Article Text

Abstract
Background Glioma-induced immune dysregulation of the hematopoietic system has been described in a limited number of studies. In this study, our group further demonstrates that gliomas interrupt the cellular differentiation programming and outcomes of hematopoietic stem and progenitor cells (HSPCs) in the bone marrow. HSPCs from glioma-bearing mice are reprogrammed and driven towards expansion of myeloid lineage precursors and myeloid-derived suppressor cells (MDSCs) in secondary lymphoid organs. However, we found this expansion is reversed by immunotherapy. Adoptive cellular therapy (ACT) has been demonstrably efficacious in multiple preclinical models of central nervous system (CNS) malignancies, and here we describe how glioma-induced dysfunction is reversed by this immunotherapeutic platform.
Methods The impact of orthotopic KR158B-luc glioma on HSPCs was evaluated in an unbiased fashion using single cell RNAseq (scRNAseq) of lineage− cells and phenotypically using flow cytometry. Mature myeloid cell frequencies and function were also evaluated using flow cytometry. Finally, ACT containing total body irradiation, tumor RNA-pulsed dendritic cells, tumor-reactive T cells and HSPCs isolated from glioma-bearing or non-tumor-bearing mice were used to evaluate cell fate differentiation and survival.
Results Using scRNAseq, we observed an altered HSPC landscape in glioma-bearing versus non-tumor-bearing mice . In addition, an expansion of myeloid lineage subsets, including granulocyte macrophage precursors (GMPs) and MDSCs, were observed in glioma-bearing mice relative to non-tumor-bearing controls. Furthermore, MDSCs from glioma-bearing mice demonstrated increased suppressive capacity toward tumor-specific T cells as compared with MDSCs from non-tumor-bearing hosts. Interestingly, treatment with ACT overcame these suppressive properties. When HSPCs from glioma-bearing mice were transferred in the context of ACT, we observed significant survival benefit and long-term cures in orthotopic glioma models compared with mice treated with ACT using non-glioma-bearing HSPCs.
- myeloid-derived suppressor cells
- immunotherapy
- dendritic cells
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The impact of tumors on hematopoietic stem and progenitor cells has been well described in non-CNS-derived tumors. However, the impact of tumors derived in the intracranial compartment is not well understood.
WHAT THIS STUDY ADDS
Our group describes a bias toward myeloid cell progenitors and their progeny using an intracranial murine glioma model. We found this bias can be overcome using adoptive cellular therapy.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
We demonstrate our adoptive cellular therapy platform is capable of redirecting cell differentiation and providing a survival benefit.
Background
In adults, glioblastoma (GBM) is the most common primary brain tumor and remains difficult to treat with a mean survival of 21 months1. Standard of care for GBM is limited to surgical resection, temozolomide, radiotherapy, and tumor-treating fields.2 Several clinical trials evaluating the safety and efficacy of immunotherapy in GBM patients are underway,3 but preliminary results largely demonstrate a lack of durable antitumor responses.4 The lack of efficacy may be due to the inherent immune cell dysfunction that occurs in newly diagnosed individuals. In fact, elegant studies have shown that the cellular composition of peripheral blood of GBM patients is altered, showing AIDS-level CD4+ lymphopenia and expansion of myeloid-derived suppressor cells (MDSCs).5–7 Furthermore, studies across several cancers have found myeloid progenitors are expanded in peripheral blood.8–10 The combination of these findings prompted us to evaluate the common origins of these cell types which are bone marrow-derived multipotent hematopoietic stem and progenitor cells (HSPCs) and determine how gliomas impact cellular homeostasis.
This study conducts an extensive characterization of the bone marrow, specifically on glioma-associated changes in progenitor populations between healthy, non-tumor bearing hosts and glioma-bearing hosts. We examined HSPCs, downstream progenitor populations and finally mature myeloid populations. We demonstrate that HSPCs from intracranial glioma-bearing mice possess an altered HSPC landscape compared with HSPCs from non-tumor-bearing control mice. Importantly, downstream granulocyte macrophage precursors (GMPs) and their progeny are expanded in the bone marrow of glioma-bearing mice, which we believe leads to an observed expansion in immune modulating MDSCs. Additionally, we demonstrate that MDSCs isolated from mice bearing intracranial gliomas are functionally more suppressive on T cell proliferation. Interestingly, we found that combining HSPCs with immunotherapy may reverse the immune modulating fate of HSPCs from glioma-bearing mice, preventing them from differentiation into MDSCs.
In our immunotherapeutic studies that combine transferred HSPCs (from non-tumor-bearing mice) with adoptive cellular therapy (ACT), HSPC-derived cells that are found within the tumor microenvironment have differentiated largely into dendritic cells (DCs).11 12 We have previously published that this is driven by antigen specific T cell-derived IFNγ and that the expression of IFNγ receptor (IFNγR) is crucial for this observation.12 This is observed when HSPCs are cotransferred with immunotherapy including ACT or anti-PD-1 blockade.11 12 HSPCs transferred without immunotherapy are typically found in the tumor microenvironment as MDSCs or tumor-associated macrophages (TAMs).12 Of importance, HSPCs from glioma-bearing mice were found to have higher expression of IFNγR1 and IFNγR2 relative to HSPCs from non-tumor-bearing mice. We believe this increase in IFNγR on HSPCs from glioma-bearing mice allows immunotherapy to alter its cell fate and drive it towards immune propagating DCs. Collectively, these data demonstrate gliomas alter the hematopoietic compartment at the progenitor level to promote expansion of suppressive myeloid cells and their progenitors and how this glioma-induced immune modulation of HSPCs is overcome by adoptive cellular therapy.
Methods
Mice
Female 7–12 week-old C57BL/6 mice (Jackson Laboratories, 000664), transgenic DsRed mice (Jackson Laboratories, 006051), and B6.129S7-Ifngr1tm1Agt/J (referred to as IFNγR−/− mice, Jackson Laboratories, 003288) were used for experiments. The investigators adhered to the ‘Guide for the Care and Use of Laboratory Animals’ as proposed by the committee on care of Laboratory Animal Resources Commission on Life Sciences, National Research Council. The facilities at the University of Florida Animal Care Services are fully accredited by the American Association for Accreditation of Laboratory Animal Care.
Tumor models
Glioma-bearing experiments were performed in syngeneic sex-matched C57BL/6 mice. KR158B-luc gliomas were supplied by Dr Karlyne M. Reilly at the National Cancer Institute, Bethesda, Maryland, USA. For experiments, 104 KR158B-luc or GL261 murine high-grade astrocytomas were implanted as previously described.12
Selection of lineage-negative HSPCs
Tibias and femurs of healthy or glioma-bearing mice 28 days after implantation were isolated, and whole bone marrow was flushed using a 25-gauge needle and 5 mL syringe filled with DMEM (Fisher Scientific cat. 11 965–118) with 10% FBS (Seradigm cat. 97 068–091). Red blood cells are lysed and mononuclear cells are counted. Lineage-negative (lin−) cells are isolated an using magnetic bead isolation kit (Miltenyi Biotec, cat. 130-090-858) as per manufacturer’s protocol.
