Article Text

Abstract
Background Immune tolerance contributes to resistance to conventional cancer therapies such as radiation. Radiotherapy induces immunogenic cell death, releasing a burst of tumor antigens, but this appears insufficient to stimulate an effective antitumor immune response. Radiation also increases infiltration of cytotoxic T lymphocytes (CTLs), but their effector function is short lived. Although CTL exhaustion may be at fault, combining immune checkpoint blockade with radiation is insufficient to restore CTL function in most patients. An alternative model is that antigen presentation is the limiting factor, suggesting a defect in dendritic cell (DC) function.
Methods Building on our prior work showing that cancer cells treated with radiation in the presence of the poly(ADP-ribose) polymerase-1 inhibitor veliparib undergo immunogenic senescence, we reexamined senescent cells (SnCs) as preventative or therapeutic cancer vaccines. SnCs formed in vitro were cocultured with splenocytes and evaluated by scRNA-seq to examine immunogenicity. Immature bone-marrow-derived DCs cocultured with SnCs were examined for maturation and activation by flow cytometry and T cell proliferation assays. Viable SnCs or SnC-activated DCs were injected subcutaneously, and vaccine effects were evaluated by analysis of immune response, prevention of tumor engraftment, regression of established tumors and/or potentiation of immunotherapy or radiotherapy.
Results Murine CT26 colon carcinoma or 4T1 mammary carcinoma cells treated with radiation and veliparib form SnCs that promote DC maturation and activation in vitro, leading to efficient, STING-dependent CTL priming. Injecting mice with SnCs induces antigen-specific CTLs and confers protection from tumor engraftment. Injecting immunogenic SnCs into tumor-bearing mice increases inflammation with activated CTLs, suppresses tumor growth, potentiates checkpoint blockade, enhances radiotherapy and blocks colonization by disseminated tumor cells. Addressing the concern that reinjecting tumor cells into patients may be impractical, DCs activated with SnCs in vitro were similarly effective to SnCs in suppressing established tumors and blocking metastases.
Conclusions Therapeutic vaccines based on senescent tumor cells and/or SnC-activated DCs have the potential to improve genotoxic and immune therapies and limit recurrence or metastasis.
- Immunotherapy
- Vaccination
- Immunogenicity, Vaccine
- Dendritic Cells
- Radiotherapy
Data availability statement
Data are available upon reasonable request. Further information and requests for resources and reagents should be directed to and will be fulfilled by Stephen Kron (skron@uchicago.edu).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Although therapy-induced senescence is considered deleterious and implicated in therapy resistance, cancer recurrence and metastasis, there is also evidence that senescent cells (SnCs) can drive antitumor immunity.
WHAT THIS STUDY ADDS
Our study shows that dendritic cells (DCs) cocultured with SnCs become mature and activated and efficiently prime T cells through a STING-dependent mechanism. Injecting SnCs into mice induces antitumor immunity and serves as a cancer vaccine that potentiates immunotherapy and radiotherapy and suppresses growth of disseminated tumor cells. Injecting DCs activated by SnCs in vitro recapitulates SnC vaccine effects, confirming the mechanism.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our observation that SnC-activated DCs can enhance therapy response and block metastatic spread in preclinical models points the way to personalized cancer vaccines formed by loading DCs with senescent patient tumor cells that will promote tumor regression and suppress distant spread.
Background
Cellular senescence, where cells remain viable but irreversibly arrested as an outcome of cellular stress, serves critical roles in diverse physiological and pathological processes, including development, healing, aging and cancer.1 2 Senescence is considered an important barrier to malignancy insofar as replicative senescence induced by telomere erosion can prevent unlimited proliferation while oncogene-induced senescence resulting from oncogenic or tumor suppressor mutations can block transformation. Although cancer cells are often considered immortal, they can undergo stress-induced senescence in response to genotoxic or other therapies,3 which may have both beneficial and deleterious effects on outcomes. Given these many links to malignancy, senescence is now considered a hallmark of cancer4 and a potential target for cancer therapies.5
Senescent cells (SnCs) typically form when proliferating cells fail to complete cell division due to a persistent DNA damage checkpoint and/or mitotic catastrophe. As these SnCs form over a few days, they may undergo endoreduplication and continue to grow, often increasing dramatically in cell size. SnCs may remain viable indefinitely, persisting until they undergo cell death or are eliminated by immune surveillance. Reflecting persistent cell stress, SnCs typically display altered metabolism and gene expression, present distinct cell surface proteins and display an altered secretome, the senescence-associated secretory phenotype (SASP), characterized by expression of inflammatory mediators.6
The SASP, like SnCs themselves, has been considered a double-edged sword in modulating immune responses.7 An immune-suppressive SASP has been reported to increase immune suppressing myeloid cells and inhibit antitumor T cell and NK cell responses. These effects may contribute to progression, resistance and recurrence, arguing for SnC elimination after therapy.8 However, the immune surveillance mechanism that limits SnC accumulation has long been considered a critical mechanism in limiting age and stress-related disorders and carcinogenesis.9 Oncogene-induced senescence facilitates the immune surveillance of premalignant cells by promoting the recruitment and activation of innate and adaptive immune cells, including monocytes, macrophages, neutrophils, NK cells and T cells.10–12 This raises the question whether senescent cancer cells might retain the ability to drive antitumor immune response even after the onset of malignancy. Indeed, previous studies from our group and others have indicated that therapy-induced senescence is capable of promoting antitumor immunity and improving treatment outcomes.13–15 For instance, SnCs formed in situ with ionizing radiation (IR) and poly(ADP-ribose) polymerase (PARP) inhibitor veliparib13 or CDK4/6 inhibition14 led to T cell-dependent antitumor immunity. Additionally, aneuploidy-associated senescence could promote NK cell-mediated tumor clearance.15 Although features of the SASP might be primary determinants of immune response, cell surface expression of major histocompatibility complex (MHC) molecules and altered antigen presentation in SnCs may also play critical roles.14
Here, we revisited the immunogenic potential of senescent tumor cells in the form of a cancer vaccine. Single-cell RNA sequencing (scRNA-seq) analysis of splenocytes cocultured with SnCs suggested that DCs may be particularly responsive to and key mediators of SnC effects. Indeed, therapy-induced SnCs could promote DC activation and maturation via STING-dependent mechanisms and DCs activated by SnCs were effective at priming T cells. Consistent with our prior studies,13 therapy-induced SnCs were effective as a preventative vaccine, inducing antigen-specific cytotoxic T cell responses and blocking engraftment of tumor cells. Examined as therapeutic vaccines, peritumoral injection of SnCs suppressed tumor growth on its own and increased the effectiveness of immune checkpoint blockade of programmed cell death ligand 1 (PD-L1) interaction with programmed cell death protein 1 (PD-1) in overcoming immunosuppression. Injecting SnCs combined with radiotherapy eliminated tumors. On their own, SnCs were able to suppress colonization by disseminated tumor cells (DTCs) in a breast cancer metastasis model. Importantly, injecting SnC-activated DCs rather than SnCs recapitulated these effects, serving as effective therapeutic vaccines to limit tumor growth and prevent colonization by DTCs. Translated to the clinic, adjuvant therapy with autologous or allogeneic DC vaccines activated with SnCs formed from transiently cultured patient tumor tissue may have significant value in enhancing local and systemic therapy to limit metastatic spread or recurrence.
Materials and methods
Cell lines and tissue culture
BALB/c-derived colon carcinoma cell line CT26 (HTB-85) and mammary carcinoma cell line 4T1 were obtained from ATCC. The cells were tested for mycoplasma contamination and authenticated by a short tandem repeat profile (IDEXX BioResearch) prior to performing experiments.
Cells were maintained in RPMI 1640 (Thermo Fisher Scientific) supplemented with 10% FBS (Atlanta Biologicals) and 1% penicillin/streptomycin (Thermo Fisher Scientific). The basic murine immune cell culture medium was RPMI 1640 (Thermo Fisher Scientific) supplemented with 10% heat-inactivated FBS (Thermo Fisher Scientific), 1% penicillin/streptomycin (Thermo Fisher Scientific) and 50 µM β-mercaptoethanol. All experiments were performed within 3 to 10 passages after thawing cells. At least three replicates were performed in all in vitro experiments.
Preparation of mouse BMDCs
Bone marrow-derived dendritic cells (BMDCs) were differentiated as previously described.16 Briefly, bone marrow was isolated from 7-week to 8-week BALB/c mice, maintained in basic immune cell culture medium supplemented with 1 ng/mL mouse recombinant GM-CSF (PeproTech) and 200 ng/mL mouse recombinant Flt-3 ligand (PeproTech) for 14–16 days, changing medium on days 6, 9 and 12.
Animal models
For animal experiments, 6-week to 8-week BALB/c wild-type (WT) mice were purchased from Harlan-Envigo. NOD SCID gamma (NSG) mice were maintained by breeding. All mice used for these studies were procured and maintained following guidelines approved by the Institutional Animal Care and Use Committee. Tumor volume was measured using calipers every 2–3 days from day 7 after subcutaneous tumor cell inoculation. Mice that did not develop palpable tumors within 10 days after inoculation were excluded from the study. For preventative cancer vaccines, mice were randomized to receive SnC vaccines. For therapeutic cancer vaccines, mice were randomized after tumor cell inoculation. At least 5 mice per group were used in all in vivo experiments reported here.
Chemical probes
Veliparib was provided by AbbVie. Etoposide, GSK461364, ABT-263, DMXAA and C178 were purchased from Cayman Chemical.
