Article Text

Abstract
Background The phase I first-in-human study ENGAGE-1 evaluated the humanized IgG1 OX40 agonistic monoclonal antibody GSK3174998 alone (Part 1 (P1)) or in combination with pembrolizumab (Part 2 (P2)) in patients with advanced solid tumors.
Methods GSK3174998 (0.003–10 mg/kg) ± pembrolizumab (200 mg) was administered intravenously every 3 weeks using a continuous reassessment method for dose escalation. Primary objectives were safety and tolerability; secondary objectives included pharmacokinetics, immunogenicity, pharmacodynamics, and clinical activity.
Results 138 patients were enrolled (45 (P1) and 96 (P2, including 3 crossovers)). Treatment-related adverse events occurred in 51% (P1) and 64% (P2) of patients, fatigue being the most common (11% and 24%, respectively). No dose-toxicity relationship was observed, and maximum-tolerated dose was not reached. Dose-limiting toxicities (P2) included Grade 3 (G3) pleural effusion and G1 myocarditis with G3 increased troponin. GSK3174998 ≥0.3 mg/kg demonstrated pharmacokinetic linearity and >80% receptor occupancy on circulating T cells; 0.3 mg/kg was selected for further evaluation. Limited clinical activity was observed for GSK3174998 (P1: disease control rate (DCR) ≥24 weeks 9%) and was not greater than that expected for pembrolizumab alone (P2: overall response rate 8%, DCR ≥24 weeks 28%). Multiplexed immunofluorescence data from paired biopsies suggested that increased infiltration of natural killer (NK)/natural killer T (NKT) cells and decreased regulatory T cells (Tregs) in the tumor microenvironment may contribute to clinical responses: CD16+CD56–CD134+ NK /NKT cells and CD3+CD4+FOXP3+CD134+ Tregs exhibited the largest magnitude of change on treatment, whereas CD3+CD8+granzyme B+PD-1+CD134+ cytotoxic T cells were the least variable. Tumor gene expression profiling revealed an upregulation of inflammatory responses, T-cell proliferation, and NK cell function on treatment with some inflammatory cytokines upregulated in peripheral blood. However, target engagement, evidenced by pharmacologic activity in peripheral blood and tumor tissue, did not correlate with clinical efficacy. The low number of responses precluded identifying a robust biomarker signature predictive of response.
Conclusions GSK3174998±pembrolizumab was well tolerated over the dose range tested and demonstrated target engagement. Limited clinical activity does not support further development of GSK3174998±pembrolizumab in advanced cancers.
Trial registration number NCT02528357.
- clinical trials as topic
- immunotherapy
- antibodies, neoplasm
- biomarkers, tumor
- costimulatory and inhibitory T-cell receptors
Data availability statement
Data are available upon reasonable request. Information about GlaxoSmithKline’s data sharing commitments and access requests to anonymized individual participant data and associated documents can be requested for further research from ClinicalStudyDataRequest.com.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- clinical trials as topic
- immunotherapy
- antibodies, neoplasm
- biomarkers, tumor
- costimulatory and inhibitory T-cell receptors
WHAT IS ALREADY KNOWN ON THIS TOPIC
GSK3174998 is an investigational humanized IgG1 OX40 agonistic monoclonal antibody. Pembrolizumab (Keytruda) is a marketed humanized monoclonal IgG4 programmed death receptor-1 (PD-1)-blocking antibody.
Preclinical data support that activating OX40 signaling may overcome primary or acquired resistance to anti-PD-(L)1 antibodies.
This phase I first-in-human study ENGAGE-1 (NCT02528357) evaluated the safety and tolerability, pharmacokinetics, immunogenicity, pharmacodynamics, and clinical activity of GSK3174998 alone and in combination with pembrolizumab in patients with advanced solid tumors.
WHAT THIS STUDY ADDS
This study provides important clinical and translational data showing that even with evidence of target engagement, combining an OX40 agonist with a PD-1-blocking antibody in an unselected patient population did not result in sufficient clinical efficacy to support further development.
GSK3174998 up to 10 mg/kg±pembrolizumab 200 mg was well tolerated.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study adds to the body of knowledge in the scientific community on the development of agonist monoclonal antibodies, notably targeting OX40. This article provides novel data and reviews important questions on why this study failed, which may be addressed by future research in this field.
Introduction
The development of immune checkpoint blockers (ICBs), such as cytotoxic T-lymphocyte-associated antigen 4, programmed cell death 1 protein (PD-1), and programmed cell death 1 ligand (PD-L1) blockers, has revolutionized advanced cancer immunotherapy.1 2 ICB therapies are currently approved for use in treating a number of cancers,1 3–7 with response rates ranging from 15% to 60% in patients with advanced solid tumors,8 depending on whether treatment is driven by the presence or absence of tumor biomarkers (ie, PD-L1), treatment line, and other tumor or immune cell characteristics. The effectiveness of ICB can be limited by immune and other cells within the tumor that promote tumor proliferation and metastases.9 10 Many patients will have disease, that is, already primarily resistant to, or becomes resistant during or after treatment with ICB, including anti-PD-(L)1 therapies.8 9 A major resistance mechanism is the presence of an immunosuppressive tumor microenvironment. Consequently, there is a need to develop novel anticancer agents and combinations that can overcome the immunosuppressive tumor microenvironment in solid tumors.
One strategy being explored to enhance anticancer T-cell responses is to augment the signaling activity of costimulatory receptors, such as OX40, with agonists.10 11 OX40 is a member of the tumor necrosis factor (TNF) receptor superfamily and is expressed on activated CD4+ and CD8+ T cells.12 13 Engagement of OX40 on T cells with OX40 ligand (OX40L) on the surface of antigen-presenting cells promotes T-cell survival, proliferation, differentiation, and memory.12 13 OX40 is also expressed by forkhead box protein 3 (FOXP3)+ regulatory T cells (Tregs) and suppresses Treg differentiation and interleukin (IL)-10 production.12 14 15 OX40 is expressed to a lesser extent on natural killer (NK) cells, thus an OX40 agonist may enhance Fc gamma receptor (FcγR)-dependent NK-mediated tumor cell killing.16 Preclinical studies have shown that OX40 agonists increase antitumor immunity and suppress tumor growth.17–20
Because tumors have a variety of mechanisms to suppress immune responses,2 combining a PD-1 blocker with a costimulatory OX40 agonist may result in additive or even synergistic antitumor activity. In preclinical studies of mouse syngeneic tumor models, the combination of an OX40 agonist with anti-PD-1 therapy improved antitumor immune responses, reduced tumor growth, and prolonged overall survival to a greater degree than monotherapy controls.21–23 Furthermore, the OX40 agonist GSK3174998 demonstrated increased inflammatory and T helper 1 cell cytokine production in combination with pembrolizumab in human peripheral blood mononuclear cells in vitro.24 Here, we report the results of a phase I study of GSK3174998 as monotherapy or in combination with the PD-1 blocker pembrolizumab in 138 patients with selected advanced solid tumors.
Methods
Study design and treatment
ENGAGE-1 was a first-in-human study designed to evaluate the safety, pharmacokinetics, immunogenicity, pharmacodynamics, and clinical activity of GSK3174998±pembrolizumab in patients with advanced solid tumors. ENGAGE-1 was conducted at eight sites in The Netherlands, France, Canada, and USA. The study included two parts: GSK3174998 monotherapy (Part 1 (P1)) and GSK3174998+pembrolizumab (Part 2 (P2)), each incorporating dose-escalation and -expansion phases; P2 began once a tolerable and biologically active GSK3174998 dose was identified in P125 (online supplemental figure 1). In the dose-escalation phases, the Fixed and Adaptive Clinical Trial Simulator was used to conduct the continual reassessment method informed by the dose-limiting toxicity (DLT) assessed over a 4-week interval.26 Dose-escalation cohorts could enroll up to 12 patients to evaluate pharmacodynamic endpoints, but the DLT incidence could not exceed 33%. GSK3174998 was dosed at 0.003, 0.01, 0.03, 0.1, 0.3, 1, 3, or 10 mg/kg±pembrolizumab 200 mg; each dose was administered as 30-min intravenous infusions every 3 weeks (Q3W) for up to 2 years.
Supplemental material
The primary objective of this study was to assess safety and tolerability and identify the maximum tolerated dose (MTD) or maximum administered dose of GSK3174998±pembrolizumab. All available safety and tolerability data were to be considered in the determination of the MTD. Key secondary endpoints included pharmacokinetic parameters of GSK3174998 and pembrolizumab, antidrug antibodies (ADAs), overall response rate (ORR), and disease control rate (DCR). Response endpoints were assessed by Response Evaluation Criteria in Solid Tumors (RECIST) v.1.1 and immune-related (ir)RECIST, with the latter driving treatment decisions. Exploratory endpoints included OX40 receptor expression and occupancy by GSK3174998, and pharmacodynamic activity in blood and the tumor microenvironment.
