Article Text

Abstract
Background To enhance the efficacy of adoptive NK cell therapy against solid tumors, NK cells must be modified to resist exhaustion in the tumor microenvironment (TME). However, the molecular checkpoint underlying NK cell exhaustion in the TME remains elusive.
Methods We analyzed the correlation between TIPE2 expression and NK cell functional exhaustion in the TME both in humans and mice by single-cell transcriptomic analysis and by using gene reporter mice. We investigated the effects of TIPE2 deletion on adoptively transferred NK cell therapy against cancers by using NK cells from NK-specific Tipe2-deficient mice or peripheral blood-derived or induced pluripotent stem cell (iPSC)-derived human NK cells with TIPE2 deletion by CRISPR/Cas9. We also investigated the potential synergy of double deletion of TIPE2 and another checkpoint molecule, CISH.
Results By single-cell transcriptomic analysis and by using gene reporter mice, we found that TIPE2 expression correlated with NK cell exhaustion in the TME both in humans and mice and that the TIPE2 high NK cell subset correlated with poorer survival of tumor patients. TIPE2 deletion promoted the antitumor activity of adoptively transferred mouse NK cells and adoptively transferred human NK cells, either derived from peripheral blood or differentiated from iPSCs. TIPE2 deletion rendered NK cells with elevated capacities for tumor infiltration and effector functions. TIPE2 deletion also synergized with CISH deletion to further improve antitumor activity in vivo.
Conclusions This study highlighted TIPE2 targeting as a promising approach for enhancing adoptive NK cell therapy against solid tumors.
- immunotherapy, adoptive
- killer cells, natural
Data availability statement
Data are available on reasonable request.
Data availability statement
Single-cell transcriptomic analysis was based on the dataset available in the Gene Expression Omnibus (GEO) with the accession code GSM4134620.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
NK cell exhaustion in the tumor microenvironment (TME) of solid tumors limits the therapeutic potential of adoptive NK cell therapy, whose molecular checkpoint remains elusive.
WHAT THIS STUDY ADDS
We found that TIPE2 expression correlated with NK cell exhaustion in the TME in both humans and mice. TIPE2 deletion endowed NK cells with elevated capacities for tumor infiltration and effector functions and promoted the antitumor activity of adoptively transferred mouse NK cells, as well as adoptively transferred human NK cells, either derived from peripheral blood or differentiated from induced pluripotent stem cells. TIPE2 deletion also synergized with the deletion of another checkpoint molecule, CISH, to further improve antitumor activity in vivo.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study suggests that targeting TIPE2 represents a promising approach for enhancing adoptive NK cell therapy against solid tumors.
Background
Adoptive NK cell therapy represents an important cutting-edge strategy against tumors.1 However, tumor-associated exhaustion limits NK cell efficacy, especially in the tumor microenvironment (TME) of solid tumors.2 3 Under exhausted conditions, NK cells poorly accumulate in the tumor tissue and display impaired cytolytic activity and reduced effector cytokine production. NK cell exhaustion is usually accompanied by upregulation of cell surface inhibitory receptors and downregulation of cell surface activating receptors. We previously identified a checkpoint for tumor-associated NK cells, the cell surface receptor TIGIT, whose protein expression correlates with the exhaustion and dysfunctional status of NK cells in the TME2 and mediates NK cell exhaustion on interaction with its ligand(s) within the TME.4 However, whether intracellular molecular checkpoints also exist and function to regulate NK cell exhaustion in the TME remains elusive.
Genetic modifications have shown great potential for improving various parameters of adoptive NK cell therapy for cancers. Chimeric antigen receptors (CARs) endow NK cells with tumor-specific targeting capability.5 A noncleavable, high-affinity CD16 facilitates ADCC by NK cells.6 IL-15 receptor fusion supports NK cell survival without the need for cytokine supplementation. CISH deletion promotes NK cell IL-15 signaling and metabolic fitness.7 8 In particular, induced pluripotent stem cell (iPSC)-derived NK cells, an important strategy for producing a large number of genetically standardized single-clonal NK cells for immunotherapy, have enabled the potential for multiplex genome editing to enhance multiple parameters of NK cells to maximize improvements.9 However, genome editing to improve adoptive NK cell resistance to the TME has not been reported.
TIPE2 (tumor necrosis factor-α (TNF-α)-induced protein-8 like-2) is a negative regulator of antitumor immunity. Global deletion of Tipe2 in mice suppressed xenografted tumor growth, accompanied by the loss of tumor-promoting effects of myeloid-derived suppressor cells.10 We have previously shown that NK-specific deficiency of Tipe2 promotes NK cell maturation and the optimal functional status in the steady state11 by elevating the mTOR response to IL-15.12 NK-specific deficiency of Tipe2 also improves the endogenous antitumor immune response. However, whether TIPE2 deficiency renders NK cells resistant to exhaustion in the TME and promotes NK cell antitumor activity has not been reported.
Here, the gene expression of TIPE2, a checkpoint molecule targeting IL-15-induced mTOR activity in NK cells, correlated with the functional exhaustion status in human tumor-associated NK cells, as shown by single-cell transcriptomic analysis. The deletion of TIPE2 in either mouse or human NK cells enhanced NK cell effector functions and rendered these NK cells more strongly controlled solid tumor growth in vivo after adoptive transfer into tumor-bearing mice. Therefore, targeting the checkpoint molecule TIPE2 might represent a promising approach for enhancing NK cell adoptive therapy against tumors.
Methods
Mice
Tipe2flox/flox mice were purchased from the Cam-Su Genomic Resource Center (Suzhou, China). Ncr1-iCre mice and NOD-Scid-γc −/−mice with transgenic human IL-15 expression (‘NDG-hIL15’) were purchased from Biocytogen (Beijing, China). NK-specific Tipe2-deficient mice (‘Tipe2 ΔNK/ΔNK mice’) were generated by crossing Tipe2flox/flox mice with Ncr1-iCre mice. Age-matched and sex-matched Tipe2flox/flox mice were used as controls (referred to as ‘Tipe2WT/WT mice’). Tipe2 reporter mice with a Tipe2-ires-Egfp knock-in cassette were generated by Cyagen (Suzhou, China). All mice used were 5–8 weeks old with a C57BL/6 background (except for NDG-hIL15) and were housed in a specific pathogen-free facility at the Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences.
Cell lines
K562 cells and HCT15 cells were purchased from CellBank (Shanghai, China). Tap2-deficient RMAS lymphoma cells were preserved in-house.