Preparation of SnC vaccine and SnC-activated DC vaccine
1×106 CT26 cells or 4T1 cells were seeded into T175 flasks, then treated with IR+veliparib (12 Gy+20 µM). After 5 days, SnCs were trypsinized and washed in phosphate-buffered saline (PBS), then resuspended in cold PBS to a final concentration of 5×106 cells/mL. To form an SnC-activated DC vaccine, naive BMDCs were cocultured with SnCs for 10–12 hours. Non-adherent BMDCs were collected and washed in PBS, then resuspended in cold PBS to a final concentration of 5×106 cells/mL. One hundred microliters of SnCs or SnC-activated DCs were injected as a vaccine dose.
Quantification and statistical analysis
Statistical significance was determined using the paired Student’s t-test or logrank test as indicated. Calculations were performed using Prism software (GraphPad) or Excel. p≤0.05 was considered statistically significant.
SA-β-Gal, SASP characterization, single-cell RNA sequencing (scRNA-seq), BMDC activation, T cell cross-priming, in vitro cell proliferation, Western blotting, immunofluorescence, AH-1 dextramer staining, in vivo vaccination, TIL analysis, lung colonization and histology assays are described in the online supplemental methods.
Supplemental material
Results
SnCs stimulate immune responses in vitro
To model the interactions of senescent tumor cells with the tumor immune microenvironment, we cocultured SnCs with splenocytes in vitro. CT26 murine colon carcinoma cells were treated with the PARP1/2 inhibitor veliparib (20 µM) along with IR (12 Gy) to induce immunogenic senescence, as has been shown in other cell lines.13 After 5 days, more than 90% of the surviving CT26 cells had developed an SA-β-Gal+ senescent phenotype (online supplemental figure S1A). Then, the SnCs or proliferating controls were cocultured for 3 days with splenocytes from CT26 tumor-bearing mice and non-adherent cells were collected for single-cell RNA sequencing (scRNA-seq), serving as a cells-as-sensors assay (online supplemental figure S1B). Seurat integration analysis resolved 11 cell clusters, including multiple types of immune cells known to contribute to the antitumor immune response (figure 1A–B and online supplemental figure S1C,D). Reanalysis after excluding the non-immune cell population and B cells allowed resolution of additional immune cell types classified in 8 clusters (figure 1C,D). Splenocytes cocultured with SnCs displayed markedly increased fractions of type 1 conventional dendritic cells (cDC1) and CD8+ T cells compared with controls (figure 1EF and online supplemental figure S1C,D). Analyzing composite gene expression in the cDC1 cell clusters revealed marked differences in differentially expressed genes (DEGs) (online supplemental figure S1E). Biological process (BG) gene ontology (GO) analysis of the cDC1 indicated that coculture with SnCs upregulated pathways such as stress response, cytokine response, T cell activation and antigen processing (figure 1G). GO analysis with other databases revealed complementary patterns, including upregulation of antigen processing and presentation and type I interferon (IFN) signals (online supplemental figure S1F,G). These results suggest that DCs may be particularly responsive to senescent tumor cells and thereby may mediate the effects of SnCs on immune response.
Supplemental material
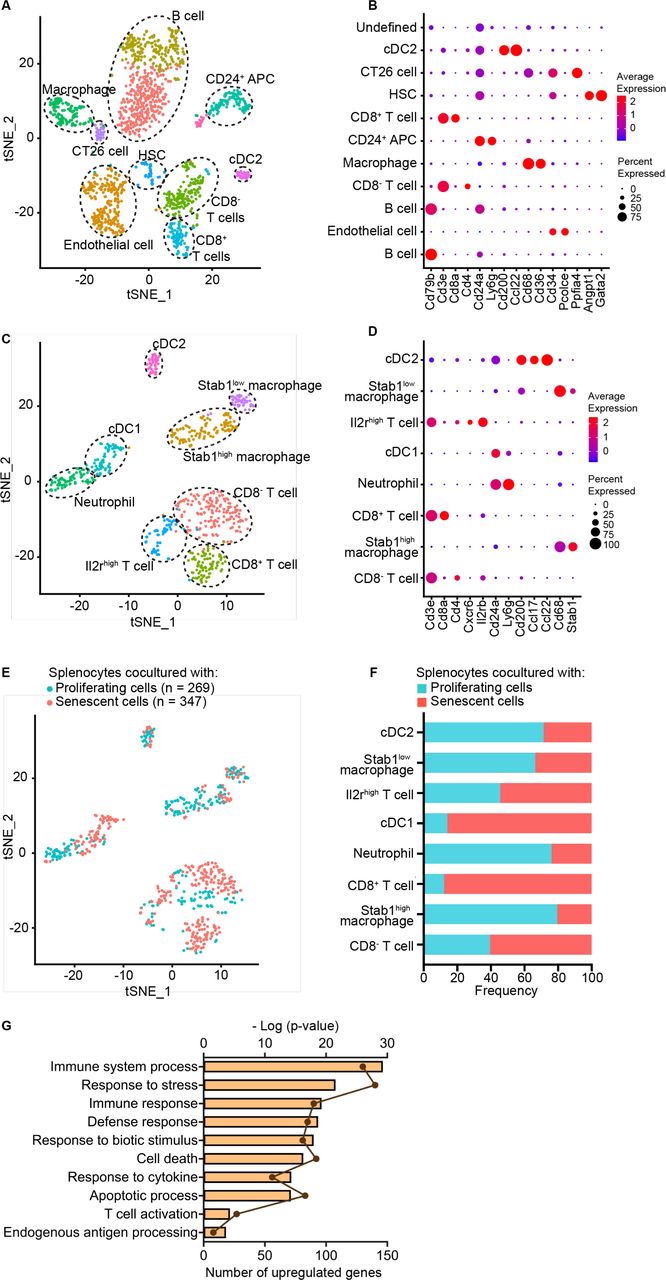
scRNA-seq cells-as-sensors assay reveals dendritic cell activation by SnCs. After coculture with CT26 SnCs or proliferating cells, splenocytes were analyzed by scRNA-seq, and the two datasets were pooled, yielding, (A) tSNE plot demonstrating 11 cell clusters from 1618 cells assigned to immune subsets as inferred from (B) dot plot indicating relative expression of marker genes in each cell cluster. (C–F) Reanalysis of pooled data after excluding cells from B cell, HSC, endothelial cell, CT26 and undefined clusters. (C) tSNE plot demonstrating 8 cell clusters from 616 cells with subsets inferred from (D) dot plot indicating relative expression of marker genes in each cell cluster. (E) tSNE plot indicating relative distributions of splenocytes cocultured with SnCs or proliferating cells. (F) Stacked bar graph indicating the relative frequency of splenocytes cocultured with SnCs or proliferating cells in each cluster, indicating enrichment of cDC1 and CD8+ T cells. (G) Biological process gene ontology analysis of differential expressed genes (DEGs) among cDC1s. Shown are enriched, upregulated signaling pathways in the cDC1 population of splenocytes cocultured with SnCs vs proliferating cells. Dots indicate the number of DEGs and bars indicate the -Log10 (p value) for each enriched pathway. APC, antigen-presenting cell; cDC1, conventional type 1 dendritic cell; cDC2, conventional type 2 dendritic cell; HSC, hematopoietic stem cell; SnCs, senescent cells.
