Article Text

Abstract
Background Pixatimod is a unique activator of the Toll-like Receptor 9 pathway. This phase I trial evaluated safety, efficacy and pharmacodynamics of pixatimod and PD-1 inhibitor nivolumab in immunologically cold cancers.
Methods 3+3 dose escalation with microsatellite stable metastatic colorectal cancer (MSS mCRC) and metastatic pancreatic ductal adenocarcinoma (mPDAC) expansion cohorts. Participants received pixatimod once weekly as a 1-hour intravenous infusion plus nivolumab every 2 weeks. Objectives included assessment of safety, antitumor activity, pharmacodynamics, and pharmacokinetic profile.
Results Fifty-eight participants started treatment. The maximum tolerated dose of pixatimod was 25 mg in combination with 240 mg nivolumab, which was used in the expansion phases of the study. Twenty-one grade 3–5 treatment-related adverse events were reported in 12 participants (21%); one participant receiving 50 mg pixatimod/nivolumab had a treatment-related grade 5 AE. The grade 3/4 rate in the MSS mCRC cohort (n=33) was 12%. There were no responders in the mPDAC cohort (n=18). In the MSS mCRC cohort, 25 participants were evaluable (initial postbaseline assessment scans >6 weeks); of these, three participants had confirmed partial responses (PR) and eight had stable disease (SD) for at least 9 weeks. Clinical benefit (PR+SD) was associated with lower Pan-Immune-Inflammation Value and plasma IL-6 but increased IP-10 and IP-10/IL-8 ratio. In an MSS mCRC participant with PR as best response, increased infiltration of T cells, dendritic cells, and to a lesser extent NK cells, were evident 5 weeks post-treatment.
Conclusions Pixatimod is well tolerated at 25 mg in combination with nivolumab. The efficacy signal and pharmacodynamic changes in MSS mCRC warrants further investigation.
Trial registration number NCT05061017.
- Drug Therapy, Combination
- Immunomodulation
- Immunotherapy
- Lymphocyte Activation
Data availability statement
No data are available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Immunologically cold tumors, such as microsatellite-stable metastatic colorectal cancer, contain limited numbers of tumor-targeting immune cells and have proven to be resistant to checkpoint inhibitor drugs such as anti-PD-1 agents.
WHAT THIS STUDY ADDS
This clinical trial evaluated a dendritic cell activating agent (pixatimod) in combination with the anti-PD-1 drug, nivolumab, to see whether this approach could demonstrate benefit to patients. The trial showed that the combination treatment was associated with manageable toxicity and signs of clinical benefit for colorectal patients and found evidence of biomarkers that correlate with clinical benefit.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The objective of this work, and the subsequent ongoing clinical trials, is to demonstrate meaningful patient benefit from the combination and, thus, make this treatment available for all patients.
Introduction
Immune checkpoint inhibition (ICI), principally by targeting programmed cell death protein 1 (PD-1) or its ligand programmed death-ligand 1 (PD-L1), have propelled PD-(L)1 antibodies into mainstream cancer therapy.1 However, while some PD-1 directed therapy has been very effective in some cancers, in other cancers PD-1 therapy is ineffective.2 3 For example, immunologically ‘cold’ cancers, that is, tumors that are not likely to trigger a strong immune response such as metastatic pancreatic ductal adenocarcinoma (mPDAC) and microsatellite stable metastatic colorectal cancer (MSS mCRC), are unresponsive to PD-(L)1 inhibitors alone.4 5 Thus, the immune desert of pancreatic cancer and the main subsets of MSS mCRC represent a major therapeutic challenge and combination approaches are underway to expand the utility of PD-(L)1 inhibitors to these cancer patients.5
Though the majority of agents approved by the Food and Drug Administration (FDA) to date are designed to enhance T cell function, there are significant efforts underway to characterize how the cross talk between the innate and adaptive immune responses influences sustained antigen-specific immunity.6 Pattern recognition receptors (PRRs) consist of five families including Toll-like receptors (TLRs), RIG-I-like receptors, nucleotide-binding oligomerization domain-like receptors, C-type lectin receptors, and cytoplasmic DNA sensors. PRR agonists activate PRR pathways stimulating tumor resident innate immune cells such as dendritic cells (DCs) to release cytokines, upregulate costimulatory molecules and promotes cross-priming of tumor antigens.7 TLR agonists have or are being evaluated in mCRC as one approach to activate the innate immune response through combination with ICI.8 9
Pixatimod is a cholestanol-sulfotetrasaccharide conjugated small molecule compound with immunomodulatory properties under development for the treatment of advanced cancer.10 It exerts an immunostimulatory effect specifically via the TLR9 pathway in DC, which leads to the activation of natural killer (NK) cells capable of eradicating established lymphoma in mice.11 In a poorly immunogenic 4T1 breast tumor model, pixatimod also enhanced the effectiveness of PD-1 inhibition12 warranting further assessment of this combination in the clinical setting.
Here, we report a phase I dose-escalation study on the safety and activity of pixatimod and nivolumab in patients with advanced cancer. In addition, we report on two expansion cohorts. These were MSS mCRC patients who had progressed following previous therapeutic regimen/s, had no standard therapy available, or who chose not to pursue standard therapies, and mPDAC patients who had received no more than one prior line of therapy. (ACTRN12617001573347). The primary objective was to define the maximum tolerated dose (MTD) of pixatimod using a once-weekly, 1-hour intravenous infusion in combination with nivolumab and to further define the safety profile of the recommended phase II dose in expansion cohorts. Secondary objectives were to evaluate safety, tolerability, clinical activity, pharmacokinetics (PK), and pharmacodynamics based on peripheral blood, plasma samples, and biopsy material.
