Article Text

Abstract
Background Mitochondrial transcription factor A (TFAM) is a transcription factor that maintains mitochondrial DNA (mtDNA) stabilization and initiates mtDNA replication. However, little is known about the immune regulation function and TFAM expression in immune cells in the tumors.
Methods Mouse tumor models were applied to analyze the effect of TFAM deficiency in myeloid cell lineage on tumor progression and tumor microenvironment (TME) modification. In vitro, primary mouse bone marrow-derived dendritic cells (BMDCs) were used in the investigation of the altered function and the activated pathway. OVA was used as the model antigen to validate the activation of immune responses in vivo. STING inhibitors were used to confirm the STING activation provoked by Tfam deficient in DCs.
Results The deletion of TFAM in DCs led to mitochondrial dysfunction and mtDNA cytosolic leakage resulting in the cGAS-STING pathway activation in DCs, which contributed to the enhanced antigen presentation. The deletion of TFAM in DCs has interestingly reversed the immune suppressive TME and inhibited tumor growth and metastasis in tumor models.
Conclusions We have revealed that TFAM knockout in DCs ameliorated immune-suppressive microenvironment in tumors through STING pathway. Our work suggests that specific TFAM knockout in DCs might be a compelling strategy for designing novel immunotherapy methods in the future.
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as online supplemental information. Data are available on reasonable request. The RNA-seq data are available in the Gene Expression Omnibus (GEO) dataset with the accession number GSE223950. All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The mitochondrial transcription factor A (TFAM) is the most abundant protein associated with mitochondrial DNA (mtDNA) encoded by nuclear genes. This protein initiates mtDNA transcription by recruiting mitochondrial RNA polymerase to the promoter. Moreover, TFAM is indispensable for maintaining mtDNA structure. Previous studies on TFAM in tumor models mainly focused on its effect on metabolism of the tumor cells and how it affects their proliferation and invasion.
WHAT THIS STUDY ADDS
In this study, we found that Tfam deficiency in myeloid cells led to promoted activation of the antigen-presenting cells. We demonstrated that TFAM modulated activation and function of dendritic cells (DCs). The enhanced antitumor humoral and cellular immunity were provoked by Tfam deficient DCs through cGAS-STING pathway elicited by mtDNA leakage. Additionally, the suppressed tumor growth and transformed immune microenvironment were characterized in myeloid-specific TFAM knockout mice.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our findings highlighted the essential role of TFAM in balancing immunity and metabolism in immune cells, and partly elucidated the distinct roles of TFAM in different cell types, suggesting that TFAM is a potential candidate target in DCs for tumor immunotherapy. Furthermore, we also complemented the metabolic reprogramming pathways of immune cells in the tumor microenvironment.
Background
The immunosuppressive tumor microenvironment (TME) represents one of the main reasons for the poor outcome of immunotherapy against cancer. Hence, exploring efficient methods to activate immune cells in immunosuppressive environment is a crucial task for the development of better immunotherapies. Dendritic cells (DCs) are a heterogeneous group of specialized antigen-presenting cells that play a vital role in initiating and regulating innate and adaptive immune responses.1 In the TME, DCs acquire, process, and present tumor-associated antigens on MHC (major histocompatibility complex) molecules, and provide co-stimulation and soluble factors to elicit T cell responses.2 In clinical settings, many therapy strategies have been developed to target DCs, such as delivering antigens, mobilizing and activating endogenous DCs using stimulant adjuvants, and generating DC-based vaccines.3 4 Nevertheless, to achieve more effective antitumor immunotherapy, strategies to accurately target DC and avoid immune tolerance of DC in the TME are needed.