SnCs promote DC activation and T cell priming in vitro
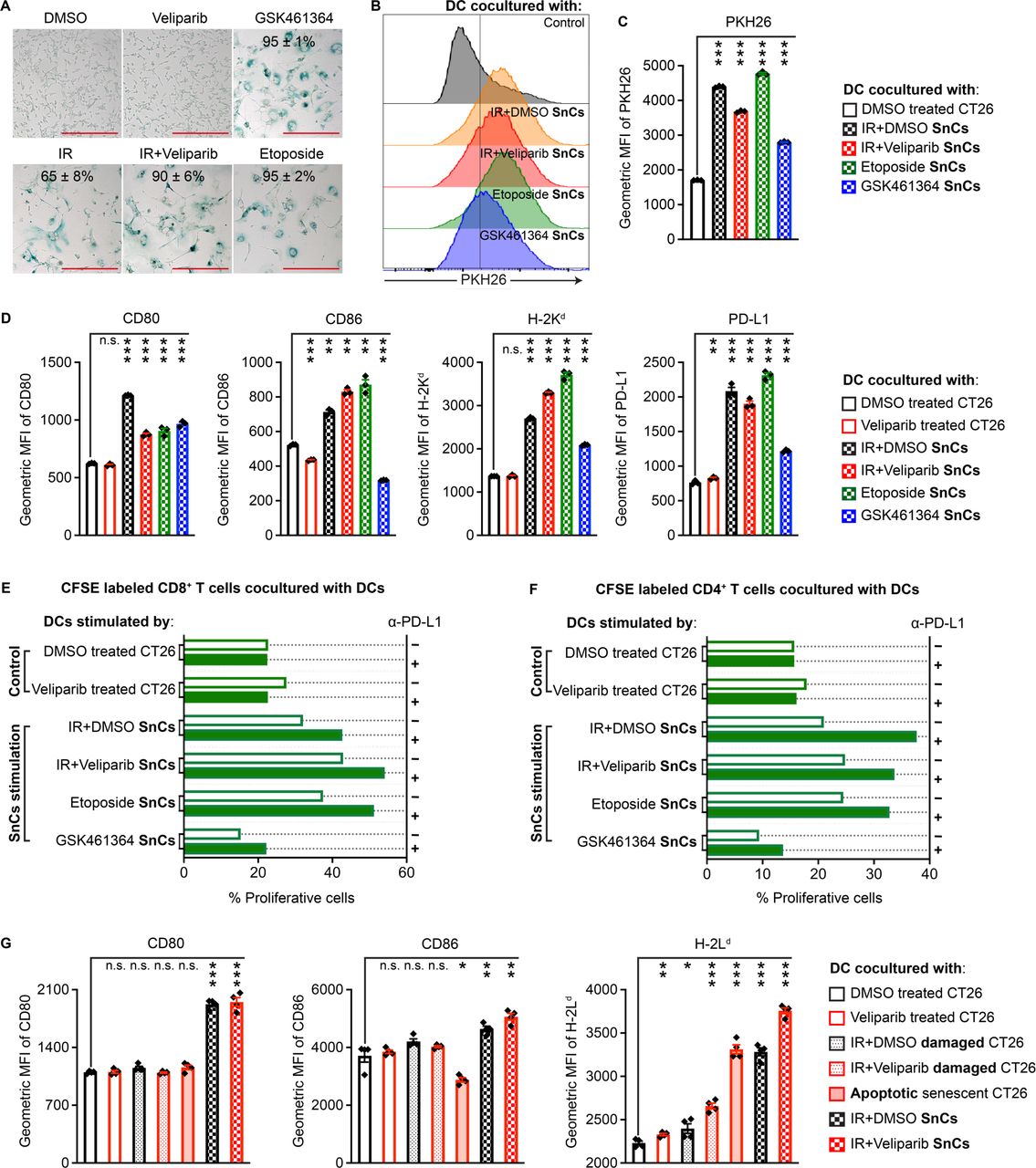
Our scRNA-seq studies encouraged us to further examine DCs to determine if SnCs might serve as a source of antigen for presentation. Considering that diverse stresses can induce cancer cell senescence, we investigated whether different senescence inducers might influence interactions with DCs. CT26 cells treated with IR (12 Gy), IR+veliparib (12 Gy+20 µM), topoisomerase II poison etoposide (2 µM) or PLK1 inhibitor GSK461364 (5 µM) displayed~65%, 90%, 95% and 95% SA-β-Gal+ SnCs, respectively, at 5 days (figure 2A). Cells treated with dimethyl sulfoxide (DMSO) or veliparib alone were used as non-senescent controls. Compared with proliferating cells, SnCs secreted higher levels of cytokines and chemokines, including those that promote DC maturation/activation, such as CCL5 (online supplemental figure S2A,B). BMDCs were formed as described16 by culturing BALB/c bone marrow in GM-CSF and Flt-3 ligand. To examine phagocytosis and/or trogocytosis, senescent or control cells were labeled with lipophilic dye PHK26 or the pH-sensitive fluorescent probe pHrodo Red and incubated with BMDCs for 6 hours. Transfer of PHK26 to DCs or uptake of pHrodo Red into DC endosomes or lysosomes were analyzed by flow cytometry (online supplemental figure S3A,B). Overall, SnCs were engulfed or nibbled more than proliferating cells and different senescence inducers appeared to influence uptake (figure 2B,C and online supplemental figure S4A). In turn, SnCs induced BMDC activation and maturation based on flow cytometric analysis of costimulatory molecules CD86 and CD80 and MHC I molecule H-2Kd (figure 2D and online supplemental figure S3C). SnCs also upregulated surface expression of PD-L1 on BMDCs (figure 2D). Similar experiments using BALB/c-derived 4T1 murine mammary carcinoma cancer cells revealed that 4T1 SnCs induced similar responses in BMDCs (online supplemental figures S4B and S5A–C).
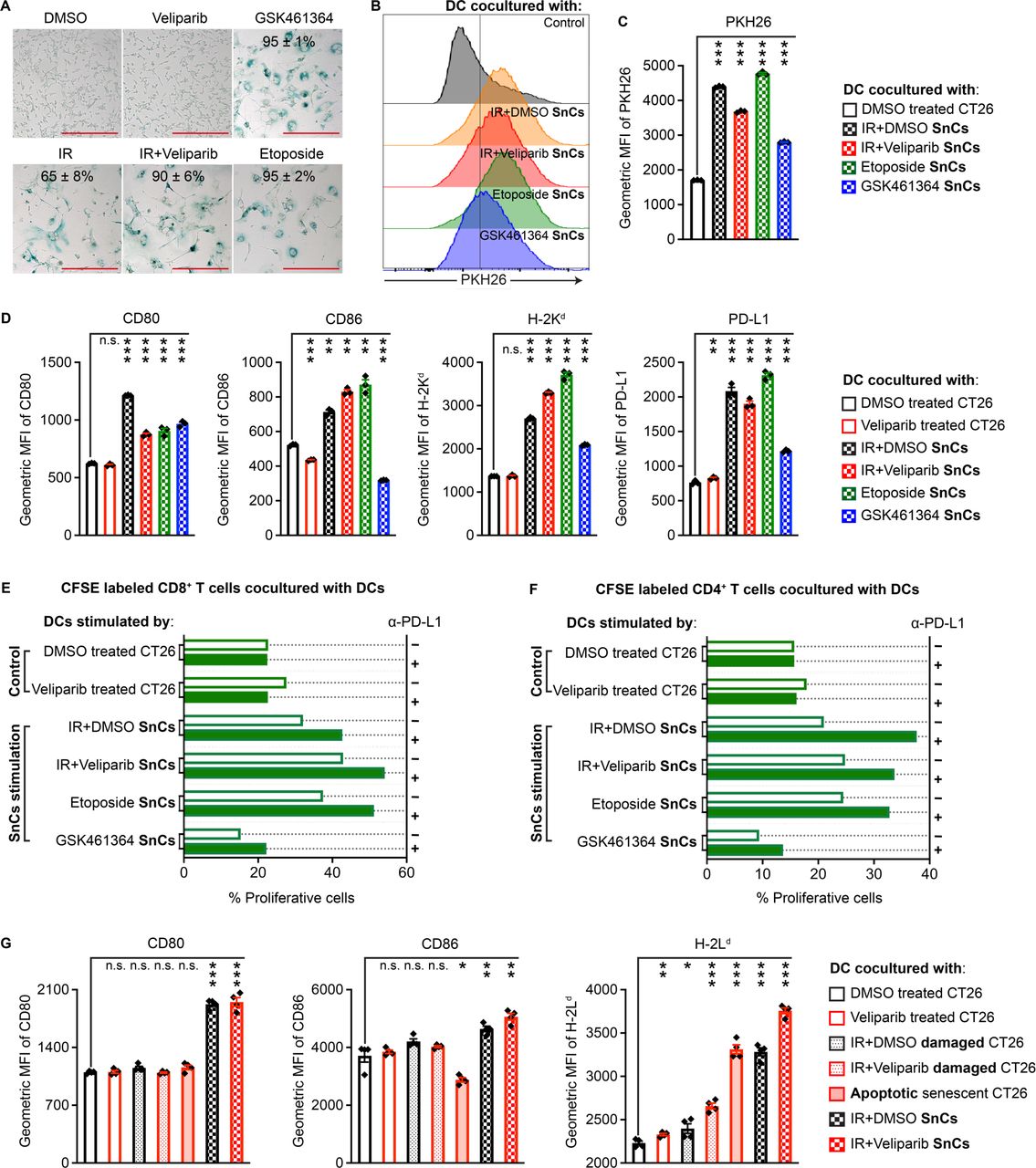
Senescent CT26 cells promote DC maturation/activation and CD8+ T cell priming in vitro. (A) Compared with CT26 cells treated with DMSO vehicle or veliparib as controls, GSK461364, IR, IR+veliparib and etoposide each induced senescence after 5 days. Indicated is mean±SD of % SA-β-Gal-positive cells from five fields. Scale bars: 200 µm. (B) Representative histograms indicating effective uptake of PHK26-labeled SnCs by BMDCs. (C) Geometric mean fluorescence intensity (MFI) bar graph demonstrating uptake from each cell population (n=3, mean±SD). Paired t-test. (D) CT26 SnCs and controls prepared as in (A) were cocultured with BMDCs overnight, followed by flow cytometry analysis for CD80, CD86, MHC I and PD-L1 maturation and activation markers. Shown are bar graphs of geometric MFI for the viable CD11c+/CD103+ DC population (n=3, mean±SD). Paired t-test. (E and F) Analysis of T cell priming by SnC-activated DCs. Shown are the % proliferative (CFSE-diluted) fraction of viable CD8+/CD4− (E) or CD8−/CD4+ (F) T cells. (G) Comparison of BMDC activation by CT26 proliferating cells, damaged cells, apoptotic SnCs or live SnCs. Shown are bar graphs of geometric MFI (n=4, mean±SD). Paired t-test. For statistical analysis, ***p<0.001, **0.001<p<0.01, *0.01<p<0.05, n.s. p>0.05. BMDCs, bone marrow-derived dendritic cells; CFSE. carboxyfluorescein succinimidyl ester; DC, dendritic cell; DMSO, dimethyl sulfoxide; IR, irradiation; PD-L1, programmed cell death ligand 1; SnCs, senescent cells.