Cell culture
Human primary NK cells were enriched from the peripheral blood mononuclear cells (PBMCs) of healthy donors by an NK cell isolation kit via a magnetic cell sorting kit from Miltenyi (Bergisch Gladbach, Germany), followed by expansion with an NK cell expansion kit from DAKEWE (Shenzhen, China), according to the manufacturer’s instructions. Alternatively, enriched and gene-edited or control NK cells were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS) and penicillin and streptomycin (100 IU/mL) plus 1000 IU/mL human IL-2 to determine the effects of gene editing on NK cell proliferation under more defined conditions. Under such conditions, the medium was half-changed every other day. iPSCs were cultured using the mTeSR1 plus kit from Stemcell Technologies (Vancouver, Canada). iPSC differentiation into NK cells was performed using the STEMdiffTM NK Cell Kit from Stemcell Technologies. K562 and RMAS cells were cultured in RPMI 1640 medium with 10% FBS and penicillin and streptomycin (100 IU/mL). HCT15 cells were cultured in DMEM with 10% FBS and penicillin and streptomycin (100 IU/mL). Mouse splenic cells were cultured in RPMI 1640 medium with 10% FBS, penicillin and streptomycin (100 IU/mL), and 10 ng/mL IL-2 (Peprotech, Cranbury, USA). When indicated, an additional 10 ng/mL IL-10 (Peprotech, Cranbury, USA), 10 ng/mL TNF-α (Peprotech, Cranbury, USA), 10 ng/mL TGF-β (Biolegend, San Diego, USA), or 10 µM H2O2 (Boster, Wuhan, China) was added. The use of peripheral blood from healthy donors was approved by the ethical committee of Shenzhen Institutes of Advanced Technology.
Tumor models
Groups of six mice per experiment were used. The group size ensured enough power to determine biological differences. No mice were excluded from this study, and no active randomization was applied to groups. Potential confounders were minimized by treatments performed in parallel. The investigators were not blinded to group allocation during the experiment and/or when assessing the outcome. Single-cell suspensions of RMAS, K562 or HCT15 cancer cells were injected subcutaneously into the indicated strains of mice (5×105 cells for RMAS per mouse or 5×106 cells for K562 and HCT15 per mouse). Tumor size, endpoint tumor weight, infiltrating NK cell numbers and effector functions were measured, among which tumor size and endpoint tumor weight were the primary outcomes measured.
Genome editing in human NK cells
For the generation of TIPE2, CISH, or TIPE2 & CISH double-deleted human NK cells, enriched NK cells from PBMCs of healthy donors were subjected to electroporation using the Lonza 4D Nucleofector as described.13 For the generation of single clonal TIPE2-deleted human iPSC-NK cells, WT human iPSCs were subjected to electroporation using the Lonza 4D Nucleofector, followed by single clonalization through limited dilution and further screening of PCR and sequencing for single clones with frame-shift mutations in both alleles.
Western blot
The following primary antibodies were used for Western blotting: TIPE2 (Proteintech, Rosemont, USA), CIS (Boster, Wuhan, China), and β-Tubulin (Proteintech, Rosemont, USA).
Flow cytometry
Single-cell suspensions were stained with the appropriate monoclonal antibody in phosphate-buffered saline containing 5% rat serum. When necessary, intracellular staining was performed using the TrueNuclear Transcription Factor Buffer Set (BioLegend) according to the manufacturer’s instructions. For detection of intracellular IFN-γ, TNF-α, or perforin expression, mouse lymphocytes were stimulated with Cell Stimulation Cocktail for 3 hours in vitro before staining with antibodies (Thermo Fisher, Waltham, USA). CytoFLEX (Beckman Coulter, Brea, USA) and FACSAria III (BD Biosciences, San Diego, USA) were used for analysis and cell sorting, with dead cells excluded by the LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Invitrogen, Carlsbad, USA). Antibodies specific for mouse CD45 (30-F11), NK1.1 (PK136), mouse CD3 (17A2), mouse IFN-γ (XMG1.2), mouse TNF-α (MP6-XT22), human CD3 (UCHT1), human CD94 (DX22), human CD2 (TS1/8), human NKG2D (1D11), human NKG2C (S19005E), human NKp44 (P44-8), human NKp46 (9E2), human NKp30 (P30-15), human TRAIL (RIK-2), human FasL (NOK-1), human CD16 (3G8), human CD57 (HNK-1), human NKG2A (S19004C), human DNAM-1 (11A8), and human 2B4 (C1.7) were purchased from Biolegend (San Diego, USA). Antibodies specific for phospho-S6 (N7-548) were purchased from BD Biosciences (San Diego, USA).
Cytolytic assays
For cytolytic assays against K562 cells, Cell Trace Violet (CTV, Invitrogen, Carlsbad, USA)-labeled target cells were cocultured with NK cells at the indicated effector:target (E:T) ratios for 4 hours. After that, cell mixtures were stained with 7-AAD to determine the percentages of 7-AAD+ dead cells among CTV+ target cells.
Single-cell transcriptomic analysis
Analysis of filtered data on sorted CD45+CD56+CD3-CD4-CD8a-CD14-CD163- NK cells from dissociated human melanoma lesions from GEO dataset GSM413462014 was performed with Seurat15 16 by selecting highly variable genes using FindVariableGenes functions with default parameters, reducing the dimensionality by PCA, and clustering the cells using FindNeighbors(dims=1:5) and FindClusters (resolution=0.1). We then applied RunTSNE (dims=1:5) for visualization.
Statistical analysis
Statistically significant differences between two groups were determined by Student’s t-tests or one-way analysis of variance (ANOVA) when appropriate, except that two-way ANOVA was used in the comparison of tumor growth. A p<0.05 was considered significant in all analyses.