In recent years, considerable attention has been attracted by the critical roles of mitochondria within the TME, including metabolism-regulation and immune-activation. On the one hand, mitochondria participate in the regulations of cellular metabolism, which produces reactive oxygen species (ROS). It has been well documented that ROS is an important factor affecting the proliferation, metastasis, and stemness of tumor cells.5 On the other hand, mitochondria could activate immune response through damage-associated molecular patterns (DAMPs). According to ‘endosymbiont hypothesis’, it is very likely that mitochondria are evolved from aerobic prokaryotes.6 They possess many features of their bacterial ancestors, including a circular genome containing CpG DNA and the ability to form N-formyl peptides.7 It has been reported that mitochondrial components could efflux into the cytoplasm or extracellular space in specific conditions to be recognized as DAMPs by the corresponding pattern recognition receptors.8 During the process, circular genome containing CpG DNA plays key role in the activation of immune response. Compelling evidence suggests that the release of mitochondrial DNA (mtDNA) from necrotic cells activates the NF-κB signaling pathway and stimulates expression of other pro-inflammatory genes by interacting with the neutrophil TLR9 receptor, resulting in a corresponding inflammatory response in vitro.9 Besides, our previous work has demonstrated that the oxidized mtDNA from tumor cells could activate STING signaling and then induce the tumor specific immune response.10
Most mitochondrial proteins are encoded by nuclear DNA, translated in the cytoplasm, and then transported to the corresponding functional sites in the mitochondria.11 The mitochondrial transcription factor A (TFAM) is the most abundant protein associated with mtDNA encoded by nuclear genes.12 This protein not only initiates mtDNA transcription and replication but also maintains mtDNA structure.13 Interestingly, the role of TFAM in mitochondria is similar to the role of histones in nucleosome. TFAM wraps mtDNA entirely to form a nucleoid structure14 that may protect mtDNA against ROS.15 Tissue-specific ablation of TFAM has been used to mimic the mitochondrial dysfunction observed in various human diseases.16 17 Previous studies on TFAM in tumor models mainly focused on how it affects metabolism, proliferation, and invasion of tumor cells.18 However, a study to reveal how TFAM in immune cells contributes to remodeling of the TME is absent.
In the current study, we used the myeloid-specific Tfam knockout mice to investigate how TFAM contributes to antitumor immunity and the underlying mechanism. Interestingly, Tfam deficiency in myeloid cells led to preferable activation of DCs. The enhanced antitumor humoral and cellular immunity were provoked by Tfam deficient DCs through cGAS-STING pathway elicited by mtDNA leakage. Additionally, the tumor growth and immune microenvironment were characterized in myeloid-specific Tfam knockout mice. This study may indicate the critical role of TFAM in balancing immunity and metabolism in DCs, which provides more insights for tumor immunotherapy targeting mitochondria.
Methods
Animals
Six-to-eight-week-old C57BL/6 mice were purchased from Beijing Vital River Laboratory Animal Technology Company, and C57BL/6 mice with Tfam-floxed alleles, OT-I, or expressing Cre-lyz2 were purchased from Jackson Lab (Stock#026123, stock#003831 and Stock#004781, respectively). The Tfamflox/flox mice were crossed with transgenic mice expressing Cre-lyz2 as lyz2-Cre/Tfamflox/flox or Tfam-/-. Tfamfl/fl littermates not expressing Cre and wild type (WT) were used as control mice in this study. The mice were housed in a specific pathogen-free environment.
Statistical analysis and reproducibility
All of the experiments (including the western blots) were repeated independently at least twice with similar results. All data were analyzed using one-way analysis of variance (ANOVA), two-way ANOVA or two-sided Student’s t-test (GraphPad InStat Software, California, USA). Log-rank (Mantel-Cox) test was used for survival curves analyses. Results were presented as the means±SEM. The statistic differences were denoted as *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, and p<0.05 was considered significant.
Other details and additional experimental procedures are provided in online supplemental methods.
Supplemental material
Results
Deletion of Tfam in myeloid lineage inhibits lung tumor progression
To investigate the role of TFAM in immune cells of cancer, we deleted TFAM in the myeloid cell lineage by crossbreeding Lyz2-Cre and Tfamfl/fl mice (online supplemental figure 1A,B), referred to as Tfam-/-. Tfamfl/fl littermates not expressing Lyz2-Cre and WT were used as control for the study. Murine pulmonary metastasis models were established by injecting LLC (Lewis lung carcinoma) or B16-F10 cells intravenously. Interestingly, we found that tumor growth and metastasis were significantly reduced in myeloid cell-specific TFAM knockout groups in both models (figure 1A–C; online supplemental figure 1C). Inhibited tumor progression was detected by measuring the lung weight, metastatic area, and metastatic nodules, which resulted in the prolonged survival of Tfam-/- tumor-bearing mice (figure 1C). Previous studies have claimed that the tumor could grow up to a diameter of 2–3 mm without their tumor microvascular system,19 and the deletion of Tfam in myeloid cells significantly reduced the metastatic nodules with diameter >3 mm (online supplemental figure 1D). Moreover, representative immunohistochemical staining of cleaved caspase-3 and CD31 were analyzed to observe tumor cell apoptosis and tumor angiogenesis. The Tfam-/- group showed higher expression of cleaved caspase-3 and lower expression of CD31 in the tumor sections (figure 1D,E; online supplemental figure 1E,F), suggesting more apoptosis tumor cells and fewer numbers of CD31+ tumor vessels compared with the control group. These results revealed that knockout of TFAM in myeloid lineage effectively inhibited tumor growth and prolonged survival in mice.