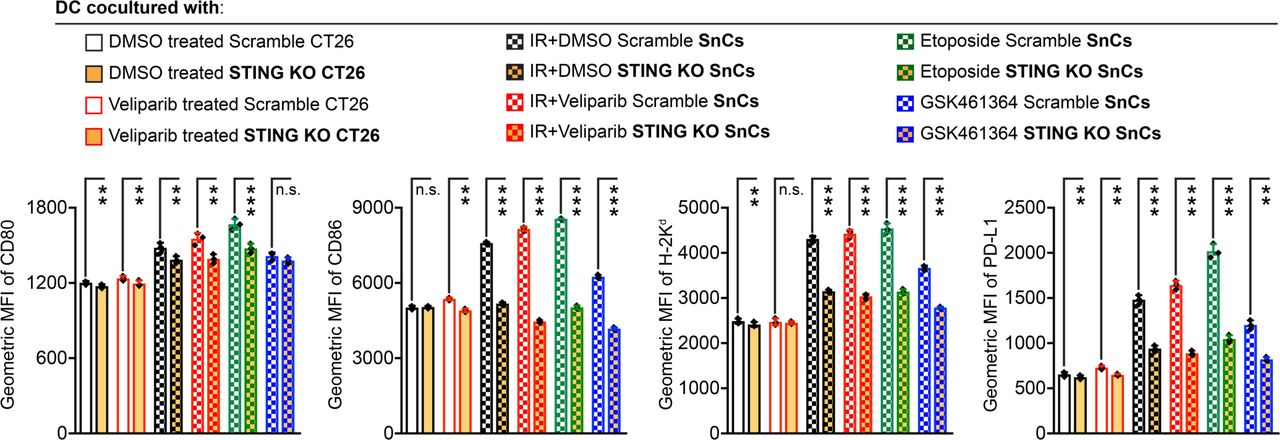
STING signaling mediates SnC activation of BMDCs. For analysis of DC activation/maturation, Cas9 STING KO and scramble control CT26 cells were treated with DMSO, veliparib, IR (12 Gy), IR+veliparib (12 Gy+20 µM), etoposide (2 µM) or GSK461364 (5 µM) for 5 days, followed by coculturing with BMDCs overnight. Then, the viable CD11c+/CD103+ DC population was analyzed for expression of CD80, CD86, H-2Kd and PD-L1. Shown are bar graphs of geometric MFI (n=3, mean±SD). Paired t-test. ***p<0.001, **0.001<p<0.01, *0.01<p<0.05, n.s. p>0.05. BMDCs, bone marrow-derived dendritic cells; DC, dendritic cell; DMSO, dimethyl sulfoxide; MFI, mean fluorescence intensity; IR, irradiation; KO, knock out; PD-L1, programmed cell death ligand 1; SnCs, senescent cells.
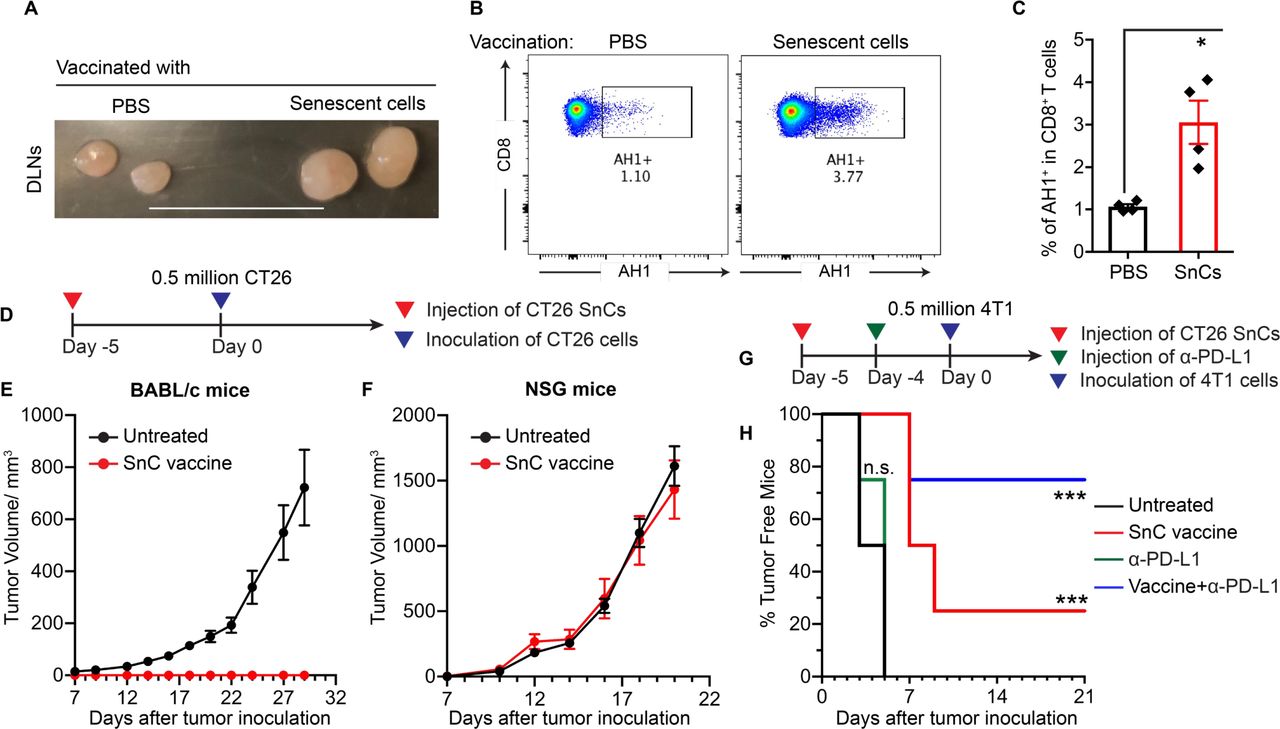
SnC vaccine induces tumor-specific CTLs and blocks tumor engraftment in immune-competent hosts. (A) Representative mouse inguinal draining lymph nodes (DLNs) collected from BALB/c mice 2 days after subcutaneous injection of 0.5×106 CT26 SnCs or PBS. Scale bar: 1 cm. (B and C) Dissociating DLNs for analysis by flow cytometry demonstrates increased CD8+ T cell binding to H-2Ld dextramer presenting AH1. (B) Representative 2D plots showing AH1-binding TCR+ cells in gate. (C) Histogram of % AH1-binding TCR+ cells. Shown by individual mice (dot) and mean±SD (bar). Paired t-test. (D) Experimental schema for CT26 SnC vaccination and CT26 cell challenge in vivo. Growth kinetics of tumors in BALB/c (E) or immunodeficient NSG mice (F) after receiving SnCs on day −5 followed by challenge with 0.5×106 CT26 proliferating cells on day 0. Shown is tumor growth plotted as mean±SEM. (G) Experimental schema for CT26 SnC vaccination and 4T1 cell challenge, with or without α-PD-L1. BALB/c mice injected with 0.5×106 CT26 SnCs at day −5, α-PD-L1 at day −4 or both treatments were challenged with 0.5×106 4T1 proliferating cells on day 0 and examined for palpable tumors at 2-day to 3-day intervals. (H) Kaplan-Meier analysis of 4T1 tumor incidence over time. Logrank test. ***p<0.001, **0.001<p<0.01, *0.01<p<0.05, n.s. p>0.05. CTLs, cytotoxic T lymphocytes; NSG, NOD SCID gamma; PBS, phosphate-buffered saline; PD-L1, programmed cell death ligand 1; SnCs, senescent cells.
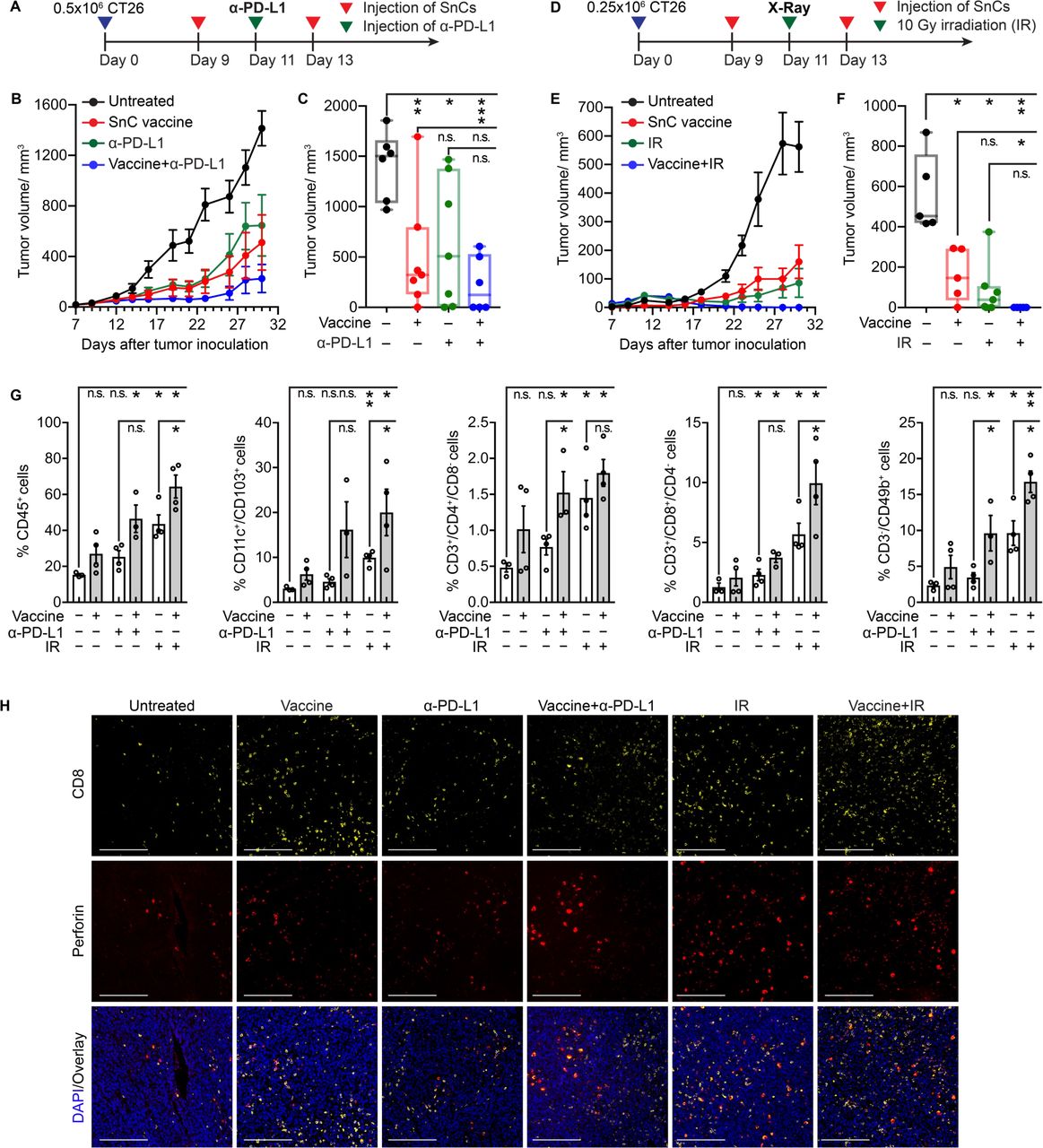
SnC vaccines suppress tumor growth, potentiate cancer therapies and promote tumor immune infiltrates. (A–C) SnC vaccine enhances checkpoint blockade. (A) Experimental schema for treating CT26 tumor-bearing mice with vaccine and/or α-PD-L1. (B) Growth kinetics of CT26 tumors untreated, treated with SnC vaccine, α-PD-L1 or combination therapy (mean±SEM). (C) CT26 tumor size at day 30. Shown by individual tumor (dot) and size range (box and whisker). Paired t-test. (D–F) SnC vaccine potentiates radiotherapy. (D) Experimental schema for treating CT26 tumor-bearing mice with vaccine and/or irradiation (IR). (E) Growth kinetics of CT26 tumors untreated, treated with SnC vaccine, IR or combination therapy (mean±SEM). (F) CT26 tumor size at day 30. Shown by individual tumor (dot) and size range (box and whisker). Paired t-test. (G) Analysis of immune infiltrate in CT26 tumors treated as in (A) and (D) reveals increased CTLs and NK cells after SnC vaccine+IR. Analyzing the viable (Zombie Yellow-) single cell population, total immune, DC, Th, CTL and NK cell were quantified. Shown by individual tumor (open circle) and mean±SD (bar). Paired t-test. (H) Detection of activated CTL infiltrate in situ in CT26 tumors confirms SnC vaccine effects. The tumors examined in (G) were evaluated by immunofluorescence staining for CD8 (yellow), perforin (red) and DAPI (blue). Scale bars: 200 µm. For statistical analysis, ***p<0.001, **0.001<p<0.01, *0.01<p<0.05, n.s. p>0.05. CTLs, cytotoxic T lymphocytes; DC, dendritic cell; NK, natural killer; PD-L1, programmed cell death ligand 1; SnCs, senescent cells.
