α. n.s. denotes p>0.05, * indicates p<0.05 *** indicates p<0.0005, and **** indicates p<0.0001 by an ordinary one-way analysis of variance with Šidák’s multiple comparison correction.
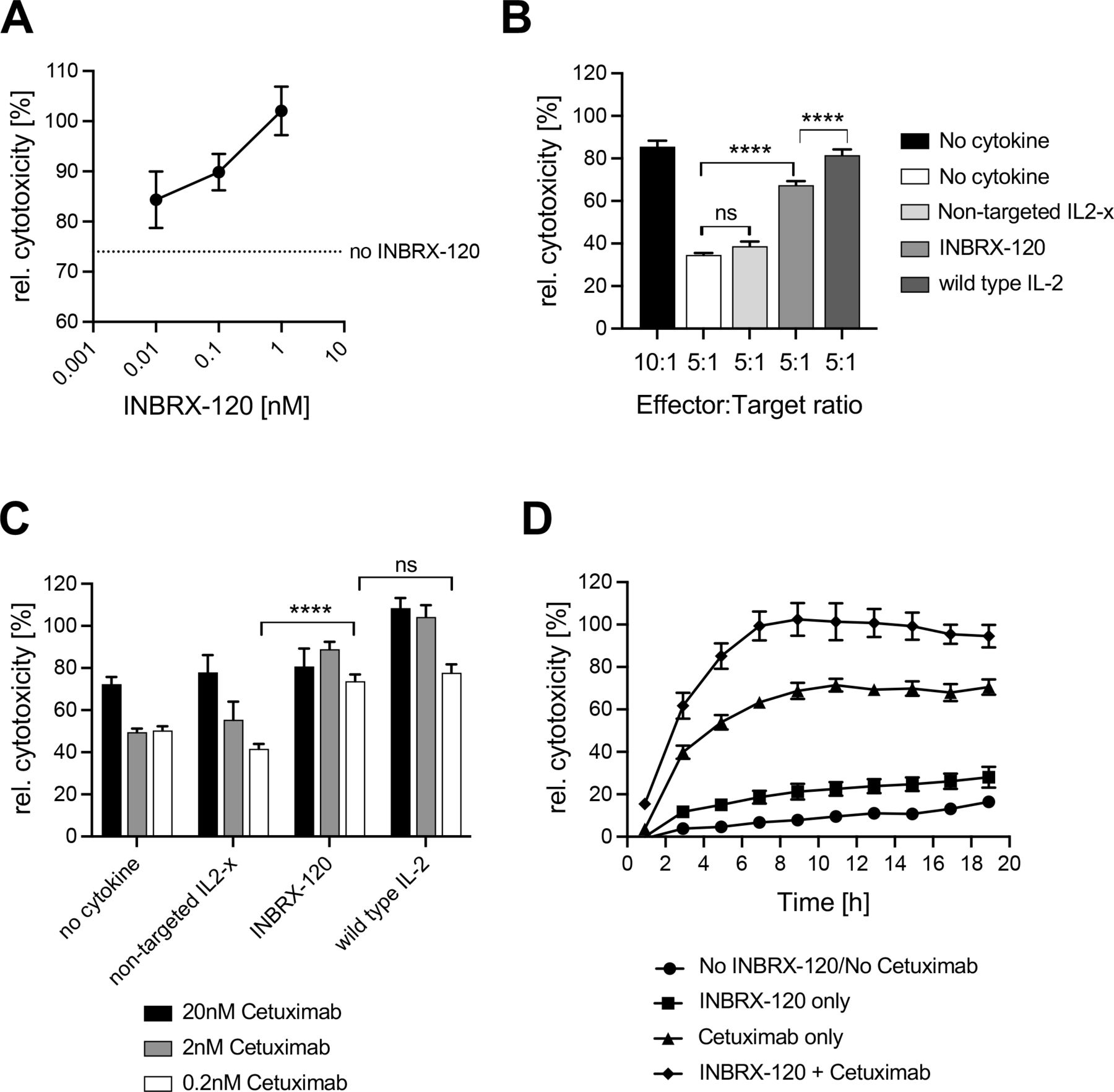
In addition to improving the antigen-specific and major histocompatibility complex mismatch-driven cytotoxic activities of CD8 T cells, INBRX-120 also increased the antibody-dependent cellular cytotoxicity (ADCC) of NK cells (figure 4). When added to a co-culture of resting PBMC and epidermal growth factor receptor (EGFR)-positive A431 target cells at an optimal effector-to-target ratio along with the EGFR-specific therapeutic antibody cetuximab, INBRX-120 enhanced the relative target cell killing in a dose-dependent manner (figure 4A). Even more significant was the ability of INBRX-120 to rescue ADCC in conditions with suboptimal numbers of effector cells (online supplemental figure 8A), indicating that INBRX-120 enhanced the per-cell killing capacity of each NK cell (figure 4B). The recovery of cytotoxicity at suboptimal effector cell numbers required targeted IL2-x signaling, as a non-targeted IL2-x did not significantly improve target cell death, again highlighting the specific cis-signaling nature of the molecule. Maximally achieved target cell killing induced by INBRX-120 was lower than that mediated by wild type IL-2, but the overall improvement was still greater than 50% compared with a no cytokine control.

INBRX-120 enhances the cytotoxicity of NK cells. (A) Dose-dependent increases of ADCC-driven target cell killing of A431 epidermoid carcinoma cells by resting PBMC from a healthy donor co-incubated for 15 hours at a 10:1 NK:target ratio in the presence of 20 nM of cetuximab and INBRX-120. (B and C) Rescue of cetuximab-driven ADCC against A431 cells by resting PBMC from a healthy donor at suboptimal effector:target ratios (B) or suboptimal cetuximab concentrations (C) by 15 hours co-incubation with 1 nM INBRX-120, non-targeted IL2-x or wild