Single-cell RNA sequencing of lineage-negative HSPCs
Tibias and femurs were collected from eight mice (four non-tumor bearing and four glioma bearing), and lineage− HSPCs were obtained using the same protocol as previously. To obtain a pure population of lineage− HSPCs, cells were stained with LIVE/DEAD Fixable Violet (ThermoFisher cat. L34963) and FITC anti-mouse Lineage Cocktail (Biolegend cat. 133302) and sorted on a Sony SH800 Cell Sorter. Lin− purity of over 95% was achieved for all samples. The bone marrow derived cells were measured directly after sorting, and a cell suspension volume equivalent to 10,000 target cells was used for further processing. Cells were diluted in ice-cold phosphate-buffered saline (PBS) containing 0.4% bovine serum albumin (BSA) at a density of 1,200 cells/µL. Cells were loaded into a Chromium NextGEM Chip G (10× Genomics, Pleasanton, California, USA) and processed in Chromium X following the manufacturer’s instructions. Preparation of gel beads in emulsion and libraries were performed with Chromium Next GEM Single Cell 3′ Kit V.3.1 (Dual Index) according to user guide provided by the manufacturer. Libraries quality and quantity were verified with 2200 TapeStation (Agilent Technologies, USA). Libraries were pooled based on their molar concentrations. Pooled library was sequencing on one high-output lane of the NovaSeq 6000 instrument (Illumina, San Diego, California, USA).
Analysis of scRNAseq results
BCL files were demultiplexed and converted to fastq files using Cellranger mkfastq (10× Genomics, V.7.0). Fastq files were then mapped to a customized mouse genome GRCm38 (mm10+luciferase), using Cellranger count (10× Genomics, V.7.0). The original mouse genome was acquired from the 10× Genomics website. After quality control and removal of dead cells, doublets, normalization, cell clustering (resolution=0.3), dimensionality reduction, differential expression, and visualization were performed using the R package Seurat.13 Clusters identified as contaminating lineage+ cells based on the expression of the mature immune cell gene sets (online supplemental table 1) were removed, leaving a total of 24,084 cells. Normalization, cell clustering, dimensionality reduction, differential expression, and visualization were then repeated with purified subset. The 2,000 most variable features identified with the variance-stabilizing transformation method were used for principal component analysis. On inspection of elbowplot and jackstraw plot, the first 30 principal components were used for downstream analysis. Pseudotime analysis were performed using the R package Monocle14 with default parameters. Cell type annotation was performed manually by assessment of cluster marker genes.
Supplemental material
Supplemental material
Flow cytometry
Flow cytometry was performed on FSC/SSC gating on the BD Biosciences LSR II or LSR Fortessa. Cells were prepared ex vivo as described previously and stained in 2% FBS (Seradigm cat. 97 068–091) in PBS (Gibco cat. 10 010–049) with TruStain FcX Antibody (Biolegend cat. 101320). Analysis was completed using FlowJo V.10 (FlowJo, LLC).
MDSC suppression assay
The spleen of a mouse primed with DCs loaded with total tumor RNA was isolated 1 week after intradermal injection of DCs. The spleen was dissociated into a single cell suspension, lysed, and suspended in buffer. CD3+ T cells were isolated using magnetic bead isolation kit (Miltenyi Biotec cat. 130-095-130) as per manufacturer’s protocol. After isolation, T cells were dyed with CellTrace Violet (CTV) cell proliferation dye (Invitrogen cat. C34557).
For MDSC isolation, tibias and femurs of non-tumor-bearing or glioma-bearing mice were isolated, and whole bone marrow was flushed using a 25-gauge needle and 5 mL syringe filled with DMEM (Fisher Scientific cat. 11 965–118) with 10% FBS (Seradigm cat. 97 068–091). Red blood cells were lysed and mononuclear cells were counted. MDSCs were isolated using the MDSC isolation kit (Miltenyi Biotec cat. 130-094-538) as per manufacturer’s protocol. CTV-labeled T cells were then cocultured with MDSCs from non-tumor-bearing or glioma-bearing mice at different ratios in a round bottom 96 well plate with 25 U/mL rmIL-2 (R&D Biosystems, cat. 402 ML-500) and Dynabeads Mouse T-Activator CD3/CD28 (Gibco cat. 11 452D) at a ratio of 10 T cells to 1 bead. After 3 days of culture, cells were collected and stained for flow cytometry.
Killing assay with MDSCs
Tumor-reactive T cells were expanded using previously published protocols.15 The 4×105 T cells were cultured in a 48-well plate with 4×104 KR158B-luc target cells. These two cell populations were then cocultured with different ratios of MDSCs derived from non-tumor-bearing or glioma-bearing mice isolated as described previously. Twenty-four hours later, cells were collected and stained with extracellular fluorescent flow cytometry antibodies, then stained with Annexin V (Biolegend cat. 640905) in Annexin binding buffer (BD cat. 556454). Both forward/side scatter discrimination and gating on CD45− population allowed for evaluation of target-specific apoptosis and cell death.
Culture of lineage-negative HSPCs
Lin− HSPCs were obtained using the previous protocol. Cells were counted and cultured in six-well plates at 200,000 cells/well in RPMI with 10% FBS for 3 days before collected and stained for flow cytometry. For T cell supernatant experiments, supernatant was collected and spun down to obtain the acellular fraction. HSPCs were then cultured at a 1:1 ratio with fresh media and supernatants.
Murine HSPC transfer experiments
DsRed mice were injected with 104 KR158B-luc cells to generate tumor-bearing donor HSPCs as described previously. In addition, a separate cohort of DsRed mice were not injected for generation of non-tumor-bearing donor HSPCs. For experiments with tumor-bearing recipients, recipient C57BL/6 mice were injected with 104 KR158B-luc cells 23 days after donor mice were injected and irradiated with 9 Gy X-ray irradiation 4 days later (X-RAD 320, Precision X-RAY Irradiation). Approximately 1×106 HSPCs were injected in the tail vein of irradiated recipient mice. Recipient mice were sacrificed 28 days after HSPC transfer to allow for adequate differentiation.
Adoptive cellular therapy
Treatment of tumor-bearing mice began with 5 Gy non-myeloablative lymphodepletion with X-ray irradiation (X-RAD 320, Precision X-ray) 4 days postintracranial injection. On day 5 post-intracranial tumor injection, mice received a single intravenous injection of 107 autologous ex vivo expanded ttRNA T cells±the inclusion of 3.5×105 lin- HSPCs (Miltenyi Biotec, 130-090-858). Beginning day 6 post-tumor injection, 2.5×105 ttRNA-pulsed bone marrow-derived DC vaccines were injected intradermally posterior to the pinna weekly for a total of three vaccines.
Statistical analysis
All experiments were analyzed in Prism 7 (GraphPad). The median survival for tumor-bearing animals is 25–42 days. Tumor-bearing survival experiments in this manuscript are n=7 and analyzed with the Mantel-Cox log-rank test. An unpaired, Mann-Whitney rank-sum test or unpaired Student’s t-test was applied for two-group comparisons for in vivo experiments or in vitro experiments, respectively. One-way analysis of variance was used for comparing experiments with multiple groups. Significance is determined as p<0.05. Animal studies were powered to include five randomized mice per group unless otherwise noted. The authors pre-established that no animals or samples were to be excluded from analysis.