Methods
Participants
Fifty-eight participants with advanced, metastatic disease were enrolled across five sites in Australia: (1) Scientia Clinical Research Pty Ltd / Prince of Wales Hospital, Sydney, New South Wales, (2) Royal Brisbane and Women’s Hospital, Brisbane, Queensland, (3) Genesis Care, North Shore Health Hub, St Leonards (formerly Northern Cancer Institute), Sydney, New South Wales (4) The Alfred Hospital, Melbourne, Victoria, and (5) Royal Adelaide Hospital, Adelaide, South Australia. To be eligible for the dose escalation stage, participants were adults (≥18 years) with a life expectancy of at least 12 weeks, adequate organ function and measurable disease according to Response Evaluation Criteria in Solid Tumors 1.1 (RECIST) and Eastern Cooperative Oncology Group (ECOG) performance status of 0–1. Participants were ineligible for entry into the study if they had clinically significant non-malignant disease or previous clinically significant bleeding from the tumor. Other exclusion criteria included uncontrolled hypertension, previous ICI, major surgery within 6 weeks of randomization, anticancer therapy within 4 weeks of cycle 1 day 1 (excluding GnRH agonists for prostate cancer), palliative radiation for bone metastases within 2 weeks of cycle 1 day 1, and participants with prior central nervous system (CNS) metastases treated with only whole brain radiation therapy, participants with a history of allergy and/or hypersensitivity and/or other clinically significant adverse drug reaction to heparin or other anticoagulant agents or a history of immune-mediated thrombocytopenia or other platelet abnormalities or other hereditary or acquired coagulopathies, or laboratory evidence of antiheparin antibodies, or any history of having tested positive for antiheparin antibodies.
In addition to the above, participants were eligible for entry into the mPDAC cohort if they had histologically or cytologically proven metastatic adenocarcinoma of the pancreas and had received no more than one prior line of chemotherapy in the metastatic setting. Participants were eligible for entry into the MSS mCRC cohort if they had histologically documented mCRC, had confirmed MSS mCRC (defined as 0–1 allelic shifts among 3–5 tumor microsatellite loci using a PCR-based assay or immunohistochemistry (IHC)).
All participants provided written informed consent. The study was conducted in accordance with the ICH Good Clinical Practice guidelines as annotated by the TGA in July 2000, the NHMRC National Statement on Ethical Conduct in Research Involving Humans (2007), the US Code of Federal Regulations, and the Declaration of Helsinki.
Study design and treatment
The study was an open-label, multicenter, phase I dose-escalation study using a 3+3 design with expansion cohorts in mPDAC and MSS mCRC patients at the recommended phase II dose. The primary objective was to determine the MTD of weekly administered pixatimod via intravenous infusion in combination with the standard dose of nivolumab (240 mg, every 2 weeks). Secondary objectives were to characterize safety, tolerability, clinical activity, PK, and pharmacodynamic biomarkers.
Prior to administration, the appropriate dose of pixatimod was diluted in 250 mL 0.9% saline infusion solution; nivolumab was prepared in line with the approved prescribing information. The initial cohort was treated at 25 mg once weekly by 1-hour intravenous infusion with dose escalation in subsequent cohorts following a predefined dose escalation scheme. No intraparticipant dose escalation was allowed. Treatment cycles were 28 days duration, with dosing occurring on days 1, 8, 15, and 22 for pixatimod and days 1 and 15 for nivolumab. The cycle continued until immediately prior to the day 29 dose (day 1 of the subsequent cycle). Participants continued treatment until disease progression, unacceptable toxicity or withdrawal (either voluntarily or investigator decision).
Safety and efficacy assessments
Safety
All participants successfully screened were assessed for safety. Participants were examined for adverse events (AEs) at every treatment visit. Vital signs and safety laboratory tests, including full blood count and liver function tests were also undertaken at each treatment visit. Safety assessment included reporting the incidence of all AEs, irrespective of relationship to study drug, according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0, and the incidence of participants experiencing dose modifications and/or premature discontinuation of study drug. Dose-limiting toxicities (DLTs) used to determine the MTD were defined as occurring during cycle 1 of treatment only (to day 29, but prior to cycle 2 dosing) and felt to be related to pixatimod, nivolumab or the combination according to investigator assessment. If no DLT occurred in the first three participants, the next three-participant cohort was enrolled at the next highest available dose level. If a single DLT occurred, an additional three participants were enrolled at the same dose level. Dose escalation continued until two participants of the cohort experienced a DLT; this was defined as the toxic dose level. The dose-escalation phase was completed and MTD determined by reducing the dose to the next lowest dose compared with the toxic dose and expanding this cohort to at least six participants.
Efficacy
Three efficacy populations were defined: the full analysis set, which consisted of all participants receiving at least one dose of study drug; the sufficient exposure population, which consisted of all participants receiving at least four doses of pixatimod; and the evaluable RECIST population, which consisted of all participants who had a RECIST assessment at least 6 weeks (≥43 days) after commencing study drug. Participants with measurable disease were assessed by CT at baseline and every 8 weeks according to RECIST, version 1.1 criteria.
Pixatimod pharmacokinetic assessment
Plasma samples for pixatimod PK analyses were obtained in cycle 1 on days 1, 4, 8, 22, 25, and 29 in the dose escalation phase as follows: Predosing (no more than 4 hours prior to infusion start); midpoint of infusion; prior to completion of infusion; 30 min postinfusion; then 2, 4, 6, 72 and 168 hours postinfusion. Samples were analyzed for pixatimod concentration using a validated liquid chromatography with tandem mass spectrometry assay with a lower limit of quantitation of 0.5 µg/mL.13 Data are presented as time versus concentration curves and basic non-compartmental analysis (NCA) for estimation of exposure parameters Cmax and AUC.
Biomarker and pharmacodynamics assessment
The inflammatory score Pan-Immune-Inflammation Value (PIV14) was calculated using data from participant blood samples collected at screening (PIV=Neutrophils×Platelets×Monocytes/Lymphocytes).
Plasma and peripheral blood mononuclear cell (PBMC) samples were obtained at various points before and during pixatimod and nivolumab treatment for pharmacodynamic assessment. Plasma samples were analyzed for the concentration of various markers, including cytokines and chemokines, using commercial multiplex arrays (Essential Immune Response Panel, BioLegend).