type IL-2. n.s. denotes p>0.05 and **** indicates p<0.0001 by an ordinary one-way analysis of variance with Šidák’s multiple comparison correction. (D) Time-course of A431 target cell death induced by PBMC from a healthy donor that were preincubated for 24 hours in the presence or absence of INBRX-120 before co-incubation with the target cells together with or without cetuximab. Shown is the average relative cytotoxicity, defined as the target cell death measured in each condition relative to the maximal target cell death observed during optimal killing conditions (10:1 effector:target cell ratio and 20 nM cetuximab) in each experiment. ADCC, antibody-dependent cellular cytotoxicity; IL-2, interleukin 2; IL2-x, low-affinity IL-2; NK, natural killer; PBMC, peripheral blood mononuclear cells.
Reduction or loss of target antigen is a common mechanism of immune evasion for tumors under the selective pressure of ADCC-enabled therapeutics.30 31 In an assay using subsaturating cetuximab concentrations (online supplemental figure 8B and 8C) mimicking a scenario of reduced EGFR surface density on target cells, INBRX-120 co-treatment recovered the relative NK cell cytotoxicity to or above the levels achieved with optimal cetuximab levels (figure 4C and online supplemental figure 8D). The loss of target cell killing capacity in this experiment was most pronounced at 0.2 nM cetuximab and INBRX-120-mediated improvement was in line with the activity of wild type IL-2. Interestingly, INBRX-120 priming of PBMC alone only mildly enhanced the killing of target cells (figure 4D). Major improvements of NK cell cytotoxicity still required the presence of cetuximab, indicating that INBRX-120 treatment primed the ADCC capacity of NK cells, but did not induce indiscriminate cytotoxicity.
These experiments demonstrate that INBRX-120 improves the per-cell cytotoxic capacity of each effector cell by lowering the antigen density requirement and necessary effector cell numbers for optimal killing.
