Article Text

Abstract
Background The success of HER2-positive (HER2+) breast cancer treatment with trastuzumab, an antibody that targets HER2, relies on immune response. We demonstrated that TNFα induces mucin 4 (MUC4) expression, which shields the trastuzumab epitope on the HER2 molecule decreasing its therapeutic effect. Here, we used mouse models and samples from HER2+ breast cancer patients to unravel MUC4 participation in hindering trastuzumab effect by fostering immune evasion.
Methods We used a dominant negative TNFα inhibitor (DN) selective for soluble TNFα (sTNFα) together with trastuzumab. Preclinical experiments were performed using two models of conditionally MUC4-silenced tumors to characterize the immune cell infiltration. A cohort of 91 patients treated with trastuzumab was used to correlate tumor MUC4 with tumor-infiltrating lymphocytes.
Results In mice bearing de novo trastuzumab-resistant HER2+ breast tumors, neutralizing sTNFα with DN induced MUC4 downregulation. Using the conditionally MUC4-silenced tumor models, the antitumor effect of trastuzumab was reinstated and the addition of TNFα-blocking agents did not further decrease tumor burden. DN administration with trastuzumab modifies the immunosuppressive tumor milieu through M1-like phenotype macrophage polarization and NK cells degranulation. Depletion experiments revealed a cross-talk between macrophages and NK cells necessary for trastuzumab antitumor effect. In addition, tumor cells treated with DN are more susceptible to trastuzumab-dependent cellular phagocytosis. Finally, MUC4 expression in HER2+ breast cancer is associated with immune desert tumors.
Conclusions These findings provide rationale to pursue sTNFα blockade combined with trastuzumab or trastuzumab drug conjugates for MUC4+ and HER2+ breast cancer patients to overcome trastuzumab resistance.
- Breast Neoplasms
- Drug Therapy, Combination
- Immune Evation
- Lymphocytes, Tumor-Infiltrating
- Macrophages
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Trastuzumab resistance is an important clinical issue. Although a plethora of resistance mechanisms that limit therapy efficacy has been characterized, few of them have been shown to be actionable.
WHAT THIS STUDY ADDS
We have identified mucin 4 as a mechanism of tumor immune evasion which causes trastuzumab treatment failure in HER2+ breast cancer; nonetheless its expression and immunosuppressive actions can be tackled by soluble TNFα blockade.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
These data support that mucin 4 should be used as a predictive biomarker to guide the administration of soluble TNFα blocking drugs in combination with trastuzumab or trastuzumab-based drug conjugates in HER2+breast cancer patients to overcome trastuzumab resistance.
Introduction
HER2-positive (HER2+) breast cancer is identified by the amplification/overexpression of the oncogene ERBB2 and affects 15%–20% of patients with breast cancer.1 These tumors have an aggressive behavior and threaten patients’ survival. Nevertheless, the administration of trastuzumab, a monoclonal antibody against HER2, has dramatically improved the outcome of patients affected by HER2+ breast cancer.2 3 Trastuzumab is administered in the adjuvant and neoadjuvant setting in early and metastatic disease.4 One of its mechanisms of action consists of inhibiting the signaling pathways downstream the tyrosine kinase receptor HER2.5 However, in vivo, the immune response takes major relevance since trastuzumab triggers an innate immune response inducing antibody-dependent cell cytotoxicity (ADCC) by NK cells and antibody-dependent cell phagocytosis (ADCP) by macrophages.6–9 In addition, an adaptive antitumor immune response driven mainly by CD8+T cells is also involved.10 11 Indeed, one of the prognostic features that indicates potential success of trastuzumab treatment is the presence of stromal tumor-infiltrating lymphocytes (TILs). High levels of TILs at the time of diagnosis are associated with better outcomes in HER2+ breast cancer patients under trastuzumab treatment.12–14 However, a plethora of resistance mechanisms that limit therapy efficacy have been characterized15 and few of them have been shown to be actionable.
Previously, we have demonstrated that TNFα is able to induce trastuzumab resistance and upregulate the expression of the transmembrane glycoprotein mucin 4 (MUC4).16 MUC4 belongs to the membrane-bound family of mucins and has two non-covalently associated subunits encoded by a single gene.17 The extracellular subunit, MUC4α, is hyperglycosylated and favors metastatic dissemination as it confers antiadhesive properties to the tumor cells. The transmembrane subunit MUC4β, contains two EGF-like domains in the extracellular portion that can interact with HER2, preventing its internalization, and therefore, enhancing its signaling.18 As trastuzumab binds to the juxtamembrane region of HER2,19 MUC4, with its heavy glycosylation patterns, shields trastuzumab epitope on the HER2 molecule hindering its binding and therapeutic effect.20 Moreover, we showed that MUC4 expression in HER2+breast cancer is an independent predictor of poor disease-free survival in patients treated with adjuvant trastuzumab.16 However, the impact of MUC4 expression on trastuzumab-induced immune response in HER2+ breast cancer remains poorly studied.
In preclinical breast cancer mouse models of de novo trastuzumab resistance, we demonstrated that TNFα blockade with etanercept, a fusion protein of TNFα receptor 2 and the human Fc portion of IgG1, induced downregulation of MUC4 expression restoring trastuzumab antitumor effect.16 It is known that TNFα is a homotrimeric cytokine that has two active isoforms: the transmembrane (tmTNFα), and the soluble TNFα (sTNFα) which is produced by cleavage of the tmTNFα isoform by the TNFα converting enzyme (TACE/ADAM17).21 Etanercept neutralizes tmTNFα and sTNFα of human and murine origin with similar affinity.22 While tmTNFα is a molecular mediator of the crosstalk between dendritic cells and NK cells and it is required for NK cell proliferation,23 sTNFα has been acknowledged to have a central role in the onset and progression of cancer.24 25 Therefore, we aimed to explore sTNFα participation in trastuzumab resistance in HER2+ breast cancer.
In this work, we demonstrated that the combination of trastuzumab and TNFα blocking drugs promote an innate immune response that involves NK cells and macrophages. Using INB03 (XPro1595), a dominant negative (DN) of TNFα able to neutralize only sTNFα,26 we proved that MUC4 was downregulated and an innate immune response was triggered upon trastuzumab treatment. DN is able to neutralize both human and murine sTNFα.26 Experiments using tumors engineered to express a doxycycline (Dox) inducible short hairpin RNA (shRNA) against MUC4 showed that macrophages are crucial for the antitumor effect of trastuzumab on MUC4 silencing. In addition, we observed that MUC4 expression is associated with HER2+breast cancer with scarce TILs, and hence is linked to poor patients’ response to trastuzumab. Here, we provide evidence that MUC4 is a key player in TNFα-induced trastuzumab resistance and promotes tumor immune evasion, favoring an immune desert tumor microenvironment (TME) with immunosuppressive characteristics that can be tackled by sTNFα blockade to achieve trastuzumab effectiveness.