Results
High TIPE2 expression closely correlates with the functional exhaustion of human tumor-infiltrating NK cells
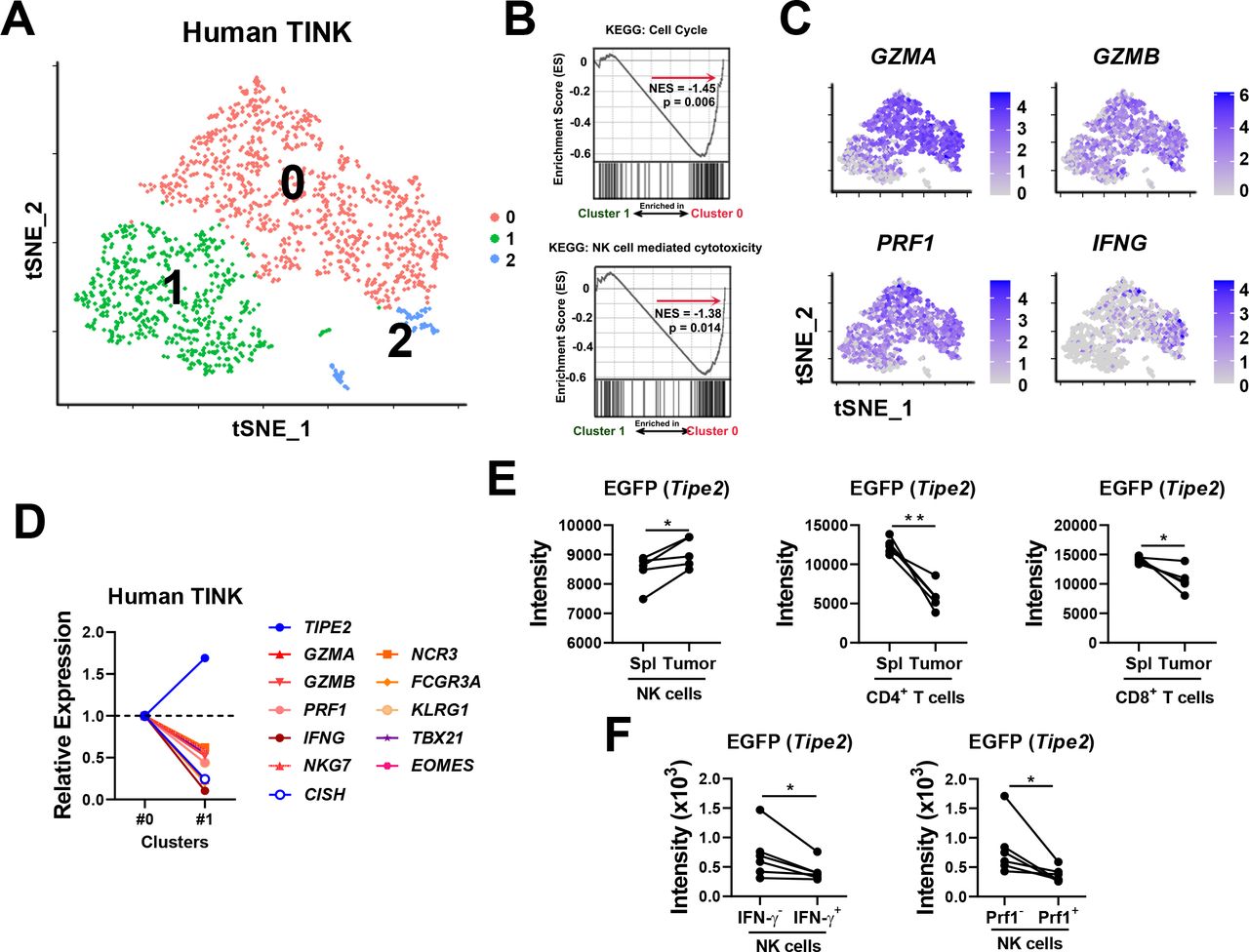
Previously, we found that TIPE2 expression correlates with NK cell maturation. To start investigating the role of TIPE2 in regulating the functional exhaustion of tumor-infiltrating NK cells, single-cell RNA sequencing data on tumor-infiltrating NK cells from human melanoma lesions were projected into two dimensions by tSNE, which revealed the clustering of tumor-infiltrating NK cells into two different major subsets with a higher or a lower level of functional status (figure 1A), as well as a minor subset. Among the two major subsets, GSEA indicated that subset #0, compared with subset #1, was enriched with gene sets related to the cell cycle and NK cell cytotoxicity (figure 1B). Subset #0 expressed higher levels of genes encoding effector molecules (eg,GZMA, GZMB, PRF1, and IFNG), maturation markers (eg,KLRG1), activating surface receptors (eg,NCR1, NCR3, and FCGR3A), and essential transcription factors governing NK cell effector functions (eg,TBX21) than subset #1 (figure 1C,D). These results indicated that subset #1 represented a more functionally exhausted subset of tumor-infiltrating NK cells with lower effector functions. Based on these analyses, we found that TIPE2 was expressed at a higher level in the more exhausted subset #1 (figure 1D), suggesting that TIPE2 expression should correlate with human NK cell exhaustion in the TME. The online software Carcinoma Ecotyper from a recent study has managed to couple subsets of tumor-infiltrating lymphocytes with survival association.17 Carcinoma Ecotyper identified five subsets within tumor-infiltrating NK cells from figure 1A that have been associated with better or poorer survival in a pan-carcinoma cohort including 6475 patients (online supplemental figure S1A,B). Analysis of TIPE2 expression in these NK cell subsets revealed that subset S1, with key features (PRF1 hi FCGR3A hi NCR3 hi) corresponding to the TIPE2 low subset in figure 1D,was significantly correlated with better survival (p=0.000021) (online supplemental figure S1A,B). Importantly, subset S1 expressed the lowest level of TIPE2 among all five subsets. In addition, subset S4, coexpressing the marker KLRG1 similar to the TIPE2 low subset in figure 1D, was mildly correlated with better survival (p=0.095). On the other hand, subset S2 was not correlated with survival (S2, p=0.55), while S3 and S5 were correlated with poorer survival (S3 p=0.0055, S5 p=0.16). Subset S3, which was most significantly correlated with poor survival (p=0.0055), expressed the highest level of TIPE2 among all five subsets (online supplemental figure S1A,B). Therefore, the TIPE2- high tumor-infiltrating NK cell subset might correlate with poorer prognosis in tumor patients. On the other hand, CISH, a checkpoint gene involved in NK cell antitumor activity,18 was expressed at a higher level in the more functional subset #0 (figure 1D). The preferential expression of TIPE2 and CISH in two different subsets with lower and higher effector functions suggests that they might function nonredundantly. Analysis of other genes involved in NK cell exhaustion, such as HAVCR2 (Tim-3),19 KLRC1 (NKG2A)20and TIGIT 4 , showed that KLRC1 (NKG2A) was expressed at higher levels in the more exhausted cluster, similar toTIPE2 (online supplemental figure S2).
Supplemental material
Supplemental material

Correlation between TIPE2 expression and functional exhaustion of tumor-infiltrating NK cells. (A) tSNE of human melanoma-infiltrating NK cells. (B) GSEA of subset #0 and subset #1 from (A) using KEGG: cell cycle and KEGG: NK cell-mediated cytotoxicity. (C) Feature plots showing the expression levels of the indicated genes in each cluster from (A). (D) Expression of the indicated genes in subset #1 relative to subset #0. (E, F) Tipe2 reporter mice were subcutaneously inoculated with RMAS tumor cells. Tumor-infiltrating lymphocytes and splenic lymphocytes were analyzed from day 9 to day 14 post tumor challenge for (E) EGFP expression in different lymphocyte populations or (F) EGFP expression in tumor-infiltrating NK cells positive versus negative for IFN-γ or perforin production. (E, F) Data are representative of two independent experiments. *p<0.05, **p<0.005 (paired Student’s t-test).
To confirm the correlation between the expression of TIPE2 and NK cell exhaustion, we generated Tipe2 reporter mice with a ‘knock-in’ IRES-EGFP downstream of the Tipe2 gene open reading frame within the Tipe2 gene expression cassette. We established the RMAS tumor model in these mice by subcutaneous injection of 5×105 RMAS cells, which developed similar tumor growth as WT mice (online supplemental figure S3). Nine days later, analysis of both splenic NK cells and tumor-infiltrating NK cells showed that tumor-infiltrating NK cells expressed significantly higher levels of EGFP than splenic NK cells in the same mice (figure 1E, online supplemental figure S4A), suggesting that Tipe2 was preferentially expressed by tumor-infiltrating NK cells. Meanwhile, such a preference was not observed in tumor-infiltrating CD4+ or CD8+ T cells (figure 1E). Moreover, IFN-γ- or perforin- tumor-infiltrating NK cells expressed a higher intensity of EGFP (figure 1F, online supplemental figure S4B), suggesting that Tipe2 expression correlated with the lower functional status of tumor-infiltrating NK cells in mice. Next, we examined several factors enriched in the TME or previously shown to upregulate TIPE2 in other cell types. We found that IL-10 increased EGFP (Tipe2) expression in NK cells (online supplemental figure S5). On the other hand, TNF-α/TGF-β/ROS did not significantly change EGFP (Tipe2) expression by NK cells. These data suggest that IL-10 might cause the upregulation of TIPE2 in the TME. Taken together, the above data indicated that TIPE2 expression correlates with NK cell exhaustion in the TME, both in humans and in mice.