Results
Glioma-bearing mice possess altered HSPC landscape relative to non-tumor bearing control mice
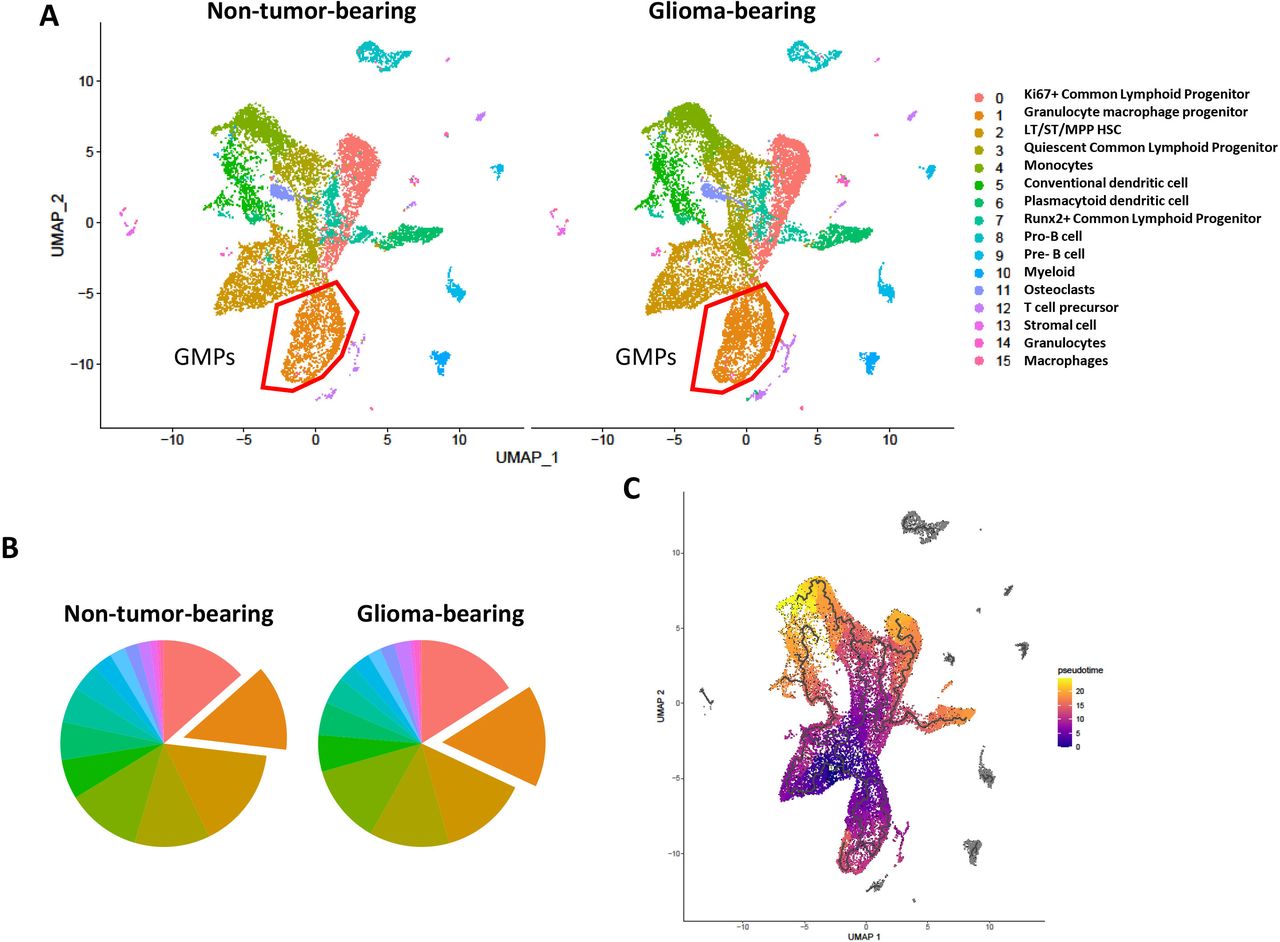
Given the alteration in progenitor phenotype in a variety of cancers derived from outside the CNS,10 we sought to determine if bone marrow-derived hematopoietic stem and progenitor cells exhibit differential expression profile in intracranial glioma-bearing mice. To evaluate this, we sorted lineage− HSPCs derived from mice with orthotopic implantation of KR158B-luc gliomas (28 days post-tumor implantation) and age-matched non-tumor-bearing mice and subjected cells to high-throughput scRNAseq using 10X Genomics platform (online supplemental figure 1A). After filtering out markers for mature immune cells, 15 unique clusters were identified in both groups (figure 1A).
Supplemental material

Differential gene expression of lineage-negative HSPCs from non-tumor-bearing and intracranial glioma-bearing mice via scRNAseq. scRNAseq analysis of HSPCs from non-tumor-bearing (n=4) and glioma-bearing (n=4) mice was performed. (A) UMAP dimensionality reduction and cluster visualization and (B) pie chart frequency depiction color coded to represent cluster ID. Clusters manually annotated based on pathway analysis of cluster marker genes. (C) Psuedotime visualization. HSPCs, hematopoietic stem and progenitor cells.
Recently, the myeloid compartment has become of great interest to other groups as well as our own. We found cluster 1, which represents a large frequency of both samples, to possess markers consistent with GMPs (online supplemental figure 1B). In addition, we show this cluster is increased in glioma-bearing mice relative to non-tumor-bearing mice (figure 1B). This is consistent with the phenotyping data shown later on in this manuscript. Of interest, we identified cluster 2 as the only multipotent, self-renewing hematopoietic stem cell (HSC) from which all other progenitors and more terminally differentiated immune cells are derived from. This cluster was found at a similar frequencies between non-tumor-bearing and glioma-bearing samples.
Pseudotime analysis by Monocle3 was used to visualize differentiation trajectories. We observed that clusters 1 (GMPs), 2 (LT/ST/MPP HSC), and 3 (Quiescent CLPs) are less differentiated than the 12 other clusters, with clusters 4 (monocytes), 5 (conventional DCs), and 6 (plasmacytoid DCs) containing the more terminally differentiated cell types (figure 1C). Since a difference in the frequency of GMPs was observed, we further investigated this finding by performing pseudotime analysis to examine the differentiation state of the GMP cluster. We found cells within this cluster existed within multiple states in pseudotime; however, no differences were seen between non-tumor-bearing and glioma-bearing cells (online supplemental figure 2A Together, these findings reveal an altered HSPC landscape in glioma-bearing mice relative to non-tumor-bearing controls.