Materials and methods
The complete experimental procedures and protocols are described in online supplemental material.
Supplemental material
Results
Trastuzumab sensitization through sTNFα blockade triggers an innate immune response in HER2+ breast cancer
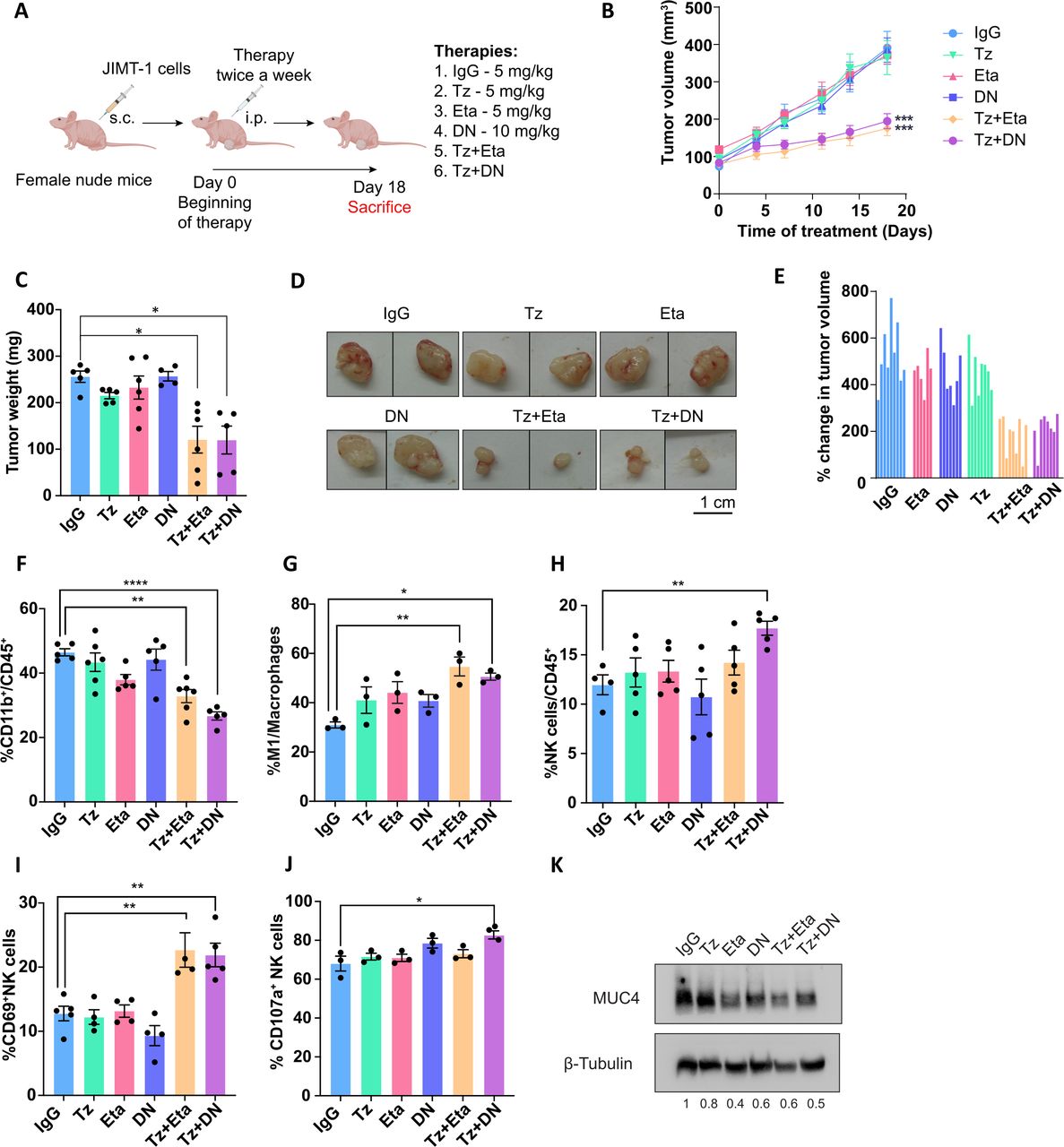
Previously, we have demonstrated that the administration of trastuzumab in combination with etanercept overcomes de novo trastuzumab resistance in JIMT-1 and KPL-4 cell lines and tumors.16 As trastuzumab administration in vivo is capable of triggering an innate immune response, we evaluated the impact of trastuzumab and TNFα blockade on this response in the TME. Female nude mice bearing JIMT-1 tumors were treated with (1) polyvalent IgG, as a control, (2) trastuzumab, (3) etanercept (which targets tmTNFα and sTNFα), (4) DN protein INB03 (that only targets sTNFα), (5) trastuzumab+etanercept or (6) trastuzumab+DN (figure 1A). Tumor growth inhibition was achieved by the combination of trastuzumab plus etanercept (55% vs IgG, figure 1B–E and online supplemental table S1) and was accompanied by a diminished proportion of infiltrating myeloid cells with respect to IgG (figure 1F). Moreover, this treatment resulted in an increased polarization of macrophages to the M1-like antitumorigenic and inflammatory subtype (figure 1G). As sTNFα has been acknowledged to have a central role in the onset and progression of certain types of cancer,24 25 we wanted to dissect its participation in the trastuzumab-mediated antitumor effect and the immune response. For that purpose, we used DN which forms inactive heterotrimers with sTNFα, preventing the binding to its receptors, without interfering with the transmembrane isoform.26 DN alone had no impact on JIMT-1 tumor growth, in line with our previous report with etanercept.16 Similar to trastuzumab plus etanercept treatment, combination of trastuzumab plus DN suppressed tumor growth by 50% in comparison with IgG (figure 1B–E, online supplemental table S1), suggesting that blocking sTNFα isoform is sufficient to overcome trastuzumab resistance. Moreover, this treatment decreased the infiltrating myeloid cells while increasing macrophage polarization to the M1-like subtype, resembling trastuzumab plus etanercept administration (figure 1F–G). NK cells only exhibited an enhanced recruitment to the TME under trastuzumab plus DN treatment, compared with the IgG (figure 1H). However, both TNFα-blocking agents combined with trastuzumab generated an increase in the activation marker CD69 on NK cells (figure 1I), while degranulation was only enhanced by the combined therapy with DN (figure 1J). Etanercept or DN administration alone has no evident immunogenicity as their administration did not alter tumor growth rate compared with IgG, nor did they modify the TME architecture or the associated innate immune response (figure 1B–J). As we previously reported, etanercept reduced MUC4 expression in tumors.16 By neutralizing only sTNFα with DN, we obtained similar results by Western blot (figure 1K). Collectively, these data suggest that blocking only the sTNFα isoform in combination with trastuzumab strongly inhibits HER2+ tumor growth and unleashes an antitumor innate immune response that radically transforms the immunosuppressive TME. Polarization of macrophages to the M1-like subpopulation, an increased activation and degranulation of NK cells and a diminished infiltration of myeloid cells are the main characteristics of this transformation.