Cryopreserved PBMC samples were analyzed by flow cytometry for changes in abundance and activation status of T cells 3 days after treatment commenced (day 4) compared with predose samples (day 1). For surface staining, PBMC were stained with a viability stain (FVS700) for 15 min at RT, then washed and stained with antibodies against various markers (CD3, UCHT1; CD4, SK3; CD8, RPA-T8; CD45, 2D1; CD45RA, 5H9; CCR7, 150503; Ki67, B56). Cells were first labeled with antibodies against CCR7 for 15 min at room temperature followed by staining with the remaining antibodies for 30 min at room temperature, then washed. For intracellular staining, cells were then fixed and permeabilized using the eBioscience Foxp3/Transcription Factor Staining Buffer Set according to the manufacturer’s protocol, and stained for Ki-67 for 30 min at room temperature. Cells were washed and fixed with Cytofix (BD Biosciences) for 20 min at 4°C, then analyzed using a 4-laser Becton Dickinson LSR Fortessa. Gating was adjusted using FMO for each surface or intracellular marker.
Fresh biopsies from metastatic lesions at baseline and 4–5 weeks after initiation of treatment were used to assess immune cell infiltrates in the tumor microenvironment. Formalin-fixed, paraffin-embedded tumor tissue sections were evaluated for viability by a qualified pathologist using H&E staining before multiplex IHC for assessment of various immune cell subsets. Sections (5 µm) were stained with DAPI and antibody panels (CD3, polyclonal; CD4, 4B12; CD8, C8/144B; Ki67, MIB-1; CD56, 123C3 from DAKO Australia; CD11c, EPR1347Y from Abcam, Australia; PD-L1, E1L3N from Cell Signaling Technology, USA) using routine laboratory methods. Slides were visualized using the Vectra 3 Quantitative Automated Imaging System equipped with a 20×UPlanApo 0.75 NA lens or a Zeiss-780 NLO point scanning microscope with a 40×Plan Apochromat V.1.4 NA lens.
Images captured using Vectra software V.3.0.5 and regions of interest were marked for multispectral image capture using Phenochart software (V.1.0.4). The resultant images were unmixed using Inform (version 2.6.0) and were segmented manually under the guidance of a pathologist. Cells were segmented using DAPI to identify individual nuclei. A cytoplasmic mask was generated extending a maximum of three pixels from the nuclear mask. The optimum threshold for positivity for each fluorophore was determined by iterative processing using the nuclear mask for nuclear dyes (Ki-67) or the cytoplasmic mask for membrane dyes (eg, CD3 and CD8). Intensity data for each fluorophore were exported to Excel for multiplex analysis and cell profiling. Images captured using Zeiss microscope analyzed using Zen software V.3.4.91.
Statistical analyses of biomarkers were performed using Prism V.6.07 and the types of comparisons are indicated.
Results
Participant demographics and baseline characteristics
Sixty-four participants were screened, of whom 58 enrolled and received at least one dose of pixatimod or nivolumab during the study. Their ages ranged from 35 to 80 years. Forty-eight participants were Caucasian, nine Asian, and one Hispanic (table 1). Forty-seven per cent males and 53% females were enrolled. 55% had an ECOG of 0 at screening and 45% had an ECOG of 1. Fifty-seven participants had received at least one line of prior chemotherapy and 15 had received prior radiotherapy. For the MSS mCRC (n=33), most participants had rectal or left-sided tumors (n=26), 16 participants were confirmed to have mutations in at least one of KRAS, NRAS or BRAF (12 KRAS, 4 NRAS, 0 BRAF), 4 were confirmed to have wild type versions of all three genes and the remaining 13 participants had unknown or wild-type status.
Participant demographics and baseline characteristics
Dose-limiting toxicities
Dosing with pixatimod began at 25 mg per participant per week (three participants) and escalated to 50 mg (three participants) but two DLTs were observed in two participants within the 50 mg cohort and this was declared the toxic dose. The first of these DLTs was a case of multiorgan failure who presented on cycle 1, day 8 with hypotension, impaired liver function, ascites and anuria and received initial resuscitation with hydrocortisone and fluids. Due to suspected immune etiology, intravenous methylprednisolone was commenced in addition to broad spectrum antibiotics for suspected but unconfirmed infection and inotropes for persistent hypotension. Despite these interventions, hepatic and renal function did not improve and he died 2 days later. The second DLT was a case of pulmonary edema, cardiomyopathy and autoimmune hepatitis. Over the course of 6 weeks, she was treated with frusemide, hydrocortisone for the initial event of pulmonary edema, followed by intravenous methylprednisolone with the addition of mycophenolate mofetil and bisoprolol for the hepatitis and cardiomyopathy respectively. Although there was recovery from these immune toxicities, she died from disease progression several months later. At that point the 25 mg cohort, which had comprised three participants, was expanded and three further participants were treated. One participant in this cohort experienced a DLT, pneumonitis, and was treated with methylprednisolone resulting in improvement and eventual resumption of alternate systemic therapy. With no further DLT observed, the 25 mg dose was declared the MTD and trial focused on two expansion cohorts in mPDAC and MSS mCRC.
Adverse events
All 58 participants (100%) had at least one treatment emergent AE (TEAE). In total, 593 TEAE were recorded in the clinical database; 369 were mild, 141 were moderate, 67 were severe, 3 were life-threatening in grading, and 13 events resulted in death. One hundred and seventy-six AEs in 46 participants (79.3% (calculated as number of participants reporting an event/total number of participants)) were considered at least possibly related to pixatimod. The most common AEs related to pixatimod in the 25 mg total cohort were diarrhea (10 participants, 18.2%), nausea (14 participants, 25.5%), fatigue (12 participants, 21.8%), and pyrexia (7 participants, 12.7%). The safety profile of nivolumab was consistent with known information about this marketed product. One hundred and sixty-six AEs in 41 participants (70.7%) were considered at least possibly related to nivolumab. The most common AEs related to nivolumab in the 25 mg pixatimod/nivolumab cohort were diarrhea (11 participants, 20.0%), nausea (8 participants, 14.5%), fatigue (13 participants, 23.6%), and pyrexia (4 participants, 7.3%) (table 2).
Treatment-related AEs (≥ 3 participants)*
Twenty-one CTCAE grade 3–5 treatment-related AEs were reported in 12 participants. These comprised (grade 3 unless otherwise stated): multiorgan failure (grade 5), pulmonary edema, cardiomyopathy, and autoimmune hepatitis (grade 4) in participants receiving 50 mg pixatimod/nivolumab; and increased AST, autoimmune hepatitis, pneumonitis, encephalopathy (grade 4), fatigue, hyponatremia, colitis, hypertension, and diarrhea in participants receiving 25 mg pixatimod/nivolumab. Four participants in the 25 mg pixatimod/nivolumab PDAC cohort (22%) and four participants in the 25 mg pixatimod/nivolumab MSS mCRC cohort (12%) had a grade 3/4 AE. All severe events occurred only in one or two participants. The most frequent event was hypertension (six events in two participants).