Study population
Eligible patients were ≥18 years old with histologically confirmed locally advanced, recurrent or metastatic cancer (bladder cancer, colorectal carcinoma displaying high microsatellite instability (CRC-MSI-H), squamous cell carcinoma of the head and neck, melanoma, non-small cell lung cancer (NSCLC), renal cell carcinoma, soft tissue sarcoma (STS), or triple-negative breast cancer whose disease had progressed after standard therapy, were intolerant to, or considered inappropriate for standard therapy and who had received ≤5 prior lines of therapy. Patients were required to have measurable disease per RECIST v.1.1, an Eastern Cooperative Oncology Group performance status of 0 or 1, life expectancy of ≥12 weeks, and adequate organ function. Key exclusion criteria included prior treatment with a TNF receptor agonist, previous bone marrow or solid organ transplant, symptomatic central nervous system metastases, or active autoimmune disease.
Study assessments
Safety assessments included evaluation of adverse events (AEs) graded by National Cancer Institute Common Terminology Criteria for Adverse Events v.4.0, laboratory parameters, vital signs, ECG, and physical examinations. AEs were assessed until 30 days and AEs of special interest and serious AEs (SAEs) until 90 days after the last treatment dose.
Blood samples for GSK3174998 pharmacokinetic evaluation were collected pre-dose and post-dose for the first six doses, then pre-dose only from dose 8 and every four dose cycles thereafter, with a final sample drawn 12±1 weeks post-treatment. Plasma levels of GSK3174998 were analyzed by liquid chromatography-tandem mass spectrometry. Blood samples for pembrolizumab pharmacokinetic evaluation were collected pre-dose and post-dose for the first dose, then pre-dose only for doses 2, 4, 6, 8 and every four dose cycles thereafter, with a final sample drawn 12±1 weeks post-treatment. Serum concentrations of pembrolizumab were analyzed using a quantitative immunocapture assay.
Blood samples for ADA testing were collected pre-dose for the first six doses, then pre-dose only from dose 8 and every four dose cycles thereafter, with a final sample drawn 12±1 weeks post-treatment. Serum samples were assayed for ADA using an electrochemiluminescence bridging acid dissociation immunoassay with a bioanalytically validated cut point.
To evaluate the binding of GSK3174998 to the OX40 receptor on CD3+T cells, the activation and proliferation status of T cells, and the enumeration of T cells and their subsets, B cells, and NK cells, blood samples were analyzed using five flow cytometry panels consisting of 24 markers: chemokine (C-C motif) receptor 7, cluster of differentiation (CD) 127, CD16, CD19, CD25, CD3, CD34, CD38, CD4, CD45RA, CD45RO, CD56, CD8, FOXP3, granzyme B, human leukocyte antigen-D related (HLA-DR), inducible T-cell costimulator (ICOS), interferon (IFN)-γ, IL-17A, IL-2, IL-4, Ki-67, OX40 total, and PD-1. Blood samples were collected pre-dose and post-dose for the first three doses, and 30 days and 12±1 weeks post-treatment. Receptor occupancy (RO) was calculated for each time point from the flow cytometry results by dividing the bound OX40 values (percentage of parent population) by the corresponding saturation control values.
An additional blood sample was collected for analysis of 27 protein biomarkers in patient plasma samples. Data generation and quality control analysis was performed at Aushon using Aushon Biosystems CiraScan Multiplexed Custom Array Testing Service. Analysis of one protein biomarker used a separate validated enzyme-linked immunoassay. Cytokines to be analyzed included IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p70, IL-13, IL-17A, IFN-γ, TNF-α trimer, transforming growth factor-β, granulocyte colony stimulating factor (G-CSF), intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), vascular endothelial growth factor (VEGF), VEGF receptor 1 (VEGFR1), growth-regulated oncogene α, monocyte chemoattractant protein 1 (MCP-1), macrophage inflammatory protein 1 α/β (MIP-1α/β), regulated on activation normal T expressed and secreted, macrophage derived chemokine MIP-3β (also known as chemokine (C-C motif) ligand 19 (CCL19)), IFN-γ–induced protein 10 (IP-10), and IL-27.
Tumor tissue samples were collected at baseline (archival or fresh) and on treatment (fresh) at week 6, where feasible. Multiplex immunofluorescence was performed on 3–5 micron thick formalin-fixed, paraffin-embedded sections of tumor biopsy tissue by NeoGenomics to evaluate expression of phenotypic and functional immune cell markers on tumor infiltrating lymphocytes and other immune cells using the MultiOmyx platform. The expression of 16 markers (CD3, CD4, CD8, granzyme B, FOXP3, Ki-67, ICOS, PD-1, PD-L1, OX40, CD16, CD56, OX40L, HLA-DR, S100, pan cytokeratin (PanCK)), and several combinations of those markers were investigated and scored using a proprietary algorithm. The anti-OX40 BER-Act35 antibody from Santa Cruz (sc20073) was used for the detection of OX40 in the MultiOmyx platform. Gene expression analysis was also performed by NeoGenomics on the same tumor samples using the NanoString PanCancer Immune Profiling panel (730 genes plus 40 housekeeping genes). The assay was run on the nCounter Analysis System (NanoString Technologies).
Disease assessments (CT scans and/or MRI) were performed at baseline and every 12 weeks thereafter until confirmed disease progression per irRECIST or withdrawal from the study; patients undergoing tumor biopsies at week 6 had an additional disease assessment at that time.
Statistical analysis
Statistical analyses were descriptive and exploratory in P1 and P2 dose escalation. For P2 expansion, the null hypothesis (H0) that the ORR of combination therapy is equal to the historical ORR of pembrolizumab alone was tested in each cohort. The sample size (10–30 patients per cohort) was chosen based on a 20% improvement in the ORR for combination therapy over the null hypothesis with power of at least 80% and a type I error rate of no more than 10%.25 In the population previously treated with pembrolizumab, the goal for the combination regimen was to observe at least a 30% response rate. The H0 for ORR of the PD-(L)1–naive STS expansion cohort was p=18%, and the alternative hypothesis (HA) was p=38%. The H0 for the ORR of pretreated PD-(L)1 expansion cohorts in melanoma and NSCLC was p=10%, and the HA was p=30%.
Biomarker analysis
For multiplexed immunofluorescence, the responses for each patient were transformed into the absolute difference before and after treatment. For every biomarker, we fit a generalized additive model27 to capture the non-linear relationship between the absolute differences and the dose. Absolute differences and doses were modeled on the inverse hyperbolic sine and logarithmic scale, respectively. All statistical analyses were conducted in R.28
Results
Patient characteristics and treatment exposure
From September 2015 to April 2019, a total of 138 patients received GSK3174998 (0.003–10 mg/kg) alone (P1, n=45) or in combination with pembrolizumab 200 mg (P2, n=96), with 3 patients participating in P1 and subsequently P2 (online supplemental figure 1). The median age was 63.0 (P1) and 63.5 (P2) years, and most patients were of white Caucasian origin (84% and 85%, respectively), (table 1). Approximately half of the patients (P1 53%, P2 48%) had received ≥3 prior anticancer therapy regimens; in P1, 29% received prior PD-(L)1 therapy (11% pembrolizumab) and, in P2, 43% received prior PD-(L)1 therapy (16% pembrolizumab) (table 1). Median duration of exposure to GSK3174998±pembrolizumab was 9.1 weeks (range: 2.7–39) in P1 and 9.1 weeks (range: 0.1–105.6) in P2, with a median of four infusions administered in each part (range: 2–14 and 1–35, respectively); the most common reason for treatment discontinuation was disease progression (89% and 79%, respectively) (online supplemental table 1).