To examine DC antigen presentation induced by SnCs, we stimulated BMDCs with SnCs or control CT26 cells and then incubated the BMDCs with carboxyfluorescein succinimidyl ester (CFSE) -labeled splenocytes collected from BALB/c mice immunized with irradiated CT26 cells. CFSE dilution was examined after 5 days by flow cytometry (online supplemental figure S3D). BMDCs cocultured with non-senescent, control CT26 cells, treated with veliparib or not, induced less than 27% of proliferating CD8+ T cells. DCs cocultured with SnCs induced by IR, IR+veliparib, etoposide or GSK461364 stimulated 32.1%, 42.9%, 37.5% and 15%, respectively, of proliferating CD8+ T cells (figure 2E and online supplemental figure S6A). After coculture with CT26 SnCs, except those induced by GSK461364, BMDCs also increased the proliferation of CD4+ T cells (figure 2F and online supplemental figure S6C). Potentially reflecting the induction of PD-L1 on BMDCs cocultured with SnCs, T cells displayed a higher proliferation rate in the presence of α-PD-L1 antibody to block PD-1 signaling (figure 2E,F and online supplemental figures S6A–D). Similar results were observed with 4T1 cells (online supplemental figures S7A–D). Interestingly, both conditioned media and cell lysate from SnCs could stimulate DCs, although with distinct patterns (online supplemental figure S8A).

SnC vaccines suppress 4T1 lung colonization by disseminated tumor cells. (A) Experimental schema for testing SnC vaccines against 4T1 lung colonization; 6×104 proliferating 4T1 cells were inoculated through tail vein (i.v.) into BALB/c mice on day 0, delivering disseminated tumor cells and lungs collected at day 21 to examine colony formation. To examine SnCs as preventative or therapeutic vaccine, 0.5×106 senescent 4T1 cells were injected subcutaneously on days −5 and −1 or days 1 and 5, respectively. (B) Effects of SnC vaccines on lung surface metastatic foci. Shown are counts from individual animals (dot), with mean±SEM (bar). Paired t-test. ***p<0.001, **0.001<p<0.01. (C) Representative H&E staining demonstrating tumor colonization in lung parenchyma, vessels and/or airways at low magnification with inset zoomed to high magnification. Scale bars: 2 mm (left) and 50 µm (right). (D) Immunofluorescence analysis of the lung CTL infiltrates at 5 days after SnC injection, staining for CD8 (yellow), perforin (red) and DAPI (blue) under 4x or 20x magnification. Scale bars: 500 µm (4x) and 100 µm (20x). CTL, cytotoxic T lymphocyte; SnCs, senescent cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
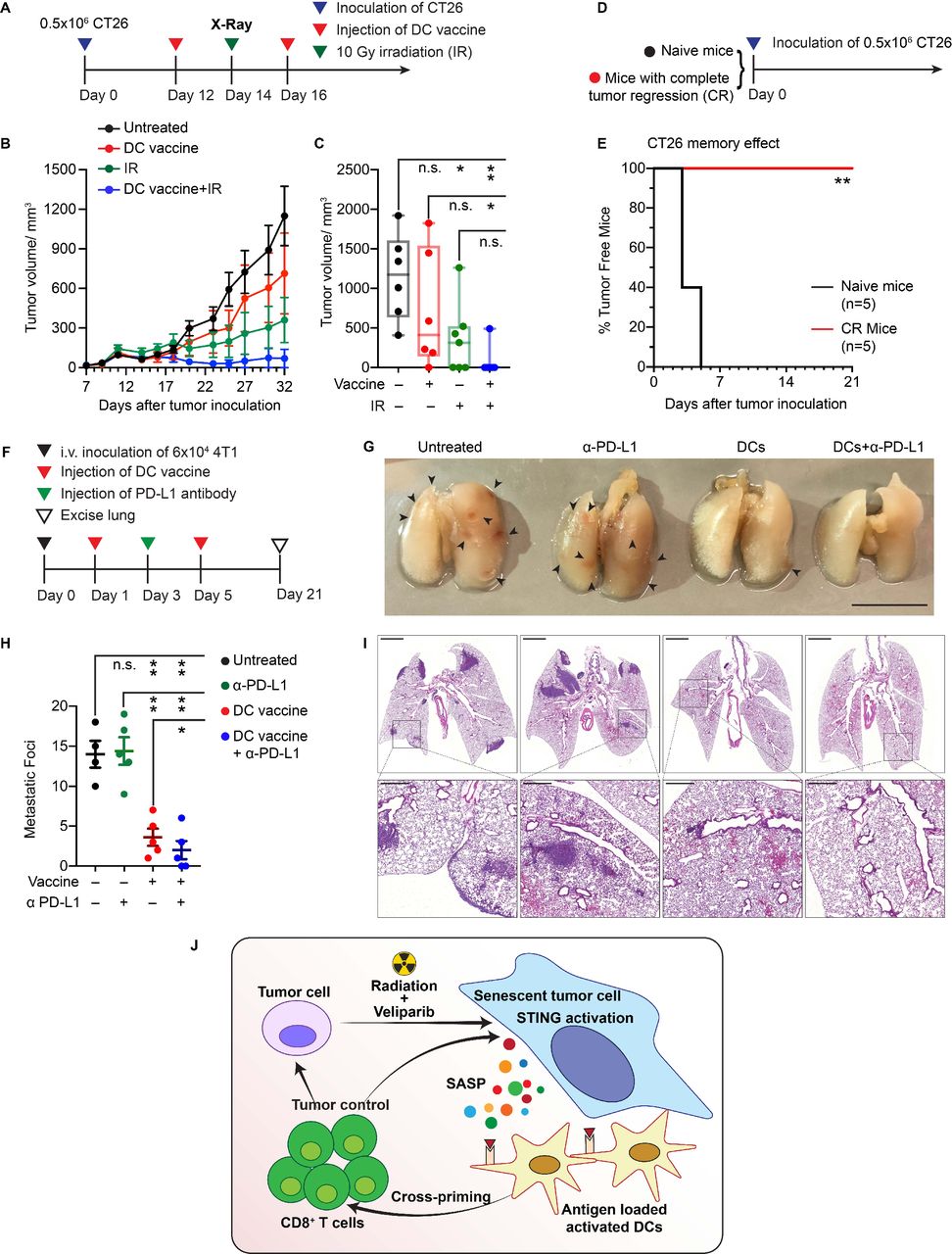
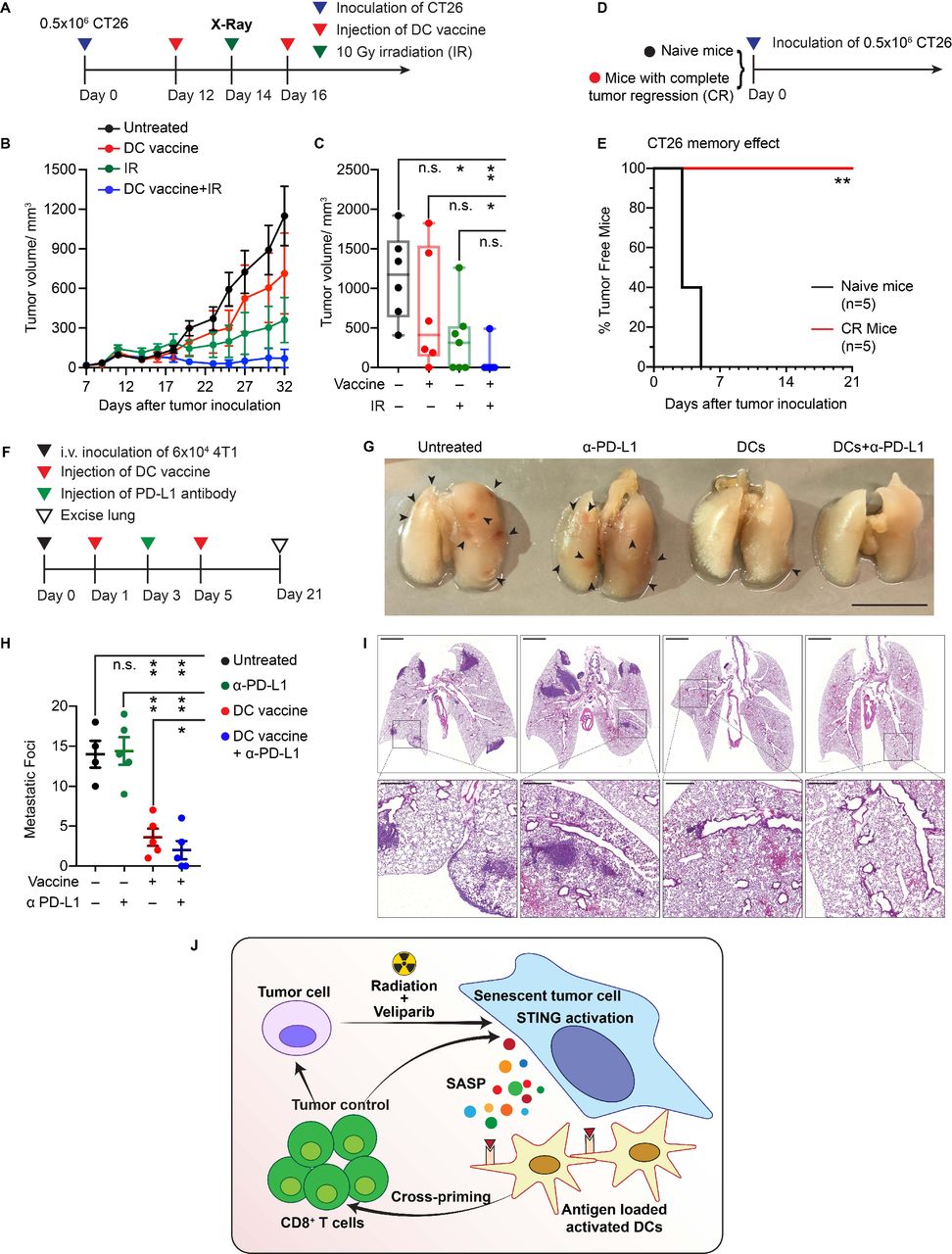
SnC-activated DC vaccine potentiates radiotherapy and suppresses lung colonization. (A) Experimental schema for treating CT26 tumor-bearing mice with SnC-activated DC vaccine and/or external beam irradiation. (B) Growth kinetics of CT26 tumors untreated, treated with vaccine, 10 Gy or combination therapy (mean±SEM). (C) CT26 tumor size at day 32 after tumor cell inoculation. Shown are individual tumor size (dot) and size range (box and whisker). Paired t-test. (D) Experimental schema for challenging naive mice or mice from the group received combination therapy and achieved a complete response. (E) Kaplan-Meier analysis of tumor development over time (n=5 per group). Logrank test. (F) Experimental schema for treating mice with SnC-activated DC vaccines to limit lung colonization. (G) Representative images of lungs from each treatment group. Scale bar: 1 cm. (H) Quantification of metastatic foci on lung surface. Shown are counts from individual lungs (dot), with mean±SEM (bar). Paired t-test. (I) Representative H&E staining of tumor colonization in lung parenchyma at low magnification with inset zoomed to high magnification. Scale bars: 2 mm (upper) and 50 µm (lower). For statistical analysis, ***p<0.001, **0.001<p<0.01, *0.01<p<0.05, n.s. p>0.05. (J) Model for how cellular damage and STING activation promotes immunogenicity of therapy-induced SnCs, leading to DC activation, stimulation of tumor antigen-reactive cytotoxic T cells and an effective adaptive immune response, eliminating surviving tumor cells. DC, dendritic cell; SnCs, senescent cells.
While SnCs can promote a DC-mediated T cell response, their immunogenicity appears to depend on the conditions of senescence induction, raising the question whether SnCs display distinct immunogenicity from other forms of damaged cells, viable or not.17 To examine viability, we treated CT26 SnCs with the senolytic Bcl-2 inhibitor ABT263, which resulted in 51% Annexin V+/PI+ and 44% Annexin V+/PI− apoptotic SnCs (online supplemental figure S3E). Alternatively, CT26 cells were treated with IR±veliparib but cocultured with BMDC after 1 day, prior to adopting a senescent phenotype, rather than after 5 days as before. Thus, BMDCs were cocultured with live senescent, apoptotic senescent, damaged or proliferating CT26 cells, followed by flow cytometric analysis of the surface expression levels of CD80, CD86 and another MHC I molecule H-2Ld. The results indicated that live SnCs are most effective at stimulating DC maturation and activation (figure 2G).