One participant in the 50 mg pixatimod/nivolumab cohort died due to treatment related multiorgan failure, attributed to both pixatimod and nivolumab. Notably, this participant had high baseline levels of IL-1α and IL-23 (online supplemental figure S1). No autopsy was performed due to cultural reasons. Twelve further deaths occurred; all were due to disease progression and were unrelated to treatment.
Supplemental material
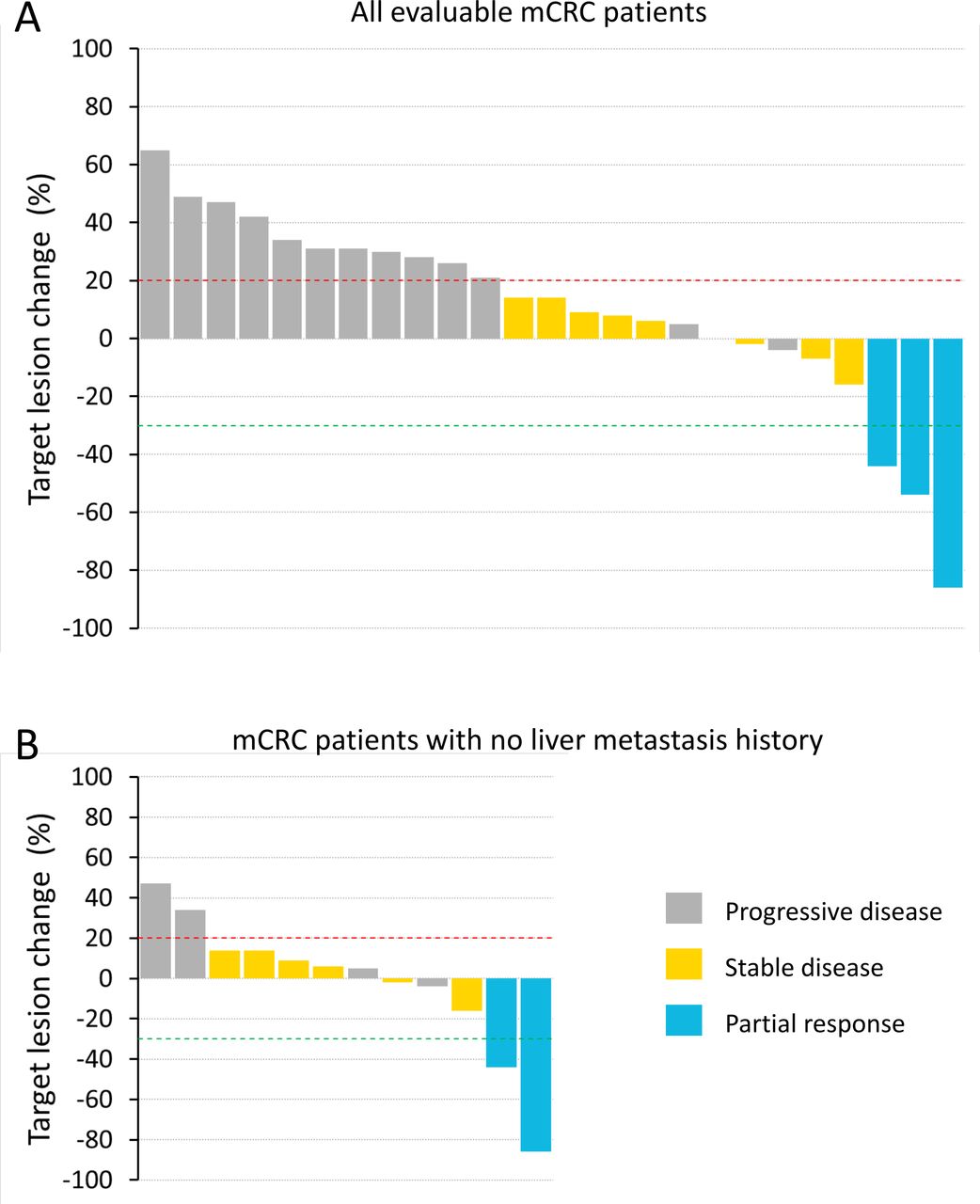
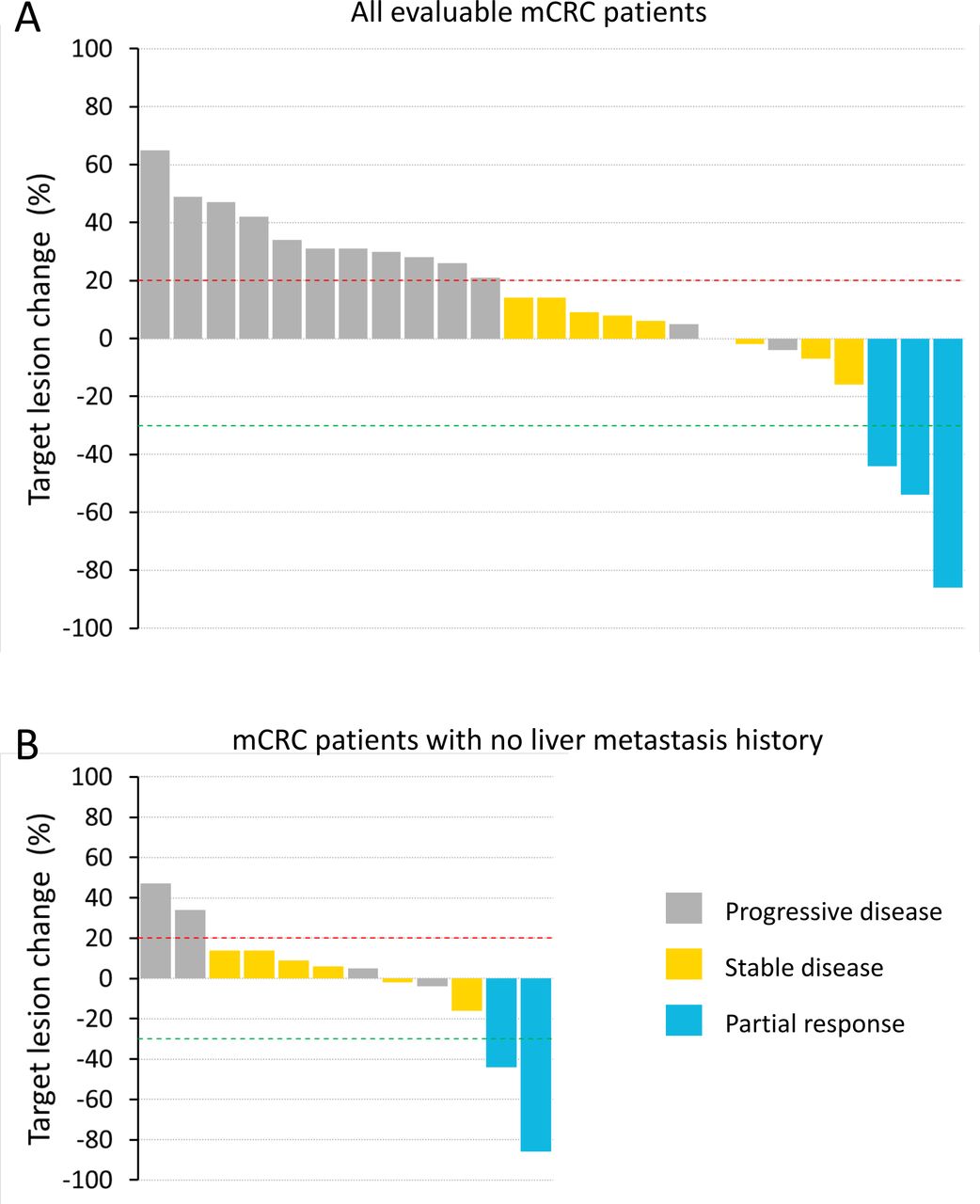
Waterfall plot of best tumor shrinkage as per RECIST for all evaluable metastatic colorectal cancer (mCRC) participants (A) and only those mCRC participants with no history of liver metastasis (B). Participants with on-study scans at 6 weeks or beyond (≥ 43 days) were included in the analyses. RECIST, Response Evaluation Criteria in Solid Tumors.
Fifty-three serious AEs were reported in 30 participants; 13 events were assessed as being at least possibly related to treatment. Treatment-related serious AEs comprised: multiorgan failure, pulmonary edema, cardiomyopathy, and autoimmune hepatitis in the 50 mg pixatimod/nivolumab cohort; autoimmune hepatitis, pneumonitis (three events in two participants), encephalopathy, diarrhea, pyrexia (two events in one participant), and transaminitis in the 25 mg pixatimod/nivolumab cohort (table 3). In total, seven participants withdrew due to treatment-related AEs, five in the 25 mg pixatimod/nivolumab cohort and two in the 50 mg pixatimod/nivolumab cohort.
Treatment-related severe AEs, AEs leading to discontinuation and serious AEs
Clinical activity
Forty-seven participants had efficacy assessments during pixatimod treatment. There were no responders in the mPDAC cohort (n=18), with the best response being stable disease (SD) in two participants with durations of 15 weeks. In the MSS mCRC cohort (n=33), 25 participants were evaluable (initial postbaseline assessment scans ≥43 