Patient and disease characteristics
Safety
In P1, AEs and treatment-related AEs (TRAEs) were reported in 100% and 51% of study patients, respectively (table 2). The most common TRAEs were fatigue (11%) and diarrhea (11%). Grade (G) 3 TRAEs were reported in 3 (7%) patients and included one event each of lymphopenia, thrombocytopenia, infusion-related reactions (IRRs), and asthenia. There were no G4 or G5 TRAEs and no DLTs. Treatment-related SAEs were reported in 2 (4%) patients; both were IRRs that occurred at cycle 3. No TRAEs leading to treatment discontinuation were reported. In P2, AEs and TRAEs were reported in 99% and 64% of study patients, respectively (table 2). The most common TRAEs were fatigue (24%) and nausea (10%). G3 TRAEs were reported in 8 (8%) patients and included fatigue (3), anemia (2), and hypoxia, hypotension, diarrhea, IRR, and increased troponin (1 each). There were no G4 or G5 TRAEs. Two DLTs occurred. One patient with triple-negative breast cancer had G3 non-malignant pleural effusion after the first dose (GSK3174998 0.03 mg/kg+pembrolizumab 200 mg); oxygen was administered, and a thoracentesis was performed with subsequent resolution of this event. Pleural effusion recurred after the second combination treatment at which time the patient was determined to have progressive disease (PD). Another patient with bladder cancer had an asymptomatic G1 myocarditis with G3 troponin increase after the first dose (GSK3174998 10 mg/kg+pembrolizumab 200 mg), which resolved after treatment with methylprednisolone 500 mg intravenous one time per day followed by a steroid taper. Treatment-related SAEs were reported in 4 (4%) patients, including fatigue (2) and increased blood creatine kinase, myocarditis, and organizing pneumonia (1 each). TRAEs leading to treatment discontinuation were reported in 3 (3%) patients, including G3 IRR (at cycle 3), G1 myocarditis with G3 increased troponin (DLT), and G2 fatigue (at cycle 1). Overall, an MTD for GSK3174998±pembrolizumab was not reached and a dose relationship for TRAEs was not observed.
Summary of AEs in Parts 1 and 2
Pharmacokinetics and RO
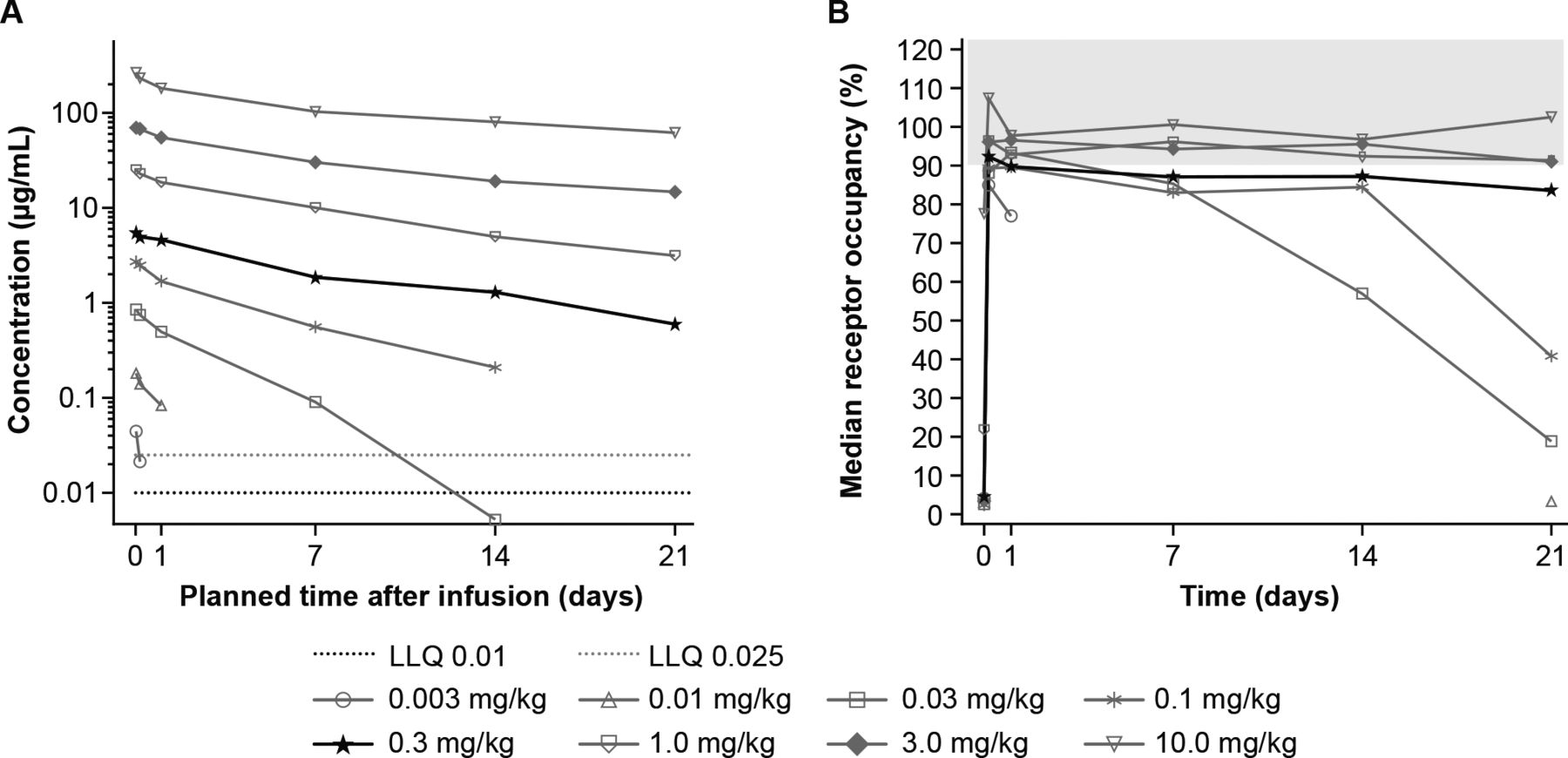
GSK3174998 demonstrated linearity over the 0.3–10 mg/kg dose range, remaining present in plasma ≥21 days (figure 1A and online supplemental figure 2A,C). At doses of 0.1 mg/kg and lower, steeper slopes at lower GSK3174998 concentrations were indicative of the impact of target-mediated clearance. Median time to maximum plasma concentration (tmax) ranged from 0.0208 to 0.0656 days in P1 and P2 (online supplemental table 2). Mean values of maximum serum concentration (Cmax) and area under the curve (AUC) (0-τ) indicated that exposure increased with increasing dose in P1 and P2. At doses ≥0.3 mg/kg, GSK3174998 demonstrated >80% RO on circulating T cells over a 3-week interval, with limited gains in RO observed at doses from 0.3 to 10 mg/kg (figure 1B and online supplemental figure 2B,D). Concomitant administration did not appear to alter the pharmacokinetics of GSK3174998 or pembrolizumab (online supplemental table 2 and online supplemental figure 2A,C,E and F). Based on these data, the 0.3 mg/kg dose was selected for further clinical evaluation in P2 expansion cohorts.

Pharmacokinetics and receptor occupancy of GSK3174998 during the first dosing cycle of GSK3174998 monotherapy. (A) Median plasma concentration-time profiles of GSK3174998 for cycle 1 displayed on a semi-logarithmic scale in Part 1. (B) Median receptor occupancy of GSK3174998 (%) on CD3+ T cells over time in Part 1. The highlighted dose level (0.3 mg/kg) was selected for further clinical evaluation in Part 2b expansion cohorts (GSK3174998+pembrolizumab). CD, cluster of differentiation; LLQ, lower limit of quantification.
Clinical activity
The P1 expansion was not initiated due to limited monotherapy activity observed during dose escalation. No confirmed responses were observed with GSK3174998 monotherapy; however, an anti-PD-(L)1 treatment-naive patient with dedifferentiated liposarcoma who received 0.3 mg/kg GSK3174998 had an unconfirmed partial response (PR; figure 2A). PR was maintained in the target lesions for this patient, but because a non-target lesion progressed, the response could not be confirmed. The DCR ≥24 weeks was 9% (online supplemental table 3); two patients (melanoma, NSCLC) with stable disease (SD) ≥24 weeks had received prior anti-PD-(L)1 treatment.