Supplemental material
Supplemental material
Supplemental material
Tipe2-deficient NK cells exert stronger therapeutic efficacy in adoptive cellular therapy against solid tumors
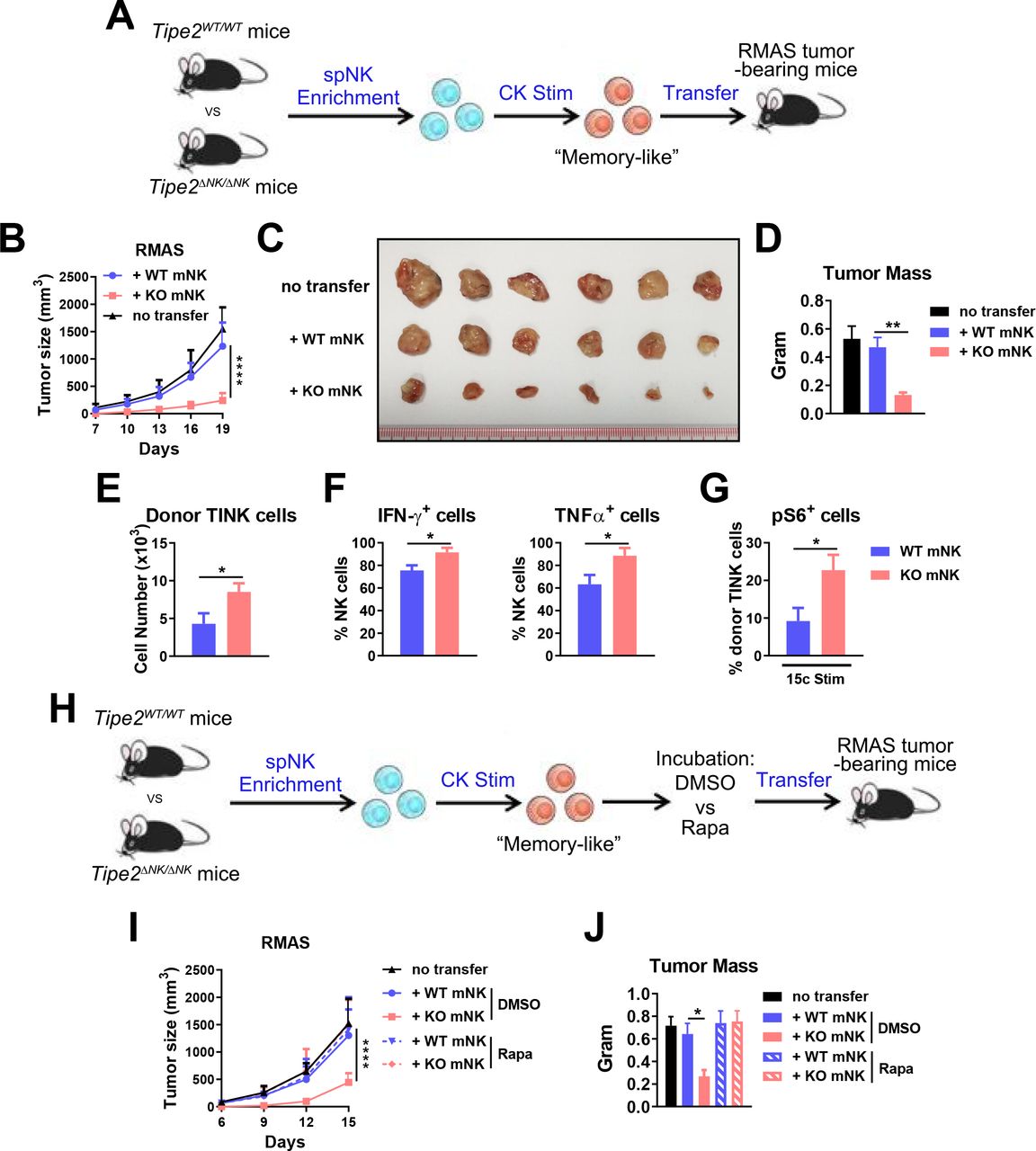
Our previous study showed that Tipe2 ΔNK/ΔNK mice display enhanced control of solid tumor growth in vivo, accompanied by increased effector functions of tumor-infiltrating NK cells. Here, we further investigated whether Tipe2 deficiency renders adoptive NK cell therapy with a better capacity for tumor control. We subcutaneously injected WT C57BL/6 mice with 5×105 RMAS cells. Three days later, we transferred a single dose of 2×105 ex vivo-stimulated WT or Tipe2-deficient mouse NK cells into the peritumoral region (figure 2A). The NK cells used in the transfer experiment were stimulated using IL-12, IL-15, and IL-18 to induce ‘memory-like’ NK cells before transfer (figure 2A). Injection of Tipe2-deficient NK cells significantly slowed RMAS tumor growth compared with injection of WT NK cells (figure 2B), resulting in a smaller tumor size and mass at the endpoint of the experiment (figure 2C,D). Analysis of donor-derived tumor-infiltrating NK cells showed significantly more Tipe2-deficient NK cells than WT NK cells (figure 2E). In addition, Tipe2-deficient NK cells produced higher levels of IFN-γ and TNF-α (figure 2F). These results indicated that Tipe2 deficiency relieved exhaustion and preserved the effector functions of adoptively transferred NK cells in the TME. TIPE2 was shown to suppress mTOR activity in NK cells. Here, we found that the coinjection of rapamycin, an inhibitor of mTORC1 activity, abolished the tumor-suppressive effects of Tipe2-deficient NK cells (figure 2G–I). These data demonstrated that Tipe2-deficient mouse NK cells display improved antitumor activity in vivo in an mTOR-dependent manner.