Supplemental material

Low expression of Tfam in myeloid inhibits lung tumor growth. (A, B) Tfam deletion in myeloid inhibited tumor growth in LLC (A) or B16-F10 (B) lung metastatic tumor models. LLC cells (5×105) or B16-F10 cells (2×105) were intravenously injected into control or Tfam-/- mice to establish experimental pulmonary metastasis models (n=6–9 mice), Tfamfl/fl littermates and wild type were used as control. Mice were sacrificed on day 24 (LLC models) or day 14 (B16-F10 models), and pulmonary physiology was evaluated, including gross images and H&E staining of lung, measurement of metastatic area (n=3 mice’s lungs were paraffin embedded) and nodules (n=6–9 mice). Scale bars represent 2 mm. (C) Tfam deletion in myeloid prolonged the survival of tumor-bearing mice. Survival statistics of mice from LLC (5×105) or B16-F10 (5×105) lung metastatic tumor models (n=10–11 mice). (D, E) Immunohistochemical staining of cleaved caspase-3 (D) or CD31 (E) in lungs of the mice described in (A, B). Scale bars represent 20 µm. Data are represented as mean±SEM. Statistical significance in (A, B) was determined by a two-sided unpaired t-test. Survival curve data in (C) were analyzed by log-rank (Mantel-Cox test). Representative results in (A–C) and pictures in (A, B, D, E) from two independent experiments are shown. *p<0.05, ***p<0.001. LLC, Lewis lung carcinoma.
Tfam deletion activates tumor immune microenvironment with increased lymphocyte infiltration
To further study the effects of Tfam deletion on tumor growth, we next investigated whether and how TFAM deficiency in myeloid lineage would modulate tumor immune microenvironment (TIME). We used flow cytometry (FCM) to analyze the lung tissues from tumor-bearing mice inoculated with LLC cells. DCs are known to critically influence the adaptive immune response against tumors. In the current study, the percentages of DCs in Tfam-/- mice were significantly increased when compared with control group (figure 2A). Interestingly, the infiltration of alveolar macrophages and tumor-associated macrophages (TAMs) was not altered (figure 2B,C). Furthermore, TFAM deficiency didn’t obviously influence the polarization of TAMs (figure 2C). In the meantime, tumor-infiltrating monocytes, as well as Ly6Cmid monocytes, and neutrophils were assessed, and the increased population of the infiltrating innate cells suggested an activated inflammatory immune response (online supplemental figure 2A).20 Furthermore, the efficiency of TFAM knockout in DCs from the TME was also confirmed by FCM, and the TFAM expressions in DCs were dramatically reduced in the lungs of Tfam-/- mice (online supplemental figure 2B).