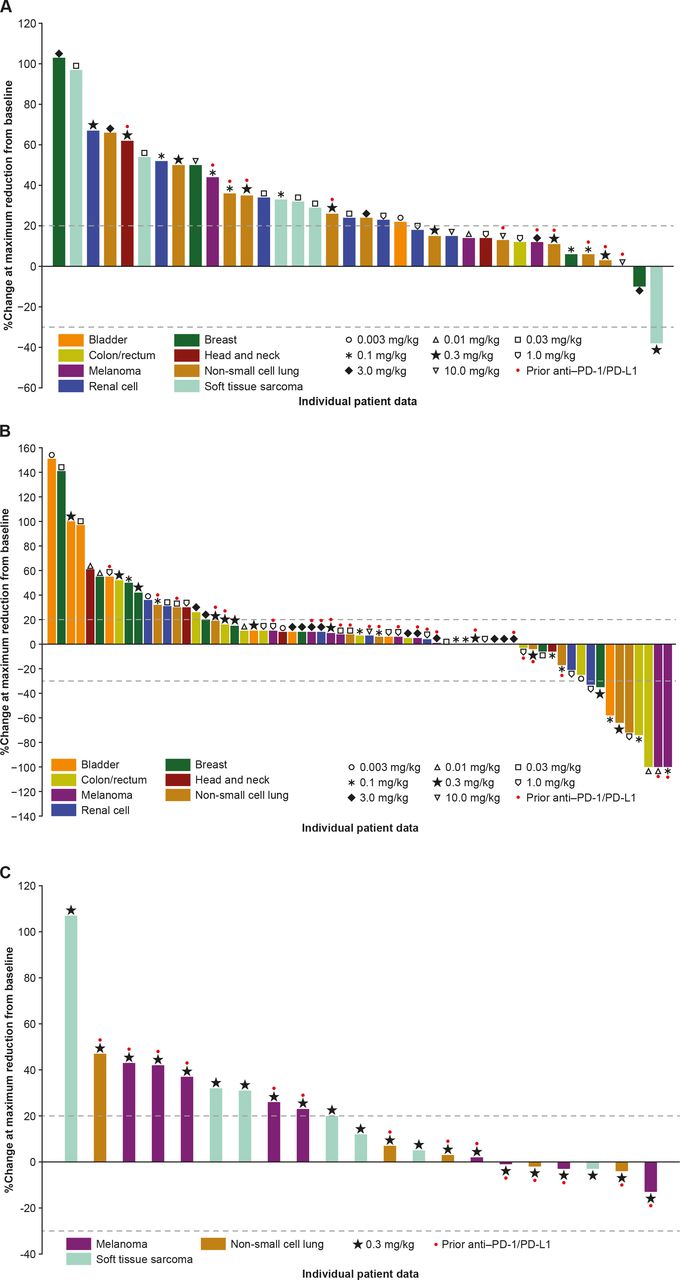
Percentage change in sum of target lesion diameters from baseline in (A) Part 1 dose escalation (GSK3174998 alone), (B) Part 2 dose escalation (GSK3174998+pembrolizumab), and (C) Part 2 dose expansion (GSK3174998+pembrolizumab; one patient with soft tissue sarcoma was not evaluable) assessed using irRECIST. irRECIST, immune-related Response Evaluation Criteria in Solid Tumors; PD-1, programmed cell death 1 protein; PD-L1, programmed cell death 1 ligand.
The ORR in P2 dose escalation was 8% (two complete responses (CRs), four PRs) and DCR ≥24 weeks was 28% (online supplemental table 3); of the responders, one male patient in his early 60s (melanoma, CR) had progressed on prior nivolumab plus ipilimumab treatment, while the remainder (one CR: CRC-MSI-H; four PR: two NSCLC; one CRC-MSI-H; and one bladder) were anti-PD-(L)1 treatment-naive (online supplemental table 3, figure 2B). Nine of 15 patients with SD ≥24 weeks had received prior anti-PD-(L)1 treatment. Based on the results from the dose-escalation phase, the P2 expansion included PD-(L)1–experienced patients with melanoma or NSCLC and PD-(L)1–naive patients with STS (dedifferentiated liposarcoma). No confirmed responses were observed in P2 expansion (figure 2C) of 22 patients enrolled (9 melanoma, 8 STS, and 5 NSCLC). Based on the lack of response and emerging data in the field, a decision was made to end enrollment in the P2 expansion cohorts early (target enrollment had been ≥10 patients/cohort). The DCR ≥24 weeks was 14% (online supplemental table 3) in P2 expansion, and all three patients with SD ≥24 weeks had received prior anti-PD-(L)1 treatment.
Immunogenicity
At baseline, 4 (2%) patients had detectable anti-GSK3174998 antibodies (1/45 (2%) patients in P1 and 3/133 (2%) patients in P2). Anti-GSK3174998 antibodies were detected in 54 (39%) patients overall, typically after the second or third dose of GSK3174998. Positivity was detected in 21 (47%) patients in P1, including 2 with IRR and 1 with an unconfirmed PR, and in 33 (35%) patients in P2, including 3 with IRR and 3 with confirmed responses (1 CR and 2 PRs). Of note, five of six patients who experienced IRR had positive ADA tests with high titers (≥8000), suggesting a possible relationship with immunogenicity at high ADA titer levels. Anti-pembrolizumab antibodies were detected in 1 of 59 (2%) of patients in P2; the single ADA-positive subject was positive pre-dose day 1, that is, immunogenicity was not treatment emergent.
Peripheral blood biomarkers
A non-parametric analysis of 44 monotherapy (P1) and 62 combination therapy (P2) plasma samples, comparing changes in cytokines from baseline to 24 hours post-treatment, identified the following statistically significant (p<0.005) increases of plasma cytokines in P1: MIP-1α/β, IP-10, soluble VCAM1, MCP-1, and soluble VEGFR1; and in P2: MIP-1α/β, IL-10, IP-10, soluble VCAM-1, IL-6, MIP-3β (or CCL19), IFN-γ, soluble ICAM1, MCP-1, G-CSF, and IL-5 (online supplemental table 4A). Overall, these data suggest that cytokine changes were generally greater for P2 compared with P1; however, while statistically significant, the changes were modest (up to ≈2-fold) and variable during the 24-hour post-dose period (online supplemental table 4B and online supplemental figure 3).
Tumor biomarkers
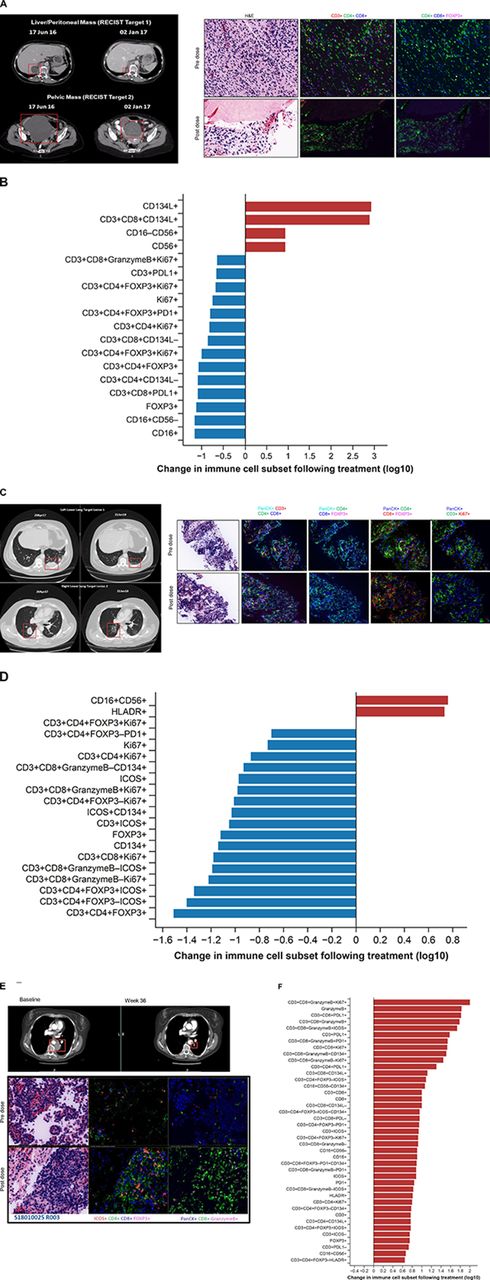
Baseline tumor tissue samples (archival and/or fresh tumor biopsies) were obtained for 48 patients, while paired fresh biopsies (baseline and week 6) were obtained for 34 patients. Multiplexed immunofluorescence revealed 0% to 1.4% of total cells in baseline tumor samples were OX40+ (figure 3A); no correlation between baseline OX40 expression and response was identified. Dose-dependent changes were observed for OX40+immune cells within the paired biopsy samples, with a biphasic dose dependence seen across the dose range tested. The largest changes were seen at 0.1 and 10 mg/kg GSK3174998 (figure 3B). Notably, the greatest changes were observed for CD134+ cells, CD16+CD56−CD134+ NK/NKT cells, and CD3+CD4+FOXP3+CD134+ Tregs, while the least change was observed for cytotoxic T cells (CD3+CD8+ granzyme B+PD-1+CD134+ and CD3+CD8+granzyme B+CD134+). CD3+CD8+, CD4+Foxp3+, and non-tumor (PanCK-neg) Ki67+ populations were reviewed before and after treatment in P1 and P2 (online supplemental figure 4); although changes were seen for some patients, there was no consistent pattern of change observed for all patients combined, or in P1 and P2 considered independently. No other consistent pattern of pharmacodynamic response related to treatment with GSK3174998 was observed across these samples. Multiplexed immunofluorescence data from paired biopsies also indicated increased infiltration of NK/NKT cells and decreased Treg cells in the tumor microenvironment in two responding patients. One of the responders was a patient with dedifferentiated liposarcoma who had an unconfirmed response with monotherapy in P1 (figure 4A,B), and the other patient with melanoma had a CR on study treatment in P2, having previously progressed on nivolumab plus ipilimumab treatment (figure 4C,D). A different immune profile, indicating activation of CD8+ T cells, was observed in a PD-(L)1 treatment-naive patient with NSCLC (tumor proportion score 1%) who responded to the combination of GSK3174998+pembrolizumab (figure 4E,F). Fresh paired tumor biopsies (baseline and week 6) from 26 patients plus baseline archival tissue samples from 48 patients were investigated using the NanoString PanCancer Immune Profiling panel. Unsupervised hierarchical clustering revealed 15 statistically significant genes involved in inflammatory response, T-cell proliferation, and NK cell function that were significantly associated with response or disease control (CR, PR, or SD vs PD; online supplemental figure 5A) and independent of the dose. Six of these genes (ANP32B, XCL2, NOS2A, RELA, RPS6, and ATG16L1) were upregulated or downregulated >2-fold (online supplemental figure 5B).