Antitumor activity of Tipe2-deficient mouse NK cells. (A) Scheme of mouse NK cell enrichment, stimulation and adoptive transfer. Mouse splenic NK cells were enriched and stimulated in vitro with 20 ng/mL IL-12, 50 ng/mL IL-15 and 10 ng/mL IL-18 for 2 days before transfer into B6 WT mice challenged with RMAS tumor cells 3 days prior. (B–D) tumor growth and endpoint tumor mass are shown for (A). (E–G) Flow cytometry analysis of tumor-infiltrating donor NK cells is shown. (E) Cell numbers per gram tumor mass. (F) IFN-γ and TNF-α production. (G) Phosphorylated S6 levels on 10 ng/mL IL-15/IL-15Rα complex stimulation ex vivo. (H) Scheme of mouse NK cell enrichment, stimulation, incubation with DMSO or rapamycin, and adoptive transfer. Mouse splenic NK cells were enriched, stimulated and transferred as in (A) but with an additional in vitro incubation with 2 ng/mL rapamycin or 0.001% (v/v) DMSO for 1 hour before the transfer. (I, J) Tumor growth and endpoint tumor mass are shown for (H). (A–J) groups of 6 mice per experiment were used. data are representative of three (A–D) or two (E–J) independent experiments and are presented as the mean±SEM *p<0.05, **p<0.005, ****p<0.0001 (two-way ANOVA (B, I), one-way ANOVA (D, J) or unpaired Student’s t-test (E–G)). ANOVA, analysis of variance.
TIPE2-deleted human NK cells display stronger functions in vitro
The above results prompted us to further explore the role of TIPE2 in human NK cells. To do so, we enriched NK cells from the peripheral blood of healthy donors and deleted the TIPE2 gene by electroporation with Cas9 protein plus a pair of guide RNAs (gRNAs) targeting the direct and complementary strands within the open reading frame of the TIPE2 gene (figure 3A). Loss of most TIPE2 protein expression in NK cells was confirmed by western blotting (figure 3B). TIPE2-deleted NK cells showed enhanced proliferation in response to IL-2 stimulation in vitro (figure 3C). Meanwhile, TIPE2-deleted NK cells expressed similar levels of typical NK cell surface antigens (CD16, NKp44, NKp46, NKG2D, NKG2A, KIR, TRAIL, and FasL) and maturation markers (CD94, CD2, CD57, and NKG2C) (figure 3D).

Phenotypic and functional analysis of TIPE2-deleted human NK cells. (A) Schema of CRISPR-Cas9-mediated TIPE2 deletion using two guide RNAs (gRNAs) targeting exon 2 of the TIPE2 gene in the direct and complementary strands. (B) Western blot analysis of TIPE2 protein expression in WT or TIPE2-deleted human NK cells. (C) WT or TIPE2-deleted human NK cells were analyzed for IL-2-triggered proliferation in vitro. (D) Flow cytometry analysis of surface receptor expression on WT or TIPE2-deleted human NK cells. (E) WT or TIPE2-deleted human NK cells were analyzed for (left) CD107a and (right) IFN-γ expression left untreated (N.T.) or on stimulation with K562 tumor cells or PMA and ionomycin. (F) WT or TIPE2-deleted human NK cells were analyzed for cytolytic activity against K562 tumor cells. (B–F) Data are representative of three independent experiments. (C, E, F) Data are presented as the mean±SEM. *p<0.05, ***p<0.001, ****p<0.0001, N.S. not significant. (C) two-way ANOVA. (E, F) one-way ANOVA. ANOVA, analysis of variance.
After feeder-free expansion, TIPE2-deleted NK cells displayed enhanced degranulation either left untreated or in response to K562 tumor cells (figure 3E). In line with this, the cytolytic activity of TIPE2-deleted NK cells against K562 tumor cells was stronger than that against WT NK cells (figure 3F). In addition, TIPE2-deleted NK cells produced significantly more IFN-γ than WT NK cells (figure 3E). These data indicated that TIPE2-deleted human NK cells display better effector function.
TIPE2-deleted human peripheral blood-derived NK cells show stronger therapeutic efficacy in solid tumor xenograft mice on adoptive cellular therapy
To evaluate the antitumor activity of TIPE2-deleted human peripheral blood-derived NK cells in vivo, NDG-hIL15 mice were injected subcutaneously with 2×106 K562 cells. Four days later, these mice received a single injection of 106 WT or TIPE2-deleted NK cells into the peritumoral region (figure 4A). Treatment with WT NK cells significantly slowed tumor growth, while injection of TIPE2-deleted NK cells further enhanced tumor-suppressive effects compared with WT NK cells (figure 4B). At the endpoint, a smaller tumor burden and weight were observed in the TIPE2-deleted group than in the WT group (figure 4C,D). Similarly, enhanced tumor-suppressive effects of TIPE2-deleted NK cells were found in the HCT15 human colon tumor model (figure 4E,F). Along with the improved tumor control by TIPE2-deleted NK cells, we detected increased NK cells infiltrating the tumors in these mice (figure 4G,H). The tumor-suppressive effects of TIPE2-deleted NK cells were dose dependent, with better effects achieved using more NK cells (figure 4I). The injection time was also important, since injection of TIPE2-deleted NK cells at earlier time points when tumors were still smaller resulted in better therapeutic effects (figure 4J). These results indicated that stronger therapeutic effects could be achieved using adoptively transferred TIPE2-deleted NK cells, which results in better antitumor outcomes in vivo.

TIPE2 deletion for adoptive peripheral blood-derived NK cell therapy. (A) Schematic of the generation and expansion of WT or TIPE2-deleted human peripheral blood-derived NK cells for adoptive transfer. NK cells were enriched from PBMCs of healthy donors and electroporated with Cas9 protein and TIPE2-specific gRNAs. NK cells were expanded in a feeder-free manner and stimulated with 20 ng/mL IL-12, 50 ng/mL IL-15, and 10 ng/mL IL-18 for 2 days before transfer into NDG hIL-15 mice challenged with K562/HCT15 tumor cells 3 days prior. (B–D) K562 tumor growth and endpoint tumor mass are shown for (A). (E, F) HCT15 tumor growth and endpoint tumor weight are shown for (A). (G) The numbers of K562 tumor-infiltrating human NK cells per gram tumor weight are shown for (A). (H) A representative immunofluorescent photo of the K562 tumor tissue is shown. CD56 (red) and DAPI (blue) were stained. (I) Different doses of TIPE2-deleted NK cells were used based on the protocol in (A). K562 tumor growth is shown. (J) Different time points of adoptive NK cell transfer post tumor challenge were selected based on the protocol in (A). K562 tumor growth is shown. (B–D) Data are representative of at least three independent experiments. (E–J) Data are representative of two independent experiments. (B–J) Groups of six mice per experiment were used. Data are presented as the mean±SEM. *p<0.05, **p<0.005, ***p<0.001. (Two-way ANOVA (B, E), one-way ANOVA (D, F), unpaired Student’s t-test (G)). ANOVA, analysis of variance; PBMCs, peripheral blood mononuclear cells.