days); of these, 3 participants had confirmed partial responses (PR) (12% objective response rate per the Evaluable RECIST population) and 8 had SD (disease control rate of 44% per the Evaluable RECIST population) (figure 1A). Of the three PR, two patients had no history of liver metastasis. Tumor mutation burden (TMB) was measured for one of the three PR and found to be three mutations per megabase (mut/Mb). TMB could not be assessed in the other two PR participants due to a lack of primary tumor material or a lack of patient consent. The durations of response for the three confirmed PR were 48, 16 and 23 weeks. The trajectory of target lesion change for the mCRC participants are shown in the form of a spider plot (online supplemental figure S2). A waterfall plot is also presented showing the 12 mCRC participants who had no history of liver metastasis (figure 1B). The objective response rate for this group of mCRC participants with no history of liver metastasis was 17%; 13 participants had a history of liver metastasis, with one responder (8% objective response rate per the Evaluable RECIST population). There were four participants with cancer types other than mPDAC or mCRC: one metastatic uterine adenosarcoma, one adrenocortical carcinoma, one metastatic squamous cell carcinoma and one metastatic endometrial carcinoma. None of these participants had a PR or SD and two developed DLT, of which one was in the 50 mg dose escalation cohort.

Pan-Immune-Inflammation Value (PIV) at screening was significantly lower in mCRC participants who received clinical benefit (PR+SD) compared with those who did not receive benefit (PD+NE). PIV=Neutrophils×Platelets×Monocytes/Lymphocytes. Median values are indicated by horizontal lines. Data failed Shapiro-Wilk normality test so comparison was performed with Mann-Whitney test (*p<0.05). mCRC, metastatic colorectal cancer; PR, partial response; SD, stable disease; NE, not evaluable.