Multiplexed immunofluorescence analysis of OX40 expression. (A) OX40 expression in baseline tumor biopsies (fresh and archival samples). Baseline tumor expression of OX40 is low (0% to 1.4% across all tested samples). (B) Dose dependence of fold changes in OX40+ cell subsets from baseline to on-treatment (week 6) paired fresh tumor biopsies. From left to right: Row 1: CD134+, PD-1+CD134+; Row 2: CD16−CD56+CD134+, CD16+CD56−CD134+, CD16+CD56+CD134+; Row 3: CD3+CD4+ FOXP3−CD134+, CD3+CD4+FOXP3+CD134+, CD3+CD4+FOXP3+PD-1+CD134+, CD3+CD4+FOXP3−PD-1+CD134+; Row 4: CD3+CD8+granzyme B−CD134+, CD3+CD8+granzymeB+CD134+, CD3+CD8+granzymeB−PD-1+CD134+, CD3+CD8+granzymeB+PD-1+CD134+. CD, cluster of differentiation; FOXP3, forkhead box protein 3; PD-1, programmed cell death 1 protein.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CT scans and multiplexed immunofluorescence analysis of immune cell infiltration in paired biopsies from responding patients. (A, B) GSK3174998 monotherapy (0.3 mg/kg dose)—dedifferentiated liposarcoma. (A) PD-(L)1-naive female patient in her mid-60s with dedifferentiated liposarcoma who remained on 0.3 mg/kg GSK3174998 monotherapy for 39 weeks. At the time her first scan was performed at week 12, her two target lesions (sum of diameters=167 mm) had decreased in size by 29% and further decreased by 38% at 24 weeks (baseline and week 24 scans are shown). The target lesion response was maintained; however, non-target lesion disease progression resulted in treatment discontinuation at week 39. Prior to receiving GSK3174998, the patient’s treatment was surgery, then metronomic doxorubicin (15 mg/kg weekly) ending 1 month before initiating treatment with GSK3174998. Multiplexed immunofluorescence analysis of baseline and week 6 paired tumor biopsy samples show decreased Treg and increased NK/NKT cell infiltration. (B) Changes in immune cell subsets in tumor tissue from baseline to week 6. Immune cells displaying >5-fold changes are highlighted. (C, D) GSK3174998 (0.1 mg/kg dose) + pembrolizumab (200 mg) – melanoma. (C) A male patient in his early 60s with melanoma treated with prior ipilimumab+nivolumab until disease progression after 14 months, ending treatment with nivolumab ≈2 months before initiating treatment with 0.1 mg/kg GSK3174998+pembrolizumab 200 mg. At week 4, the patient developed organizing pneumonia and treatment was held until week 9, when CT scans showed an 8% reduction in target lesions (sum of diameters=98); 12 weeks later, the target lesions had achieved CR, which was maintained for the duration the patient remained on study, completing the 2-year treatment period and remaining in follow-up until the study was closed (baseline and week 57 scans are shown). Multiplexed immunofluorescence analysis of baseline and week 6 paired tumor biopsy samples show decreased Treg and increased NK/NKT cell infiltration. (D) Changes in immune cell subsets in tumor tissue from baseline to week 6. Immune cells displaying >5-fold changes are highlighted. (E, F) GSK3174998 (0.3 mg/kg dose) + pembrolizumab (200 mg) – NSCLC. (E) PD-(L)1-naive female patient in her mid-50s with NSCLC (TPS=1%) with no activating mutations and who had received prior treatment with carboplatin, pemetrexed, and bevacizumab. She received 0.3 mg/kg GSK3174998+pembrolizumab 200 mg treatment for the maximum duration of 2 years. At her week-6 CT scan, the target lesion (54 mm) had decreased in size by 37% and continued to decrease to a maximum of 64%, maintaining a PR for the duration of the study (baseline and week 36 scans are shown). Multiplexed immunofluorescence analysis of baseline and week 6 paired tumor biopsy samples show increased infiltration of T helper cells, cytotoxic T cells, granzyme B–expressing CD8+ T cells, ICOS+ T cells, and proliferating cytotoxic T cells. (F) Changes in immune cell subsets in tumor tissue from baseline to week 6. Immune cells displaying >5-fold changes are highlighted. CD, cluster of differentiation; CR, complete response; FOXP3, forkhead box protein 3; H&E, hematoxylin and eosin; HLA-DR, human leukocyte antigen D related; ICOS, inducible T-cell costimulatory; NK, natural killer; NKT, natural killer T cell; NSCLC, non-small cell lung cancer; PanCK, pan cytokeratin; PD-1, programmed cell death 1 protein; PD-L1, programmed cell death 1 ligand; PR, partial response; Treg, regulatory T cell; TPS, tumor proportion score.
Discussion
The OX40 agonist monoclonal antibody (mAb) GSK3174998 was well tolerated up to the maximum administered dose of 10 mg/kg when administered alone and in combination with pembrolizumab 200 mg Q3W in this phase I first-in-human study. No dose relationship was observed for the incidence of TRAEs, and an MTD was not identified for GSK3174998±pembrolizumab. The safety profile observed in this study appears similar to reports from other OX40 agonist mAbs,22 29–32 and when considering the well-established safety profile of pembrolizumab,3 no new safety signals were observed with the combination.
GSK3174998 demonstrated target engagement in the periphery as evidenced by pharmacokinetic and RO analyses. The 0.3 mg/kg dose was the threshold for linear pharmacokinetic and peripheral RO saturation (>80%) over the 3-week dose interval, suggesting sufficient target saturation, and thus was selected for further clinical evaluation in the P2 cohort expansion. Observed median pharmacokinetic profiles at dose levels ≥0.3 mg/kg GSK3174998 exposure appeared to be dose proportional with two-compartment behavior expected for mAbs and with target-mediated clearance apparent at lower doses, similar to data reported for another OX40 agonist, MOXR0916.32 While pharmacodynamic effects were observed in the peripheral blood, including upregulation of some inflammatory cytokines, none were dose related.
No confirmed response was observed for GSK3174998 monotherapy. However, there were signs of limited clinical activity with prolonged SD lasting at least 6 months in four patients, two of whom were PD-(L)1 experienced and one (with dedifferentiated liposarcoma) who had an unconfirmed PR and remained on treatment at the 0.3 mg/kg dose level for 39 weeks. Multiplexed immunofluorescence data from paired biopsies of this patient indicated increased infiltration of NK/NKT cells and decreased Treg cells in the tumor microenvironment. Prior to receiving GSK3174998, this patient had received metronomic doxorubicin (15 mg/kg weekly), ending 1 month before initiating treatment with GSK3174998. Low-dose metronomic chemotherapy has been explored as a potential immunostimulatory mechanism to improve the efficacy of immunotherapies33–36; it is possible that this recent treatment was sufficient to prime the tumor microenvironment and increase the opportunity for GSK3174998 to have an effect.