TIPE2-deleted human iPSC-derived NK cells show stronger therapeutic efficacy in solid tumor xenograft mice on adoptive cellular therapy
To test whether TIPE2 deletion would also benefit iPSC-NK cell therapy, we generated a single clonal human iPSC cell line through the electroporation of iPSCs with Cas9 protein plus the paired gRNAs of TIPE2, followed by screening for single clones with deletions in genomic DNA that would result in a frameshift mutation and inactivation of TIPE2 protein (figure 5A,B). WT or TIPE2-deleted iPSC-NK cells were generated by in vitro differentiation from WT or TIPE2-deleted iPSC cells to CD34+ hematopoietic progenitor cells and then to CD3-CD56+CD45+ NK cells, followed byfeeder-free expansion.8 106 Adoptively transferred TIPE2-deleted iPSC-NK cells significantly suppressed tumor growth and displayed superior antitumor effects compared with WT iPSC-NK cells (figure 5C), resulting in a significantly smaller endpoint tumor mass (figure 5D,E). Therefore, TIPE2 deletion also enhanced the antitumor activity of human iPSC-NK cells in vivo.
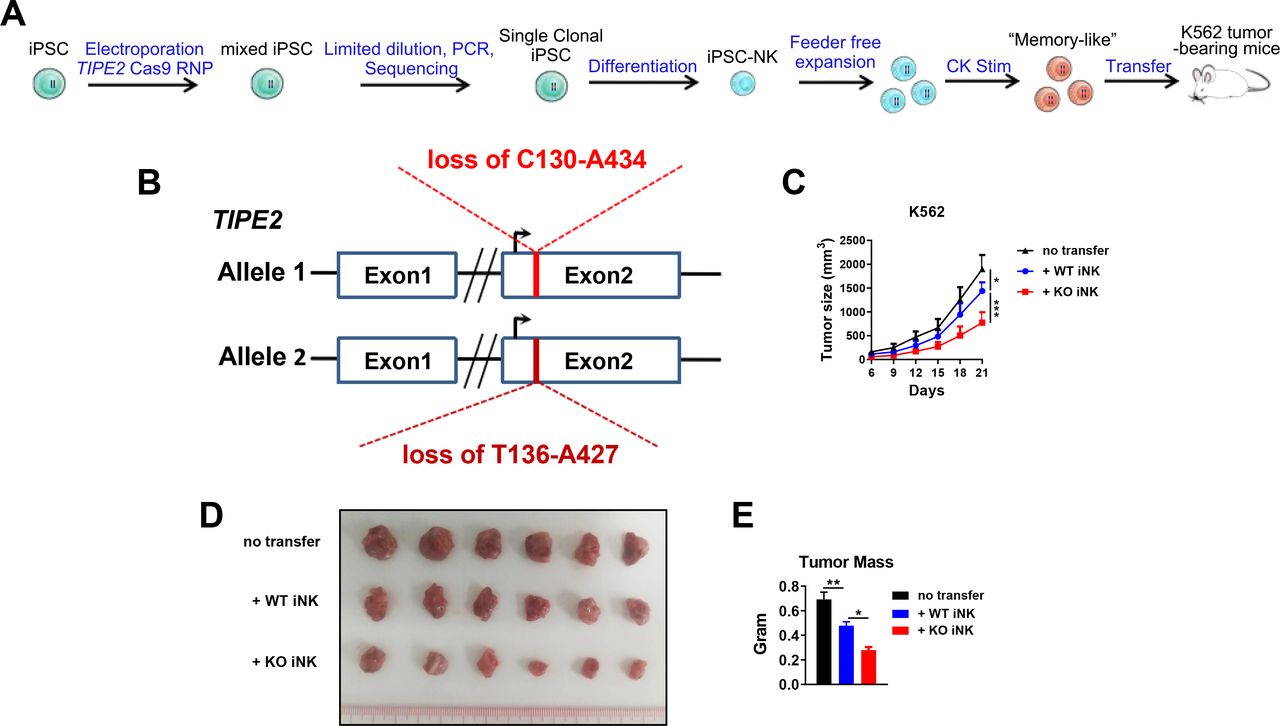
TIPE2 deletion for adoptive iPSC-derived NK cell therapy. (A) Schematic ofthe generation and expansion of WT or TIPE2-deleted human iPSC-derived NK cells for adoptive transfer. iPSCs were electroporated with Cas9 protein together with TIPE2-specific gRNAs before single clonalization by limited dilution. Single clones were screened for TIPE2-deletion mutants with frame shift mutations/fragment loss. WT or TIPE2-deleted iPSCs were differentiated into NK cells, expanded in a feeder-free manner, and subsequently stimulated with 20 ng/mL IL-12, 50 ng/mL IL-15, and 10 ng/mL IL-18 for 2 days before transfer into NDG hIL-15 mice challenged with K562 tumor cells 3 days prior. (B) An iPSC clone with fragment loss in two TIPE2 alleles was used in the study. Numbers indicate the position