Supplemental material
Expansion of immature and mature myeloid cells in bone marrow of intracranial glioma-bearing mice
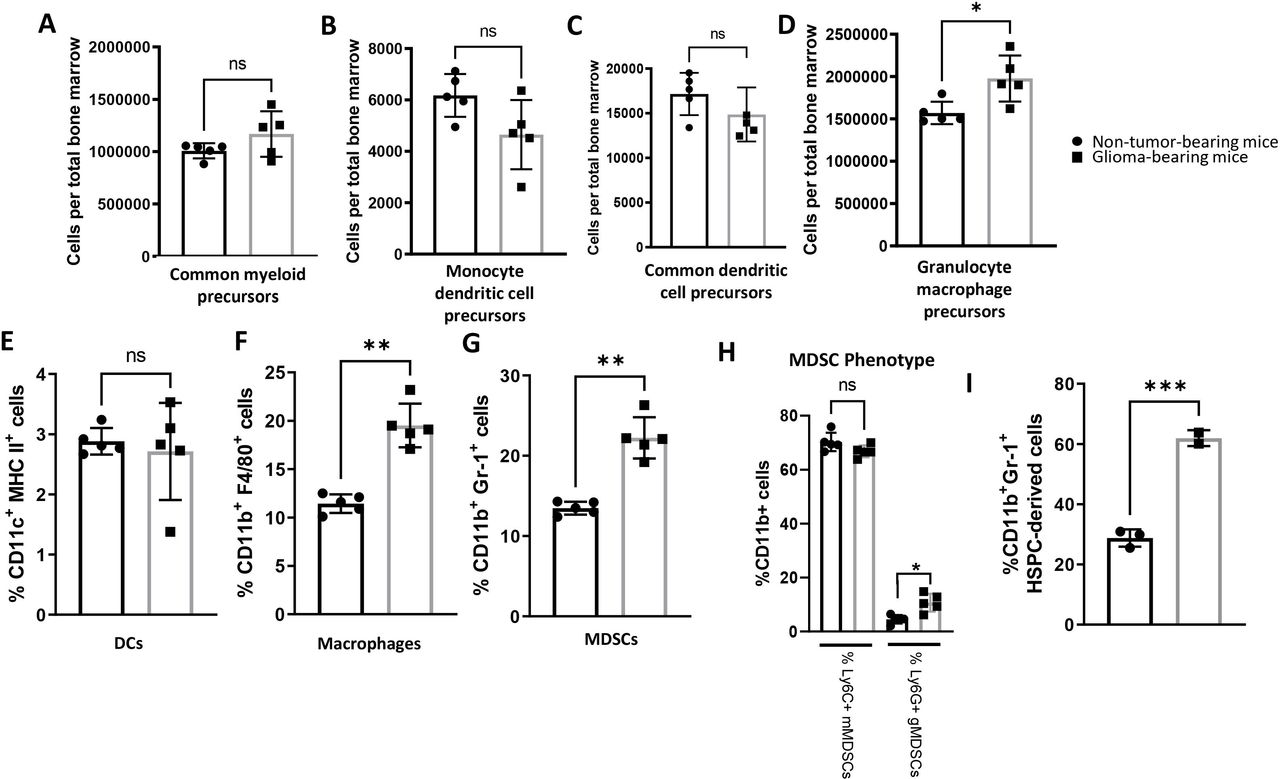
Investigators have found alterations in progenitor populations in a variety of solid tumors, including the sleeping beauty glioma model containing NRA G12V, shATRX, and shp53 with or without IDH1R132H.8–10 These studies, in conjunction with our scRNAseq findings discussed previously led us to hypothesize that disproportional frequencies of immune progenitor populations might be present in the bone marrow of glioma-bearing hosts. To determine if glioma-bearing mice experienced a phenotypic alteration of bone marrow-derived HSPCs and progenitor populations, we assessed various bone marrow populations (online supplemental figure 3A) in mice 28 days post-implantation of orthotopic KR158B-luc glioma cells using flow cytometry (example gating strategies shown in online supplemental figure 3B). Interestingly, a significantly lower frequency of total lineage− HSPCs were observed in glioma-bearing mice compared with non-tumor-bearing age-matched control mice (online supplemental figure 4A). There were no observed differences in the frequency or absolute counts of lin−cKit+Sca-1−CD16/32lo common myeloid precursors (CMPs) (figure 2A and online supplemental figure 4B). We next found that the relative frequency of lin−Sca-1−M-CSFR+Flt3+cKithi monocyte dendritic cell precursors (MDPs) were higher in glioma-bearing mice as measured by percent of total live cells (online supplemental figure 4C). However, there was no significant difference in MDPs as measured by cells per total bone marrow (figure 2B). There also were no observed differences in the relative frequencies or cell count of downstream lin−Sca-1−M-CSFR+Flt3+cKitlo common dendritic cell precursors (CDPs) between glioma-bearing and non-tumor-bearing mice (figure 2C and online supplemental figure 4D). Importantly, compared with their non-tumor-bearing counterparts, glioma-bearing mice were found to have a significantly higher frequency and absolute counts of lin−cKit+Sca-1−CD16/32hi GMPs (figure 2D and online supplemental figure 4E), which give rise to granulocytic MDSCs (gMDSC). gMDSCs contribute to cancer growth and are correlated with poor prognosis in subsets of glioma-bearing hosts.15 We next describe relative frequencies of myeloid populations in the bone marrow.
Supplemental material
Supplemental material

Expansion of immature and mature myeloid cells in bone marrow of intracranial glioma-bearing mice. (A–D) Bone marrow was collected from non-tumor-bearing, age-matched mice and glioma-bearing mice 28 days after implantation. Glioma-bearing mice have higher absolute counts of lin−cKit+Sca-1−CD16/32hi GMPs relative to non-tumor-bearing, age-matched mice, while the frequencies of lin−cKit+Sca-1−CD16/32lo CMPs, lin−cKit+M-CSFR+Flt3+ CDPs, and lin−cKit+Sca-1+M-CSFR+Flt3+ MDPs were similar between groups. Data represent mean±SD. *P<0.05 by Mann-Whitney t-test (n=5 biological replicates). (E–G) Bone marrow was collected from non-tumor-bearing, age-matched mice and glioma-bearing mice 28 days after implantation. Glioma-bearing mice have higher frequencies of CD11b+F4/80+ macrophages and CD11b+Gr-1+ MDSCs but possess similar frequencies of CD11c+MHC II+ DCs as non-tumor-bearing mice. (H) CD11b+Ly6G+ gMDSCs are expanded in glioma-bearing mice, while no difference in CD11b+Ly6C+ mMDSCs are observed. Data represent mean±SD. * **p<0.01, by Mann-Whitney t-test (n=5 biological replicates). I) % CD11b and Gr-1 expression on HSPC-derived cells after in vitro culture. ***p<0.001 by unpaired tudent’s t test (n=3 technical replicates). CDPs, common dendritic cell precursors; CMPs, common myeloid precursors; HSPCs, hematopoietic stem and progenitor cells; MDSCs, myeloid-derived suppressor cells.
A study by Raychaudhuri et al6 previously characterized an expansion of MDSCs in the peripheral blood of patients with GBM. This previous report along with our findings in the bone marrow as described previously prompted us to evaluate the relative frequency of myeloid populations within the bone marrow of glioma-bearing mice. Although no difference in the relative frequency of CD11c+MHC-II+ DCs was observed (figure 2E), we did observe significant expansion of CD11b+F4/80+ macrophages in glioma-bearing mice relative to non-tumor bearing controls (figure 2F). Additionally, an expansion of CD11b+Gr-1+ MDSCs was found in the bone marrow of glioma-bearing mice relative to non-tumor-bearing controls (figure 2G). In addition, phenotypic analysis revealed most MDSCs were of the Ly6C+Ly6G− monocytic population in both non-tumor-bearing and glioma-bearing mice. However, significantly more Ly6C-Ly6G+ gMDSCs were found in the bone marrow of glioma-bearing mice relative to healthy controls (online supplemental figure 5A and figure 2H). Although we are primarily interested in myeloid populations found in the bone marrow, a primary lymphoid organ, we also desired to evaluate the frequency within a secondary lymphoid organs. Within the splenic compartment, we found these myeloid cell population expansions did not hold true, as no difference in the frequencies of macrophages, DCs, or MDSCs were observed in the spleens of glioma-bearing mice relative to non-tumor-bearing controls (online supplemental figure 5B–G), suggesting that these alterations specifically impact the bone marrow primary lymphoid organ but not secondary lymphoid organs. Taken together, these data suggest glioma bearing mice have a remodeling specific to myeloid bone marrow populations with expansions in the macrophage and gMDSC subpopulations.
Supplemental material
To determine if the cell fate differentiation outcomes are altered in steady state conditions, HSPCs from non-tumor-bearing mice and glioma-bearing mice (21 days after tumor implantation) were cultured in RPMI with 10% FBS for 3 days. We found HSPCs derived from glioma-bearing mice had a higher frequency of CD11b+Gr-1+ MDSCs relative to HSPCs derived from non-tumor-bearing mice (figure 2I). These results suggest glioma-bearing HSPCs are predisposed to differentiate into a suppressive cell type.