Pharmacokinetics
Systemic exposure of pixatimod (Cmax and AUC0-last) in combination with nivolumab was determined using plasma concentrations from nine participants in the dose escalation cohorts (25 mg=6, 50 mg=3) using NCA. Time versus concentration profiles and pixatimod exposure as measured by Cmax and AUC (online supplemental figure S3) were considered similar as previously reported in a monotherapy study.10

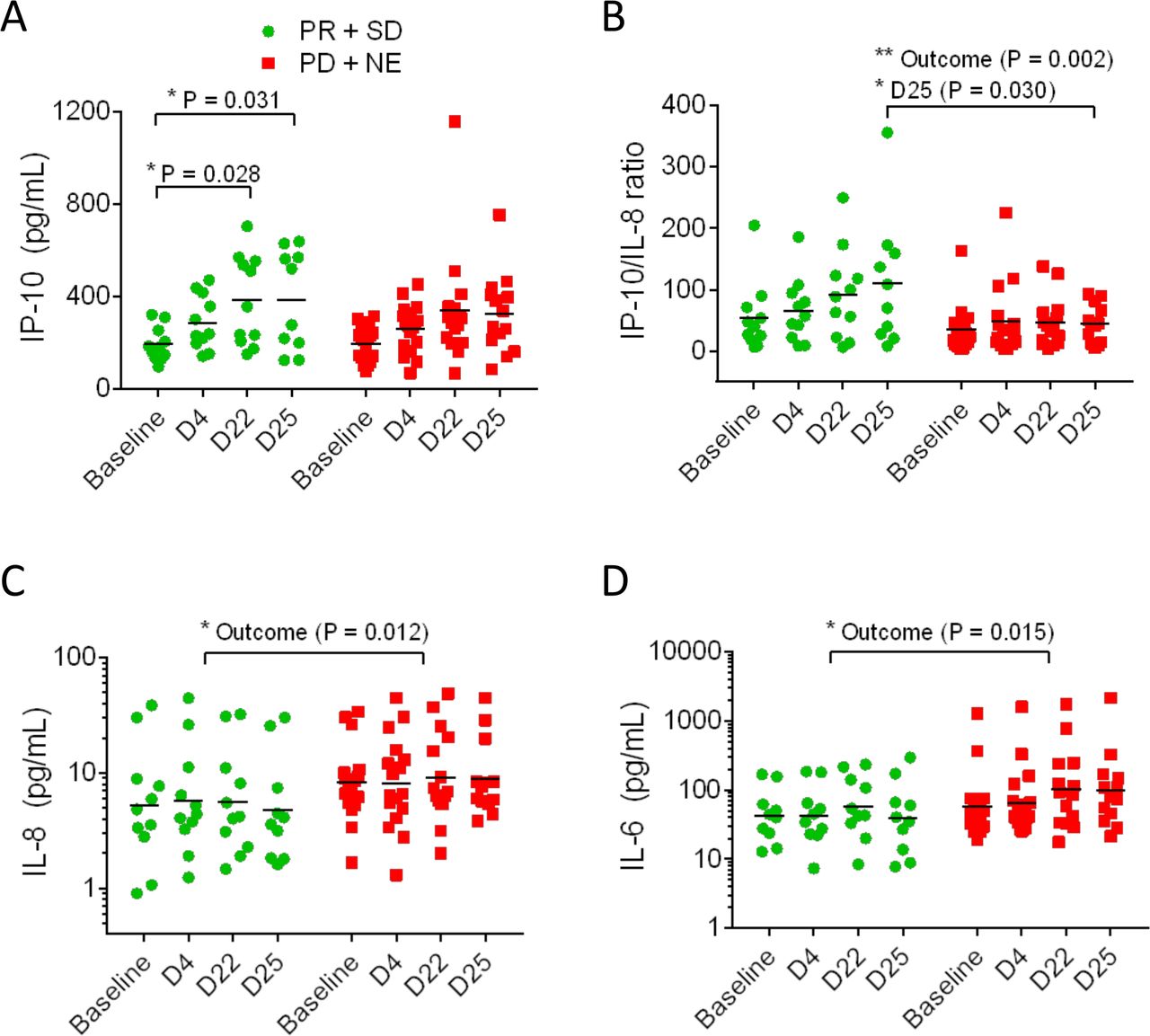
Plasma protein biomarkers IP-10, IP-10/IL-8 ratio, IL8, and IL-6 showed differences in the clinical benefit metastatic colorectal cancer (mCRC) participants (PR+SD) compared with the non-clinical benefit mCRC participants (PD+NE). IP-10 levels significantly rose by D22 and D25 in the benefit group but not the non-benefit group (A). The IP-10/IL-8 ratio was higher overall in the benefit group and on D25 (B). IL-8 was lower overall in the benefit participants compared with the non-benefit participants but there were no significant differences during treatment (C). IL-6 was higher overall in the benefit group but there were no significant differences during treatment (D). Comparisons performed with ANOVA (*p<0.05, **p<0.01). ANOVA, analysis of variance ; PR, partial response; SD, stable diseas.
Biomarkers and pharmacodynamics
Evaluation of PIV revealed that mCRC participants receiving clinical benefit (PR+SD) had significantly lower PIV at screening than participants who did not receive benefit (figure 2).
Analysis of plasma cytokines and chemokines revealed that the plasma concentration of the cytokine IP-10 increased during treatment in mCRC participants receiving benefit but did not increase in those participants not receiving benefit (figure 3A). The ratios of IP-10 to IL-8 were higher (figure 3B), and conversely, the levels of IL-8 were lower in participants receiving benefit (figure 3C). Plasma IL-6 levels were higher in participants not receiving benefit (figure 3D). None of these cytokines showed consistent changes in mPDAC patients. Global trends in plasma cytokines are presented as heat maps (online supplemental figure S4). These data indicate that there was little difference in the global cytokine responses of the mCRC participants receiving benefit from those who did not benefit (online supplemental figure S4A). In contrast, there was evidence of more reductions in cytokines in PDAC participants (24% of samples, online supplemental figure S4B) compared with mCRC participants (6%).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
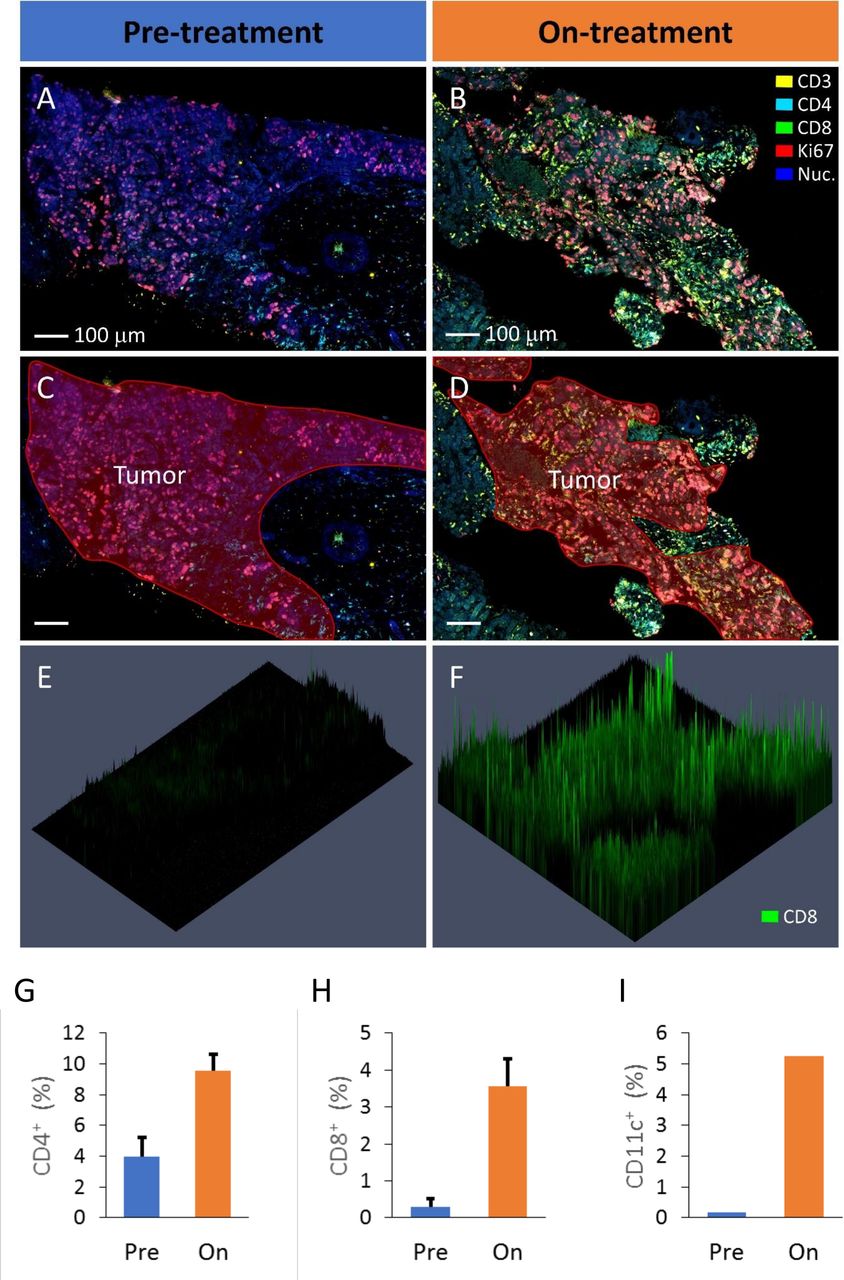
Analysis of pretreatment and on-treatment liver lesion biopsies of a PR mCRC participant (Pt 059) by fluorescence multiplex microscopy. The on-treatment lesion was biopsied 5 weeks after treatment commenced. Representative areas of pretreatment (A) and on-treatment (B) lesions are shown with the regions of tumor tissue indicated in red (C,D). In this example the markers CD3 (yellow), CD4 (cyan), CD8 (green), Ki67 (red) and nuclei are stained with DAPI (blue). To exemplify CD8 staining of these biopsy regions, three-dimensional presentation of the CD8 label fluorescence intensity is presented (E,F). Identification of positively staining cells in the tumor regions of the biopsies was performed for various markers, including CD4+ cells (G), CD8+ cells (H) and CD11c+ cells (I) which were all elevated in the on-treatment biopsy compared with the prebiopsy. The CD4 and CD8 data were collected from two separately stained sections (of each biopsy) allowing the data to be averaged and errors bars included (±SE). mCRC, metastatic colorectal cancer; PR, partial response.
