Article Text

Abstract
Background Oncolytic immunotherapy represents a unique therapeutic platform for the treatment of cancer. Here, we evaluated the safety and efficacy of the combination of pexastimogene devacirepvec (PexaVec) plus durvalumab (anti-programmed death ligand 1) with and without tremelimumab (anti-cytotoxic T-lymphocyte associated protein 4) in patients with standard chemotherapy refractory mismatch repair proficient (pMMR) metastatic colorectal cancer (mCRC) in a phase I/II trial.
Methods Adult patients with histologically confirmed advanced pMMR mCRC, who had progressed on at least two prior lines of systemic chemotherapy were studied in four cohorts. Patients received four doses of PexaVec IV at a dose of 3×108 plaque forming units (pfu) (dose level 1) or 1×109 pfu (dose level 2) every 2 weeks. Twelve days after the first PexaVec administration, patients received either 1500 mg of durvalumab every 28 days alone or an additional single dose of 300 mg tremelimumab on day 1. Responses were assessed every 8 weeks by CT or MRI. AEs were recorded. The primary endpoints were safety and feasibility. Secondary endpoints included progression-free survival (PFS) and overall survival. Paired tumor samples and peripheral blood were collected to perform immune monitoring.
Results Thirty-four patients with mCRC enrolled on to the study: 16 patients in the PexaVec/durvalumab cohorts and 18 patients in the PexaVec/durvalumab/tremelimumab cohorts. Overall, the combination of PexaVec plus immune checkpoint inhibitors did not result in any unexpected toxicities. Most common toxicities observed were fever and chills after PexaVec infusion. Two cases of grade 3 colitis, one case of a grade 2 myositis and one case of grade 3 hypotension resulted in discontinuation of immune checkpoint inhibitor and PexaVec treatment, respectively. The median PFS in the PexaVec/durvalumab/tremelimumab cohorts was 2.3 months (95% CI: 2.2 to 3.2 months) vs 2.1 months (95% CI: 1.7 to 2.8 months; p=0.57) in the PexaVec/durvalumab cohorts. Flow cytometry analysis of peripheral blood mononuclear cells revealed an increase in Ki67+CD8+ T cells on treatment.
Conclusion PexaVec in combination with durvalumab and tremelimumab is safe and tolerable. No unexpected toxicities were observed. The combination of PexaVec/durvalumab/tremelimumab demonstrated potential clinical activity in patients with pMMR mCRC, but further studies are needed to identify the predictive biomarkers.
Trial registration number NCT03206073.
- immunotherapy
- oncolytic viruses
- clinical trials, phase II as topic
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Oncolytic virotherapy acts by directly killing tumor cells, thereby promoting in situ vaccination causing tumor-specific immune responses. Both intra-tumoral and intravenous injection of PexaVec, a genetically engineered oncolytic vaccinia virus expressing granulocyte macrophage colony-stimulating factor, have been previously tested in various types of cancer.
WHAT THIS STUDY ADDS
This study demonstrates that it is safe and feasible to combine intravenous PexaVecc with tremelimumab (anti-CTLA4) and durvalumab (anti-PD-L1) as systemic treatment in patients with mismatch repair proficient colon cancer. There is preliminary data of potential efficacy, but further studies are needed to confirm.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study provides safety and feasibility data for future studies combining oncolytic virus therapy such as PexaVec with combined immune checkpoint inhibitors in immunological cold tumors.
Background
Colorectal cancer (CRC) remains the third most common cause of cancer death in both men and women in the USA.1 The treatment of metastatic disease remains challenging even with the development of targeted therapies and immunotherapy that improve the outcomes for small subgroups of patients with certain tumor characteristics. Therapeutic options in the third-line setting after treatment with fluoropyrimidine, oxaliplatin and irinotecan-based chemotherapy, anti-vascular endothelial growth factor (VEGF) agents and anti-epidermal growth factor (EGFR) therapies, if applicable, provide only minimal improvement in overall survival (OS) and progression-free survival (PFS). For example, the oral multi-kinase inhibitor regorafenib has demonstrated an increase of 1.4 months in OS while the oral cytotoxic agent trifluridine–tipiracil (TAS-102) has shown an increase of 1.8 months in OS.2–4
Immunotherapy has been successful in the treatment of multiple solid tumors. However, this benefit has not translated into the metastatic colorectal cancer (mCRC) arena. Only a small percentage of patients with mCRC whose tumors present MMR gene abnormalities respond to immunotherapy.5–8
Pexastimogene devacirepvec (PexaVec, JX-594) is a genetically engineered oncolytic vaccina virus. Oncolytic immunotherapy represents a novel therapeutic platform for the treatment of cancer with unique attributes compared with conventional chemotherapy. Oncolytic viruses preferentially replicate in malignant tumor cells inducing immunogenic cell death, while sparing normal cells. As a result of modification, the virus has disruption of the viral thymidine kinase gene while expressing the human granulocyte macrophage colony-stimulating factor (hGM-CSF) and β-galactosidase transgenes under control of the synthetic early/late and p7.5 promoters. It has been designed and used to promote an antitumor immune response.9–11 Vaccinia virus, when administered intravenously (IV), shows an inherent selectivity toward tumors. Activation of the EGFR pathway has been shown to drive vaccina replication and VEGF stimulates PexaVec replication in tumor cells and tumor-associated endothelial cells ultimately leading to preferential replication in tumors.9 12–14 So far, PexaVec has only been studied as single agent in a small cohort of patients with CRC.15 16
PexaVec treatment induces a proinflammatory tumor environment through tumor cell death and subsequent release of tumor antigens, which is optimal for combination with immunotherapy.17 Data with immunomodulatory agents demonstrates a high correlation of tumor-infiltrating T cells and immunotherapy targets such as PD-L1 as a response to this treatment combination.18 Furthermore, vaccinia has shown to ligate toll-like receptors (TLRs) 2 and 8, initiating an acute inflammatory response and promoting immune cell infiltration into tumors.19 Finally, GM-CSF produced by PexaVec activates mature dendritic cells, thereby promoting and priming the activation of cytotoxic T cells.16
Here, we reported the results of a phase I/II study of PexaVec oncolytic virus in combination with immune checkpoint inhibition (ICI) in refractory mCRC.
Methods
Patient characteristics and trial design
Primary eligibility criteria were age 18 years or older with histologically confirmed microsatellite stable (MSS) mCRC by the Laboratory of Pathology of the National Cancer Institute (NCI). Patients who had a known KRAS wild-type (WT) tumor must have progressed or been intolerant to cetuximab or panitumumab-based chemotherapy. Additional key eligibility criteria included disease not amenable to curative resection and progression on chemotherapy. Eastern Cooperative Oncology Group (ECOG) performance status score of 0 or 1; adequate hepatic, renal and hematological function; at least one focus of intolerance to at least one line of oxaliplatin or irinotecan containing, fluorouracil-based, chemotherapy regimen. ECOG performance status score of 0 or 1; adequate hepatic, renal and hematological function; at least one focus of measurable metastatic disease per Response Evaluation Criteria in Solid Tumors (RECIST) V.1.1; and at least one focus of metastatic disease amenable to pretreatment and on-treatment biopsies. The biopsied lesions were not one of the target measurable lesions. Patients with prior immune checkpoint inhibitor treatment or with brain metastasis were excluded.