MDSCs from glioma-bearing mice suppress T cell proliferation and T cell-mediated tumor cell killing
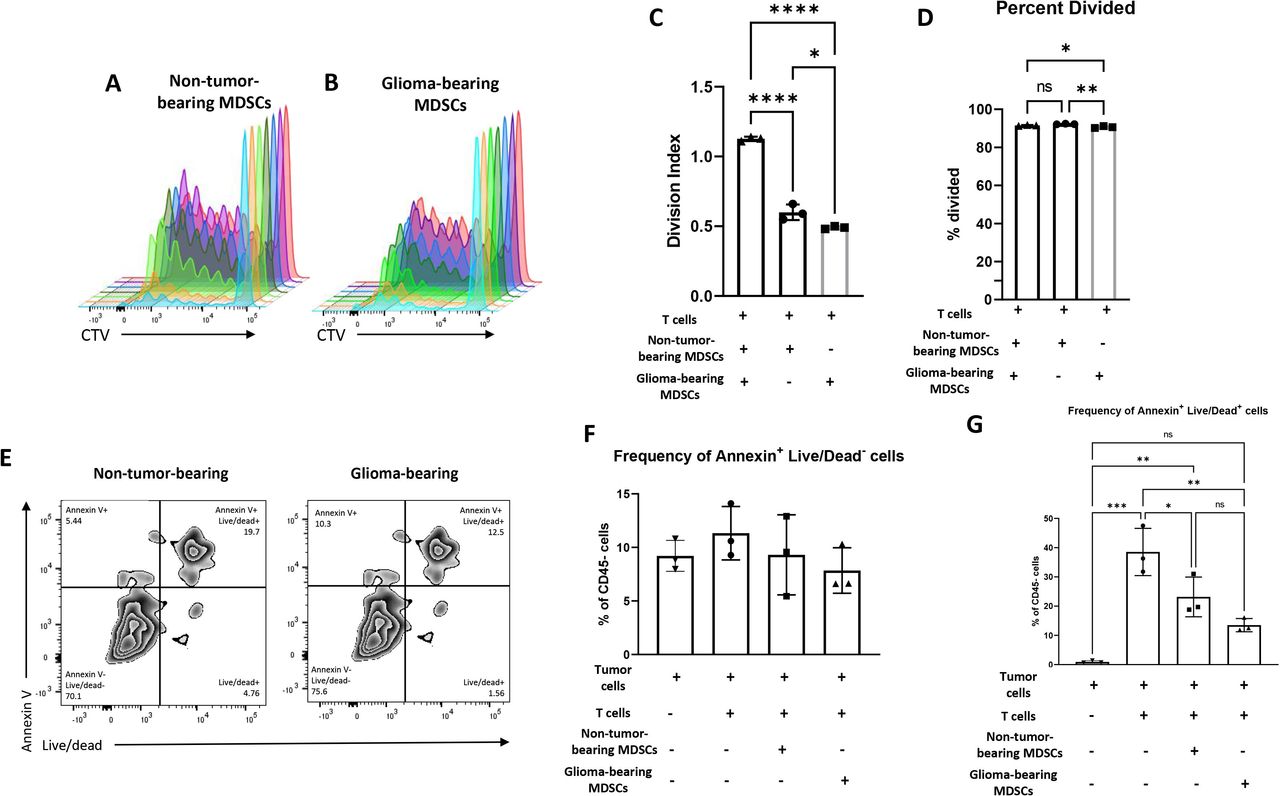
The role of MDSCs in tumor progression and anti-tumor immune suppression has become a focus in the immuno-oncology field.16–21 Our findings thus far have demonstrated disproportional changes in the relative frequencies of the myeloid compartments in glioma-bearing hosts. We then sought to determine if there were functional differences between the MDSCs from glioma-bearing mice and non-tumor-bearing mice. We chose to examine differences in the MDSCs’ capacity to suppress proliferation and killing capacity of tumor-specific T cells. To generate tumor-specific T cells, splenocytes were harvested from C57BL/6 mice that were previously immunized with tumor-RNA-pulsed DCs as previously described.22 2×105 T cells were selected and were cocultured with 6.25×103 MDSCs isolated from either glioma-bearing mice or non-tumor-bearing mice (online supplemental figure 6A,B). Analysis of proliferating cells using CTV (figure 3A,B) revealed that the T cells cocultured with MDSCs from glioma-bearing mice had a significantly lower division index (figure 3C) as well as fewer cells that underwent division (figure 3D) compared with activated T cells that were cocultured with MDSCs from non-tumor bearing mice. This strongly suggests that MDSCs from glioma-bearing mice significantly inhibit T cell proliferation compared with MDSCs from healthy mice.
Supplemental material

MDSCs isolated from glioma-bearing mice possess greater suppressive capacity on T cell proliferation but similar T cell mediated tumor cell killing relative to MDSCs from non-tumor-bearing mice. (A–C) The 6.25×103 bone marrow MDSCs were isolated from non-tumor-bearing (ntMDSC) and glioma-bearing mice (gMDSCs) and cocultured with 2×105 T cells derived from mice primed with KR158B-luc RNA-pulsed DCs. T cells were gated on using CD3+ cells. When cocultured with non-tumor-bearing MDSCs, T cells possessed higher division indices than T cells cocultured with glioma-bearing MDSCs. (D and E) Splenocytes were cocultured with DCs electroporated with KR158B-luc RNA to expand tumor-specific T cells. On expansion, T cells were cultured at a 1:10 ratio with target KR158B-luc glioma cells and a 1:1 ration with ntMDSCs or gMDSCs for 24 hours. Similar frequencies of Annexin+Live/dead- and Annexin+Live/dead+ tumor cells were observed s when cocultured with glioma-bearing MDSCs relative to co-culture with non-tumor-bearing MDSCs. Cells were gated on CD45− tumor cells. Data represent mean±SD. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by one-way analysis of variance (n=3 technical replicates). DCs, dendritic cells; MDSCs, myeloid-derived suppressor cells.
Next, we assessed whether MDSCs from glioma-bearing mice inhibit T cell-mediated tumor cell killing more than MDSCs from non-tumor bearing mice. In this experiment, tumor-specific effector T cells were cultured against target KR158B-luc glioma cells in a 10:1 (effector:target) ratio alone or with MDSCs from glioma-bearing mice or non-tumor-bearing mice (online supplemental figure 6C). Tumor cell killing was determined by expression of Annexin V and Live/Dead on CD45− tumor cells. Flow cytometric analysis revealed no differences in CD45−Annexin+Live/Dead− early apoptotic tumor cells between conditions with either non-tumor-bearing or glioma-bearing MDSCs (figure 3E,F). We also assessed CD45−Annexin+Live/Dead+ late apoptotic tumor cells and observed a non-statistically significant decrease in tumor cells cocultured with glioma-bearing MDSCs relative to tumor cells cocultured with non-tumor-bearing MDSCs (figure 3G). These results suggest that MDSCs from glioma-bearing hosts have a similar capacity to impede T cell mediated tumor cell killing relative to MDSCs derived from non-tumor-bearing mice.
IFNγ secretion from tumor-specific T cells overcomes MDSC skewing in glioma bearing model toward DCs
The use of ACT, including host irradiation and transfer of tumor-reactive T cells, first demonstrated anti-tumor responses in non-CNS-derived malignancies.23–26 In addition, our own ACT platform, which uses tumor RNA-pulsed DCs, tumor-reactive T cells, and irradiation has shown promising results in models of malignant brain tumors.27–29
In murine models, our previous studies demonstrate that HSPCs migrate to the tumor microenvironment of orthotopic gliomas and differentiate into DCs when transferred with immunotherapy.11 12 22 30 In addition, the HSPC-derived cells are capable of inducing T cell activation and had significant amounts of T-cell derived IFNγ at the tumor site.12 Given that bone marrow-derived HSPCs also give rise to MDSCs, we analyzed relative frequencies of HSPC-derived CD11b+ Gr-1+ MDSCs after culture in either media alone or activated T cell supernatants (figure 4A). We found a reduction in the expansion of CD11b+ Gr-1+ MDSCs when glioma-bearing HSPCs were cultured in the activated T cell supernatants relative to when cultured in media alone (figure 4B). In addition, we observed that HSPC differentiation into DCs is driven by activated tumor-reactive T cell-derived supernatant (figure 4C,D).