sTNFα blockade overcomes trastuzumab resistance. (A) Female nude mice were injected s.c. with 3×106 JIMT-1 cells. When tumors reached~100 mm3, animals were treated twice a week i.p. with 5 mg/kg of IgG, Tz or Eta, 10 mg/kg of DN or the com-bination therapies Tz+Eta or Tz+DN. (B) Tumor volume was monitored routinely for 18 days (n=6–9 per group). (C) At the end of the experiment, tumors were weighed and (D) photographed; scale bar 1 cm and (E) the percentage change in tumor volume was calculated for each tumor with respect to the pre-treatment volume. (F–J) JIMT-1 tumor infiltrating innate immune cell populations were determined by immunofluorescence staining and flow cytometry analysis. (F) Myeloid cells were determined by CD11b staining from total leukocytes (CD45+). (G) Macrophage polarization to the M1 and M2 subtype was determined by CD86 or CD206 staining, respectively, and referred to the total macrophage population. (H) NK cells were identified as the CD3-/CD49b+ population and referred to total leukocytes. (I) NK cell activation and (J) degranulation were determined by staining with anti-CD69 and anti-CD107a respectively, on the NK cell population. (K) Immunoblot of MUC4 from representative tumor lysates are shown. β-tubulin was used as loading control. Quantification of MUC4/β-tubulin ratio is shown below the panel. Data are presented as mean±SEM and represent one of two independent experiments (B–D, F–J). P values were calculated using two-way ANOVA test (B) or by one-way ANOVA coupled with a Tukey post hoc test (C, F–J). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 vs IgG. Tz: trastuzumab, Eta: etanercept, DN: INB03, soluble TNFα dominant negative, ANOVA, analysis of variance.
MUC4 is the main player in sTNFɑ-induced trastuzumab resistance in vivo
Given that sTNFα may be driving trastuzumab resistance, we evaluated the precise contribution of MUC4 expression in this scenario. We engineered JIMT-1 and KPL-4 cell lines to stably express a doxycycline (Dox)-inducible shRNA targeting human MUC4 and obtained JIMT-1-shMUC4 and KPL-4-shMUC4 cells. Both cell lines were transduced with the corresponding Control shRNA (JIMT-1-shControl and KPL-4-shControl). Western blots showed that MUC4 downregulation was achieved by Dox induction in both cell lines (online supplemental figure S1A). First, we examined the effect of MUC4 silencing on JIMT-1 and KPL-4 cell proliferation in vitro. We found that trastuzumab alone was able to inhibit proliferation when cells were silenced for MUC4 expression, and the addition of TNFɑ blocking drugs did not further enhance this effect (online supplemental figure S1B-E). In contrast, in the absence of Dox, both cell lines remained resistant to trastuzumab and only the combination of trastuzumab and TNFα blocking drugs was able to decrease their proliferation (online supplemental figure S1B-E). Similar results were also obtained with JIMT-1-shControl and KPL-4-shControl cells regardless of Dox addition (online supplemental figure 1F,G), and with the parental lines (online supplemental figure 1H,I).
Next, to evaluate the role of MUC4 expression in TNFα-induced trastuzumab resistance in vivo, we established JIMT-1-shMUC4 and KPL-4-shMUC4 tumors in female nude mice (figure 2A). Animals were divided into two groups which were administered or not with Dox, and treated as described before (figure 1A). On MUC4 silencing, trastuzumab treatment alone was able to reduce JIMT-1-shMUC4 and KPL-4-shMUC4 tumor growth at comparable levels to its combination with TNFα blocking drugs with respect to IgG (figure 2B,C and online supplemental tables S2 and S3). Tumor growth inhibition in the absence of Dox was observed only with the combined regimes, when compared with the IgG group (figure 2B,C and online supplemental tables S2 and S3), in agreement with our previous findings (figure 1B). JIMT-1-shControl tumors behaved similarly to JIMT-1 parental tumors, regardless of Dox administration (online supplemental figure S2A,B and figure 1B, respectively). Noteworthy, in tumors silenced for MUC4 expression, DN treatment alone partially inhibited tumor growth (figure 2B,C). We confirmed MUC4 silencing in vivo through Western blot of the harvested tumors (figure 2D,E). These results are the first straightforward in vivo demonstration that MUC4 is a crucial mediator of TNFα-induced trastuzumab resistance. We proved that neutralizing both TNFɑ isoforms or just the sTNFɑ rendered similar antitumor results and did not further enhance the therapeutic effect of trastuzumab in MUC4-silenced tumors.

MUC4 plays a key role in TNFɑ-induced trastuzumab resistance in vivo. JIMT-1 and KPL-4 cells were transduced with lentiviral particles carrying a doxycycline (Dox)-inducible MUC4 shRNA as described in Materials and Methods. (A) Female nude mice were injected s.c. with 3×106 JIMT-1-shMUC4 or 5×106 KPL-4-shMUC4 cells, respectively. When tumors reached~100 mm3, animals were randomly assigned to the control group (-Dox) or the induced group (+Dox), and treated twice a week i.p. with 5 mg/kg of IgG, Tz or Eta, 10 mg/kg of DN or the combination therapies Tz+Eta or Tz+DN. (B–C) Tumor volume was monitored routinely for 22 (B) or 18 (C) days (n=5–7 per group). All data shown are representative of two independent experiments. (D–E) The efficiency of the shRNA was determined by an immunoblot analysis of MUC4 from representative tumor lysates, ran and developed simultaneously. β-tubulin was used as a loading control. Quantification of MUC4/β-tubulin ratio is shown below the panel. Data are presented as mean±SEM P values were calculated by one-way ANOVA coupled with a Tukey post hoc test (B–C). *p<0.05, ***p<0.001, ****p<0.0001 vs IgG. ANOVA, analysis of variance.