The study consisted of four patient cohorts treated with PexaVec oncolytic virus at two dose levels in combination with either durvalumab or tremelimumab plus durvalumab (online supplemental figure 1). All patients received durvalumab at a dose of 1500 mg IV every 28 days. Cohort A1 also received treatment with PexaVec IV at a dose of 3×108 plaque forming units (pfu) (DL1), whereas cohort A2 received PexaVec IV at a dose of 109 pfu (DL2). Patients in cohorts B1 and B2 received a single dose of tremelimumab treatment on day 1 plus durvalumab at a dose of 1500 mg IV every 28 days. Cohort B1 received treatment with PexaVec IV at a dose of 3×108 pfu (DL1), whereas cohort B2 received PexaVec IV at a dose of 109 pfu (DL2). PexaVec treatment was started on day −12 followed by administration on D1, 15 and 29 (four planned doses in total). The dosing of PexaVec every 2 weeks was based on the prior study of PexaVec in this patient population that showed safety of this dosing.15 The cycle length was 28 days and treatment continued until disease progression, unacceptable toxicity or withdrawal of consent. Continuation of treatment after radiographic disease progression was not permitted. Patients were imaged after 2 cycles of ICI treatment (ie, 10 weeks after the first PexaVec dose) and every 8 weeks thereafter. Patients were considered evaluable for response to treatment if they had received at least three of the four planned PexaVec doses. The imaging could be done prior to the 10-week first imaging timepoint if needed. Staging was performed by radiographic assessment of contrast-enhanced CT scan at baseline, after 2 cycles of ICI treatment (ie, 10 weeks after the first PexaVec dose) and every 8 weeks thereafter. Objective response was evaluated using the RECIST V.1.1 criteria.
Supplemental material
Research biopsies of the same lesion were performed at baseline (before D −12) and either after one dose of PexaVec treatment only (D1) or after treatment with PexaVec and ICI (D29). Safety and tolerability were assessed from the first dose of study treatment by the incidence of treatment-related adverse events (AEs) and by severity and type of AE per the National Cancer Institute Common Terminology Criteria for Adverse Events V.4.0.
Bulk RNA-seq and data analysis
RNA was extracted from fresh-frozen needle biopsy tumor samples at baseline and post-treatment from 13 patients. One patient (P7) had a third biopsy taken at the time of progression (11 months after enrollment). RNA integrity and quantitation were assessed using the RNA Nano 6000 Assay Kit of Bioanalyzer 2100 system (Agilent Technologies, California, USA). Sequencing libraries were prepared using the NEB next Ultra RNA Library Prep Kit for Illumina (NEB, USA) following manufacturer’s recommendations and index codes were added to attribute sequences to each sample. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using PE Cluster Kit cBot-HS (Illumina) according to the manufacturer’s instruction. After cluster generation, the library preparations were sequenced on an Illumina platform and paired-end reads were generated. The quality of sequenced reads was assessed per sample using FastQC (V. 0.11.5), Preseq (V. 2.0.3),20 Picard tools (V. 1.119)21 and RSeQC (V. 2.6.4).22 Low-quality bases and adapter sequences were removed from reads using Cutadapt (V. 1.18).23 The trimmed reads were aligned to the GRCh38 human genome (GENCODE hg38, V. 30). Gene and isoform expression levels are quantified using RSEM (V. 1.3.0). Distribution of read counts per gene revealed that samples P12_Bx and P4_Bxp had a higher number of lowly expressed genes and were not included in further analysis. Genes with a log2 (CPM)>3 in at least 10 samples were selected for analysis. Immune cell signatures for each patient were identified using quanTIseq24 from the Immunedeconv R package.25
Whole exome sequencing and data analysis
DNA was extracted from fresh-frozen needle biopsy tumor samples. The genomic DNA sample was randomly fragmented by sonication (Covaris, Massachusetts, USA) to the size of 180–280 bp fragments. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities and enzymes were removed. Library preparation was performed using the Agilent SureSelect XT Human All Exon v7+UTR kit. All samples were sequenced on an Illumina platform (PE150) according to effective concentration of library and data amount needed per sample. Low-quality and adapter sequences were trimmed from the raw sequencing reads using Trimmomatic (V. 0.39).26 Trimmed reads were then aligned to the human hg38 reference genome using BWA mapping software (V. 0.7.17).27 Duplicate reads were marked using Samblaster (V. 0.1.25)28 and sorted using samtools (V. 1.8). Finally, base quality score recalibration was performed as indicated in the GATK4 (V. 4.2.2.0) best practices. Sequence, mapping and variant statistics were computed using FastQC (V. 0.11.9), Qualimap (V. 2.2.1)29 and SNPeff (V. 4.3t). Somatic variant calling (SNPs and Indels) was performed using Mutect (V. 1.1.7),30 Mutect2 (GATK V. 4.2.0), Strelka2 (V. 2.9.0)31 and VarDict (V. 1.4)32 in tumor–normal mode. Variants from all callers were merged using the CombineVariants tool from GATK (V.3.8.1). Genomic, functional and consequence annotations are added using Variant Effect Predictor (VEP V. 99)33 and converted to Mutation Annotation Format (MAF) using the vcf2maf tool (V. 1.6.16). Variant signatures and Oncoplots were generated using maftools package in R.34
Flow cytometry immunophenotyping
Peripheral blood mononuclear cells (PBMCs) were isolated from patient blood samples collected at baseline and on D1 of C1 and C2. Multi-color flow cytometry was performed to study different immune cell subsets using the following antibodies: CD19 (HIB19), CD11c (B-ly6), CD27 (O323), PDL1 (MIH1), HLA-DR (L243), CD16 (3G8), CD11b (ICRF44), CD38 (S17015A), TCR γ/δ (B1), CD3 (UCHT1), CD14 (M5E2), CD33 (WM53), CD56 (QA17A16), FoxP3 (259D), CD8 (SK1), TIM3 (F38-2E2), CD45RA (HI100), CD4 (RPA- T4), CCR7 (150503), ICOS (DX29), PD1 (EH12.1), CD25 (M-A251), CTLA4 (L3D10), Ki67 (Ki67), CD127 (A019D). Data for all samples were collected on a CytoFLEX LX flow cytometer (Beckman Coulter CytoFLEX Flow Cytometer) and analyzed using FlowJo software (FlowJo). Gating strategies were performed as previously described35 36 and are shown in online supplemental figure 2. For unsupervised analysis of high-dimensional flow cytometry data, .fcs files were imported into FlowJo and gated on either live, CD45+ leukocytes (for immune profiling panel, online supplemental figure S4) or live, CD3+ lymphocytes (for T cell profiling). These populations were then down sampled and exported as .csv files. After import of these .csv files, graph-based clustering and tSNE projections could be generated using the PartekFlow software pipeline (Build V.10.0.22.0703, Partek, St. Louis, Missouri, USA).