STING signaling is required for senescence-mediated DC activation
Therapy-induced SnCs displayed nuclear DNA double-strand breaks and cytoplasmic DNA accumulation, resulting in the activation of cGAS/STING/TBK/IRF3 signaling and the production of type I IFNs (online supplemental figures S8B–E). It has long been considered that the STING signaling axis plays a critical role in antitumor immunity.18 Recent work has demonstrated that activated STING and IRF3 are required for CD8+ T cell priming in response to tumor antigens.19 To examine the effects of STING activation on the immunogenicity of therapy-induced SnCs, we generated a population of STING knockout (KO) CT26 cells using CRISPR/Cas9/gRNA RNP to target the STING ORF (online supplemental figure S9A). Electroporation with a scrambled gRNA was used as control. After passaging the cells, cellular senescence was induced by IR, IR+veliparib, etoposide or GSK461364. Although STING KO did not affect cell proliferation, senescence induction, γH2AX foci persistence or cytoplasmic DNA accumulation (online supplemental figures S9B–F), it decreased MHC I surface expression and CCL5 secretion in SnCs (online supplemental figures S10A–C). Notably, the ability of CT26 SnCs with STING KO to stimulate the maturation and activation of BMDCs was also diminished (figure 3). Similarly, we pretreated SnCs formed from CT26 or 4T1 cells and proliferating controls with 4 µM covalent STING inhibitor C178. As with the STING KO SnCs, STING inhibition decreased the ability of SnCs to induce BMDC maturation and activation (online supplemental figures S11A–C and S12A–C), indicating that STING signaling in SnCs mediates DC activation.
To explore whether STING activation alone is sufficient to explain the immunogenicity, we treated proliferating CT26 cells for 2 days with DMXAA, a STING agonist. Despite showing robust activation of TBK1, the DMXAA-treated proliferating CT26 cells appeared unable to activate DCs (online supplemental figure S13A,B). Additionally, the DC activation ability of SnCs, whether STING WT or KO, was not affected by DMXAA (online supplemental figure S13B). These results indicate that STING signaling may be necessary, but not sufficient, for SnCs to display immunogenicity.
SnCs induce antigen-specific anti-tumor immune response in vivo
SnCs formed with IR+veliparib appeared to be particularly effective at activating DCs to prime T cells in vitro. To evaluate the immune response to these SnCs in vivo, we injected BALB/c mice subcutaneously (SQ) with 0.5×106 senescent CT26 cells or PBS. Injection of SnCs did not result in tumor growth. However, compared with controls, mice inoculated with SnCs on the dorsum displayed swollen inguinal draining lymph nodes (DLNs) 2 days after injection (figure 4A). CT26 is a BALB/c-derived tumor that expresses the endogenous MuLV retroviral envelope glycoprotein 70 (gp70) and presents the MHC I H-2Ld-restricted immunodominant peptide SPSYVYHQF (AH1, residues 423–431).20 Flow cytometric analysis confirmed a significant increase in AH1-specific CD8+ T cells in the DLNs after SnC injection (figure 4B,C).
Considering that AH1-specific CTLs are sufficient to eliminate CT26 tumors,20 we examined SnCs as a preventative vaccine. To test protection against tumor engraftment, BALB/c mice were left untreated or inoculated with CT26 SnCs and then, after 5 days, injected with 0.5×106 proliferating CT26 cells (figure 4D). While control BALB/c mice developed tumors that grew to the humane endpoint within 4 weeks, no tumors formed in mice that had been treated with SnCs (figure 4E). Notably, STING KO SnCs were less effective at preventing tumor engraftment in BALB/c mice (online supplemental figure S13C). SnC injection did not affect tumor formation or growth in immunodeficient NSG BALB/c mice (figure 4F). Consistent with the shared expression of gp70,21 injection of CT26 SnCs also displayed modest suppression of tumor formation after SQ injection of 0.5×106 proliferating 4T1 cells. While anti-PD-L1 antibody (α-PD-L1) offered no protection on its own, treatment with CT26 SnCs and then α-PD-L1 significantly increased protection against engraftment of 4T1 cells (figure 4G,H). Overall, these results suggest that SnCs formed with IR+veliparib with intact STING signals display significant immunogenicity in vivo.