Supplemental material

TFAM deletion in myeloid transforms tumor immune microenvironment and increases lymphocyte infiltration. (A–F) LLC cells (5×105) were intravenously injected into control or Tfam-/- mice to establish experimental pulmonary metastasis models. Mice were sacrificed on day 24 to collect the lungs. Then the single-cell suspension of the whole lungs with metastatic tumors was prepared and subjected to FCM analysis. (A) Representative scatterplots of the gated DCs (CD45+ CD3- CD11b+ CD11c+ MHCII+ CD24+) are shown in the left panel and quantified in the right panel (n = 4 mice). (B) The percentages of alveolar macrophages (CD45+ CD3- CD11b- CD11c+) are quantified (n = 4 mice). (C) The percentages of TAMs (CD45+ CD3- CD11b+ F4/80+), M1 TAMs (CD45+ CD3- CD11b+ F4/80+ MHCII+), and M2 TAMs (CD45+ CD3- CD11b+ F4/80+ CD206+) are quantified (n = 4 mice). (D) The percentages of CD3+ CD8+ CTLs gated from CD45+ cells (left panel) and activated CD69+ CTLs gated from CD45+ CD3+ CD8+ (right panel) are quantified (n=5 mice). (E) The percentages of GzmB+ or IFN-γ+ T cells are quantified. Cells are gated from CD45+ CD3+ CD8+ subpopulation (n=5 mice). (F) The percentages of CD25+ or PD-1+ T cells are quantified. Cells are gated from CD45+ CD3+ CD4+ subpopulation (n=5 mice). (G, H) Immunofluorescence staining of CD45 (green), CD3 (red), CD8 (green) and DAPI (blue) in lungs of the mice from pulmonary metastasis models of LLC (G) or B16-F10 (H). Scale bars represent 20 µm. (I) B16-F10 cells (2×105) were intravenously injected into control or Tfam-/- mice to establish experimental pulmonary metastasis models. Mice were sacrificed on day 14, then the single cell suspension of the whole lung with metastatic tumor was prepared and subjected to FCM analysis. Representative scatterplots of the p15E-specific CD8+ T cells are shown in the left panel and quantified in the right panel. Cells are gated from CD45+ CD3+ subpopulation (n=4 mice). Data are presented as mean±SEM. Statistical significance in (A–F, I) was determined by a two-sided unpaired t-test. Representative results in (A–F, I) and pictures in (G, H) from two independent experiments are shown. *p<0.05, ***p<0.001, NS, not significant. LLC, Lewis lung carcinoma. FCM, flow cytometry; DC, dendritic cell; MHC, major histocompatibility complex; TAMs, tumor-associated macrophages.
Besides immune myeloid cells, the populations of T cells play dominant roles in activating antitumor immunity. We assessed how TFAM knockout in myeloid cells affects the tumor-infiltrating T cells in LLC lung metastasis models. As shown in figure 2D, the elevation in the percentages of both CD8+ cytotoxic T lymphocyte (CTL) and activated CTLs (CD8+ CD69+ T cells) were observed. Furthermore, Tfam-/- mice also exhibited a significant increase in the percentages of CD8+ IFN-γ+ T cells and CD8+ GzmB+ T cells (figure 2E). Elevated expression of IFN-γ and granzyme B (GzmB) in activated T cells are generally considered as the favorable condition to provoke a more effective antitumor immunity.21 As for T helper cells, CD4+ CD25+ and CD4+ PD-1+ T cells were significantly diminished in Tfam-/- mice when compared with that of the control group as detected (figure 2F). Moreover, immunofluorescence staining of lung sections also confirmed the significant increase of tumor-infiltrating CD45+, CD3+, CD8+ lymphocyte cells in LLC or B16-F10 lung metastasis models (figure 2G,H), which did not undergo apoptosis (online supplemental figure 2C,D). We further investigated whether the CD8+ T cells could specifically recognize p15E, an endogenously expressed antigen in B16-F10. As shown in figure 2I, we found that the induction of p15E-specific CD8+ T cells was significantly increased in Tfam-/- group in B16-F10 lung metastasis tumor models (figure 2I). These results demonstrated that TFAM knockout in myeloid cells might significantly enhance immune cell infiltration and T cell activation, which might result in a reshaped TME for boosting host immune responses to kill tumor cells.
TFAM deficiency activates DCs in vitro and in vivo
To investigate how Tfam deficiency in myeloid linage affects the cell characteristics and how they stimulate TME, we further investigated the effect of TFAM deletion in a population of myeloid-derived cells, especially DCs, which are responsible for the antigen presentation and antitumor immunity activation. It is known that DCs express several surface receptor costimulatory molecules after the exposure to antigens or inflammatory stimulants, which induces the maturation, migration of DCs and enhances the antigen-presenting process to primary T cells in secondary lymphatic organs.22 To simulate the tumor condition in vitro, we used a conditioned medium containing tumor supernatant (TS) of LLC cells to stimulate primary mouse bone marrow-derived DCs (BMDCs), whose phenotypes and TFAM recombination efficiency were analyzed by FCM (online supplemental figure 3A–D). The identified studies indicate that the TS could mediate DC activation.23We found that the expression of costimulatory molecules on DCs including CD40, CD86, and MHCII were significantly elevated in the Tfam-/- group when compared with the control (figure 3A). This process partially relied on nucleic acid, because SuperNuclease repressed the maturation of DCs induced by TS stimulation (online supplemental figure 4A–C). Moreover, cytokine levels such as TNF-α, IL-6, IL-1β, and IL-12 p40 were also heightened in the Tfam-/- mice derived BMDCs after TS stimulation (figure 3B). In addition, TFAM deletion also notably up-regulated the phagocytosis capacity of the BMDCs after TS stimulation as assessed by the uptake of FITC-dextran (figure 3C,D).