Statistical methods
The primary objective of this study was to determine the safety and tolerability of the combination of PexaVec with ICI in patients with refractory mCRC. Secondary objectives were response rate based on RECIST V. 1.1, PFS and OS. Patients who progressed prior to receiving ICI were considered non-evaluable. Kaplan-Meier method reported with 95% CIs was used to calculate PFS and OS. The significance of the difference between the Kaplan-Meier curves was determined by a log-rank test. PFS was defined as the time from the date of consent to the date of first documentation of disease progression or death. OS was defined as the time between the date of consent and last follow-up or death. Statistical analysis was performed using SAS V.9.4 (SAS Institute) and Graph Pad Prism 9 (San Diego, CA).
Results
Trial layout and safety
Between December 2017 and August 2020, a total of 34 patients with advanced MMR proficient CRC were enrolled into the study: 16 patients in the PexaVec/durvalumab cohorts and 18 patients in PexaVec/durvalumab/tremelimumab cohorts (online supplemental figure 3). The baseline characteristics of the patients are shown in online supplemental table 1. The median age of the population was 55 years (range 28–76), and 59% of patients were female. Twenty-seven (79%) of 34 patients had an ECOG performance status score of 0, with the remaining having a score of 1. The most common primary site of disease in both treatment groups was the left colon, and the most frequent sites of metastasis were the liver and the lung. Most patients had received two or more systemic chemotherapy regimens in the metastatic setting.
The median duration of therapy was 1.9 months (range 0.8–2.9) in the PexaVec/durvalumab cohort and 1.0 month (range 0.8–1.2) in the PexaVec/durvalumab/tremelimumab cohort; 87.5% of the patients in the PexaVec/durvalumab cohort received four doses of PexaVec, while only 50% of the patients in the PexaVec/durvalumab/tremelimumab cohort received the planned four doses (table 1). The most common reason for early discontinuation of treatment in both groups was radiologically confirmed disease progression (four patients). Three patients in the PexaVec/ durvalumab/tremelimumab cohort discontinued treatment early because of treatment-related toxicity (in two cases (colitis and myositis) possibly related to ICI therapy and in one case (hypotension) possibly related to PexaVec treatment). The median follow-up in the PexaVec/durvalumab cohort was 26.3 months (range 1.1–26.3) and 14 months (range 1.1–18.8) in the PexaVec/durvalumab/tremelimumab cohort. There were two disease-related deaths on study.
Patient exposure and disposition
The combination of PexaVec plus ICI did not cause any unexpected AEs. The most common treatment-related AEs of all grades in both cohorts were fever (n=34) and chills (n=32). The most common grade 3 and 4 AEs were fever (n=9) and decreased lymphocyte count (n=4) in the PexaVec/durvalumab cohort and decreased lymphocyte count (n=6), anemia (n=4) and fever (n=3) in the PexaVec/durvalumab/tremelimumab cohort (table 2). Four patients presented with toxicities that led to discontinuation of treatment. This includes the three patients who discontinued treatment before the first treatment evaluation and another cases of treatment-related colitis. As expected, we observed more irAEs (including grade 3 colitis and grade 2 myositis) in patients treated with the combination of anti-CTLA and anti-PD-L1 than in the group treated with anti-PD-L1 alone. Overall, it appears that treatment with PexaVec plus ICI was feasible in most patients.
Treatment Related Adverse Events
Clinical outcome
Secondary endpoints of the study included response, PFS and OS. Patients were assessed for response to treatment by imaging with CT or MRI 10 weeks after the first dose of PexaVec. Patients were considered evaluable for response to treatment if they had received at least three of the four PexaVec doses. A total of 25 patients were evaluable for response. In the PexaVec/durvalumab cohort, 14 out of the 16 patients received the four doses of PexaVec (1 patient had rapid progression of disease and 1 was non-compliant). In the PexaVec/tremelimumab/durvalumab cohort, only 11 out of 18 patients received at least three doses of PexaVec (3 demonstrated early progression, 1 patient withdrew consent and 3 patients developed treatment-related toxicities leading to treatment discontinuation prior to imaging). In the PexaVec/durvalumab cohort, one patient had a partial response (PR) with a 51% tumor shrinkage, which occured after 4 months of treatment and lasted 9 months (figure 1C), and one patient had stable disease (SD) per RECIST V. 1.1 lasting more than 3 months. Twelve patients had progressive disease (PD). In the PexaVec/durvalumab/tremelimumab cohort, three patients had SD and eight patients had PD (figure 1A,B). The disease control rate (complete response (CR), PR or SD) was 12.5% in the PexaVec/durvalumab cohort and 16.7% in the PexaVec/durvalumab/tremelimumab cohort (online supplemental table 2).