While significant monotherapy activity was not anticipated for GSK3174998 (as with other OX40 agonist mAbs), expectations for efficacy with GSK3174998±pembrolizumab were much greater based on data from multiple preclinical models.23 24 37 Despite the promising preclinical data, the increase in efficacy for OX40 agonist and PD-(L)1 blocker combinations did not translate to the clinic; responses reported for the GSK3174998+pembrolizumab combination were not considered to be greater than those expected for pembrolizumab alone.3 38 Since the majority (5/6) of the responses observed were in PD-(L)1–naive patients, one cannot rule out that these responses were largely driven by pembrolizumab alone. Preliminary responses in dedifferentiated liposarcoma, NSCLC, and melanoma in the dose-escalation cohorts supported the selection of these tumor types for P2 cohort expansion. Disappointingly, no patients treated in these expansion cohorts developed an objective response and with this, along with data emerging from ongoing OX40 trials, a decision was made to stop further enrollment in the expansion cohorts.
Although anti-GSK3174998 antibodies were present in almost half (39%) of the patients treated with GSK3174998 in both P1 and P2, the presence of these antibodies did not appear to prevent responses. The single patient that responded in P1 (an unconfirmed PR) exhibited a modest titer of anti-GSK3174998 antibodies during treatment, and 3 (1 CR and 2 PRs) of the 6 confirmed responders (2 CRs and 4 PRs) in P2 were also ADA-positive with high titers (up to 163,840) during treatment. Thus, it is unlikely that the presence of ADAs played a role in failure of the OX40 agonist monoclonal antibody GSK3174998 to elicit tumor responses.
The favorable safety profile of GSK3174998 and other OX40 agonist antibodies administered both as a monotherapy and in various combinations has been surprising (online supplemental table 5).20 29–32 39–41 The low starting dose selected for dose escalation (0.003 mg/kg GSK3174998) was based on the anticipation that elevated cytokine levels may increase the incidence of immune-related AEs and possibly lead to cytokine release syndrome. However, changes in plasma cytokines were modest, not dose-related, and none of these events manifested. It is notable that some cytokines demonstrated statistically significant changes for both P1 and P2 (MIP-1α, MIP-1β, IP-10, VCAM1, MCP-1), perhaps suggesting a role for GSK3174998, whereas others were only statistically significant in P2 (MIP-3β, IL-10, IFN-γ, IL-6, soluble ICAM1, G-CSF, IL-5), suggesting they are modulated by pembrolizumab or potentially an interaction between GSK3174998 and pembrolizumab. However, the modest changes observed (up to ≈2-fold during the 24-hour post-dose period) did not appear to be sufficient to result in antitumor efficacy. These findings, along with the absence of a robust efficacy signal, raise important questions about our understanding of the complex biology of this target and the engagement of OX40 agonist mAbs in the context of the tumor microenvironment.
Several potential mechanisms of action have been described for OX40 agonist antibodies, including stimulation of CD4+ and CD8+ effector T cells, suppression of CD4+ Treg cells, increased NK cell activation, and potential for depletion of intratumoral Tregs in an activating FcγR-dependent manner. This makes interpretation of tumor biomarker data somewhat complex, with few responders in mixed tumor histologies. Evaluation of paired tumor biopsies by multiplexed immunofluorescence analysis in the current study suggested a potential role for increased infiltration of NK/NKT cells and decreased Treg cells in the tumor microenvironment in the responses of two patients. The first being the patient with dedifferentiated liposarcoma on monotherapy who had an unconfirmed PR and the second being the PD-1–experienced patient with melanoma on combination treatment who had a CR. In contrast, a different immune profile indicating activation of CD8+T cells was observed in a PD-(L)1 treatment–naive patient with NSCLC (tumor proportion score 1%) who responded to the combination of GSK3174998+pembrolizumab, a durable PR perhaps driven solely by pembrolizumab. The immune-focused NanoString panel on pretreatment and on-treatment biopsies revealed 15 statistically significant genes involved in inflammatory response, T-cell proliferation, and NK cell function that were significantly associated with response or disease control. However, the ORR was unfortunately low, and paired biopsies were not obtained for all responding patients; thus, data informing biomarker-response relationships remained anecdotal. More samples were available for the evaluation of pharmacodynamic effects in the tumor microenvironment. Dose-dependent changes were observed only for selected OX40+ immune cells. The greatest changes being observed for NK/NKT cells (CD16+CD56−CD134+) and Tregs (CD3+CD4+ FOXP3+ CD134+), and the least for cytotoxic T cells (CD3+CD8+ granzyme B+PD-1+ CD134+ and CD3+CD8+ granzyme B+CD134+). Thus, a role for Fcγ receptor–dependent activity with this IgG1 OX40 mAb is possible but still not sufficient to drive increased clinical activity. Since no direct competition assays were conducted with the anti-OX40 BER-Act35 antibody used in the multiplexed immunofluorescence assay, interference cannot be ruled out. However, the presence of Act35 staining in samples from patients on treatment with GSK3174998, together with very different functional activity of GSK3174998 and Act35, suggests they bind different epitopes on OX40.15 Furthermore, the shape of the dose-response curves shown in figure 3B peaking at both 0.1 and 10 mg/kg do not indicate an exposure-related interference between GSK3174998 and Act35.
Baseline OX40 expression in the tumor was low (0% to 1.4%) in this study, an observation also reported in the BMS-986178 trial, in which 82% of tumor samples tested had <1% expression of OX40.29 Given the transient expression of the OX40 receptor on activated CD4+ and CD8+ T cells, it is possible that, in patients with advanced cancer, the T cells in the tumor microenvironment are not in an activated state. An additional priming step may be needed before administration of an OX40 agonist, perhaps, for example, strategies that are currently under investigation via CD40 activation, use of toll-like receptor agonists, vaccine administration to augment OX40 receptor expression42 or perhaps low dose metronomic chemotherapy.33–36 With so few responders in the current trial, it was not possible to identify robust markers predictive of response. High baseline OX40 expression may have been the obvious candidate but, given its inducible expression and limited variability in magnitude of expression, it is likely not an ideal selection biomarker.
Beyond expression of the target, the optimal engagement of the OX40 agonist mAbs with the OX40 receptor is not well understood. Unlike antagonist mAbs, a higher dose and constant engagement of the mAb and receptor may not be optimal for agonist mAbs. The rationale for the Q3W dosing frequency was based on clinical data with MEDI6469, where a key biomarker, Ki-67 expression on T cells, exhibited maximal stimulation at about 14 days after the first dose.19 The Ki-67 stimulation declined by about 28 days after the first dose (or 23 days after the last dose in the cycle).19 Guided by these biomarker dynamics and the expectation of standard IgG1 mAb pharmacokinetic (ie, terminal elimination half-life longer than 2 weeks), a dosing frequency of Q3W for GSK3174998 was chosen. With so few patients responding to OX40 monotherapy, it was not possible to identify exposure-response relationships. Both pharmacokinetic and RO data provided a clear framework for dose-related changes. Therefore, pharmacokinetics and RO were used to inform dose selection of the 0.3 mg/kg dose. This dose represented the inflection below which target-mediated clearance was observed and provided ≥80% RO in the periphery, which would potentially be lower in the tumor tissue. Additionally, the one unconfirmed response for monotherapy was observed at the 0.3 mg/kg dose level. Doses of 0.1–0.3 mg/kg have been shown to have more prominent effects on T-cell activation with other OX40 antibodies, such as ivuxolimab and BMS-986178, although preclinical modeling with BMS-986178 suggested optimal peripheral blood RO of 20% to 50% for maximal effector T-cell function.43 In addition to understanding the dose and schedule for the OX40 agonists, questions remain about the optimal dosing schedule for combinations.44 45 with a suggestion from preclinical data that sequential administration of an OX40 agonist followed by PD-1 therapy may be better.44
Given the need for trimerization of the OX40 receptor to activate the signaling pathway,46 it is possible that the conventional bivalent mAbs that engage with only two OX40 receptors may not induce optimal agonistic activity. Additional secondary crosslinking is necessary to promote clustering via Fc-FcγR interactions. Thus, novel strategies are needed to enhance the Fc region of mAbs to boost Fc receptor clustering and enhance effector function, including antibody-dependent cellular cytotoxicity. Additional strategies include developing novel drug designs to increase antibody valency, for example, a hexavalent OX40 mAb that can bind six OX40 receptors per antibody molecule. Furthermore, the development of agonists that mimic the OX40L may also address the need for trimerization.47 48
Multiple early clinical trials of OX40 agonist mAbs have now reported trial outcomes (online supplemental table 5).20 29–32 39–41 To date, all have shown limited or no monotherapy activity and combination responses that do not appear better than the single-agent activity of the combination partner. Within these reported trials and the current trial of GSK3174998, a total of ≈900 patients were treated with OX40 agonist agents. Notably, these studies have largely been conducted in patients with advanced disease and with treatment schedules similar to those developed for checkpoint blockers. Biomarker analyses performed in the current study and the aforementioned published studies clearly document target engagement and pharmacodynamic effects, yet these effects did not translate to clinical benefit, providing an important negative result in the evaluation of this target. Furthermore, clinical responses and available tumor material were too scarce to formally prove biological activity of GSK3174998 or robustly identify any biomarker predictive of clinical efficacy. Consequently, one may conclude that targeting OX40 alone or combined with PD-1 blockade is not an effective treatment strategy for patients with advanced cancer. While multiple proposals to optimize OX40 interaction might be considered (eg, through multimerization), such development should proceed cautiously with an emphasis on early futility analyses to protect patients who are enrolling in these studies.