MUC4 expression generates an immunosuppressive TME
To investigate the impact of MUC4 expression on the innate immune response triggered by trastuzumab alone or in combination with DN, we studied the infiltrating innate immune cells in JIMT-1-shMUC4 and KPL-4-shMUC4 tumor bed, with or without Dox induction. Regardless of MUC4 expression, we observed a general decrease in myeloid cell infiltration in the TME of tumors treated with trastuzumab plus DN, as well as with trastuzumab alone in the MUC4-silenced groups (figure 3A). Moreover, in the absence of Dox both tumor types revealed a reduced infiltration of myeloid-derived suppressor cells (MDSCs) under the combined therapy (figure 3B). Strikingly, when MUC4 was silenced in JIMT-1-shMUC4 tumors, MDSCs decreased regardless of the administered treatment. However, in KPL-4-shMUC4 tumors this effect was only observed in animals treated with trastuzumab alone or combined with DN (figure 3B).
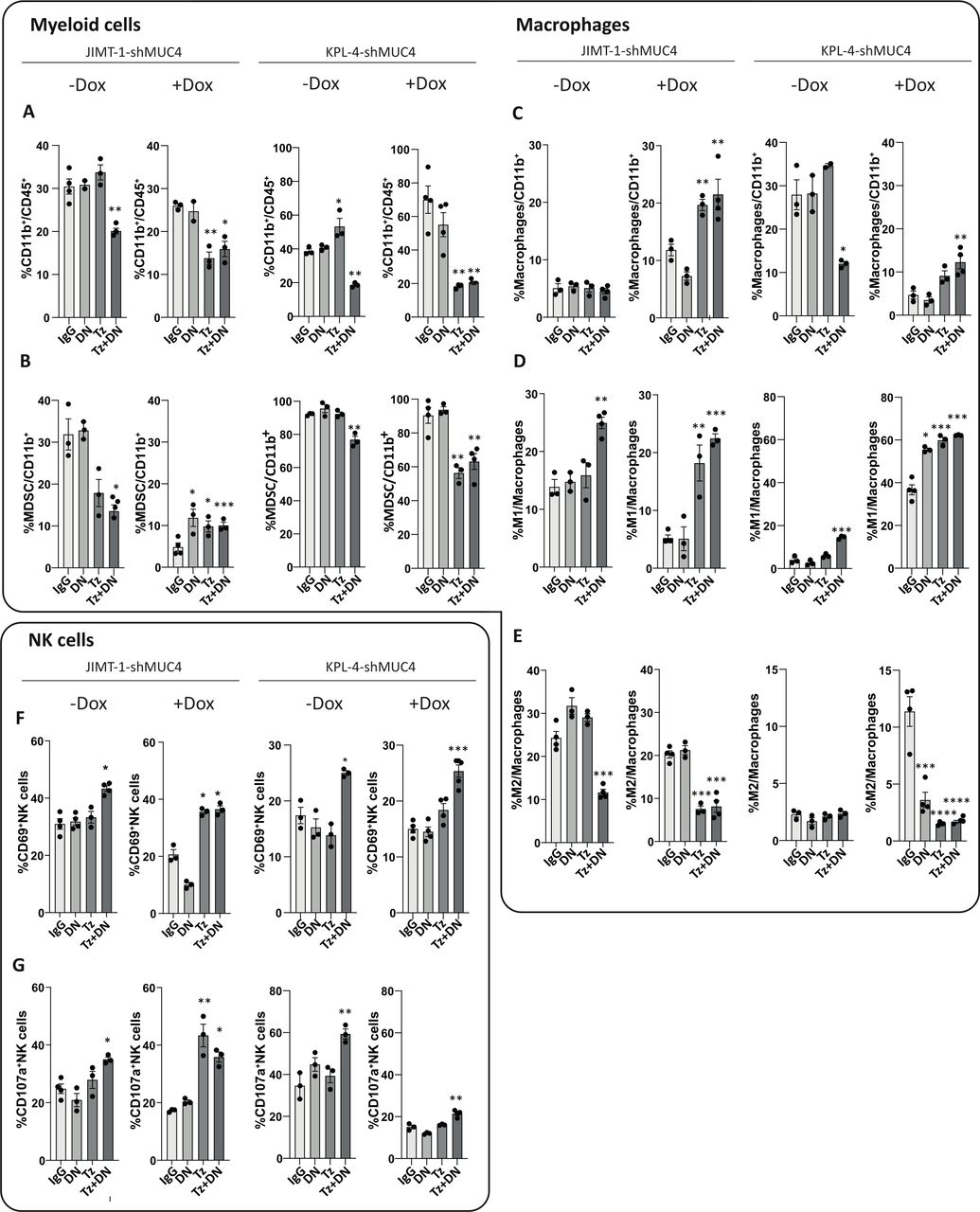
MUC4 expression generates an immunosuppressive tumor microenvironment. JIMT-1-shMUC4 and KPL-4-sh-MUC4 tumor infiltrating innate immune cell populations from the control group (-Dox) or the induced group (+Dox) were determined by immunofluorescence staining and flow cytometry analysis at day 22 (for JIMT-1-shMUC4) or 18 (for KPL-4-shMUC4). (A) Myeloid cells were determined by CD11b staining from total leukocytes. (B) MDSCs were determined by anti-Ly6C+ staining in the CD11b+ gate. (C) Total macrophages were determined by F4/80 staining and referred to total CD11b+ leukocytes. (D) Macrophage polarization to M1 or (E) M2 subtype were determined by CD86 or CD206 staining respectively, and referred to the total macrophage population. (F) NK cell activation and (G) degranulation were determined by staining with anti-CD69 and anti-CD107a respectively, on the NK cell population. Data are presented as means±SEM All data shown are representative of two independent experiments. P values were calculated by one-way ANOVA coupled with a Tukey post hoc test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 vs IgG. ANOVA, analysis of variance.
While total macrophages showed no changes among treatments in JIMT-1shMUC4 tumors in the absence of Dox, a decrease was observed in the combined therapy group in KPL-4-shMUC4 tumors (figure 3C). In addition, an increase in the macrophage M1-like subtype was observed on treatment with the combined therapy with respect to IgG in both JIMT-1-shMUC4 and KPL-4-shMUC4 tumors, regardless of Dox administration (figure 3D). In a MUC4-silenced scenario, an increase in M1-like and a decrease in M2-like macrophages were evident in both tumor types, either under trastuzumab regime alone or administered in combination with DN (figure 3D,E). Notably, in Dox(-) induced KPL-4-shMUC4 tumors treated with DN alone, a polarization of macrophages to an M1-like subtype and a decrease in the M2-subtype was also observed (figure 3D,E). These results go in hand with the remarkable antitumor effect shown before (figure 2C). Finally, in both tumor models, mice receiving the combined therapy exhibited an increase in NK cell activation and degranulation, regardless of MUC4 silencing (figure 3F,G). These changes were also evident in Dox-induced JIMT-1-shMUC4 tumors under trastuzumab treatment alone (figure 3F,G). Similar results on all the above-mentioned innate immune cell populations were obtained with etanercept (data not shown).