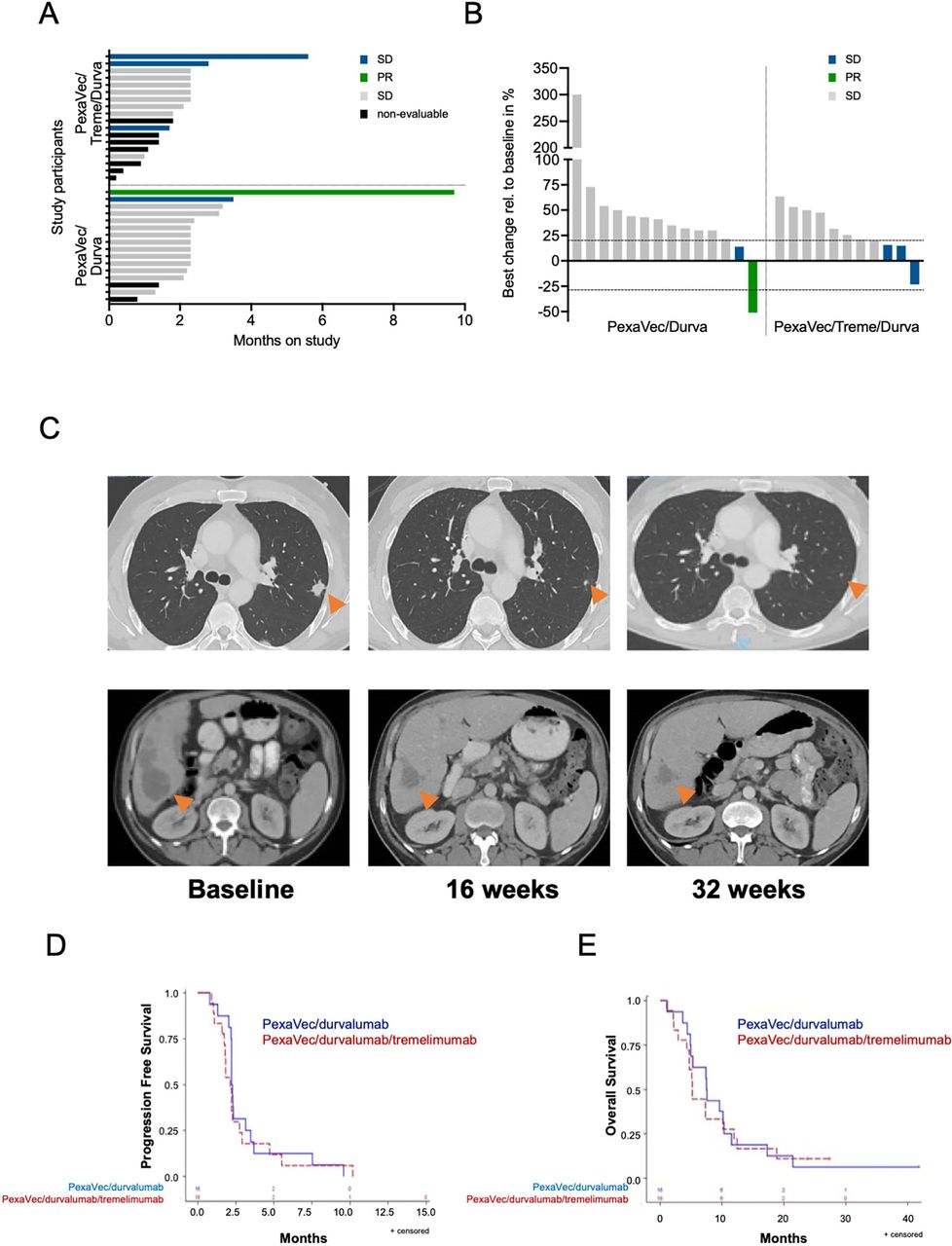
Clinical outcome. (A) Swimmer plot showing time on study and response status. Months on study; colored bars indicate confirmed responses assessed by RECIST 1.1. (B) Waterfall plot of radiographic responses. Colored bars indicate confirmed responses assessed by RECIST 1.1. (C) Representative CT images of the abdomen and lung baseline and on treatment at 16 and 32 weeks of patient presenting with a PR to treatment. (D) Kaplan-Meier estimates of progression-free survival: PexaVec/durvalumab median 2.3 months (95% CI 2.2 to 3.2) vs PexaVec/durvalumab/tremelimumab 2.1 months (95% CI:1.7 to 2.8); p=0.57. (E) Kaplan-Meier estimates of overall survival; PexaVec/durvalumab median 7.5 months (95% CI:4.9 to 10.3) vs PexaVec/durvalumab/tremelimumab 5.2 months (95% CI:4.3 to 10.2); p=0.80. PD, progressive disease; PR, partial response; RECIST 1.1, Response Evaluation Criteria in Solid Tumors V.1.1; SD, stable disease.
The median PFS in the PexaVec/durvalumab/tremelimumab group was 2.1 months (95% CI: 1.7 to 2.8 months) and the median PFS in the PexaVec/durvalumab group was 2.3 months (95% CI: 2.2 to 3.2 months) (figure 1D). The median OS in the PexaVec/durvalumab/tremelimumab group was 5.2 months (95% CI: 4.3 to 10.2 months) vs 7.5 months (95% CI: 4.9 to 10.3 months) in the PexaVec/durvalumab group (figure 1E), with three patients remaining alive at 23.8, 27.4 and 41.8 months after first dose of PexaVec administration at the time of preparation of the current report. No statistically substantial differences in PFS and OS were observed between the two cohorts. Tumor progression was the primary cause of death in all cases.
Immune correlatives
As an exploratory trial endpoint, pharmacodynamic effects and potential underlying immune mechanisms were studied via profiling of serial patient blood samples and paired tumor biopsies obtained pretreatment and on-treatment (online supplemental figure 1). Tumor samples were profiled with RNA sequencing, whereas blood samples were profiled with flow cytometry. A total of 13 patients had paired needle biopsies from tumor lesions at baseline and either on D1 (ie, after one dose of PexaVec alone) or on D 29 (ie, after receiving ICI and PexaVec); one patient with PR had a third biopsy at the time of disease progression. Tumor biopsies underwent bulk RNA sequencing analysis and a total of 11 major immune cell subsets were quantified. Due to the small sample number, cohorts A and B (PexaVec/durvalumab and PexaVec/durvalumab/tremelimumab) were combined for immune correlative analysis, but samples were grouped according to treatment (PexaVec only vs PexaVec plus ICI). Very few CD8 T cells were identified in the analyzed samples from all patients. CD4 cells were found in most patients, but there was no correlation with clinical response noted. More than half of the samples contained regulatory T cells. B cells, M1 and M2 macrophages, monocytes, dendritic cells and neutrophils were found in almost all samples and their presence did not correlate with outcome or type of treatment (PexaVec vs PexaVec plus ICI) (figure 2A). Next, we performed a pairwise analysis of changes in the cell composition before and after treatment in all samples (with and without ICI treatment). All biopsy pairs were used for quantification of immune cells and the results were submitted to unsupervised clustering. Based on the overall transcriptome, biopsies primarily clustered by patient rather than by treatment status (figure 2B). No clear differences are seen in pairwise sample correlation as far as cell composition.

(A) Estimated immune cell fraction from the QuanTIseq deconvolution of sample transcriptome profiles. (B) Immune signature in patients after treatment: Pairwise Pearson’s correlations of gene expression between individual patient samples. Samples are organized by hierarchical clustering based on their Pearson’s correlation coefficients (cor). The column to the right of the correlation matrix indicate when the biopsy was taken during treatment for each patient. The bar plots to the left of the correlation matrix reflect the estimated immune cell fraction from the QuanTIseq deconvolution of sample transcriptome profiles for each sample. Immune cell types are summarized into myeloid (neutrophil, eosinophil, monocyte, macrophage, M1 and M2 macrophage, myeloid dendritic cell) and lymphoid (B cell, NK cell, CD4+ and CD8+ T cell) cells. PD, progressive disease; PR, partial response; SD, stable disease
Next, we analyzed the presence of different immune cells (B cells, monocytes, dendritic cells, macrophages, monocytes, neutrophils, NK cells, CD4+ and CD8+ T cells as well as regulatory T cells) in tumors individually before and after treatment. The relative fraction of individual immune cells in the paired biopsies varied among individuals, and in general, paired biopsies did not show uniform changes in relative fractions of the immune cells studied between the ones collected at baseline and on treatment (D1 and D29, respectively) (figure 3, online supplemental figure 4). These results suggest that intratumoral immune responses from oncolytic virus in combination with ICI varied between patients, and a more granular analysis of immune cells will be needed in future studies.