in the TIPE2 gene open reading frame. (C–E) K562 tumor growth and endpoint tumor mass are shown for (A). Groups of six mice per experiment were used. (C, E) Data are presented as the mean±SEM. *p<0.05, **p<0.005, ***p<0.001. (Two-way ANOVA (C), one-way ANOVA (E)). ANOVA, analysis of variance; iPSCs, induced pluripotent stem cell
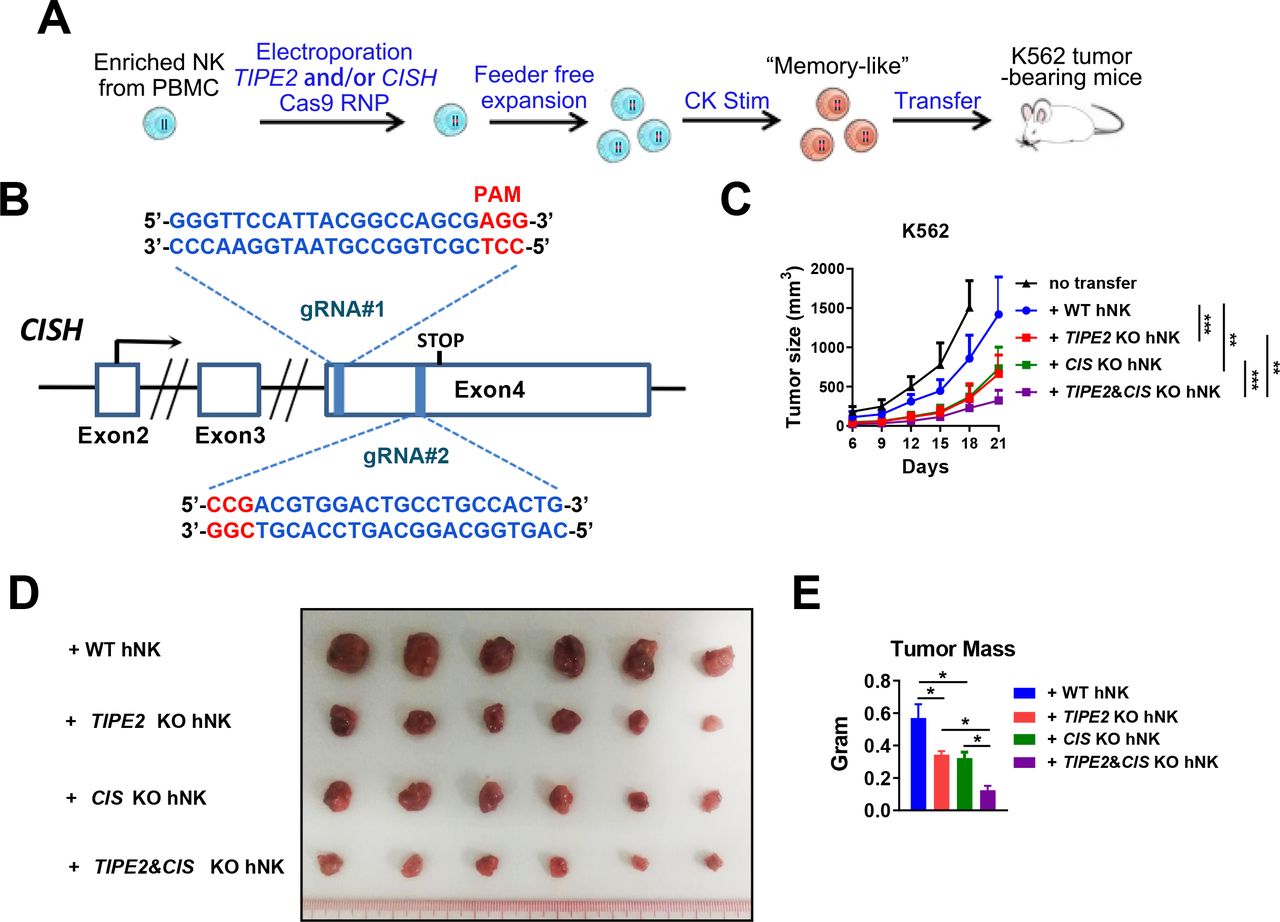
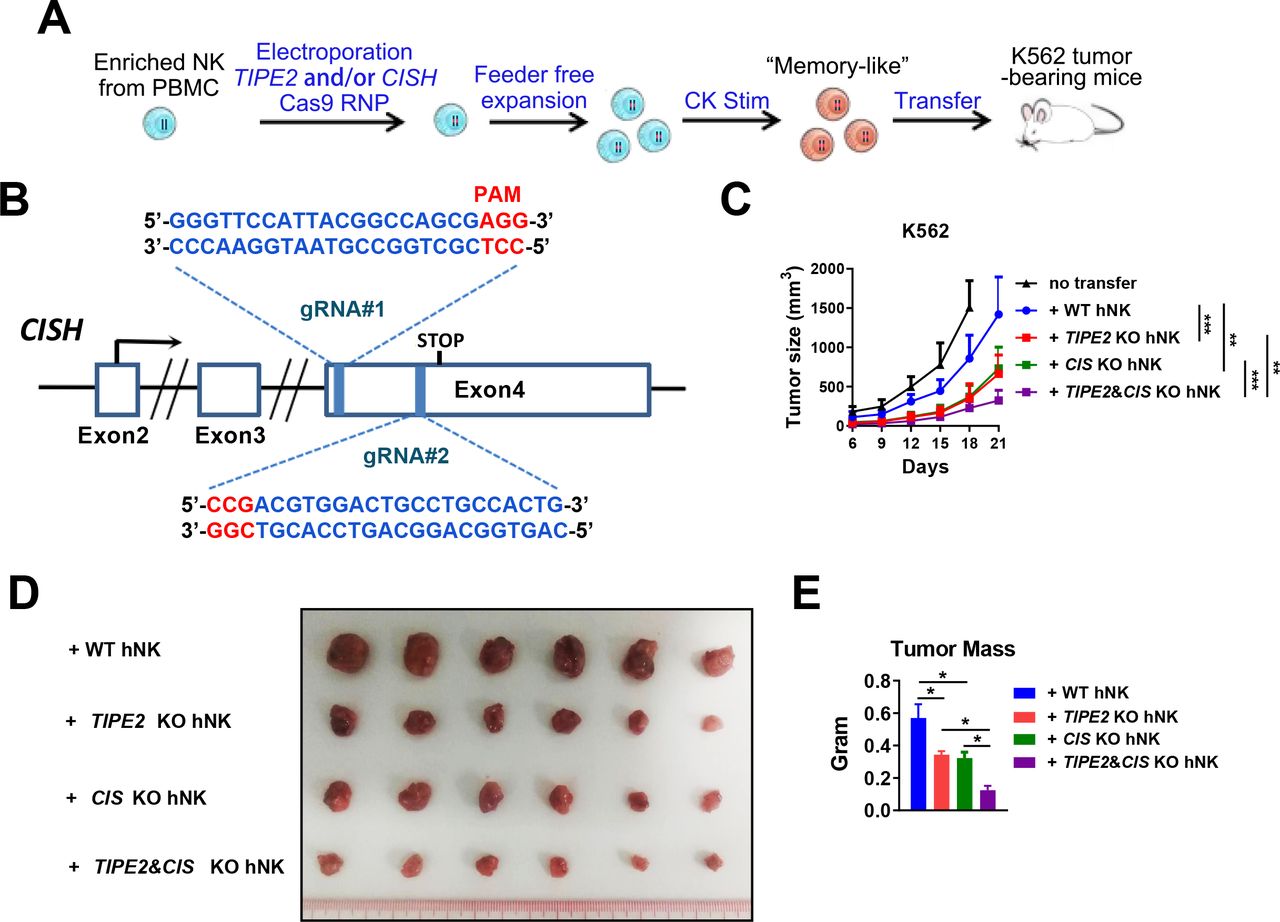
TIPE2/CISH double deletion greatly enhances the therapeutic efficacy of human peripheral blood-derived NK cells in solid tumor xenograft mice on adoptive cellular therapy
We showed that TIPE2 and CISH were preferentially expressed by two major subsets of human tumor-infiltrating NK cells (figure 1A). Analysis of Cish gene expression in Tipe2 -/- NK cells in the GSE168256 dataset we recently published showed that the Cish expression level was similar between wild-type andTipe2 -/- NK cells (online supplemental figure S6), suggesting that both genes might be regulated independent of each other. We hypothesized that the deletion of both genes might relieve the functional inhibition in a larger fraction of tumor-infiltrating NK cells and further enhance their antitumor activity. Therefore, we designed a pair of gRNAs targeting the direct and complementary strands within the open reading frame of the CISH gene (figure 6A,B). Deletion of either TIPE2 or CISH genes was performed by electroporation with Cas9 protein plus the paired gRNAs of TIPE2, CISH or both. A total of 106 adoptively transferred NK cells with double deletion of TIPE2 and CISH further slowed K562 tumor growth compared with NK cells with single gene deletion (figure 6C) and resulted in a significantly smaller endpoint tumor mass (figure 6D,E). These data indicated that TIPE2 deletion synergizes with CISH deletion to promote stronger antitumor activity of human NK cells than the deletion of either in vivo.