Supplemental material
Supplemental material

TFAM deficiency activates DC both in vitro and in vivo. (A) Tfam deficiency promotes the maturation of DCs before or after TS stimulation in vitro. BMDCs from control or Tfam-/- mice were stimulated with or without LLC tumor supernatant (TS) for 24 hours, then subjected to flow cytometry analysis to detect the expression of costimulatory molecules. The upper panel shows the representative histograms of the gated CD11c+ DCs (n = 3 biologically independent samples). The quantitative data of flow cytometry results are shown in the lower panel. (B) Tfam deficiency promotes the secretion of inflammatory cytokines of DCs after TS stimulation for 24 hours in vitro. Levels of TNF-α, IL-6, IL-1β, and IL-12 p40 in the supernatant from BMDCs treated as in (A) were detected by ELISA (n = 3 biologically independent samples). The original levels of cytokines in TS were subtracted. (C) Tfam deficiency promotes the antigen uptake of DCs in vitro. BMDCs were stimulated with or without TS for 24 hours, followed by incubation with 1 mg/mL FITC‐Dextran for 1 hour at 37°C and then analyzed under a fluorescent microscope. Scale bars represent 10 µm. (D) The mean fluorescence intensity (MFI) of CD11c+ BMDCs in (C) was further analyzed by flow cytometry (n = 3 biologically independent samples). The control group was performed by co-culturing BMDCs with FITC-dextran at 4°C. (E) Tfam deficiency promotes the antigen presentation of DCs in vitro. Representative scatterplots of the gated CD8+ T cells from OT-I mice are shown in the left panel and quantified in the right panel. Numbers indicate the percentage of proliferated CFSE-negative CD8+ T cells from OT-I mice (n = 3 biologically independent samples). (F) Tfam deficiency promotes the migration and maturation of DCs in vivo. Single-cell suspension of lymph nodes from control or Tfam-/- mice were subjected to flow cytometry analysis. The percentages of CD197+, MHC II+, CD40+, or CD80+ of CD11c+ DCs are quantified from CD45+ gated subpopulation (n=6 mice). Data are presented as mean±SEM. Statistical significance was determined by two-way ANOVA in (A, B, F) or a two-sided unpaired t-test in (D, E). Representative results in (A, B, D, F) and pictures in (C) from three independent experiments are shown. Representative results in (E) from two independent experiments are shown. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance; BMDCs, bone marrow-derived dendritic cells; MHC, major histocompatibility complex. CFSE, carboxyfluorescein succinimidyl ester.
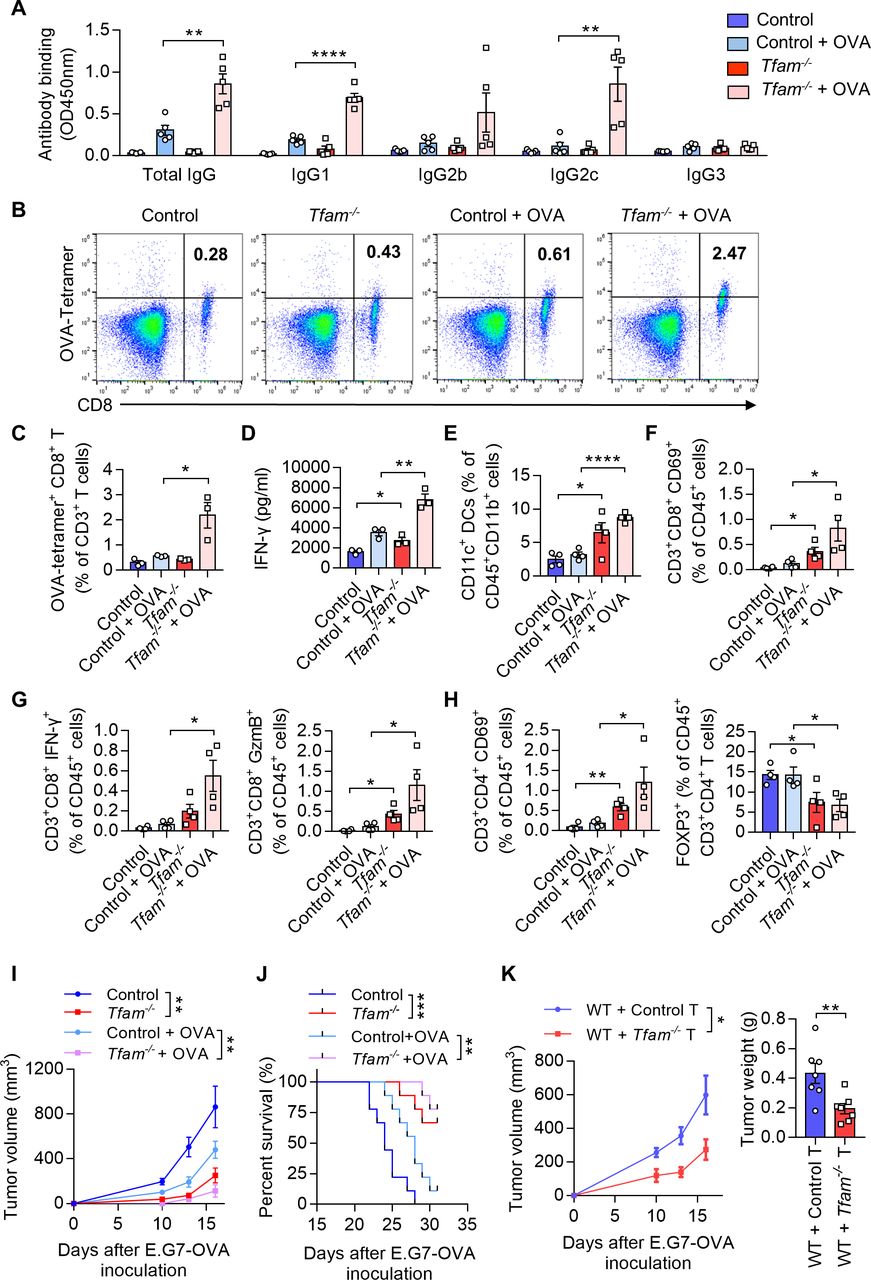