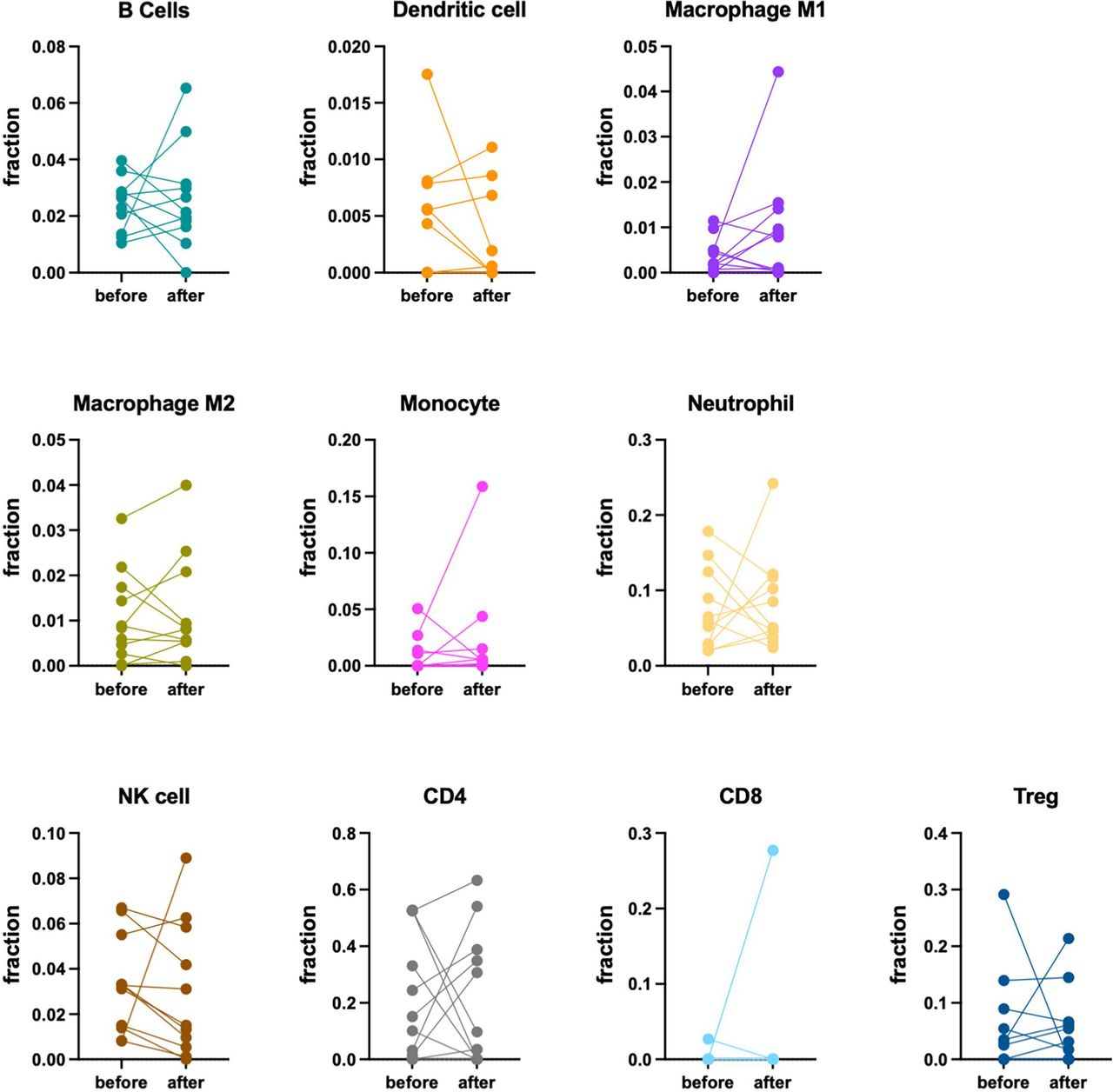
Immune signature in patients after treatment. Scatter plot for immune cell fraction determined by QuanTIseq. Samples for each cell type are grouped according to timeline of biopsy collection during treatment. Biopsies taken from the same patient during treatment are connected by a line.
Transcriptome and exon profiling
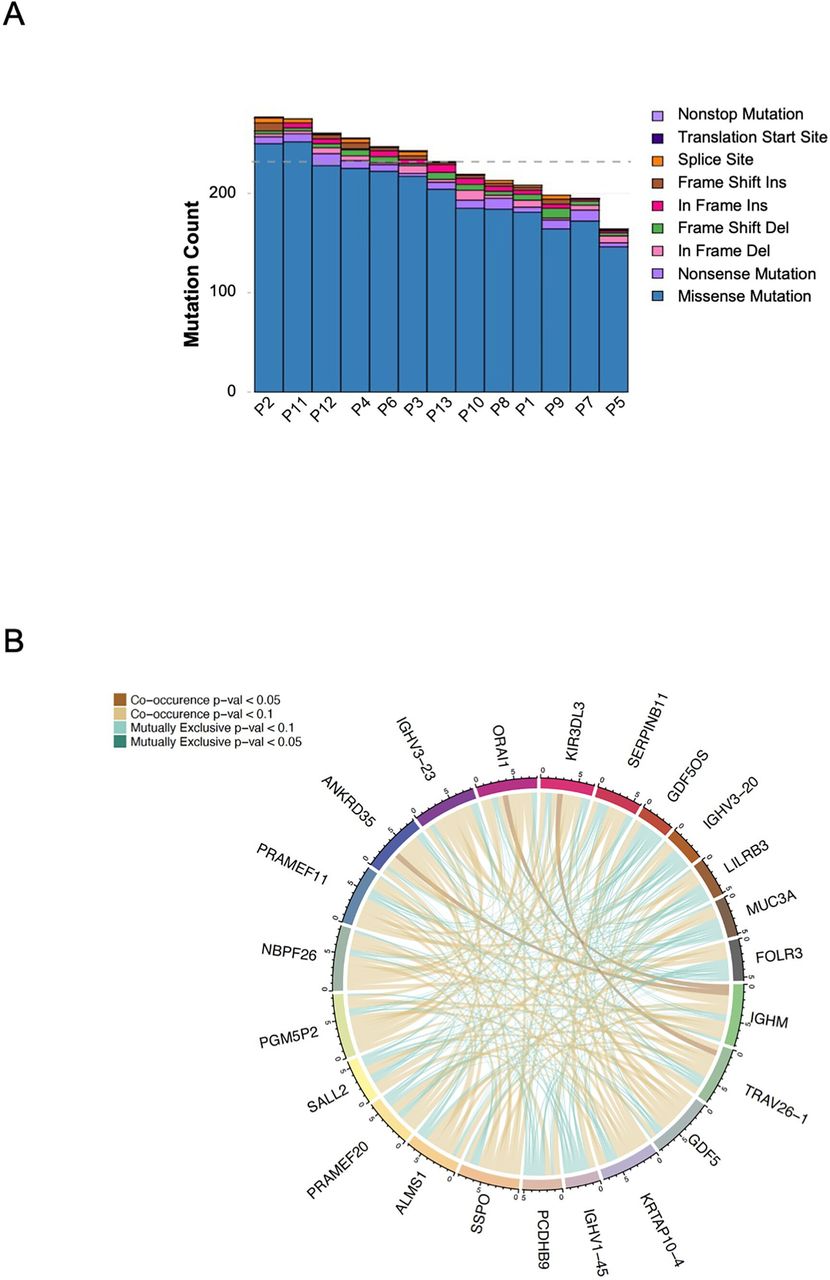
We determined the mutational burden (TMB) in the tumor samples available. As expected, patients with MSS mCRC were found to have a low TMB (figure 4A). Figure 4B depicts the relationship between the tumor samples in terms of single nucleotide variation, somatic insertions and deletions in a circus plot. Given the small sample size and variable clinical response as well as known interindividual variability, identifying gene pathways that change consistently in response to treatment or according to clinical results was unlikely.