Components of ACT platform are capable of redirecting HSPC differentiation outcomes. (A and B) Expression of CD11b+ Gr-1+ cells after 3 days of in vitro culture in RPMI with 10% FBS either alone or conditioned with supernatants of activated T cells. Conditioned media was a 1:1 mix of 1 mL RPMI with 10% FBS and 1 mL of acellular supernatants generated from activated T cell cultures with cognate antigen-pulsed DCs (Wildes et al 201812). (C and D) Expression of CD11c+MHC II+ cells on HSPC-derived cells after in vitro culture. (E) HSPCs derived from C57BL/6 or IFNyR−/− mice were cultured in RPMI with 10% FBS alone or supplemented with recombinant mouse IFNy and differentiation of CD11c+MHC II+ cells was evaluated 3 days later via flow cytometry.(F) scRNAseq expression of IFNyR1 from non-tumor-bearing and glioma-bearing HSPCs. (G) Visualization of IFNyR1 expression overlayed on UMAP projections. (H and I) HSPCs from non-tumor-bearing or glioma-bearing mice were harvested 28 days after implantation and flow cytometry was used to evaluate expression of IFNyR1 and IFNyR2 on lineage− cells. All data represent the mean±SD. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by one-way analysis of variance (ANOVA) (n=3 technical replicates) for in vitro studies and Mann-Whitney t-test (n=10 biological replicates) for ex vivo studies. ACT, adoptive cellular therapy; HSPC, hematopoietic stem and progenitor cell.
We next hypothesized that IFNγ derived from T cell supernatant was driving DC expansion. To determine this, we isolated HSPCs from either wild-type mice or IFNγ receptor one knockout (IFNγR1−/−) mice and cultured with recombinant IFNγ (figure 4E). Only the HSPCs from wild-type mice upregulated CD11c and MHCII, confirming that DC differentiation in these populations are driven by IFNγ.
We next probed our scRNAseq data and found higher IFNyR1 expression in glioma-bearing cells relative to cells from non-tumor-bearing mice (figure 4F). Interestingly, we found IFNyR1 to be most highly expressed in cluster 2 (GMPs), the population we found to be expanded in glioma-bearing hosts (figure 4G). In addition, we found high levels of IFNyR1 in clusters 4 (monocytes) and 5 (conventional DCs), two antigen-presenting cell populations. Given this finding, we sought to determine expression of IFNγR1 and IFNγR2 in HSPCs from non-tumor-bearing and glioma-bearing mice. We found HSPCs from glioma-bearing mice possess a higher frequency of IFNγR1+ (figure 4H) and IFNγR2+ lineage- cells (figure 4I) relative to HSPCs from non-tumor-bearing mice (online supplemental figure 7A). In addition, glioma-bearing mice possessed higher gMFI of IFNγR1 and IFNγR2 (online supplemental figure 7B,C). Furthermore, glioma-bearing mice possessed more IFNγR1+ and IFNγR2+ cells (online supplemental figure 7D,E) on cKit+Sca1− myeloid precursors relative to non-tumor-bearing mice. These results suggest the higher expression of IFNγR on HSPCs from glioma-bearing mice may drive DC expansion in the context of IFNγ-containing T cell supernatants.
Supplemental material
Irradiation abrogates glioma-induced myeloid precursor and mature myeloid cell expansion
Our studies have demonstrated that irradiation is required for the efficacy of ACT.11 12 22 27 28 30 Earlier, we observed glioma-bearing mice experience relative expansions of MDSCs and macrophages in their bone marrow (figure 2). Given these results, we sought to determine if irradiation impacts IFNγR1 and IFNγR2 expression in glioma-bearing mice. To evaluate this, glioma-bearing mice were irradiated with 5 Gy total body irradiation (TBI), and 7 days later, expression of IFNγR1 and IFNγR2 on HSPCs was evaluated via flow cytometry in irradiated and non-irradiated control mice (figure 5A). We found mice subjected to TBI had significant upregulation of IFNγR1+ (figure 5B) and IFNγR2+ (figure 5C) relative to non-tumor-bearing mice on lin− bone marrow cells. In addition, we found upregulation of IFNγR1 and IFNγR2 (online supplemental figure 8A,B) by gMFI in irradiated glioma-bearing mice compared with non-irradiated glioma-bearing controls. Furthermore, frequencies of IFNγR1 and IFNγR2 were found to be higher on cKit+ Sca-1− myeloid precursors (online supplemental figure 8C,D). These results indicate that irradiation induces IFNγR1 and IFNγR2 upregulation in glioma-bearing hosts.
Supplemental material
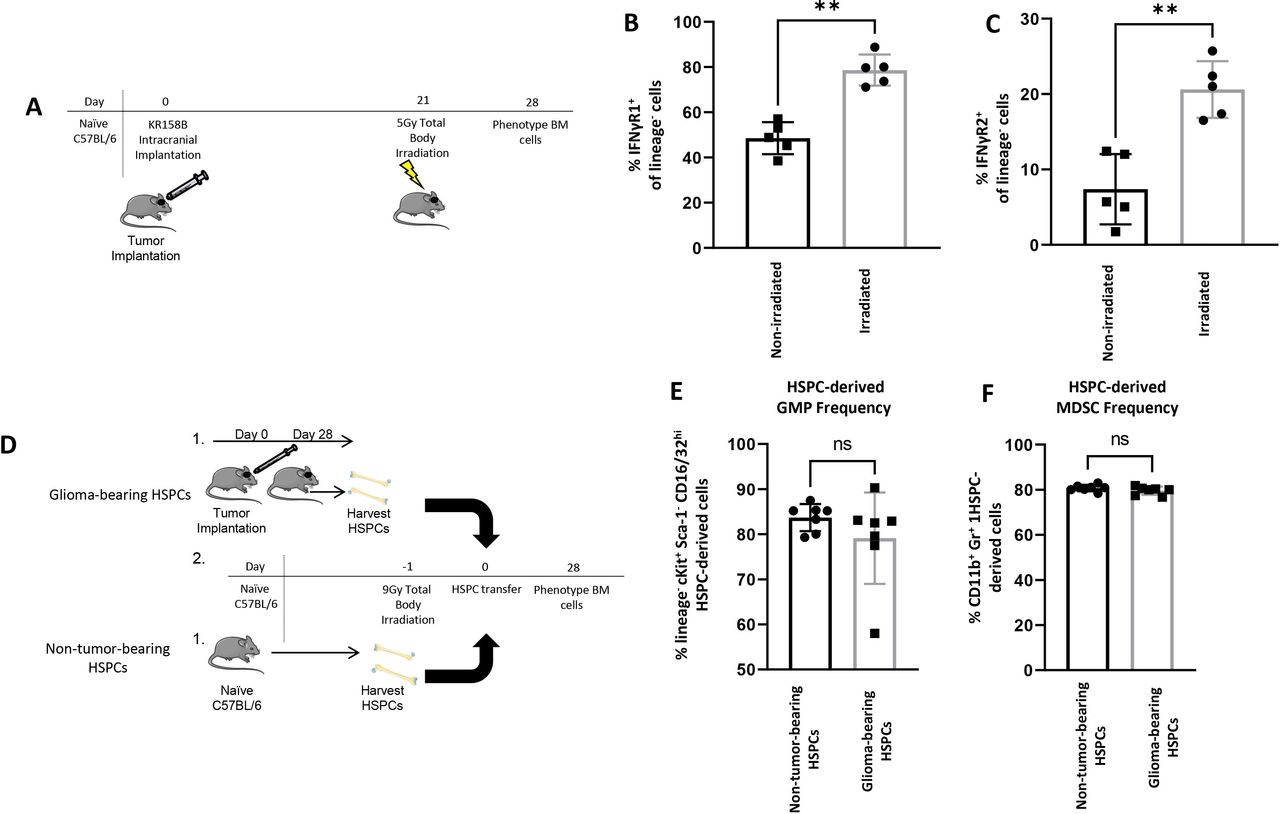
Irradiation abrogates myeloid cell precursor and mature myeloid cell expansion and impacts IFNγR1 and IFNγR2 expression. (A–C) Mice were injected with KR158B-luc cells and 21 days later, half of the mice were subjected to 5 Gy total body irradiation. Seven days later, bone marrow was obtained from both groups and frequencies of IFNyR1+ and IFNyR2+ cells were evaluated on lineage− HSPCs. Mice subjected to total body irradiation were found to possess more IFNyR1+ and IFNyR2+ lineage- cells via flow cytometry. (D–F) A cohort of non-tumor-bearing mice were injected with KR158B-luc cells while a separate cohort remained non-tumor-bearing. Twenty-eight days after implantation, both cohorts were sacrificed and lineage− HSPCs were FACS sorted and then either non-tumor-bearing or glioma-bearing HSPCs were transferred into C57BL/6 that were subjected to 9 Gy total body irradiation the day prior. Twenty-eight days after HSPC transfer, bone marrow was collected and stained for flow cytometry. No significant differences in the frequencies of lin−cKit+Sca-1−CD16/32hi GMPs and CD11b+Gr-1+ MDSCs between mice that received non-tumor-bearing HSPCs and those that received glioma-bearing HSPCs. All data represent the mean±SD. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by Mann-Whitney t-test (n=5 biological replicates). HSPCs, hematopoietic stem and progenitor cells.