TFAM deficient DC enhanced specific humoral and cellular immune responses. (A) Tfam deficiency enhances the specific anti-OVA humoral immunity. Control and Tfam-/- mice were vaccinated subcutaneously three times with or without 10 µg OVA antigen in PBS on days 0, 14, and 21. The mouse serum were collected on day 28 and levels of the total IgG and IgG subclasses were determined by ELISA. Serum antibody binding was determined by absorbance at 450 nm (n=5 mice). (B–D) Tfam deficiency enhances the specific anti-OVA cellular immunity. Splenic lymphocytes from immunized mice in (A) were isolated on day 28 and further incubated in vitro with CD8+ specific OVA257–264 peptides (10 µg/mL) for 72 hours. The generation of CD8+ CTLs was determined by FCM using PE-conjugated H-2Kb/OVA257–264 tetramer. Representative scatterplots of the OVA-specific CD8+ T cells gated from CD3+ cells are shown in (B) and quantified in (C). Level of IFN-γ in the supernatant was measured by ELISA (D) (n=3 mice). (E) Frequency of CD11c+ DCs in the spleen gated from CD45+ CD3- CD11b+ are determined (n=4 mice). (F) Frequency of CD3+ CD8+ CD69+ CTLs in the spleen gated from CD45+ are determined (n=4 mice). (G) Frequency of CD3+ CD8+ IFN-γ+ CTLs in the spleen gated from CD45+ are determined in the left panel, and frequency of CD3+ CD8+ GzmB+ CTLs in the spleen gated from CD45+ are determined in the right panel (n=4 mice). (H) Frequency of CD3+ CD4+ CD69+ T cells in the spleen gated from CD45+ are determined in the left panel, and frequency of FOXP3+ T cells in the spleen gated from CD45+ CD3+ CD4+ are determined in the right panel (n=4 mice). (I) Tfam deficient potentiates the antitumor effect of OVA vaccine in vivo. In the prophylactic model, control or Tfam-/- mice were immunized as in (A) (n=5 mice) and then injected subcutaneously with E.G7-OVA cells (5×105) 1 week after the third immunization. Tumor growth was monitored at the indicated times. (J) Tfam deficiency potentiates the survival of OVA-vaccinated tumor-bearing mice. Control and Tfam-/- mice were immunized with OVA and injected with E.G7-OVA (5×105) as previously described. Survival of mice was monitored daily (n=9 mice). (K) In the cellular adoptive therapy model, CD8+ T lymphocytes were isolated from immunized mice as in (A) on day 28 and subsequently injected intravenously into recipient mice, which were wild type mice subcutaneously inoculated with E.G7-OVA (5×105) cells. Tumor growth was monitored at the indicated times (left panel) and tumor weight was recorded after sacrifice on day 16 post-transplantation (right panel) (n=7 mice). Data represent the mean±SEM. Statistical significance was determined by two-way ANOVA in (I), left panel of (K) or a two-sided unpaired t-test in (A, C–H, right panel of K). Survival curve data in (J) were analyzed by log-rank (Mantel-Cox test). Representative results in (A–K) from two independent experiments are shown. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance; DC, dendritic cell; i.v., intravenous.