In conclusion, this study demonstrated that treatment with GSK3174998 up to the maximum planned dose of 10 mg/kg alone and in combination with pembrolizumab 200 mg was well tolerated in patients with advanced solid tumors. While target engagement was evidenced by robust RO with evidence of pharmacologic activity in peripheral blood and tumor, these findings did not translate into clinical efficacy for GSK3174998 monotherapy or the anticipated increased efficacy when dosed in combination with pembrolizumab. Overall, these data are consistent with available reports from other OX40 agonist antibodies (eg, MOXR0916, BMS-986178, MEDI0562, and PF-04518600) and do not support the continued development of GSK3174998±pembrolizumab in advanced solid tumors; thus development of GSK3174998 has been discontinued.
Data availability statement
Data are available upon reasonable request. Information about GlaxoSmithKline’s data sharing commitments and access requests to anonymized individual participant data and associated documents can be requested for further research from ClinicalStudyDataRequest.com.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by IntegReview IRBIRB00008463, IRB00003657, IRB00004920, IRB00001035, IRB000060751. Princess Margaret Hospital: The University Health Network Research Ethics Board - Renewal (16-6261.4)2. Netherlands Cancer Inst: Stichting Beoordeling Ethiek Biomedisch Onderzoek - ICON code 9018/00013. Gustave Roussy Cancer Campus:Ile-de-France 3 EC Hôpital Tarnier-Cochin - N° EUDRACT : 2015-000152-14Réf. CPP Dossier N° : Am8419-13-3336US IRBs: IRB00008463, IRB00003657, IRB00004920, IRB00001035, IRB00006075US Institutions -Sarah Cannon (SCRI): IntegReview IRB - -Dana Farber Cancer Inst (DFCI): DFCI Institutional Review Board - -NYU Langone Medical Center: NYU School of Medicine’s Institutional Review Board (IRB) Office of Science and Research Institutional Review Board-MDACC: UT MD Anderson Cancer Center Institutional Review Board 5-MSKCC: MSKCC Institutional Review Board/Privacy Board-B Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors acknowledge the following individuals for their contributions and critical review during the development of this manuscript: Yolanda Alvarez, Sara Brett, Bob Gagnon, and Hoang Tran, and Vinod Kumar from GlaxoSmithKline (GSK). The authors also wish to acknowledge the patients who participated in this study, their families and caregivers, site personnel, and the GSK, Merck, ICON, and Neogenomics teams. Editorial support was provided by Brittany Woodby, PhD, at MediTech Media and was funded by GSK.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @OsamaRahma2
Contributors Conceptualization: FSH, JHMS, EMP, CMA, HZ, HS, MW, NY, EVS, AH, AM, JSW, JVH. Data acquisition: SP-V, VKL, WR, TMB, ARH, DCC, FSH, JHMS, JKL, SA, KAA, FLO, MM, NS, SC, MA, AS, OR, AM, JSW, JVH. Data analysis/interpretation: SP-V, VKL, WR, TMB, ARH, DCC, FSH, JHMS, JKL, SA, KAA, FLO, MM, NS, SC, MA, AS, OR, EMP, CMA, HZ, HS, SAG, MW, KMY, NY, MJC, EVS, AH, AM, JSW, JVH. Writing, review, and editing: SP-V, VKL, WR, TMB, ARH, DCC, FSH, JHMS, JKL, SA, KAA, FLO, MM, NS, SC, MA, AS, OR, EMP, CMA, HZ, HS, SAG, MW, KMY, NY, MJC, EVS, AH, AM, JSW, JVH. All authors read and approved the final version of the manuscript. SP-V accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Funding Study sponsored by GlaxoSmithKline in collaboration with Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, USA.
Competing interests SP-V is a principal/subinvestigator of clinical trials for AbbVie, Adaptimmune, Adlai Nortye USA Inc, Aduro Biotech, Agios Pharmaceuticals, Amgen, Argenx BVBA, Arno Therapeutics, Astex Pharmaceuticals, AstraZeneca A, Aveo, Basilea Pharmaceutica International Ltd, Bayer Healthcare AG, BBB Technologies BV, Beigene, BicycleTx Ltd, Bioalliance Pharma, Blueprint Medicines, Boehringer Ingelheim, Boston Pharmaceuticals, Bristol Myers Squibb, Celgene Corporation, Chugai Pharmaceutical Co, Cullinan-Apollo, Curevarc, Daiichi Sankyo, Debiopharm, Eisai, Eisai Limited, Eli Lilly, Exelixis, Faron Pharmaceuticals Ltd, Forma Tharapeutics, GamaMabs, Genentech, GSK, H3 Biomedicine, Hoffmann La Roche AG, Imcheck Therapeutics, Innate Pharma, Institut De Recherche Pierre Fabre, Iris Servier, Iteos Belgium SA, Janssen Cilag, Janssen Research Foundation, Kura Oncology, Kyowa Kirin Pharm. Dev, Lilly France, Loxo Oncology, Lytix Biopharma AS, Medimmune, Menarini Ricerche, Merck Sharp & Dohme Chibret, Merus, Molecular Partners AG, Nanobiotix, Nektar Therapeutics, Novartis Pharma, Octimet Oncology NV, Oncoethix, Oncopeptides, Onyx Therapeutics, Orion Pharma, Oryzon Genomics, Ose Pharma, Pfizer, Pharma Mar, Pierre Fabre Medicament, Plexxikon, Roche, Sanofi Aventis, Seattle Genetics, Sotio A.S, Syros Pharmaceuticals, Taiho Pharma, Tesaro, Turning Point Therapeutics, and Xencor outside the submitted work; non-financial support (drug supplied) from AstraZeneca, Bayer, Bristol Myers Squibb, Boehringer Ingelheim, GSK, Medimmune, Merck, NH TherAGuiX, Pfizer, Roche; research funding from Boehringer Ingelheim, AstraZeneca, Roche, and Merck KGaA for projects unrelated to this manuscript. VL reports consulting for Takeda, Seattle Genetics, Bristol Myers Squibb, AstraZeneca, Guardant Health; research funding from GSK, Bristol Myers Squibb, Merck, Seattle Genetics. WR reports nothing to disclose. TMB reports consulting for Guardant Health, Loxo Pharmaceuticals, Pfizer, Exelixis, Blueprint Medicines, Foundation Medicine, Bayer, AstraZeneca, Ignyta, Moderna Therapeutics, Pfizer. Speakers Bureau: Bayer, Bristol Myers Squibb, and Lilly; research funding from Daiichi Sankyo, Medpacto, Inc., Incyte, Mirati Therapeutics, MedImmune, AbbVie, AstraZeneca, Leap Therapeutics, MabVax, Stemline Therapeutics, Merck, Lilly, GSK, Novartis, Pfizer, Genentech/Roche, Deciphera, Merrimack, Immunogen, Millennium Pharmaceuticals, Ignyta, Calithera Biosciences, Kolltan Pharmaceuticals, Principa Biopharma, Peleton, Immunocore, Aileron Therapeutics, Bristol Myers Squibb, Amgen, Moderna Therapeutics, Sanofi, Boehringer Ingelheim, Astellas Pharma, Five Prime Therapeutics, Jacobio, Top Alliance BioScience, Loxo, Janssen, Clovis Oncology, Takeda, Karyopharm Therapeutics, Onyx, Phosplatin Therapeutics, Foundation Medicine, and ARMO BioScience; personal expenses from Astellas Pharma, AstraZeneca, Celgene, Clovis Oncology, EMD Serono, Genentech, Lilly, Merck, Novartis, Pharmacyclics, Sysmex, and Pfizer. AHa reports research funding from Genentech/Roche, Merck, GSK, Bristol Myers Squibb, Novartis, Boston Biomedical, Boehringer Ingelheim, AstraZeneca/Medimmune Eisai; personal fees from Merck and GSK. DCC is a consultant for Nektar, Pfizer, Werewolf, and HUYA. FSH reports research funding from Bristol Myers Squibb and Novartis; receives personal fees from Bristol Myers