Next, we determined if irradiation impacts how transferred HSPC-derived cells from either glioma-bearing or non-tumor-bearing mice differentiate in the bone marrow. To determine this, naïve C57BL/6 mice were subjected to myeloablative TBI and the following day, injected with sorted lin− HSPCs derived from non-tumor-bearing DsRed mice or from glioma-bearing DsRed mice 28 days after tumor implantation (figure 5D). HSPCs were allowed to engraft in non-tumor-bearing hosts and differentiate for an additional 28 days after transfer before the phenotype of BM cells was evaluated via flow cytometry. No difference in the frequency of GMPs (figure 5E) or MDSCs (figure 5F) were observed between mice that received non-tumor-bearing HSPCs and mice that received glioma-bearing HSPCs. In addition, no difference in the frequency of HSPC-derived CMPs (online supplemental figure 8C), macrophages (online supplemental figure 8D), or DCs (online supplemental figure 8E) were observed between mice that received non-tumor-bearing and glioma-bearing HSPCs. Together, these data suggest myeloablative TBI abrogates the myeloid precursor and mature myeloid cell expansion that is observed in glioma-bearing mice.
ACT induces GMP reduction
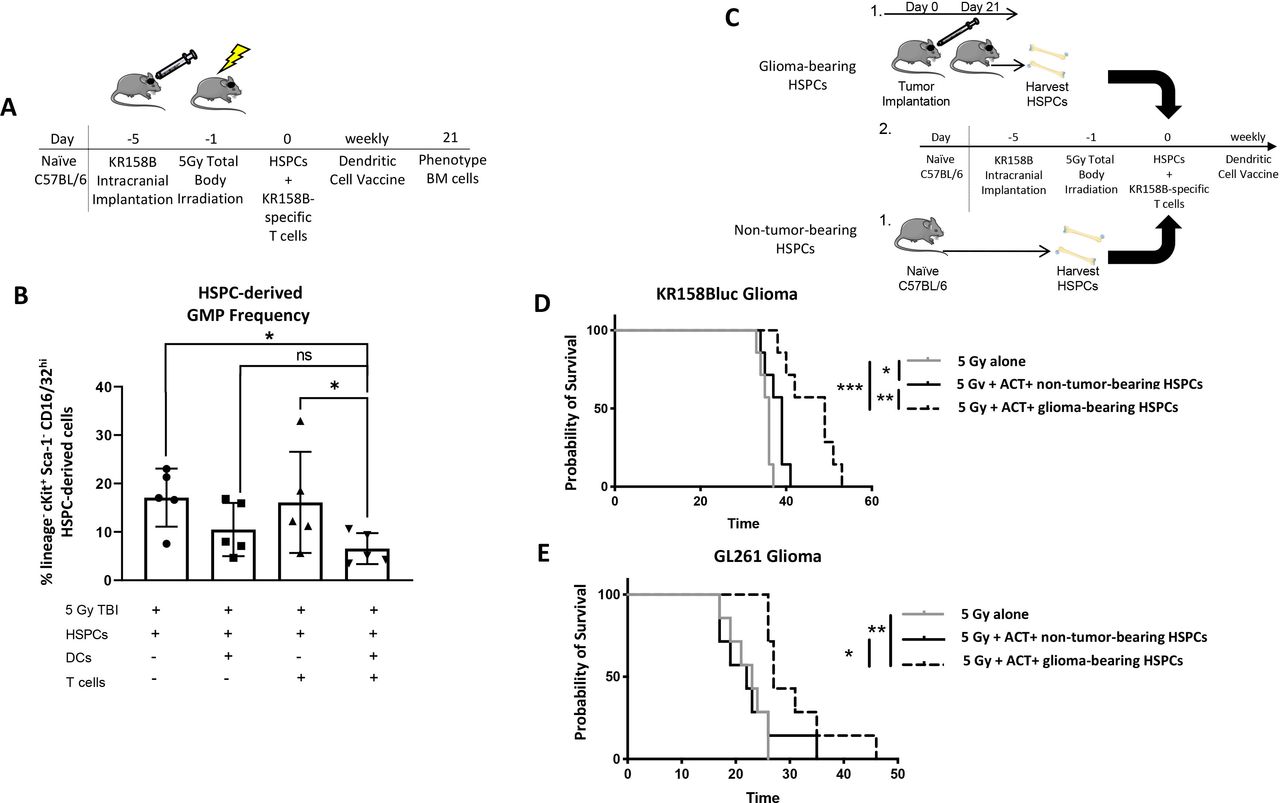
We next sought to determine if ACT could reduce the frequency of MDSC precursors, the GMP population (figure 6A). We found, relative to the control cohort that received lymphodepletive TBI and HSPCs alone, mice that received ACT had a significantly lower frequency of GMPs (figure 6B and online supplemental figure 9A). Interestingly, we found no difference in the frequency of myeloid precursors or CMPs (online supplemental figure 9B,C). These data suggest the ACT platform reverses the myeloid cell expansion observed in glioma-bearing mice.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACT abrogates GMP expansion and in context of glioma-bearing HSPCs, provides superior survival benefit relative to non-tumor-bearing HSPCs. (A) Glioma-bearing mice were treated with components ACT, and 21 days later, GMP frequency was evaluated via flow cytometry. (B) Mice treated with TBI, HSPCs, tumor RNA-pulsed DCs, and tumor-specific T cells were found to have significantly less GMPs compared with mice treated with TBI and HSPCs as well as mice treated with TBI, HSPCs, and tumor-specific T cells. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by Mann-Whitney t-test (n=5 biological replicates). (C) After glioma-bearing mice were treated with our ACT platform using HSPCs from either non-tumor-bearing mice or glioma-bearing mice, survival was evaluated. (D). Survival curve of KR158B-luc-bearing animals treated with 5 Gy, ACT, and non-tumor-bearing or glioma-bearing HSPCs. (E). Survival curve of GL261-bearing animals treated with 5 Gy, ACT, and non-tumor-bearing or glioma-bearing-HSPCs. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by Mantel-Cox log-rank test for survival experiments (n=7 biological replicates). ACT, adoptive cellular therapy; DCs, dendritic cells; GMP, granulocyte macrophage precursor; HSPCs, hematopoietic stem and progenitor cells; TBI, total body irradiation.