CD8α-targeted IL2-x has in vivo antitumor efficacy alone and in combination with PD-1 blockade
IL-2 improves immunological anticancer activity by increasing the number of tumor-specific effector cells and enhancing their cytotoxic capacity.2 5 In the same way, INBRX-120 increased the proliferation of CD8 T cells and improved their individual cytotoxic potential.
INBRX-120’s dual activity translated to in vivo mouse models of different genetic backgrounds, in which the INBRX-120 surrogate delivered a specific signal to CD8 T cells to induce dose-dependent cell expansion and increased expression of granzyme B (figures 2D, 5A and B). Notably, mouse NK cells do not express CD8α, thus INBRX-120 had no direct effects on mouse NK cells.32 A small population of CD19/T cell receptor-β/CD49b triple-negative mouse PBMC (<1 cell per µL of blood) also expressed CD8α and expanded after stimulation with the INBRX-120 surrogate (online supplemental figure 9A and 9B).

CD8α-targeted IL2-x has in vivo antitumor efficacy alone and in combination with PD-1 blockade. (A and B) Dose-dependent changes in peripheral blood T cell counts (A) and granzyme B expression (B) in healthy BALB/c mice (n=3) 3 days after a single dose of an INBRX-120 surrogate. (C) Complete tumor clearance achieved by each individual treatment arm in the CT-26 and MC-38 tumor mouse models. (D and E) Antitumor efficacy against syngeneic CT-26 (D) or MC-38 (E) tumors induced by INBRX-120 surrogate treatment alone or in combination with an anti-PD-1 antibody. Shown are average tumor volumes of 10 animals per group (left) and individual animal tumor volumes comparing the effects of anti-PD-1 blockade alone to that of the combination with INBRX-120 surrogate (right). (F) Average tumor volumes after rechallenge of animals with complete clearance of CT-26 tumors on treatment with a combination of anti-PD-1 blockade and INBRX-120 surrogate compared with tumor growth in naïve animals. n.s. denotes p>0.05, * indicates p<0.05, ** indicates p<0.005, *** indicates p<0.0005 and **** indicates p<0.0001 by an ordinary one-way ANOVA with Dunnett’s multiple comparison correction (A–B) and by a Mann-Whitney unpaired t-test on the final time point shown in each figure at which the vehicle group maintained at least 50% of the group’s animals (D, E, F). GrzB, granzyme B; IL-2, interleukin 2; IL2-x, low-affinity IL-2; PBS, phosphate-buffered saline; PD-1, programmed cell death protein-1.
In the syngeneic colon cancer models CT-26 and MC-38, a single dose of INBRX-120 showed antitumor efficacy with an average tumor growth inhibition comparable to six bi-weekly doses of a PD-1 blocking antibody (figure 5C–5E and online supplemental figure 10). In CT-26, significant single agent activity of the INBRX-120 surrogate resulted in 30% of animals achieving complete and durable tumor clearance (figure 5C and D and online supplemental figure 10A). Most strikingly, INBRX-120 combinatorial activity with PD-1 blockade significantly exceeded the antitumor efficacy of both individual treatments. In total, 9/10 animals in the CT-26 model completely cleared their tumors after a combination therapy of INBRX-120 surrogate and anti-PD-1 compared with only 4/10 in the arm dosed with anti-PD-1 monotherapy (figure 5C and D and online supplemental figure 10A). Even in MC-38, a model that poorly responds to checkpoint blockade, combination therapy with INBRX-120 surrogate resulted in significant tumor growth inhibition with 3/10 animals achieving complete tumor clearance (figure 5C and E and online supplemental figure 10A). Tumor clearance in the CT-26 model was not only durable, but likely also resulted in the formation of immunological memory. When animals from the combination group that had achieved complete tumor clearance were rechallenged with CT-26 cells in their contralateral flanks no tumor take was observed in 8/9 animals (figure 5F and online supplemental figure 11). All naïve control animals, however, grew tumors as expected. Treatment of mice with efficacious doses of INBRX-120 was generally well tolerated and did not lead to meaningful body weight loss or other noteworthy clinical observations (online supplemental figure 12).