Cumulatively, these data reveal that MUC4 expression favors an immunosuppressive tumor milieu in the context of trastuzumab treatment. When trastuzumab accomplishes its antitumor effect, either after downregulation of MUC4 by sTNFɑ blockade or by silencing MUC4 using a shRNA, a TME remodeling proceeds. This remodeling is characterized by a strong polarization of macrophages to the M1-like subtype, a decrease in myeloid infiltration and by a potent activation and degranulation of NK cells, all of them probably promoting an effective antitumor innate immune response.
Macrophages are necessary for trastuzumab antitumor effect in the absence of MUC4 expression
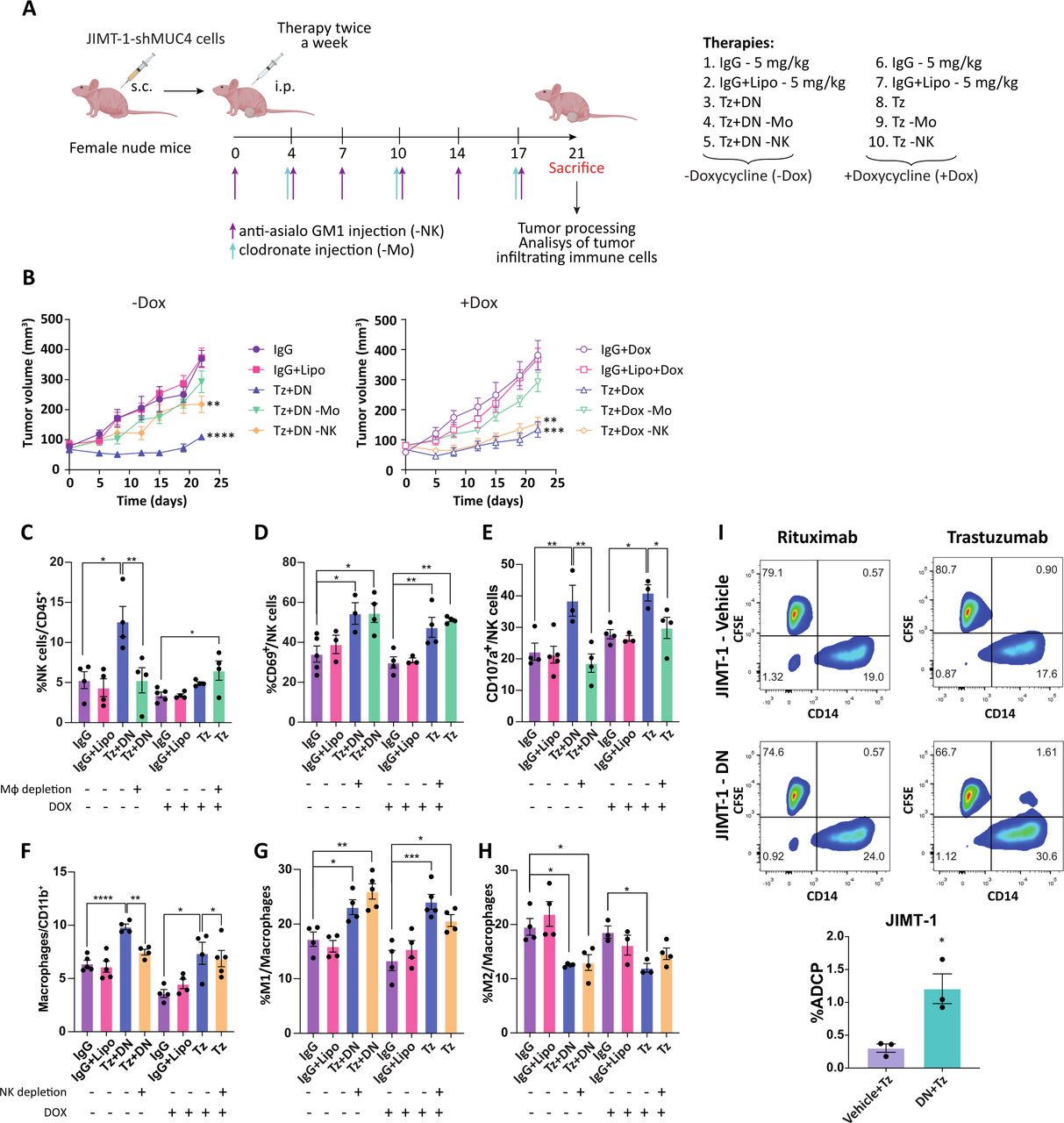
Taking into account our results, which point out that macrophages and NK cells are the possible drivers of the antitumor innate immune response, we evaluated their contribution to the effect of trastuzumab alone in MUC4-silenced tumors, or in combination with DN in the absence of Dox. When JIMT-1-shMUC4 tumors were established, we depleted NK cells with the anti asialo-GM1 antibody or macrophages with liposomes-containing clodronate (figure 4A). In the absence of Dox, the antitumor effect of the combination therapy was blunted when macrophages were depleted, and only partially impaired in the case of NK cells depletion (figure 4B). Moreover, we observed that removing macrophages from the TME restricted NK cell recruitment and degranulation but had no effect on their activation (figure 4C–E). Likewise, NK cell depletion prevented recruitment of macrophages to the tumor bed but did not affect their polarization to the M1-like phenotype (figure 4F–H). When MUC4 was silenced, trastuzumab antitumor effect was lost by macrophage depletion, but it was preserved when NK cells were absent (figure 4B). While exploring the changes in the infiltrating leukocytes in a MUC4 silenced context, we found that NK cells were barely recruited to the tumor bed in the presence of trastuzumab (figure 4C). Again, in the absence of macrophages, NK cells were able to achieve activation but not degranulation (figure 4D,E). In contrast, NK cells depletion did not modify either macrophage recruitment or polarization to the M1-like phenotype (figure 4F–H). Macrophage and NK cell depletion efficacy is shown in online supplemental figure S3. These results suggest that MUC4 expression prevents trastuzumab-induced macrophage recruitment and its antitumor effect, while sTNFα blockade allows a cooperative cross-talk between NK cells and macrophages, thus achieving an effective antitumor innate immune response mediated by trastuzumab.