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TIPE2/CISH single or double deletion for adoptive NK cell therapy. (A) Scheme ofthe generation and expansion of WT or TIPE2/CISH-single or TIPE2/CISH double-deleted human peripheral blood-derived NK cells for adoptive transfer. NK cells were enriched from PBMCs of healthy donors and electroporated with Cas9 protein and either one or both TIPE2/CISH-specific gRNAs. NK cells were expanded in a feeder-free manner and stimulated with 20 ng/mL IL-12, 50 ng/mL IL-15, and 10 ng/mL IL-18 for 2 days before transfer into NDG hIL-15 mice challenged with K562/HCT15 tumor cells 3 days prior. (B) Schema of CRISPR-Cas9-mediated CISH deletion using two guide RNAs (gRNAs) targeting exon 4 of the CISH gene in the direct and complementary strands. (C–E) K562 tumor growth and endpoint tumor mass are shown for (A). Groups of six mice per experiment were used. (C, E) Data are presented as the mean±SEM. *p<0.05, **p<0.005, ***p<0.001. (Two-way ANOVA (C), one-way ANOVA (E)). ANOVA, analysis of variance; PBMCs, peripheral blood mononuclear cells.
Discussion
In this study, we revealed that TIPE2 is a checkpoint molecule of tumor-infiltrating NK cells. TIPE2 expression correlates with NK cell exhaustion in tumors. Targeting TIPE2 in either peripheral blood-derived NK cells or iPSC-derived NK cells benefits adoptive NK cell therapy with enhanced antitumor immunity. Therefore, our study has revealed a novel checkpoint molecule of NK cell exhaustion and has shed light on resistance to immune exhaustion in tumors, a new aspect of gene modifications to improve adoptive NK cell therapy. Targeting TIPE2 could not only synergize with targeting other checkpoint molecules (such as CISH) but also might be combined with genome modifications, such as the expression of CAR, including stable CD16 expression, IL-15 receptor fusion, and chemokine receptors, to fully release the antitumor potential of adoptive NK cell therapy.
The expression profile of TIPE2 in tumor-infiltrating NK cells underlies its unique potential as a therapeutic target. TIPE2 expression in tumor-infiltrating NK cells correlates with NK cell exhaustion in the TME, suggesting that TME-associated immunosuppressive factors might favor TIPE2 expression. TNF-α, reactive oxygen species, IL-6, and L-arginine are abundant in the TME and have been reported to induce TIPE2 expression.10 21 Here, we identified IL-10 as a positive regulator of TIPE2 expression in NK cells. Alternatively, the dysregulated balance of the inhibitory and activating surface receptors on NK cells in the TME that mediate NK cell exhaustion might also be TIPE2-inducing factors. Importantly, targeting CISH has been shown to improve adoptive cell therapy by NK cells from multiple sources.7 8 However, TIPE2 and CISH seem to be expressed by NK cells from functionally distinct subsets. CISH expression in NK cells increases on IL-15 stimulation in vitro.18 In the TME, CISH was preferentially expressed in the more functional NK cell subset. On the other hand, TIPE2 maintains steady expression in NK cells on IL-15 stimulation12 and is expressed at a higher level by a more exhausted NK cell subset. These results suggest that TIPE2 and CIS might function nonredundantly to maintain/promote exhaustion/tolerance preferentially by TIPE2 and prevent overactivation preferentially by CIS. This nonredundancy offers us an opportunity to target TIPE2 and CISH at the same time to relieve the suppression of NK cells in both states to maximize the improvement in NK cell therapy.
The IL-15-mTOR axis is essential for NK cell effector functions.22 Although IL-15 is expressed in the TME,23 the activity of IL-15 signaling in tumor-associated NK cells is impaired,24 resulting in the impaired effector functions of tumor-associated NK cells. Here, the absence of TIPE2 renders adoptively transferred NK cells with elevated expression of effector functions, as well as a higher phosphorylation level of S6, indicative of higher mTORC1 activity. Inhibition of mTORC1 activity by rapamycin abrogates the improved antitumor activity of adoptively transferred TIPE2-deficient NK cells. These data suggest that, similar to the mode of action of TIPE2 in the steady state, TIPE2 might suppress NK cell activity in the TME by inhibiting the IL-15-mTORC1 axis and that preserving the activity of this axis might be essential for promoting the antitumor activity of adoptively transferred NK cells.
In this study, we employed a K562 xenograft mouse model and adoptive NK cell therapy as experimental approaches to test the therapeutic potential of TIPE2-deleted NK cells. In this model, K562 tumor sizes usually reach endpoints (over 1000 mm3) approximately 15–18 days post tumor injection. Therefore, we chose day 3 post tumor challenge as the NK cell transfer time when the tumor tissue started to form. Adoptive transfer of NK cells in tumor mouse models on day 3 post tumor inoculation has also been reported previously.25 26 In addition to transferring NK cells 3 days post tumor injection, we also tested later time points (day 4 and day 6) for NK cell transfer. Day 6 post tumor injection is the time when tumor sizes usually reach approximately 150–200 mm3 (diameters ranging from 6 to 8 mm), which represents significant tumor mass and TME formation in this model, mimicking the tumor burden of patients with late-stage cancers who might receive cancer therapies. Our results showed that transferring TIPE2-deleted NK cells even on day 6 post tumor injection led to an approximately 50% reduction in tumor size, demonstrating the benefits of TIPE2 deletion on adoptively transferred NK cell therapy. Alternatively, later time points were reported using a tumor model, usually with much slower tumor growth. For example, investigators transferred NK cells on day 13 post tumor inoculation in the MDA-MB231 model in one study27 when the tumor size was smaller (below 50 mm3) than that in our study. Taken together, our study supports further investigations using animal models more closely mimicking clinical conditions to evaluate the performance of TIPE2-deleted NK cell therapy in various tumor conditions to help us determine suitable indications.
In summary, we report that deletion of the intracellular checkpoint molecule TIPE2 in primary NK cells from either mice or humans enhances their effector functions and antitumor activity in adoptive NK cell therapy against solid tumor models. Our study supports further clinical validation and suggests that the resistance to exhaustion in the TME through TIPE2 deletion could be combined with tumor sensors (eg, NKR or CAR), immune checkpoint blockade (eg, KIR, TIGIT) and synthetic gene circuits for future NK cell immunotherapy.
Data availability statement
Data are available on reasonable request.
Data availability statement
Single-cell transcriptomic analysis was based on the dataset available in the Gene Expression Omnibus (GEO) with the accession code GSM4134620.
Ethics statements
Patient consent for publication
Ethics approval
All animal experiments were approved by the Institutional Animal Care and Use Committee with the approval reference number as belowed: SIAT-IACUC-20220607-HCS-MYZX-BJC-A2020-01.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conceptualization: JB, ZT and HS; Methodology: JB, CH, XJ, CZ, YH and XZ; Investigation: JB, CH, XJ, ZT and HS; Writing: JB, ZT and HS; Supervision: JB, ZT and HS; Guarantor: JB.
Funding The study was supported by grants from theNationalKey R&D Program of China 2020YFA0710802 (JB) and 2019YFA0508502/3 (HS) and by grants from the Natural Science Foundation of China 82071768 (JB) and 81788101 (HS).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.