Analysis of PBMC at baseline and on day 4 of treatment indicated that there were increased numbers of CD4+ effector memory (EM) T cells (online supplemental figure S5A) and CD8+ EM T cells (online supplemental figure S5B) in many mCRC participants although the changes were only significant for CD4+ EM cells. Moreover, there were also significant increases in proliferating CD4+ (online supplemental figure S5C) and CD8+ (online supplemental figure S5D) T cells in CRC peripheral blood following treatment as indicated by the expression of Ki67. In contrast, there were no significant increases in EM nor in proliferating T cells in patients with PDAC and the other cancer types on trial (online supplemental figure S5E–H).
Pretreatment and on-treatment biopsies were available for one MSS mCRC participant with best response as PR (Pt 059). Biopsies could not be collected from the other PR participants because they did not have accessible lesions. These samples were analyzed using multiplex fluorescence microscopy to assess immune infiltrate with typical images of tumor regions provided for the pretreatment (figure 4A,C) and on-treatment biopsies (figure 4B,D). The marker CD8 is provided to exemplify the analysis of the biopsies (figure 4E,F). These three-dimensional graphs show the biopsy section in the horizontal plane with fluorescence intensity measured vertically. Similar datasets were collected for several relevant markers and, using DAPI staining of nuclei to identify individual cells, the frequency of cells that were positively expressing these markers was quantified.
When comparing the pretreatment and on-treatment biopsies of Pt 059, there was a marked increase in immune infiltration of tumor regions of the on-treatment sample characterized by increases in both CD4+ (figure 4G) and CD8+ cells (figure 4H). Also increased following treatment was the number of CD11c+ cells which likely would be indicative of an influx of DC into the tumor regions of the on-treatment biopsy (figure 4I). This on-treatment immune infiltrate was also proliferating as indicted by the higher expression of Ki67 by the CD4+ and CD8+ cells (online supplemental figure S6A,B). Furthermore, there was evidence of increased numbers of cells expressing PD-L1 (online supplemental figure S6C) and to a lesser extent, increased NK cell (CD56+, CD3−) infiltration (online supplemental figure S6D) in the on-treatment tumor regions compared with the presample.
Discussion
Pixatimod is a novel immunomodulatory agent, which has been demonstrated to possess potent antitumor activity in multiple preclinical models of cancer, including pancreatic and colorectal cancer, either as a monotherapy,11–13 15–23 or in combination with chemotherapy,11 13 15 molecularly targeted agents17 and ICI.12 Here, we report on a phase Ib study of pixatimod in combination with the PD-1 inhibitor nivolumab in advanced cancer with expansion cohorts in mPDAC and MSS mCRC. The objectives of the study were to identify the MTD, define the safety and tolerability profile, and report on the clinical activity, pharmacokinetics and pharmacodynamics of this new combination regimen in immunologically ‘cold’ tumor types.