Together these data show that targeting IL2-x to CD8α-expressing cells in vivo induces potent antitumor activity and improves the efficacy of PD-1 blockade to achieve complete and durable tumor clearance. An INBRX-120 surrogate further improves the therapeutic activity of anti-PD-1 blockade in responsive models, but also confers activity in non-responsive tumors.
CD8α-targeted IL2-x is tolerated in NHP and has a favorable PK/PD profile
Cynomolgus monkeys are a common species to model the toxicology profile, as well as the PK/PD behavior of biologics because of their close evolutionary relationship to humans.33 Due to high sequence homology between CD8α from human and cynomolgus origin (online supplemental figure 5) and the conserved IL-2 signaling pathway between the two species we used cynomolgus monkey as the NHP species to model the toxicity and PK/PD of INBRX-120.
The molecule used in the NHP study, INBRX-120-NHP, differed from INBRX-120 in that it contained a different humanized variant of the CD8α-binding sdAb. Additionally, INBRX-120-NHP contained a mutation (P329G) known to disrupt Fc gamma receptor binding and disable downstream effector function,25 however, P329G in the context of the xELL modification was redundant. The sdAb clone used in the NHP study (B7v15) bound cynomolgus monkey CD8 T cells with similar affinity (Kd=25 pM) as the clone (B7v31) in INBRX-120 (Kd=39 pM) (online supplemental figure 13A). This was also in line with the apparent affinity observed for INBRX-120 on human CD8 T cells (figure 1B). Consequently, the functional activity of INBRX-120-NHP as measured by its ability to specifically induce STAT5 signaling in cynomolgus monkey CD8 T cells was comparable to the activity of INBRX-120 on human CD8 T cells (EC50 range 25–72 pM) (online supplemental figure 13B). A single intravenous dose of INBRX-120-NHP induced specific and dose-dependent expansion of CD8 T cells and NK cells (figure 6A). For most animals, peak expansion was observed on day 7 post dose and cell numbers were still mildly elevated after 14 days. Tregs and conventional CD4 T cells, as well as other PBMC subpopulations like B cells did not expand at a dose of 0.3 and 1 mg/kg of INBRX-120-NHP (figure 6A and online supplemental figure 14A and 14B). The highest dose-level (3 mg/kg) induced a moderate expansion of Tregs, but with a delayed peak indicating that this was likely a secondary compensatory effect. The maximal fold-expansion of effector cells at the different dose levels is summarized in table 1. INBRX-120-NHP also induced a strong expansion of NK-T cells with a kinetic profile comparable to that of CD8 T cells and NK cells (online supplemental figure 14C).
Maximal fold-expansion of CD8 T cells and Tregs in cynomolgus monkeys

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CD8α-targeted IL2-x expands cytotoxic effector cells in non-human primates. Shown are the dose-dependent pharmacodynamic effects in cynomolgus monkeys after a single dose of INBRX-120-NHP. (A) Fold-increase in cell counts in the peripheral blood of study animals compared with pre-dose numbers and (B) granzyme B expression in cytotoxic effector cell populations (CD8 T cells and CD16+NK cells) were determined by flow cytometry. (C) Dose-dependent increases in soluble CD25 levels in the serum of each animal and corresponding serum PK levels (D) were quantified by ELISA. GrzB, granzyme B; IL-2, interleukin 2; IL2-x, low-affinity IL-2; INBRX-120-NHP, INBRX-120 variant used in non-human primate study; LLOQ, lower limit of quantitation; MdFI, median fluorescence intensity; NK, natural killer; PK, pharmacokinetic.