Macrophages are necessary for trastuzumab antitumor effect in the absence of MUC4 expression. (A) 3×106 JIMT-1-sh-MUC4 cells were injected s.c. in female nude mice. When tumors reached~100 mm3, animals were randomly assigned to the control group (-Dox) or the induced group (+Dox), and treated twice a week i.p. with 5 mg/kg of IgG or Tz for the+Dox group or the combined therapy Tz+DN (5 mg/kg Tz, 10 mg/kg DN) for the -Dox group. To deplete macrophages, lysosomes containing encapsulated clodronate were administered once a week i.p. (green arrows). Empty lysosomes were administered once a week i.p. to mice treated with IgG to serve as a control group. To deplete NK cells, an anti-asialo GM1 antibody was administered twice a week i.p. (purple arrows). (B) Tumor volume was monitored routinely for 21 days (n=5–7 per group). Tumor infiltrating innate immune cell populations from each experimental group were determined by immunofluorescence staining and flow cytometry analysis. (C) NK cells were identified as CD3-/CD49+ population and referred to total leukocytes. (D) NK cell activation and (E) degranulation were deter-mined by staining with anti-CD69 and anti-CD107a respectively, on the NK cell population. (F) Total macrophages were determined by F4/80 staining and referred to total CD11b+ leukocytes. (G) Macrophage polarization to M1 or (H) M2 subtype were determined by CD86 or CD206 staining respectively, and referred to total macrophage population. (I) ADCP was performed using JIMT-1 cells pre-cultured for 48 hours with DN or vehicle and then labeled with CFSE. JIMT-1 cells were incubated with either rituximab or trastuzumab and then co-cultured with human macrophages. The percentage of phagocytosis was calculated as the population of phagocytized macrophages among the total macrophages, shown in the bar graph. Data are presented as means±SEM P values were calculated by one-way ANOVA coupled with a Tukey post hoc test (B-H) or by t-test (I). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance.
As macrophages are essential for the antitumor immune response when trastuzumab is combined with DN (figure 4B), we wanted to address macrophages capacity to perform ADCP in vitro in this setting. JIMT-1 cells were treated or not with DN for 48 hours and then co-cultured with trastuzumab and human macrophages. figure 4I shows that macrophages exhibited increased trastuzumab-mediated ADCP in DN-treated JIMT-1 cells. These results highlight the key role of sTNFα blockade in tumor cells to enhance trastuzumab-dependent cellular phagocytosis.
MUC4 is correlated with poor lymphocyte infiltration in HER2+ breast cancer
It is known that the presence of TILs in HER2+breast cancer correlates with better trastuzumab outcome.12–14 To address the clinical relevance of our findings, we analyzed TILs and MUC4 expression by H&E staining and immunohistochemistry respectively, in a cohort of 91 patients with HER2+ breast cancer treated with adjuvant or neoadjuvant trastuzumab. Patients’ clinico-pathological characteristics are shown in online supplemental table S4. We confirmed that MUC4 expression is associated with poor outcome in patients receiving adjuvant trastuzumab in this cohort (online supplemental figure S4A), as we described before.16 Furthermore, we found that MUC4 expression is also associated with poor prognosis for patients treated with trastuzumab in the neoadjuvant setting (online supplemental figure S4B). When scoring TILs infiltration in terms of absence (<10%), low (10%–30%), intermediate (30%–50%) and high presence (>50%), we observed that the majority of MUC4 positive tumors exhibit either absence or low TILs (figure 5A). At diagnosis, tumors exhibited an inverse correlation between presence of TILs (>50% or >30%) and MUC4 expression (figure 5B). In line with this result, figure 5C shows representative cases of tumors with MUC4 positive staining and scarce TILs, and MUC4 negative staining with abundant TILs. To validate our findings, we took advantage of the MIXTURE immune-cell-type deconvolution algorithm27 with the LM22 molecular signature, to infer the immune cell types proportion from RNA-seq data derived from The Cancer Genome Atlas Breast Invasive Carcinoma (TCGA-BRCA) dataset. We studied the immune infiltrate according to MUC4 low and high expression (defined by the median expression of MUC4) in 119 primary tumor HER2-enriched samples confidently assigned by PAM50 using the PBCMC algorithm.28 We observed that high levels of CD4+ activated memory T cells and gamma delta (γδ) T cells were associated with low MUC4 expression (figure 5D). The rest of the immune cell populations showed no correlation with MUC4 levels (online supplemental figure S5). Considering the key role of M1-like macrophages in the antitumor immune response described before (figure 4), we performed the analysis of M1 and M2 signatures according to high or low TNFα levels, and stratifying the samples according to high or low MUC4 expression. In the ‘MUC4 high’ scenario, the recruitment of M0 is impaired when TNFα levels are elevated, and no polarization is induced regardless of TNFα expression. However, in the ‘MUC4 low’ scenario, macrophages are prone to differentiate to the M1 subtype and not to the M2 subtype in the presence of high TNFα levels (online supplemental figure S6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MUC4 impairs human T cell recruitment in HER2+breast cancer. A total of 91 HER2+breast cancer specimens were analyzed for MUC4 expression by inmmunohistochemistry (IHC) and for the presence of TILs at diagnosis. (A) Graph showing MUC4 negative and MUC4 positive samples according to their TILs percentage. (B) Correlation between MUC4 expression and TILs with cut-offs of 30% and 50%. *χ2 test, **Fisher's exact test. (C) Representative cases of HER2+breast cancer with MUC4 negative expression and abundant TILs (case 1) and MUC4 positive expression and poor TILs (case 2). Scale bar 20 µm. TILs are marked with black arrows. (D) CD4+ activated memory T cells and gamma-delta T cells from HER2-enriched samples of TCGA-BRCA were analyzed via MIXTURE algorithm according to the median of MUC4 expression using Wilcoxon test. (E) Activated human T-lymphocytes were exposed to conditioned mediums from JIMT-1-shMUC4 and KPL-4-sh-MUC4 cells with (-Dox) or without (+Dox) MUC4 expression. Conditioned mediums from shControl cells served as a control. Data are presented as mean±SEM and are representative of two independent experiments. P values were calculated by one-way ANOVA coupled with a Tukey post hoc test. *p<0.05, **p<0.01, ***p<0.001 vs shControl (-Dox). (E) Working model. TNFα-induced MUC4 expression prevents trastuzumab binding to HER2 and promotes an immunosuppressive TME with abundant MDSC and M2 macrophages. TNFα blockade with DN downregulates MUC4 expression, unmasking trastuzumab epitope on HER2 and promotes macrophage polarization to M1, ADCP, ADCC and recruits TILs. ANOVA, analysis of variance.