Treatment with pixatimod and nivolumab was well tolerated although a case of multiorgan failure was reported in one mPDAC participant as a DLT at 50 mg dose. Of potential relevance here was the finding that baseline plasma IL-1α and IL-23 were significantly higher in this participant vs the 50 mg cohort (see online supplemental figure S1 for details), suggesting that pre-existing immune activation contributed to the outcome. The other DLT at 50 mg was a case of pulmonary edema and two immune-related toxicities, cardiomyopathy and hepatitis. TLR9 is expressed on human lung tissue24 and AEs such as pulmonary edema/pneumonia/dyspnea are possibly related to TLR9 agonism in patients with cancer.25 26 The most common toxicities were either associated with events previously reported for nivolumab such as autoimmune hepatitis, pneumonitis, encephalitis27 28 or on-target effects of pixatimod such as influenza-like symptoms and infusion reactions which are consistent with TLR9 agonists.29 The 25 mg dose was associated with limited grade 3, 4 toxicity. Thus, the MTD of pixatimod was determined to be 25 mg when administered in combination with nivolumab.
There were three PRs in the MSS mCRC participants, with two of these participants on trial for at least 12 months. The objective response rate in the evaluable participant MSS mCRC population (n=25) was 12% and the disease control rate (PR+SD) was 44% (three PRs and eight SD), representing an encouraging signal of efficacy in a heavily pretreated refractory patient population. Of note, the objective response rate in mCRC with no history of liver metastasis was 17%. Of the three responders, two subjects had no history of liver metastases, while one subject only had target liver metastases. Immunotherapy has been reported to be more active in subjects without liver metastases30 31 and other combination trials have proposed further studies in this subset of MSS mCRC patients.32 33 As SD up to 15 weeks was the best response reported in two participants in the mPDAC cohort, further development in this indication is not warranted.
In a previous monotherapy study of pixatimod, the best response of SD was observed in 37.5% of evaluable participants, though only a proportion of those participants had mCRC.10 As nivolumab or other ICIs have not led to PRs in monotherapy studies of MSS mCRC,8 the antitumor activity reported in the MSS mCRC cohort warrants further investigation.
Clinical benefit (PR+SD) was associated with several markers. Baseline PIV is a strong predictor of survival outcomes in participants with mCRC treated with first-line therapy.14 More recently, PIV also appears to be a strong predictor of outcomes in MSI-high mCRC participants receiving ICIs.34 Here, we report for the first time that lower PIV at screening is associated with improved clinical outcomes in a population of MSS mCRC participants receiving pixatimod and nivolumab. Exploration of predictive biomarkers may enrich the patients most likely to benefit.
IP-10 (CXCL10) is considered a biomarker for activation of the innate immune system and TLR9 agonists have been previously used to induce a rapid, sustained IP-10 response in animals and humans.35 TLR9 activation promotes the production of IP-10 by cancer cells, which could further improve T cell recruitment and the efficacy of immunotherapy.36 IP-10 is established as a surrogate biomarker for other TLR9 agonists and following repeated doses of one such agonist, vidutolimod, a trend to higher serum levels of IP-10 was observed in PD-1 refractory/relapsed melanoma patients with better clinical outcomes.37 Furthermore, increased plasma IP-10:IL-8 ratio also correlated with clinical benefit following pixatimod and nivolumab. The potential value of change in this ratio was highlighted in patients with lung cancer receiving anti-PD-1 inhibitors in combination with chemotherapy, offering a new approach for biomarkers of response to combination therapy.38 IP-10 plays a key role in T cell distribution and migration and our data from one PR participant provided preliminary evidence that the combination treatment leads to increases in T cell infiltration into the tumor microenvironment (in addition to increased numbers of DCs and NK cells). In contrast, participants with improved clinical outcomes were found to have significantly lower plasma levels of IL-6 and IL-8 after treatment compared with those participants with progressive disease. Elevation in IL-6 and IL-8 have been associated with shortened survival following CD40-based treatment and these soluble factors are known to inhibit or interfere with DC function.39 40 Thus, in patients with cancer receiving DC-targeted therapies such as pixatimod, TLR9 or CD40 agonists, reduced production of these soluble factors could be critical in mediating the fate of DCs and ultimately clinical outcomes.
Finally, the PK profile of pixatimod in advanced cancer participants in combination with nivolumab was consistent with the previous data from the monotherapy trial,10 indicating that addition of nivolumab had no impact on the pharmacokinetics of pixatimod.
In conclusion, the results from this phase Ib study report on pixatimod and nivolumab demonstrate that the combination is well tolerated, particularly in patients with MSS mCRC where clinical benefit was encouraging. Clinical outcome was associated with increases in some pharmacodynamic markers such as increases in plasma IP-10 and an increase in infiltration of T cells, DC, and to a lesser extent NK cells into tumor specimens of a responding participant. These data support the ongoing development of pixatimod, a unique activator of the TLR9 pathway in phase II development (ClinicalTrials.gov Identifier: NCT05061017), to enhance the innate immune system in tandem with checkpoint blockade in immunologically cold tumors.
Supplemental material
Data availability statement
No data are available.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Bellberry Human Research Ethics Committee. Application No: 2018-08-695Royal Adelaide Hospital Human Research Ethics Committee. HREC reference number: HREC/17/RAH/195. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank the participants and their families who participated in this study, and the study teams at the participating clinical sites.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @keithdredge
Contributors CL, KD, DB and DG conceived the idea and designed the study. CL, AH, NP, MB, MPB and DG conducted the clinical trial. AC, NJW, ACS, LL-EM, EH and GMC performed analysis of samples from the clinical trial. All authors participated in the discussion of the data. KD, DB, EH and DG wrote the manuscript. All authors approved the manuscript. CL, KD and DG supervised the entire study, and jointly controlled the decision to publish. KD accepts responsibility as guarantor for the publication.
Funding The authors also thank Zucero Therapeutics and BMS Australia for funding the study.
Competing interests KD, EH, DB are employees or consultants at Zucero Therapeutics. All other authors declare no potential conflicts of interest.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.