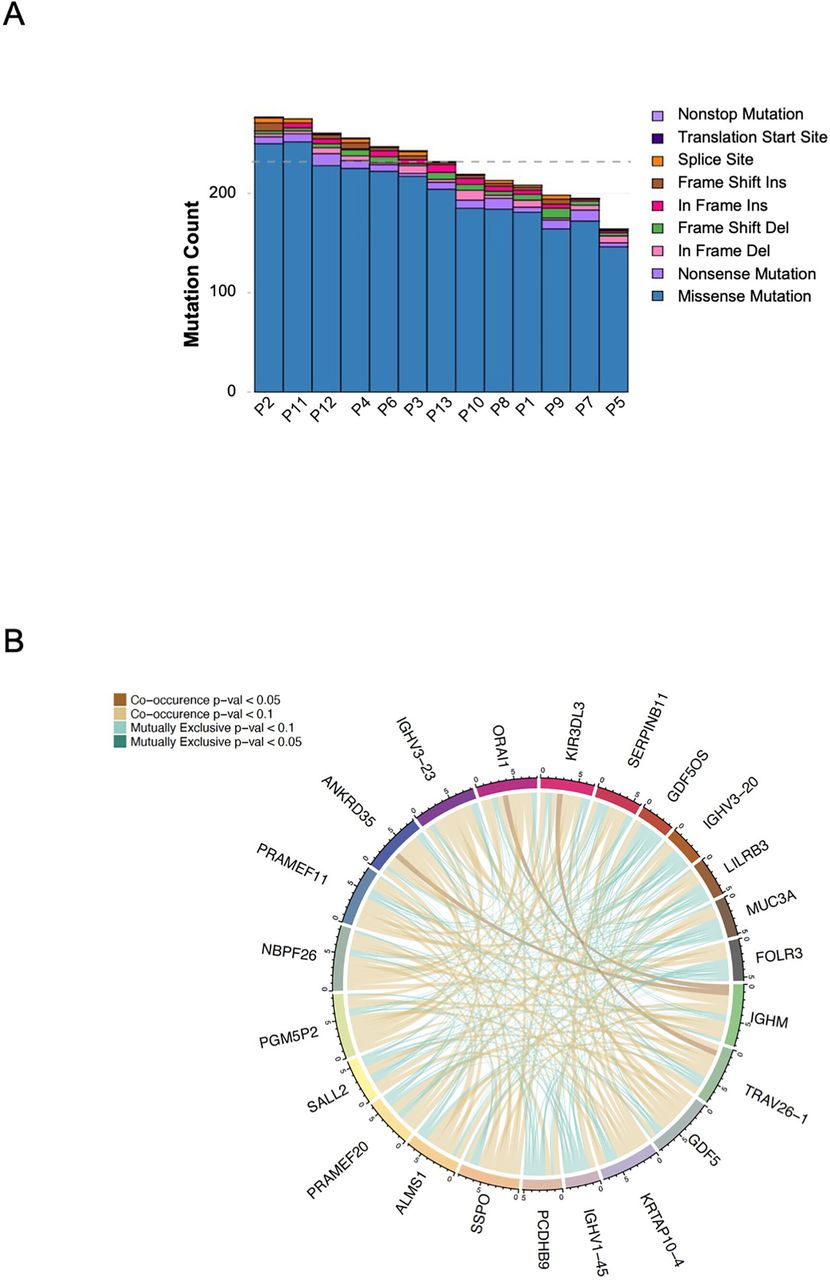
(A) Column plot for the variant count for each sample in this study. Variant counts are colored by variant classification. (B) Whole exome sequencing: ribbon plot of pairwise mutation patterns between top mutated genes. Lines between genes show co-occurrence (brown) or mutual exclusivity (green) between genes and are shaded by p value (calculated by pair-wise Fisher’s Exact test). Connections are shown for each sample in top genes.
Flow cytometry analysis of PBMCs
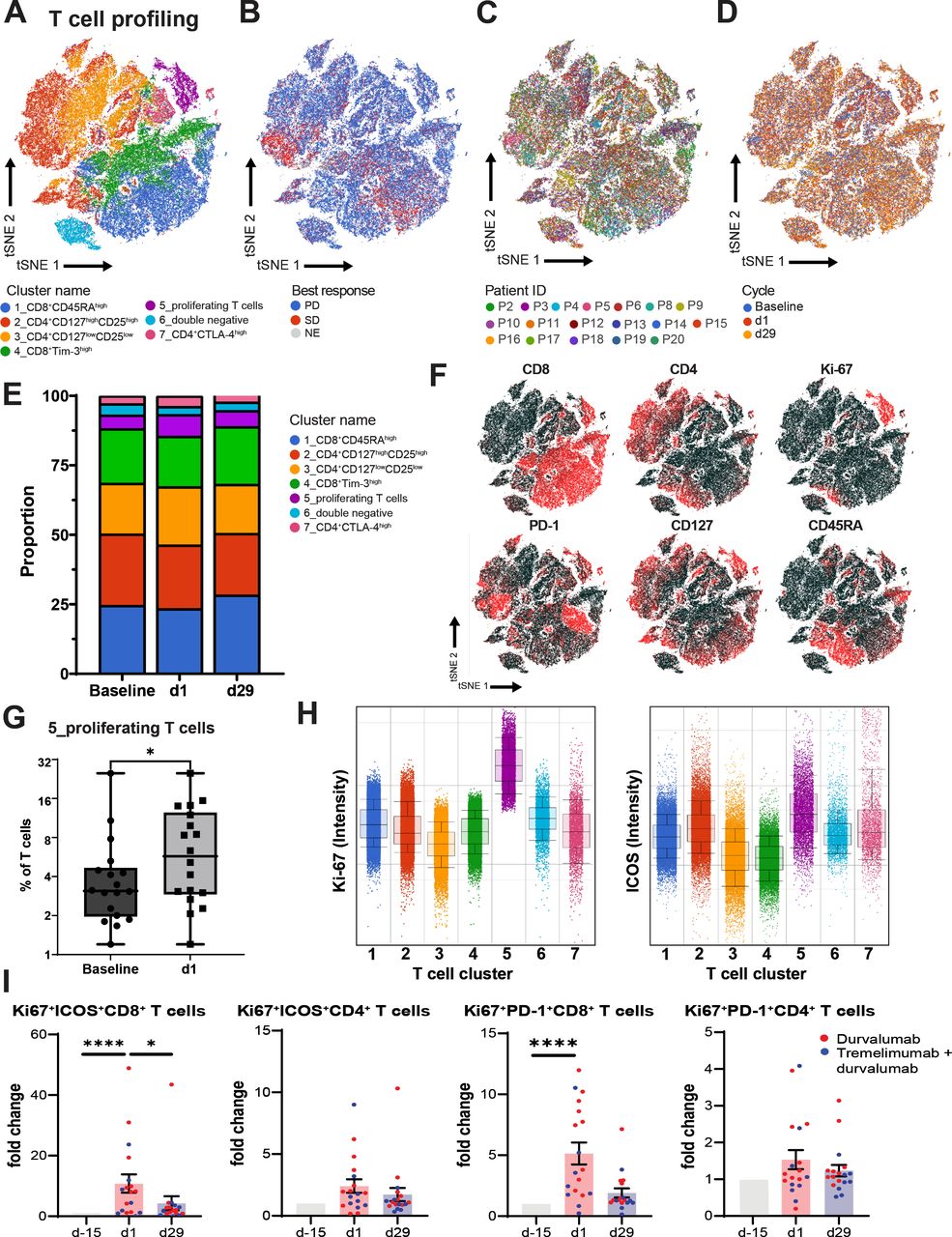
PBMCs, collected at baseline and after the first dose of PexaVec (D1) and on day 29 after receiving further ICI, were analyzed. High-plex multicolor flow cytometry was performed to study different immune cell subsets and activation status. Unsupervised analysis of immunoprofiling data revealed nine distinct immune cell clusters (containing four clusters for T cells, B cells, myeloid cells, granulocytes and NK cells), which did not vary by patient, time of analysis (before/after treatment) or treatment response (online supplemental figure 5). To better characterize potential effector cells of the combined PexaVec plus ICI therapy, a separate analysis for T cells was done. In-depth analysis of T cells revealed seven distinct T cell clusters (figure 5A–D). T cell cluster 5 representing proliferating T cells expanded after treatment (figure 5E–G). T cell cluster 5 was characterized by high expression of Ki-67 and ICOS (figure 5H). To validate findings from unsupervised analysis, a conventional supervised analysis was done. In line with findings from the unsupervised analysis, we observed an increase of the frequency of Ki67+ICOS+CD8+ T cells on PexaVec treatment (figure 5I). In addition, we observed an increase of frequency of Ki67+PD1+CD8+ T cells after the first dose of PexaVec, which decreased on D29 after tremelimumab and durvalumab treatment (28 days after the first dose of durvalumab/tremelimumab) (figure 5I). Analysis of tumor-infiltrating immune cells by mRNA analysis indicated an upward trend for M1 macrophages in some patients (online supplemental figure 4), so we also studied the group of patients with an upward trend of M1 macrophages (six patients) and those without this trend (five patients) separately. Interestingly, a statistically significant increase from D-15 to D1 was only seen in the group of patients who did not show an increase in M1 macrophages. This was the case for both PD1+Ki67+CD8+ T cells and ICOS+Ki67+CD8+ T cells (online supplemental figure 5). Finally, the frequency of other immune cells studied (CD3, NK, B cells and myeloid cells) did not change significantly in the paired blood samples (online supplemental figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
T cell profiling of peripheral blood mononuclear cells (PBMCs). PBMCs were isolated from patients at baseline and the first day of first (d1) and second (d29) course. Analysis of the T cell profiling using high-dimensional flow cytometry is shown. (A) tSNE plot, showing clustering for T cell subsets (CD3+ gated) in CRC patient PBMC samples; (B) by best response; (C) by patient (n=18); (D) by treatment cycle (baseline, d1 and d 29) (D). (E) Stacked bar graph shows the frequency of distinct T cell clusters across treatment cycles. (F) tSNE projections showing expression of indicated surface markers used to identify distinct immune cell clusters (corresponding to A–E). (G) Box-and-whiskers plot showing frequency of T cell cluster 5 as a frequency of all T cells comparing baseline and d1, n=18, *p<0.05. Wilcoxon signed rank test. (H) Bar graphs showing fold change of indicated T cell subsets as determined by conventional hierarchical gating. (I) Comparison between baseline, d1 and d29 is shown. N=18, *p<0.05, ****p<0.0001. Friedman test.