Intracellular granzyme B levels in CD8 T cells and cytotoxic NK cells were elevated 7 days after dosing and indicated that INBRX-120-NHP treatment of cynomolgus monkeys induced effector cell activity and cytotoxic capacity as observed in vitro and in mouse models (figure 6B). Shedding of CD25 from activated T cells is a biomarker that can act as measure for the bioactivity of INBRX-120.34 The concentration of soluble CD25 in the serum of treated monkeys increased in a dose-dependent manner but reached peak levels as early as day 3 post dosing (figure 6C).
Upregulation of PD-1 serves as negative feedback loop to curb T cell activation and is associated with an exhausted, dysfunctional phenotype on continuous T cell receptor stimulation.35 IL-2 has both been shown to overcome T cell dysfunction after continued antigen stimulation, but can itself drive CD8 T cell exhaustion within the tumor microenvironment.36 37 PD-1 levels on cynomolgus monkey CD8 T cells treated with INBRX-120-NHP increased in a dose-dependent manner on day 3 post dosing, but returned to baseline or below by day 7 for all groups (online supplemental figure 14D). This seems to indicate that a single dose of INBRX-120 is not enough to drive T cell exhaustion, but future studies with repeated INBRX-120 doses and continued monitoring of target cell phenotypes, particularly in the context of tumor antigen-stimulation will help to conclusively confirm this.
CD8α expressing cell types make up a large portion of cells in the peripheral blood and CD8α expression levels on individual cells are among the highest of all lymphocyte surface receptors (online supplemental figure 4E).26 While this enables the delivery of a high number of IL2-x molecules per cell, it also causes high target-mediated drug disposition (TMDD). Despite this pre-existing TMDD sink, INBRX-120-NHP still had a favorable PK profile with serum concentrations above the level required to fully saturate effector cell CD8α for up to 3 days at all dose levels tested (figure 6D). The accelerated clearance on day 3 was in line with the augmentation of the CD8α-mediated TMDD sink caused by the increasing numbers of proliferating effector cells. This type of drug-induced TMDD has been reported for several molecules that induce upregulation of their target or the expansion of cells expressing the target.38 39
INBRX-120-NHP was well-tolerated at 0.3 and 1 mg/kg with only minor clinical observations including transient redness and swelling at 1 mg/kg. In these groups body weight loss was only transient and did not exceed 8% (online supplemental figure 15). Body temperatures remained normal over the course of the study. Animals in the 3 mg/kg dose group developed diarrhea between day 1 and day 3 after dosing and then more severely starting at day 7 along with a whole-body rash. No signs of skin toxicity were observed at the lower two dose levels. Animals in group 3 were euthanized on study day 12. Analysis of serum samples from these animals drawn the day before euthanasia showed drops of globulin, albumin and total protein levels below the published normal range and increases in aspartate aminotransferase levels above the normal range for one animal.40 Alkaline phosphatase levels were also below normal in both monkeys (online supplemental table 1). Interestingly, the dose-limiting toxicities (DLT) in the 3 mg/kg group did not correlate with the amount of CD8 T cell or NK cell expansion observed across groups, indicating that DLT were not driven by effector cell expansion, but likely involved other secondary mechanisms.
Together these findings highlight that INBRX-120-NHP potently expands CD8α-expressing effector cells in NHP and enhances their cytotoxic potential up to a maximal tolerated dose of 1 mg/kg.