SnC vaccines slow tumor growth and potentiate therapy
To have broad practical value, tumor cell-derived vaccines must display benefits in treating established tumors. To evaluate SnCs as a therapeutic vaccine, BALB/c mice bearing subcutaneous CT26 tumors were treated with two peritumoral injections of 0.5×106 CT26 SnCs twice in a 5-day interval with or without a single intravenous (IV) injection of α-PD-L1 in between. Considering that the immune profile changes according to tumor size,22 we initiated treatment either on day 9 (average tumor volume ~60 mm3) (figure 5A and online supplemental S14A) or day 12 (average tumor volume ~150 mm3) (online supplemental figure S15A) after tumor inoculation. For treatment beginning on day 9, SnCs and α-PD-L1 treatment each suppressed tumor growth, while the SnCs+α-PD-L1 combination resulted in complete regression of three of six tumors (figure 5B,C and online supplemental figures S14B–E). We observed a similar pattern with 4T1 tumors when treatment was initiated on day 9 using a 4T1 SnC vaccine and/or α-PD-L1, with both SnCs and SnCs+α-PD-L1 leading to tumor elimination (online supplemental figures S16A–G). When treatment of CT26 tumors was initiated on day 12, α-PD-L1 had little effect on its own, but SnCs slowed tumor growth appreciably and the combination treatment displayed moderate tumor control, though no tumor elimination (online supplemental figures S15B–G).
As an alternative to α-PD-L1, we examined combining the SnC vaccine with radiation to treat established tumors. Here, we again formed subcutaneous CT26 tumors and treated them starting on day 9 or 12 with two peritumoral injections of 0.5×106 SnCs over a 5-day interval, with or without a single dose of 10 Gy irradiation (IR) in between. As before, SnCs alone suppressed tumor growth, but the effect was far greater on smaller tumors (figure 5D–F and online supplemental figures S15H–J). On its own, a single dose of 10 Gy IR significantly delayed tumor growth, but most tumors recovered and resumed growth within 10 days. However, combining SnCs with radiation significantly enhanced the effects of radiation, resulting in tumor elimination by 2 weeks post-treatment for both smaller and larger tumors (online supplemental figures S14F-J and S15K-N).
To investigate the effects of peritumoral injection of SnCs on the tumor microenvironment, we obtained tumors 5 days after treatment with SnCs, α-PD-L1, SnCs+α-PD-L1, IR or SnCs+IR, and used flow cytometry to evaluate total CD45+ immune infiltrates, CD11c+/CD103+ DCs, CD3+/CD4+ helper T cells (Th), CD3+/CD8+ cytotoxic T lymphocytes (CTLs) and CD3−/CD49b+ natural killer (NK) cells (online supplemental figures S3G–H). Compared with control, peritumoral injection of SnCs led to a significant increase in tumor-infiltrating lymphocytes (TILs), and this effect was amplified by both α-PD-L1 and 10 Gy (figure 5G). Similarly, immunofluorescence analysis of tumor sections demonstrated increased levels of activated CTLs (CD8+/Perforin+ or CD8+/Granzyme B+) after SnC vaccine, whether alone or in combination with α-PD-L1 or 10 Gy (figure 5H and online supplemental figure S17A,B). A similar pattern of enhanced immune response was observed in 4T1 tumors treated with 4T1 SnCs alone or in combination with α-PD-L1 (online supplemental figures S18A–D).
SnC vaccines suppress lung colonization after tail vein injection of tumor cells
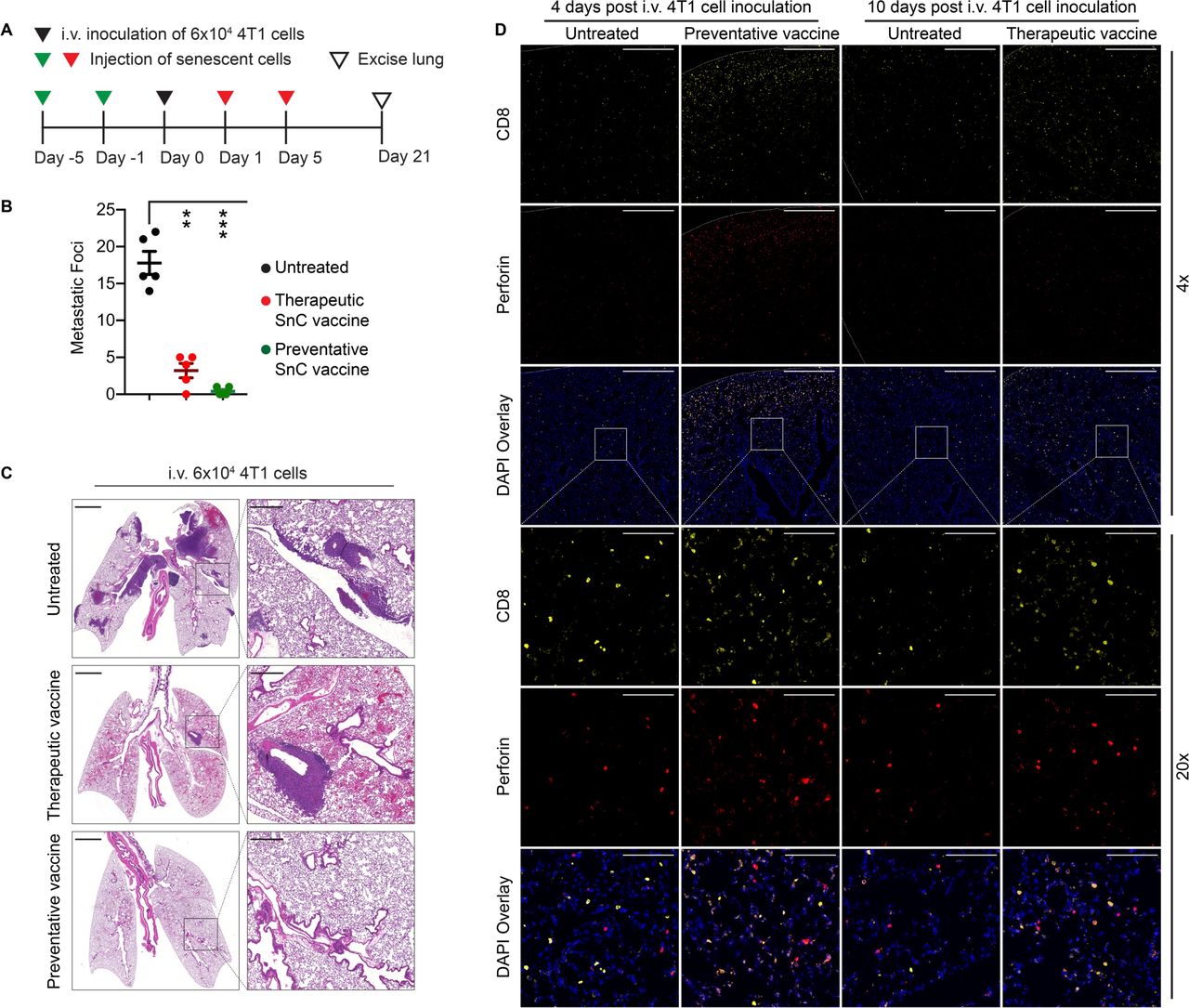
While conventional therapies may be sufficient to eradicate primary tumors, distant spread via metastasis lacks effective treatments. Used in the adjuvant setting, immunotherapies including tumor cell vaccines offer the potential to eliminate DTCs and micrometastases and thereby suppress distant recurrence.23 Toward modeling the spread of breast cancer from primary tumors to the lungs, we injected 6×104 proliferating 4T1 cells via the tail vein to colonize the lungs of BALB/c mice (day 0). Tail vein injection appears to offer a faster and simpler model to form lung metastases with similar biology to those produced by orthotopic tumors that spread via spontaneous metastasis.24 To examine preventative or therapeutic vaccination, 0.5×106 4T1 SnCs were injected subcutaneously into mice on days −5 and −1 or on days 1 and 5, respectively (figure 6A). To examine the effects of SnC vaccine on DTCs and resulting metastases, we collected the lungs on day 21 for microscopic and histological examination. While the untreated mice developed an average of 17 metastatic nodules on the lung surface, the preventative SnC vaccine protected three out of five mice from developing any visible metastatic nodules and the therapeutic SnC vaccine reduced lung surface metastases to an average of four (figure 6B). Histological analysis of lung sections confirmed that the SnC vaccine displayed a similar protective effect against metastases formed in the lung parenchyma (figure 6C). Similar protection was observed for IV injection of 8×104 proliferating cells, which otherwise produced significantly higher numbers of metastatic foci (online supplemental figurea S19A–C). Immunofluorescence analysis of lungs, whether collected on day 4 (4 days after 4T1 cell inoculation and 5 days after preventative SnC vaccine) or on day 10 (10 days after 4T1 cell inoculation and 5 days after therapeutic SnC vaccine) revealed a similar increase in activated CTLs (CD8+/perforin+) in pulmonary tissue (figure 6D).