We also confirmed the suppressive role of MUC4 expression in the differentiation of T cells to the central memory phenotype via the culture of human T cells with conditioned medium of JIMT-1-shMUC4 and KPL-4-shMUC4 or the shControl counterparts. Knockdown of MUC4 expression in tumor cells produced a secretome able to enhance CD3/CD28-activated T cells differentiation to central memory CD4+ and CD8+ T cells (figure 5E). Taken as a whole, these data reinforce the idea that MUC4 plays an important role in tumor immune evasion and suggests that its expression in HER2+ breast cancer is associated with immunologically ‘cold’ tumors and an immunotolerant TME.
Discussion
In this work, we uncovered a new layer of complexity on the role that MUC4 has in driving trastuzumab resistance in HER2+ breast cancer: not only it shields the trastuzumab epitope on the HER2 molecule but also promotes tumor immune evasion, enhancing myeloid recruitment to the TME and, under trastuzumab treatment, preventing the polarization of macrophages to the antitumoral M1-like phenotype. In this way, the presence of MUC4 in tumor cells orchestrates an immunosuppressive TME. Using samples from HER2+ breast cancer patients, we demonstrated that MUC4 expression is associated with low TILs, the so-called immunologically ‘cold’ tumors. In addition, we demonstrated that MUC4 is the central player of TNFα-induced trastuzumab resistance in HER2+ breast cancer, and that sTNFα isoform blockade is able to tackle MUC4 expression that, together with trastuzumab, triggers an effective antitumor immune response in preclinical models (figure 5E).
Previously, we reported that TNFα induces MUC4 expression, which drives trastuzumab resistance by masking the trastuzumab epitope on the HER2 molecule.16 Our in vitro findings have shown that trastuzumab, in the trastuzumab resistant models JIMT-1 and KPL-4, the monoclonal antibody decreases cell proliferation when MUC4 is downregulated, either by shRNA against MUC4 or by sTNFα neutralization. This reduction in cell proliferation could be caused by the ability of trastuzumab to block HER2 signaling pathways. However, the antitumor effect of trastuzumab in a MUC4-silenced scenario in vivo is much more complex, as it is dependent on macrophage recruitment to the tumor bed. Particularly, when MUC4 was downregulated by sTNFα blockade, both macrophage and NK cell infiltration were necessary for recruiting each other, and for achieving trastuzumab efficacy as well. This finding suggests a cooperation between NK cells and macrophages that occurs when sTNFα is present at low-to-null concentrations. This poses an explanation for trastuzumab sensitivity in HER2+breast cancer, where scarce TNFα production and poor MUC4 expression occur. The importance of trastuzumab-induced immune response on its antitumor effect has already been reported. For example, studies performed with the Fcab H10-03-6 antibody, which recognizes the trastuzumab epitope but does not inhibit HER2 signaling, demonstrated its ability to curb BT-474 tumor growth.29 In addition, it is known that trastuzumab induces ADCC through its binding to CD16 on NK cells.6 Moreover, Park et al demonstrated that both an innate and adaptive immune response are necessary for the proper inhibition of tumor growth.11 They demonstrated that cytokines and/or danger signals are released from NK cells activated by trastuzumab. This, in turn, would be able to increase cross-priming and activation of dendritic cells in both mice and humans.30 31 Taking this evidence into account, we highlight the relevance of our results neutralizing only sTNFα and thus preserving NK cell-dendritic cell interaction promoted by tmTNFα.23
In MUC4+ tumors treated with trastuzumab plus DN, MUC4 is downregulated allowing trastuzumab binding. In this scenario, macrophage recruitment is necessary for achieving NK cells infiltration and degranulation. However, NK cells are not necessary to polarize macrophages to the M1-like subtype, which would be more likely to perform trastuzumab-mediated ADCP. It is known that after ADCP, macrophages undergo a phenotypic modification toward an immunosuppressive phenotype by the upregulation of the immune checkpoints PD-L1 and B7-H4, inhibiting NK cell-mediated ADCC.32 In particular, seminal work of Lim et al demonstrated that TNFα is able to induce overexpression of PD-L1 in tumor cells by stabilization of PD-L1 protein by upregulation of COP9 signalosome 5, which prevents its ubiquitination and degradation.33 In addition, tumor-associated macrophages have higher levels of PD-L1 than circulating monocytes due to the presence of TNFα in the tumor milieu.34 We speculate that sTNFα blockade impairs the expression of the above-mentioned inhibitory immune checkpoints, allowing NK cell infiltration and the induction of ADCC. The results obtained in MUC4 silenced tumors, where only macrophages are recruited, suggest that the presence of sTNFα compromises NK cell infiltration and degranulation, forcing the antitumor effect to depend only on the ADCP by the recruited macrophages.6–8 Tumor-associated macrophages are frequently related to poor survival in several solid tumor types, as they are immersed in an immunosuppressive TME.35 36 In this regard, there are immunotherapy approaches that either pursue to reduce the number of tumor-associated macrophages or to reprogram them toward an antitumor phenotype.37 In our case, we took advantage of the macrophages intrinsic plasticity38 and, by blocking sTNFα, we achieved polarization to an M1-like phenotype.