ACT reverses bone marrow immunosuppression in glioma-bearing mice
Our previous publications describe that treatment of gliomas with ACT leads to decreased intratumoral MDSCs.11 12 This combined with the above data stating that: (1) irradiation allows upregulation of IFNγR1 and IFNγR2 and (2) ACT also leads to overall reduced GMPs, which are MDSC precursors prompted us to determine if ACT using glioma-bearing HSPCs would provide similar survival outcomes to mice treated with ACT using non-tumor-bearing HSPCs. To evaluate this, mice were intracranially implanted with glioma cells and treated with ACT with either non-tumor-bearing or glioma-bearing HSPCs (figure 6C). We found in both KR158B-luc and GL261 murine glioma models, animals treated with ACT using glioma-bearing HSPCs possessed a significant survival benefit relative to animals treated with ACT using non-tumor-bearing HSPCs (figure 6D,E). These data suggest ACT using glioma-bearing HSPCs provides superior survival benefit relative to ACT using non-tumor-bearing HSPCs.
Discussion
HSPCs are canonically known for their ability to rescue bone marrow after high dose chemotherapy or irradiation and their ability to provide a graft versus tumor response in the setting of an allogeneic transplant. Our group and others are capitalizing on an additional function of HSPCs, as immune effectors when combined with immunotherapeutic strategies. We are actively investigating these combinatorial therapies in children and young adults with high grade and brain stem gliomas (NCT03334305 and NCT03396575).
The expansion of GMPs described in peripheral cancers, and more recently in the sleeping beauty model of murine glioma,8–10 along with the observed immune cell dysfunction in GBM patients in both myeloid and lymphoid compartments5 6 prompted us to investigate the impact intracranial gliomas on multipotent stem and progenitor cells in the bone marrow. In this study, scRNAseq and flow cytometry are used to demonstrate that the presence of intracranial gliomas alters the HSPC landscape. Specifically, we found an increase in GMPs in glioma-bearing mice relative to non-tumor-bearing controls.
However, we believe this investigation into the role of CNS malignancies on HSPCs is becoming more appreciated. There have been recent studies evaluating the bone marrow within the skull compartment31 32 and although our studies were specifically interested in the HSPCs in the tibia and femur, future investigations into the skull bone marrow contributions in the context of brain tumors should be conducted. Furthermore, recent research has shown HSPCs are able to populate human gliomas, can promote glioma cell proliferation and expression of suppressive molecules and are negatively associated with overall and progression-free survival.33 In addition, one group of researchers identified GMPs as a functionally suppressive cell population.34 Similar studies should be conducted to delineate the function of these expanded myeloid precursors in the context of malignant gliomas.
Previously, most research on MDSCs has been in the context of tumor microenvironment.35–37 More recently, studies have focused on the role of MDSCs in the periphery.9 15 In addition, several reviews have been published that evaluate the role of myeloid cells in the context of cancer.17 38 39 These reports provoked us to evaluate the frequency of mature myeloid cells in the bone marrow of glioma-bearing mice. We found macrophages and MDSCs are expanded in glioma-bearing mice relative to non-tumor-bearing controls. Phenotypic analysis of MDSCs revealed while majority of cells belong to the Ly6C+ mMDSC population, glioma-bearing mice possess a higher frequency of Ly6G+ gMDSCs than non-tumor-bearing control mice. Functional analysis demonstrated bone marrow MDSCs isolated from glioma-bearing mice possess greater ability to suppress T cell proliferation but similar capacity to suppress T cell-mediated tumor cell killing. We hypothesize this impact of gliomas on myeloid cell frequency and function within the bone marrow compartment is due to secreted cytokines and growth factors, such as G-CSF, derived from the tumor or supporting cells within the tumor microenvironment.9 Further examinations are underway to determine the exact factors that cause MDSC expansion and enhanced suppression.
We next shifted our focus to evaluating the impact of components of our ACT platform on HSPC differentiation. We found treatment with activated T cell/DC supernatants redirected glioma-bearing HSPCs differentiation from MDSCs in favor of DCs, which was found to be driven by IFNγ. scRNAseq analysis revealed increased expression of IFNγR1 expression in glioma-bearing HSPCs relative to non-tumor-bearing HSPCs, and expression was highest in GMPs, monocytes, and DCs. Given the known function of IFNγ to mediate antitumor effects,40–43 we wanted to determine if irradiation could induce IFNγR expression on glioma-bearing HSPCs. We found irradiation induced IFNγR expression on glioma-bearing HSPCs relative to non-tumor-bearing HSPCs. Together, these results suggest glioma-bearing mice may possess higher IFNγR expression on HSPCs that is further induced by irradiation.
Furthermore, we found mice transferred with HSPCs from glioma-bearing mice had similar frequencies of GMPs and MDSCs compared with mice transferred with non-tumor-bearing HSPCs. We then evaluated the frequency of HSPCs within treated mice when all four components of our ACT platform (TBI, HSPCs, tumor RNA-pulsed DCs, and tumor-specific T cells) were used in conjunction. We found relative to control mice, mice treated with ACT had significantly reduced expansion of GMPs.
Finally, we evaluated the efficacy of our ACT platform using HSPCs isolated from glioma-bearing mice versus non-tumor-bearing mice. Using the KR158B-luc model and lymphodepletive TBI, we found mice treated with glioma-bearing HSPCs had superior survival relative to mice treated with non-tumor-bearing HSPCs and mice treated with irradiation and HSPCs or irradiation, HSPCs, and tumor-reactive T cells. In addition, using the GL261 model, we again found mice treated with glioma-bearing HSPCs had significantly prolonged survival relative to mice treated with non-tumor-bearing HSPCs and untreated control mice. Together, these results suggest glioma-bearing mice treated with ACT can overcome the inherent MDSC bias to experience superior survival. However, further investigations are needed to thoroughly evaluate the mechanism behind the enhanced survival efficacy observed in the context of ACT with glioma-bearing HSPCs.
Supplemental material
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
All studies were approved by the University of Florida Institutional Animal Care and Use Committee (IACUC # 201910891).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @BayliDean, @TheMatSebastian
BDD and TW contributed equally.
Contributors BDD and TW designed the study and analyzed the data. JD and CY contributed analysis tools and performed analysis. BDD, TW, OY, DS, CPF, JWF, LFF, GM, BF, CK and BW performed the experiments. BDD, TW and MS wrote the paper with input from all authors. CTF and DM conceived and supervised the study. CF serves as the guarantor.
Funding This work was supported by the National Cancer Institute (T32CA257923 to BDD) and the National Institute of Neurological Disorders and Stroke (R01NS112315 and R01NS111033 to CF). Additionally, this work was supported by Alex’s Lemonade Stand Foundation Young Investigator grant (to CF).
Competing interests CTF and DM hold interest in iOncologi, Inc, a biotechnology company focused on immuno-oncology. Other authors declare no conflicts of interest.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.