Clinical vignette
One of the study participants (P7) demonstrated an exceptional PR lasting 9 months. This was a male patient in his 50s diagnosed 2 years prior to study enrollment with stage IV rectal cancer with metastasis to bone and liver. Pathology at the time of diagnosis was consistent with metastatic adenocarcinoma; MMR stable, PDL-1 expression of 5% and KRAS/NRAS WT. Prior to treatment on study, he underwent palliative radiotherapy to control rectal bleeding, systemic capecitabine, oxaliplatin, bevacizumab and FOLFIRI. A repeat rectal biopsy showed a KRAS (Q22K and V14I) mutation. At the time of study start, the patient had additionally developed pulmonary metastasis. The patient received treatment with PexaVec plus durvalumab. His first staging scans on study showed a reduction in tumor size of 2%, then decreasing further in the next three imaging studies showing a PR of −35.7%, −41% and finally to −51% 9 months after starting on study. His disease progressed including a right inguinal lesion after 10 cycles of treatment. He then received treatment with TAS-102 at another institution. Flowcytometry analysis of his PBMC revealed an increase in the Ki67+ICOS+CD4+ T cells, Ki67+PD1+CD8+ T cells and Ki67+ICOS+CD8+ T cells on treatment (online supplemental figure 7).
Discussion
Immunotherapy for MSS mCRC has been largely unsuccessful so far. Previously, PexaVec has been tested as single agent in CRC and shown a modest efficacy with 10 out of 15 (67%) heavily pretreated patients (mean 4.5 lines of therapy) presenting with SD as best response in early phase I trials.15 16 In the current study, we were able to demonstrate feasibility and safety of PexaVec plus durvalumab and PexaVec plus tremelimumab plus durvalumab. One patient had a durable PR and several patients presented with SD suggesting that PexaVec might enhance antitumor efficiency of ICI in patients with MSS mCRC. However, this resulted in a median PFS of 2.1 and 2.3 months, which is not very different from results of FDA-approved systemic treatment options for mCRC in the third-line setting.37
Variant factors may have contributed to this disappointing result. Neutralizing antibodies causing a subsequent clearance of the virus represent a well-known efficacy limiting factor. Although we did not specifically measure neutralizing antibodies in this study, it has previously been shown that they can arise as early as 2 weeks after PexaVec treatment,16 and in another study, 50% of treated subjects developed detectable titers of neutralizing antibodies by day 29.38 However, previous studies also did not reveal correlation between antibody titers, virus replication, safety, GM-CSF expression and antitumor activity.16 38
We decided to treat patients with PexaVec every 14 days based on a prior study in CRC which demonstrated limited clinical efficacy,15 but future studies are needed to test whether a more frequent treatment may be more effective. Also, the best timepoint for the first ICI treatment is not clear. We chose a 2-week delay after the first PexaVec dose because this schedule allowed us analyze the changes in the tumor microenvironemnt accordingly. An earlier CPI treatment may have aligned better with early virus-induced TME modification and potential cell infiltration.