ADCP recently emerged as an important mechanism of trastuzumab antitumor effect.6–9 In our work, we have demonstrated that sTNFα blockade and consequently MUC4 downregulation, enhances ADCP. First, we have shown that trastuzumab-resistant cells pretreated with DN were more prone to be engulfed by macrophages (figure 4I). These findings are in line with our previous data demonstrating that MUC4 downregulation through TNFα blockade increases trastuzumab binding on tumor cells, which were more susceptible to ADCC exerted by NK cells.16 This evidence highlights the importance of MUC4 downregulation in allowing NK cells cytotoxic activity and macrophages phagocytic effect in the presence of trastuzumab. Second, MDSC recruitment was reduced on MUC4 downregulation. MDSCs express FcγRIIB, which is further upregulated when these cells infiltrate tumors. FcγRIIB is an inhibitor of ADCP and it is required for MDSCs differentiation and immunosuppressive activity.39 Finally, our results show that increased polarization to M1-like subtype in the tumor tissue was associated with an enhanced trastuzumab efficacy, either in the MUC4-silenced tumor or in the ones treated with DN. In both scenarios, macrophage depletion resulted in abolishment of the antitumor efficacy that trastuzumab provides. Additionally, M1-like macrophages elicit a more efficient trastuzumab ADCP than the M2-like subtype.8 40 These findings are in line with others that demonstrated the central role of macrophage presence to achieve the antitumor effect of trastuzumab in several preclinical models.8 Particularly, Tsao demonstrated that the antitumor effect of trastuzumab relies strongly on ADCP, and a complete tumor regression can be achieved when trastuzumab is administered with antibodies against CD47, a ‘do not eat me’ signal expressed in tumor cells that impairs macrophage phagocytosis.41 Tumor cell phagocytosis by macrophages is able to boost the priming of tumor-specific CD4+ and CD8+ T cells.42 43 We; therefore, speculate that the higher proportion of M1-like macrophages in tumors treated with trastuzumab plus DN will be capable of triggering an effective adaptive immune response. Trastuzumab antitumor effect relies on both innate and adaptive immune response. One limitation of our study concerns the fact that only the innate immune response against tumors can be analyzed by the nude mouse model. However, it is worth noting that our experiments were performed during the time frame where the innate, but not the adaptive immunity exerts tumor growth inhibition in immunocompetent mice (18–21 days of treatment).11 We speculate that in a HER2+/MUC4+TME treated with trastuzumab and DN infiltrated with antitumor innate immune cells (figure 1), sTNFα neutralization would not affect costimulatory signals for NK cells and dendritic cells, nor impact on CD8+ T cell development and action, since tmTNFα functions are being preserved.7 24 Future studies using humanized mice will shed light on the benefits of sTNFɑ blockade on T cell recruitment to the tumor bed.
Our interpretation of the in silico results is that, in the MUC4 high scenario, the abundance of sTNFα is inducing MUC4 expression. This reinforces our proposal that MUC4+/HER2+ breast cancer patients should undergo sTNFα blockade to downregulate MUC4 expression, rendering in a TME suitable for M1-like macrophage differentiation. On the other hand, in the MUC4 low scenario, we speculate that the high levels of TNFα are due to high expression of tmTNFα, which can help macrophages to differentiate to the M1-like subtype.
Currently, HER2+ breast cancer is treated mainly with trastuzumab, its biosimilars, or trastuzumab-based antibody-drug conjugates. As we have previously demonstrated, T-DM1 (trastuzumab-emtansine) effectiveness is also impaired by MUC4 expression in tumor cells.16 Therefore, we envision that the administration of TNFα blocking agents in the clinical setting will be able to enhance the effect of trastuzumab-based conjugates, such as T-DM1 and trastuzumab-deruxtecan, antitumor effects.44
Immune checkpoint inhibitors (ICI) have been recently introduced for triple-negative breast cancer treatment, and several trials are now running for HER2+ breast cancer. The current body of evidence points out that MDSCs, M2-like macrophages and poor TILs within the TME are associated with failure of ICI treatment.37 The combination of TNFα antagonists with ICI is currently an active topic of research. Seminal work of Melero and coworkers demonstrated that TNFα blockade not only prevents immune-related adverse effects (irAEs) of ICI, but also enhances the antitumor immune response.45 The administration of the anti-TNFα antibody infliximab is part of the ASCO guidelines to treat irAEs produced by ICI.46 The TICIMEL trial (NTC03293784) is an open-label, two-arm phase Ib clinical trial in which stage IIIc and IV melanoma patients were treated with nivolumab and ipilimumab (3 mg/kg) combined with the TNFα-blocking agents infliximab or certolizumab. This trial proved the safety of both combinations and high response rate in the certolizumab-treated cohort.47 In addition, several phase I or II clinical trials reported the use of anti-TNFα antibodies in numerous types of cancers such as pancreatic, breast, chronic lymphocytic leukemia and small lymphocytic lymphomas, demonstrating their safety.48–50 Here, we proved that MUC4 expression is associated with immunologically ‘cold’ tumors. Since these tumors are the ones recognized for being poorly responsive to ICI, sTNFα blockade could be a feasible therapeutic tool to tackle the immunosuppressive TME and allow an effective ICI response in HER2+breast cancer. In conclusion, our work reveals an unexpected role of sTNFα blockade in the TME when trastuzumab is present, involving MUC4 downregulation in tumor cells and a tumor immune microenvironment remodeling. In this scenario, MDSCs recruitment is decreased, and macrophages are polarized to an M1-like phenotype, presumably presenting reduced PD-L1 and B7-H4 expression, which favors ADCP and their cooperation with NK cells. Our results provide proof-of-concept for stratifying HER2+ tumors according to MUC4 expression, and a strong preclinical rationale to pursue sTNFα blockade combined with HER2-targeted standard of care therapies for treatment of HER2+/MUC4+ breast cancer patients. Evidence of phase 1b open label dose escalation trial with DN (NCT03943264) showed its safety and tolerance.51 Therefore, the administration of trastuzumab or trastuzumab-based therapies with DN, warrants further investigation in clinical trials.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
Patient samples were collected with the patient’s informed consent and with Helsinki approval from Hospital Fernández (CEI # 201629) and Henry Moore Institute of Oncology, (Buenos Aires, Argentina) and from Hospital Oncológico Provincial de Córdoba (Cordoba, Argentina). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
In memory of EH Charreau, our mentor and colleague. We thank A Molinolo (UCSD, USA) for his constant help and support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors RS and SB conceived and designed the experiments. SB, FM, MFM, RS performed the experiments and acquired the data. SB, FM, RICR, MAR, GI, AD, DR, EGD, DLDV, SB, SF, DL, AF, MCS, VG, GL, CL, FA, TC, MFM and RS analyzed and interpreted the data. EAF and DR performed biostatistics analysis. SB prepared the figures. SB, MFM and RS wrote the original draft manuscript. SB, FM, CJP, RICR, PVE, MFM and RS revised the manuscript. RS supervised the study and acts as the guarantor responsible for the overall content of this article. All authors read and approved the final manuscript.
Funding This work was supported by: The National Agency of Scientific Promotion of Argentina IDB/PICT 2017-1517, PICT 2018-2086, PICT aplicación intensiva 2021-023 awarded to RS. IDB/PICT 2015-1587, IDB/PICT 2017-1072, and PICT 2019-0288, awarded to PVE. IDB/PICT 2017-0419 and PICT 2020-2315 awarded to RICR. The National Cancer Institute of Argentina INC 2018-2020 research grant, awarded to RS and INC 2016 research grant, awarded to PVE. The Florencio Fiorini Foundation Research Grant awarded to MFM and AD. The Alberto J. Roemmers Foundation Research Grants awarded to RICR and to RS.
Competing interests RS is a consultant for and a research grant from INmune Bio. All other authors declare they have no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.