SnC-activated DC vaccine recapitulates the effects of SnC vaccine and induces antitumor memory responses
While the vaccine-like effects of SnCs may reflect multiple mechanisms, our in vitro and in vivo results suggest a model where the injected SnCs are taken up by DCs to induce maturation and activation, leading to expansion of tumor-responsive CTLs in the DLNs. Thus, we examined whether peritumoral injection of ex vivo activated DCs could recapitulate the therapeutic vaccine effects of injected SnCs. To prepare the DC vaccine, the BMDCs were cocultured with IR+veliparib-induced CT26 SnCs for ~12 hours. The resulting activated, antigen-loaded DCs were then injected adjacent to CT26 tumors, twice over 5 days starting on day 12, with or without a single X-ray dose of 10 Gy in between (figure 7A). SnC-activated DCs caused only modest tumor growth inhibition on their own but the combination with IR led to the elimination of most tumors (figure 7B,C and online supplemental figurea S20A–E). To test immunological memory, mice whose tumors had been eliminated by the SnC-activated DCs+IR treatment were re-challenged with 0.5×106 proliferating CT26 cells (figure 7D). No tumor development was observed over 3 weeks in the treated mice versus 100% tumor take within 1 week in naive mice (figure 7E).
To confirm systemic effects, we investigated whether SnC-activated DCs could suppress 4T1 lung metastasis. BALB/c mice were injected IV with 6×104 4T1 cells on day 0 and then DCs were injected SQ on days 1 and 5, with or without IV α-PD-L1 on day 3 (figure 7F). Examining the lungs on day 21, α-PD-L1 had no effect on its own while the SnC-activated DCs significantly reduced the development of lung metastases, with or without α-PD-L1 (figure 7G,H). Histology analysis revealed a similar pattern, where the SnC-activated DCs markedly suppressed tumor formation in the lung parenchyma, with or without α-PD-L1 (figure 7I).
Discussion
While the first observation of tumor elimination in mice after vaccination with irradiated tumor tissue was made over a century ago25 and studies showing enhanced radiation response after injection of irradiated tumor tissue were published over five decades ago both in rodents26 and in patients with breast cancer,27 irradiated autologous tumor cell-based vaccines have yet to enter clinical practice. The dramatic success of immune checkpoint blockade immunotherapy has revived interest in cancer vaccines, which might provide a complementary means to help eliminate primary and distant tumors and then maintain durable surveillance against recurrence.28 29 Most recent efforts to increase immunogenicity of lethally irradiated tumor cell vaccines have relied on engineering the cells to express GM-CSF or other cytokines toward promoting antigen uptake and presentation, with several examples under clinical investigation such as GVAX for pancreatic and prostate cancers30 and Vigil for ovarian cancer.31 Among many alternative strategies to produce immunogenic whole tumor cell vaccines, preclinical studies have reported encouraging results with early ferroptotic cells,32 necroptotic cells33 and etoposide-injured cells.34 Senescent tumor cells have multiple attractive features as cancer vaccines insofar as SnCs can no longer proliferate but persist indefinitely, offering the potential for prolonged release of cancer antigens and immune-stimulating cytokines.13 35 Here, we have reexamined injecting SnCs as preventative and therapeutic vaccines, finding them effective in limiting tumor formation and growth, sensitizing tumors to immunotherapy and radiation, and inhibiting tumor metastasis. Further, we find that injecting SnC-activated DCs recapitulates the effects of SnCs, providing a realistic path to the clinic.
Immune checkpoint blockade immunotherapy directed at maintaining CTL activity in the face of immunosuppressive signals in the tumor microenvironment such as PD-L1 has impacted the treatment and outcomes for many cancers, but overall response rates remain close to 25%, similar to adverse event rates.36 Resistance to checkpoint blockade may be as simple as a lack of tumor-reactive CTLs to reactivate or protect, suggesting a need for complementary approaches to enhance tumor antigen presentation. Conventional cancer therapies including radiation can be effective triggers for immunogenic cell stress and cell death (ICD),17 providing both antigen and adjuvant to serve as a vaccine in situ that may help drive antitumor immune responses. In particular, ICD can encourage DCs to collect tumor antigens and carry them into secondary lymphoid organs for antigen presentation, leading to antigen-specific T cell responses.29 Although originally linked to cells succumbing to genotoxic stress as by necrosis, the definition of ICD has broadened to include diverse forms of cell death such as necroptosis, ferroptosis and parthanatos. Nonetheless, radiation-induced ICD on its own does not appear to be sufficient to overcome immunosuppression or to dramatically improve the response to checkpoint blockade.
Cellular senescence occurs when cellular damage is irreversible but not sufficient to force outright cell death.17 37 Accordingly, immunogenic senescence and ICD share multiple driving mechanisms, including persistent DNA damage signaling, mitochondrial dysfunction, oxidative damage and deregulated autophagy. The resulting stress signaling may enhance immunogenicity by changing cell surface ligands, factors released from the cells and/or contents of exosomes or other vesicles. In both senescence and ICD, the increased antigenicity can be at least partially ascribed to the upregulation of MHC class I-dependent antigen-presenting machinery. Additionally, both senescence and ICD provide potent adjuvant signals through display or release of damage-associated molecular patterns (DAMPs) and secretion of numerous bioactive factors, including cytokines, chemokines and metabolic intermediates. While release of adjuvant factors from ICD can either be active or passive due to the change of membrane permeability and/or integrity, it is usually considered an active process in SnCs via the SASP. Along with some differences, ICD and senescence share specific features that influence immunogenicity, including but not limited to release of type I IFNs and the alarmin HMGB1.37
Much like immunosuppressive DAMPs, SASP factors can recruit myeloid-derived suppressor cells or have other effects that limit the cytotoxicity of NK cells and CD8+ T cells.38 39 Other dark sides of the SASP include paracrine signals that may promote tumor cell survival and dormancy. As such, therapy-induced senescence may play a detrimental role in the tumor microenvironment, contributing to resistance and recurrence. In order to maximize tumor cell clearance without incurring adverse consequences, a one–two punch strategy for cancer therapy has been proposed,8 40–42 where treatment with genotoxic cancer therapy that promotes therapy-induced senescence is followed by selective elimination of the senescent cancer cells. The second punch might be mediated by senolytic small molecules or immunotherapies.
Our results cannot answer the question whether senescent tumor cells are predominantly beneficial or detrimental. An attractive hypothesis is that when SnC formation is acute, beneficial effects and potential for immune clearance may be the greatest, while persistent SnCs mediate chronic, deleterious inflammation and other toxic effects.43 An added benefit of greater SnC immunogenicity is that it may potentiate rapid immune clearance and limit SnC persistence, reducing the deleterious effects of the SASP in the tumor microenvironment. Taken together with this work, prior studies13 35 along with exciting new results published while this paper was under consideration44 45 provide strong evidence that SnCs can be immunogenic, inducing a cytotoxic T cell response with potential to not only clear SnCs but also prevent or eliminate tumors.
A critical pathway linked to the immunogenicity of radiation-induced ICD and senescence is STING signaling, resulting from cGAS binding to cytoplasmic DNA, activation of STING and TBK1, and increased expression of type I IFNs, cytokines and class I MHC, which facilitates immune response and recognition.46 STING activation in tumors also promotes CTL infiltration. When STING was knocked out or inhibited, SnCs lost the ability to stimulate DCs, consistent with previously defined roles for STING in DC maturation and activation.19 Although the specific mechanism remains to be studied, increased secretion of CCL5 and/or expression of MHC I may underlie STING-mediated immunogenicity in SnCs.
A concern that may prevent personalized SnC vaccines from reaching the clinic is the potential of reintroducing viable tumor cells that may contaminate the SnC preparation and/or arise due to escape from senescence.47 Considering that SnCs efficiently activate DCs to prime T cells in vitro, we explored whether we could simply replace injecting SnCs with SnC-pulsed DCs. DC vaccines48 are already practical with several preparations pulsed with peptide or protein antigens having advanced to the clinic, where most have displayed modest benefits. Vaccines based on autologous or allogeneic DCs fused or loaded with tumor cells or pulsed with lysates have also been studied. Recent interest has shifted to exploiting ICD to enhance tumor cell antigenicity and DC functionality.49 Here, we have described a novel approach for activating and loading DCs by coculture with immunogenic senescent tumor cells in vitro. Subcutaneous vaccination with SnC-activated DCs elicits both local and systemic effects, leading to growth suppression of established tumors and DTCs. Injection of SnC-activated DCs enhanced the effects of anti-PD-L1 checkpoint blockade, and most mice treated with both radiation and SnC-activated DCs displayed tumor elimination and antitumor memory. Personalized therapies based on SnC-activated DCs may be both safe and practical. Loading by coculture with patient tumor-derived SnCs appears compatible with ongoing advances such as off-the-shelf DCs and other emerging strategies to improve DC production and function.50 Overall, our results suggest the clinical potential to develop senescent tumor cells and SnC-activated DC vaccines for immunotherapy to enhance conventional therapy and prevent recurrence and metastasis.
Data availability statement
Data are available upon reasonable request. Further information and requests for resources and reagents should be directed to and will be fulfilled by Stephen Kron (skron@uchicago.edu).
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
We thank Ani Solanki from the University of Chicago Animal Resource Center for assistance with tail vein injections, Elena Efimova for scientific advice and our colleagues at The University of Chicago and AbbVie for helpful support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
YL and JP contributed equally.
Contributors Conceptualization: YL, JP, SJK; methodology: YL, JP; visualization: YL, JP, DW; writing and editing: YL, JP, DW, KDB, JVG, SJK; funding acquisition: SJK; supervision: KDB, JVG, SJK; project administration and guarantor: SJK.
Funding These studies were supported by: AbbVie/UChicago collaboration award (SJK, KDB, JVG). METAvivor Translational Research Award (SJK). NIH R01 CA199663 (SJK). NIH R01 CA217182 (SJK). NIH R01 CA258737 (SJK). This work utilized service cores supported by: University of Chicago Comprehensive Cancer Center support grant NIH P30 CA014599.
Competing interests SJK is a co-founder of OncoSenescence. KDB and JVG are employees of AbbVie. The other authors declare that they have no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.