Discussion
Since the first approval of therapeutic IL-2, a variety of different strategies have been explored to effectively harness the powerful antitumor activity of this cytokine.2 41 Unfortunately, many of the strategies have fallen short of this goal and the clinical development has been discontinued for several initially promising therapeutic candidates. Most recently, Bempegaldesleukin, a pegylated variant of wild type IL-2 and part of a class of agents often referred to as non-α IL-2s that do not bind CD25, was discontinued after late-stage trials in renal cell carcinoma and bladder cancer in combination with PD-1 blockade not only failed to show improvement over control arms but seemed to lower the response rate to checkpoint inhibition.42 43 The non-α cytokine IL-2v, an IL-2 mutein in which the binding of CD25 was significantly lowered, was clinically tested as a fusion protein tethered to tumor-specific antibodies with the goal of targeting IL-2 activity to the tumor microenvironment. This strategy has been abandoned in favor of an effector cell targeting approach.41 In all these cases reducing or removing binding to CD25 improved the acute toxicities of IL-2 and lowered the affinity differential between Tregs and effector T cells, enabling a more balanced access to the cytokine. However, these engineering approaches also came at the cost of lower cytokine activity on antitumor effector cells like CD25 positive activated T and NK cells. Furthermore, Tregs still express CD122 and through this receptor subunit can bind and respond to non-α IL-2. This may result in immunoregulatory activity that could explain the lack of significant antitumor activity in the clinic. Closely related to IL-2, the cytokine IL-15 may provide a more natural way of avoiding the toxicity and immunosuppressive effects mediated by IL-2 binding to CD25. Like in the case of IL-2, IL-15 signaling on effector cells requires the IL-2Rβ/IL-2Rγ receptor complex. However, IL-15 is predominantly presented to these cells in trans, bound to the IL-15Rα domain expressed on monocytes and dendritic cells.44 This prevents binding to CD25-containing IL-2R complexes and prevents a natural bias to cells expressing this isoform like in the case of IL-2. IL-2 and IL-15 activate similar signal transduction cascades, however, IL-15 has been reported to promote a memory response in T and NK cells rather than facilitate the potent expansion and enhanced cytotoxicity induced by IL-2.45 Several groups are testing IL-15 or IL-15-derived therapeutics in preclinical and clinical programs, and it remains to be seen if the biological differences between the two cytokines shift the balance between cytotoxicity and efficacy in the right direction.46–50
Our engineered IL2-x enables the selective activation of cell populations based on the expression of a target receptor, creating a novel therapeutic strategy that overcomes the limitations of aldesleukin and more recent IL-2 engineering approaches. With the right choice of targeting surface antigen, IL2-x activity can be fully focused on cells with the desired antitumor activity and is not shared between immunostimulatory and immunosuppressive populations like in the case of non-α IL-2s. Expression of the co-stimulatory receptor CD8 is naturally biased to cell types mediating the strongest antitumor responses.51 52 Importantly, targeting CD8α to deliver IL2-x enables activation of the widest possible range of tumoricidal effectors compared with CD8β, which is restricted to CD8 T cells.21 26 Furthermore, CD8α is expressed constitutively on most of these cells and receptor levels are high enough to enable maximal IL-2R saturation and signaling.
Consequently, the CD8α-targeted Cisleukin molecule, INBRX-120, strongly enhanced proliferation and improved the cytotoxic capacity of major tumoricidal cell populations, including CD8α positive T cells, NK cells, γ/δ T cells and NK-T cells, but did not meaningfully signal on CD8α negative Tregs or lung endothelial cells. The specificity of the therapeutic approach translated well into animal models, strongly enhancing CD8α positive cell populations in mice and NHP. Importantly, INBRX-120 showed promising single agent activity and potently enhanced the activity of PD-1 blockade in multiple mouse tumor models. Despite the high pre-existing CD8α antigen sink, INBRX-120-NHP had a favorable PK profile with serum exposures likely sufficient to deliver saturating levels of the drug to target cells for multiple days. This is an important improvement over the short serum half-life of aldesleukin and likely enables less frequent dosing.2 However, optimal dosing will need to be determined through future multidose studies and will depend on additional factors beyond optimal target receptor occupancy. Several reports link continuous IL-2 stimulation to the development of an exhausted, functionally inert phenotype.36 53 Conversely, IL-2 has been shown to restore the functionality of exhausted T cells in the context of repeated antigen-stimulation, including cancer.37 54 It remains to be seen whether INBRX-120 stimulation over an extended period causes an exhausted CD8 T cell phenotype that would lose its ability to efficiently control tumor growth. Extending the dosing interval beyond that required to achieve continuous exposure and applying a strategy that includes exposure-free periods between doses may mitigate this possibility.