Neoadjuvant IV PexaVec was shown to be well tolerated in patients in a small single center, non-randomized biological end point study which included nine patients with colorectal liver metastasis (CRLM) or metastatic melanoma. On surgical resection, PexaVec was detected in three out of a total of four CRLM specimens, showing tumor-specific replication as expected as well as transient innate and long-lived adaptive anticancer immunity.39
The combination of anti-CTLA4 plus anti-PD1/PD-L1 has previously been tested in patients with pMMR CRC. Neoadjuvant treatment with a single dose of ipilimumab and two doses of nivolumab resulted in pathological responses in 4/15 patients. Treatment was well tolerated, and all patients underwent radical resections within the predefined 6 weeks after study inclusion.40 Furthermore, results from a phase 1b study (NCT03860272) have recently been reported. The investigators enrolled 41 patients with heavily pretreated MSS mCRC and treated these patients with the combination of botensilimab (anti-CTLA-4) and balsitimab (anti-PD-1) leading to an overall response rate of 24% (95% CI, 14% to 39%); including a 24% PR rate. Forty-nine per cent of patients achieved SD with the regimen while 27% had progression of disease. The disease control rate was 73% (95% CI, 58% to 84%). Of note, patients without liver metastases had a better response to treatment, which varies from our patient population.41
In this study, we were able to conduct correlative studies on peripheral blood and paired tumor biopsies from patients. We obtained tumor biopsies at baseline and either after PexaVec only or combination treatment, with the aim to dissect immune responses to PexaVec from immune responses to the combination treatment. All tumor samples collected at baseline in our study showed a low level of CD8 T cell infiltration, which is consistent with previous reports, that MSS CRC usually exhibits an immune desert immunophenotype.42 There was no significant difference observed in all tested immune cells including CD8 T cells in the biopsied tumor samples either after the infusion of one dose of PexaVec (day 1) or three doses of PexaVec in combination with one cycle of ICI (day 29). We also did not detect a difference in GM-CSF expression. There was clinical evidence of PexaVec selectively infecting cancer tissue after intravenous administration,15 16 but we have limited information to specifically address this issue in patients with MSS CRC in our study. The abovementioned findings were inconsistent with clinical outcome where some patients had disease control evidenced by PR and SD.
Interestingly, the tumor samples from P7, who had a durable PR, did show a reduction of the fraction of Treg along with an increase in TAM1 and neutrophils, while an increased fraction of Treg and TAM2 and a decreased fraction of TAM1 were observed on recurrence. These matched patient’s disease responses indicate that the combination of PexaVec/ICI mobilizes immune cells in the tumor microenvironment in a specific patient population. The analysis of immune cells from PBMC demonstrated that PexaVec infusion only (C1D1) was able to induce proliferation of ICOS+CD8+ and PD-1+CD8+ T cells and this change attenuated after co-exposure with ICI, suggesting systemic CD8+ T cell reactivation by oncolytic virus infusion.43 44 The increase of proliferative PD-1+CD8+ T cells in PBMC has been linked with positive clinic outcome in patients with lung cancer treated with anti-PD-1,45 however, we cannot say whether these T cells recognized tumor antigens or were virus specific. In our study, the increase of proliferative CD8+ T cells did not sustain during the subsequent PexaVec infusion and ICI exposure (C2D1). This suggests that proliferative CD8+T cells likely consist largely of virus-specific CD8+ T cells rather than tumor-specific CD8+ T cells, which indicates a successful induction of antiviral immunity and this can be further studied with TCR sequencing. These findings warrant further research to identify biomarker characteristics of patients who may benefit from this treatment option. The underlying mechanism of the attenuated fraction of active CD8+ T cells with subsequent PexaVec infusions is unknown. It is probably related to the rapid development of neutralizing antiviral antibodies46 and subsequent clearance of the virus, which may contribute to a lower efficacy observed in our study and a need to be overcome in the future.47
Our study demonstrated that the combination of PexaVec and ICI is safe and well tolerated in patients with MSS mCRC. Except for PexaVec-induced fevers and chills, the AE profile on study were comparable to the safety profile of durvalumab and tremelimumab which includes possible irAEs such as colitis and myositis as were observed in three patients on our study. As previously described, PexaVec caused grade 2/3 fever in all patients and chills in most patients. Interestingly, patients presented more frequently with grade 3 fever in the PexaVec/durvalumab cohort when compared with the ones in PexaVec/durvalumab/tremelimumab cohort which was unexpected. This may be due to our increasing experience (patient cohorts were enrolled consecutively) in the preemptive management of fever and chills with acetaminophen, non-steroidal anti-inflammatory medication and other supportive measures. Similar AEs were described in a phase Ib trial of biweekly IV PexaVec in patients with treatment refractory CRC in which 93% out of the 15 patients treated on study were reported to have chills and fever.15
This is the first clinical trial of this size to describe a group of patients with MSS mCRC who received treatment with PexaVec oncolytic virus in combination with ICI. The correlative immune studies from tumors and PBMCs included in this trial further elucidate the immune changes caused by the combination of oncolytic virotherapy and immunotherapy in a tumor typically not sensitive to immunotherapy. Further studies are needed to elucidate the potential predictive biomarkers that would identify patients who may benefit from this combination.
Data availability statement
Data are available upon reasonable request.
Ethics approval
This study involves human participants and was approved by NCI Institutional Review Board (17C0092).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @Cecilimonge4, @DavidKleiner12, @BenniRuf
CM, CX and YM contributed equally.
Contributors Study concept and design: AGD and TFG; analysis and interpretation of data: CM, CX, YM, DM-H, SMH, PH, BR and TFG; statistical analysis: SMS; administrative, technical or material support: KC, IJ, SW, WDF and MC; patient enrollment and clinical care: CM, CX, DM-H, JMH, BJW, EBL, DEK, BR, AGD and TFG; writing: CM, CX, YM and TFG. Guarantor author: TFG. All authors reviewed and approved the manuscript.
Funding The author(s) disclose receipt of the following financial support for the research, authorship, and/or publication of this article: TFG is supported by the Intramural Research Program of the NIH, NCI (ZIA BC 011343). The study was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and a Cooperative Research and Development Agreement between NCI, Sillajen and between NCI and AstraZeneca.
Competing interests AD reports participation of Advisory Boards from AstraZeneca, outside the submitted work. All other authors have nothing to disclose.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.