Squibb, Merck, EMD Serono, Novartis, Surface, Compass Therapeutics, Apricity, Sanofi, Pionyr, Torque, Bicara, Checkpoint Therapeutics, Genentech/Roche, Bioentre, Gossamer, Iovance, Trillium, Catalym, Immunocore, Amgen, Kairos, Eisai, and Rheos. JHMS is a shareholder and part-time employee of Modra Pharmaceuticals BV and patent holder of oral taxanes; reports consulting for Debiopharm. JKL reports other from GSK, Novartis, Medivation/Pfizer, Genentech, EMD-Serono, AstraZeneca, Medimmune, Zenith, Ayala, UpToDate review panels for NCCN, ASCO, NIH PDQ, Medlearning, Physicians Education Resource, Prime Oncology, Medscape, Clinical Care Options, Medpage. SA reports advisory board participation for MSD, Sanofi, Roche, Bristol Myers Squibb, Pfizer; research support from Sanofi. KAA reports research funding (to institution) from Pfizer, AstraZeneca, Amgen, Trishula, GSK, Merck, and Eli Lilly. FO is a principal/subinvestigator of clinical trials for Amgen, AstraZeneca, Cytovation, GSK, Lilly, MSD, Revmed, Incyte, Boehringer Ingelheim, Bristol Myers Squibb, Roche/Genentech, Exelexis, Relay, and InterRNA. MM reports research funding from Alpine Immune Sciences, Arcus Biosciences, Arvinas, Ascentage Pharma Group, Bayer, Bicycle Therapeutics, BioMed Valley Discoveries, BioNTech, Dragonfly Therapeutics, EMD Serono, Epizyme, Erasca, Exelixis, Foghorn Therapeutics, Genentech, Gilead Sciences, GSK, IDEAYA Biosciences, Ikena Oncology, ImmVira Pharma, Infinity Pharmaceuticals, Jacobio Pharmaceuticals, Kechow Pharma, Kezar Life Sciences, Kinnate BioPharma, MedImmune, Mereo BioPharma, Metabomed, Moderna, NBE Therapeutics, Nektar, Novartis, Oncorus, PACT Pharma, Pfizer, Plexxikon, Prelude Therapeutics, Pyramid Biosciences, Regeneron, Sapience Therapeutics, Scholar Rock Seattle Genetics, Synthrox, Takeda Pharmaceuticals, Teneobio, Tempest Therapeutics, Tizona Therapeutics, TMUNITY Therapeutics, TopAlliance Biosciences, and Xilio. M; consulting/advisory role for Astellas Pharma, AstraZeneca, BicycleTX Limited, Castle Biosciences, Eisai, Ideaya Biosciences, iTeos, Pfizer, and Regeneron Pharmaceuticals. NS reports consulting/advisory role for Deciphera, AADi, Blueprint Medicines, Bayer, Epizyme, and Boehringer Ingelheim; research funding from GSK, Karyopharm, Deciphera, Ascentage Pharma, Daiichi Sankyo/Lilly, and AstraZeneca/MedImmune. SC is the principal/subinvestigator of clinical trials for AbbVie, Adaptimmune, Adlai Nortye USA Inc, Aduro Biotech, Agios Pharmaceuticals, Amgen, Astex Pharmaceuticals, AstraZeneca AB, Aveo, Basilea Pharmaceutica International Ltd, Bayer Healthcare AG, BBB Technologies BV, Beigene, BicycleTx Ltd, Blueprint Medicines, Boehringer Ingelheim, Boston Pharmaceuticals, Bristol Myers Squibb, Casi Pharmaceuticals, Inc, Celgene Corporation (a Bristol-Myers Squibb company), Cellcentric, Chugai Pharmaceutical Co, Cullinan-Apollo, Curevarc, Cytovasion, Daiichi Sankyo, Debiopharm, Eisai, Eli Lilly, Exelixis, Faron Pharmaceuticals Ltd, Forma Tharapeutics, GamaMabs, Genentech, GSK, H3 Biomedicine, Hoffmann La Roche AG, Imcheck Therapeutics, Incyte Corporation, Innate Pharma, Institut De Recherche Pierre Fabre, Iris Servier, Iteos Belgium SA, Janssen Cilag, Janssen Research Foundation, Janssen R&D LLC, Kura Oncology, Kyowa Kirin Pharm. Dev, Lilly France, Loxo Oncology, Medimmune, Menarini Ricerche, Merck, Merck Sharp & Dohme Chibret, Merrimack Pharmaceuticals, Merus, Molecular Partners AG, Nanobiotix, Nektar Therapeutics, Novartis Pharma, Octimet Oncology NV, Oncoethix, Oncopeptides, Orion Pharma, Genomics, Ose Pharma, Pfizer, Pharma Mar, Pierre Fabre Medicament, Relay Therapeutics, Inc, Roche, Sanofi Aventis, Seattle Genetics, Sotio, Syros Pharmaceuticals, Taiho Pharma, Tesaro, Transgene SA, Turning Point Therapeutics, and Xencor; reports research grants from AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, GSK, INCA, Janssen Cilag, Merck, Pfizer, Roche, and Sanofi; reports non-financial support (drug supplied) from AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, GSK, Medimmune, Merck, NH TherAGuiX, Pfizer, and Roche; reports honoraria from Amgen, Astellas, AstraZeneca, Bristol Myers Squibb, Eisai, Genmab, Janssen, Merck, Novartis and Roche; reports participation in advisory boards for Alderaan Biotechnology, Amgen, AstraZeneca, Avacta, Ellipses Pharma, Oncovita, Seagen, UltraHuman8; reports travel and congress support from AstraZeneca, Bristol Myers Squibb, Merck, Ose Pharma, Roche, Sotio. MA reports research funding (to institution) from Genentech, Nektar Therapeutics, Merck, GSK, Novartis, Jounce Therapeutics, Bristol Myers Squibb, Eli Lilly, Adaptimmune, Shattuck Lab, and Gilead. Advisory boards: GSK, Shattuck Lab, Bristol Myers Squibb, AstraZeneca; reports speaker fees from AstraZeneca, and Nektar Therapeutics. AS reports participation in advisory boards for Merck, Bristol Myers Squibb, Novartis, Oncorus, Janssen, Medison, and Immunocore; reports research support: Novartis, Bristol Myers Squibb, Symphogen AstraZeneca/Medimmune, Merck, Bayer, Surface Oncology, Northern Biologics, Janssen Oncology/Johnson & Johnson, Roche, Regeneron, Alkermes, Array Biopharma/Pfizer, GSK, Treadwell, and Amgen. OR reports employment with AstraZeneca; reports research funding from Merck; reports speaker for activities supported by educational grants from Bristol Myers Squibb and Merck; reports consulting for Merck, Celgene, Five Prime, GSK, Bayer, Roche/Genentech, Puretech, Imvax, Sobi, Boehringer Ingelheim; reports pending patent for “Methods of using pembrolizumab and trebananib”. EMP, CMA, HZ, HS, SAG, MW, KMY, NY, and AHo report employment with GSK. MJC is an employee of Merck and owns stock. EVS is an employee of Merck. AM has been a principal or coinvestigator of clinical trials using OX40-targeted agents and/or has provided consulting services for Roche/Genentech, AstraZeneca, Pfizer, GSK, HiFiBiO, Bristol Myers Squibb, Shattuck Labs. JW reports consulting for Merck, Genentech, AstraZeneca, GSK, Novartis, Nektar, Celldex, Incyte, Biond, ImCheck, Sellas, Evaxion and EMD Serono; reports participation in advisory boards for Bristol Myers Squibb (compensated), CytoMx, Incyte, ImCheck, Biond, Sellas, Instil Bio, OncoC4, and Neximmune; reports equity holdings in Biond, Evaxion, Instil Bio, OncoC4, and Neximmune; reports research support (to institution) from Bristol Myers Squibb, Merck, GSK, Moderna, Pfizer, Novartis, and AstraZeneca, Moffitt Cancer Center; reports patent for ipilimumab biomarker and tumor-infiltrating lymphocytes preparation and a PD-1 patent (Biodesix). JVH reports other from GSK, AstraZeneca, Checkmate Pharmaceuticals, Brightpath Biotherapeutics, Eli Lilly & Co, Kairos Venture Investments, Triptych Health Partners; reports patent with Spectrum Pharmaceuticals.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.