The clinical dosing strategy will also depend on the toxicology profile of multiple INBRX-120 doses. Single doses of the therapeutic were well tolerated in NHP at efficacious doses up to 1 mg/kg, suggesting that INBRX-120 can be dosed safely in NHP at levels considerably higher than other IL-2 therapeutics.2 16 55 Future toxicokinetic studies will need to further define the maximally tolerated dose of INBRX-120, particularly when multiple doses are administered, and assess the molecule’s toxicology profile in more detail. This will help to place the toxicity induced by high doses of INBRX-120 in the context of previously described adverse events driven by cytokine or T cell therapeutics.12 14 INBRX-120 activity is not limited to tumor-reactive CD8 T cells, but also leads to the expansion of bystander CD8 T cells. While the activity of these cells may prove invaluable in efficiently eliminating tumor cells, indiscriminate stimulation of a large number of cytotoxic effector cells may cause related adverse events.56 57 However, the toxicity observed in NHP did not seem to be connected to the expansion of effector cells, since maximal fold-changes were comparable between individuals from the tolerated 1 mg/kg group and the toxic 3 mg/kg dose. Furthermore, in vitro killing assays showed that cells treated with INBRX-120 still required a primary mode of recognition via the T cell receptor or a tumor cell redirecting monoclonal antibody for efficient target cell clearance, indicating that INBRX-120 served to boost existing tumor recognition rather than broadly causing unspecific cytotoxicity.
The generation of anti-drug antibodies (ADA) is a common problem for therapeutic agents of biologic origin and clearance of drugs via ADA can significantly affect their PK profile and efficacy, as well as pose a toxicity risk.58 59 Future repeat-dose studies in NHP will also help to further characterize the risk of ADA formation for INBRX-120. While ADA observed in NHP studies does not always translate into clinical trials, an assessment of the molecule’s immunogenicity will nevertheless help to predict possible impacts on the pharmacological and toxicological profiles after repeated doses.
Taken together, we show that specific cis-targeting of IL2-x to cytotoxic effector cells expressing CD8α may provide a safer and more effective way to harness the potent intrinsic antitumor activity of IL-2 compared with other previous and current approaches. Our novel therapeutic strategy has the potential to significantly widen the patient population that could benefit from cytokine-based immunotherapy. By enhancing the patient T cell pool, INBRX-120 could improve response rates to checkpoint inhibitors in indications with low immune cell infiltration. Enhancing NK cell, γ/δ T cell and NK-T cell numbers and cytotoxicity further promises activity in indications in which traditional T cell therapeutics failed, including tumors with low tumor mutational burden.60 INBRX-120 shows promising single agent activity, but its activity may be best combined with additional therapeutic modalities like checkpoint inhibitors, ADCC-enabled therapeutic antibodies, or adoptive T and NK cell therapy to maximize the therapeutic benefit and have a meaningful impact on patients’ lives.
Supplemental material
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
Graphical abstract was created with BioRender.com.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors FJS designed studies, interpreted data, wrote the manuscript and acts as guarantor for this work. NK executed experiments, analyzed data, and wrote the manuscript. SJA, AP, JH, AH, GC, CM, RP and WC executed experiments, analyzed data, and reviewed the manuscript. ER, JCT and BPE designed studies, interpreted data, and reviewed the manuscript.
Funding Studies were funded by Inhibrx, San Diego, California, USA. Grant number N/A.
Competing interests FJS, NK, SJA, AP, JH, AH, GC, CM, RP, WC, and BPE are employees of Inhibrx. ER and JCT are former employees of Inhibrx. All authors declare competing financial interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.