To analyze the ability of presentation in BMDCs, primary BMDCs from mice immunized with OVA were co-cultured with CFSE (Carboxyfluorescein succinimidyl ester) -labeled CD8+ T lymphocytes from OT-I mice, whose TCR can specifically recognize OVA via MHCI molecule. CFSE is a fluorescent dye that is equally partitioned during cell division. FCM analysis showed an elevated rate of CFSE which indicated TFAM deficiency in BMDCs could significantly contribute to the antigen presentation and the proliferation of specific CD8+ T cells (figure 3E). In the next set of experiment, we examined the migration and activation of DCs in the lymph nodes in Tfam-/- and control mice in vivo. The chemokine receptor CCR7 (CD197) connects innate and adaptive immunity and plays a vital role in the homing of DCs to secondary lymphatic organs, enabling rapid proliferation and differentiation of T cells.24 The results suggested an elevated expression of CD197 in DCs harvested from the lymph nodes of Tfam-/- mice when compared with that of the control groups by FCM (figure 3F). In addition, the lymphoid DCs also demonstrated the increased levels in CD40, CD80, and MHCII expression in Tfam-/- group, which indicated that deletion of TFAM could effectively promote the maturation and migration of DCs in vivo (figure 3F). These results suggest that the TFAM deletion in DCs leads to enhanced maturation and activation of DCs, promoting the priming process and the proliferation of T cells.
TFAM deficient DCs enhance humoral and cellular immune responses in vivo
As TFAM deletion resulted in enhanced maturation and activation of the DCs in vitro, we next investigated whether the humoral and cellular immune responses were promoted in Tfam-/- mice. To better model tumor responses, OVA was used as the model antigen to validate the activation of immune responses in vivo. Tfam-/- and control mice were immunized with OVA for three times and the sera were collected. The levels of total IgG and its subclasses were measured. The results showed that the antibody titers of IgG, IgG1, and IgG2c were all elevated in immunized Tfam-/- mice when compared with that of the control group (figure 4A), which suggested a potent activation of humoral immunity in mice Tfam-/- BMDCs. When it comes to the cellular immune response, we demonstrated that Tfam-/- deletion simulated more effective OVA-specific CD8+ T cell responses as evidenced by the increase in spleen lymphocytes from Tfam-/- and control mice after the stimulation with OVA257-264 peptides (figure 4B,C). In accordance, the ELISA results showed the increased production of IFN-γ by CD8+ T lymphocytes in Tfam-/- cells after OVA immunization when compared with the control (figure 4D). Furthermore, the immune-microenvironment and the critical cell populations in the spleen of Tfam-/- and control mice were characterized by FCM. The increased populations of DCs (CD11c+ DCs, figure 4E), CD8+ CD69+ T cells (figure 4F), CD8+ IFN-γ+ T cells, CD8+ GzmB+ T cells (figure 4G), CD4+ CD69+ T cells (figure 4H) and the decreased percentages of CD4+ FOXP3+ cells were also detected (figure 4H).
Next, we used the prophylactic model to identify the anti-cancer immunity activation in Tfam-/- and control group. Mice were challenged with subcutaneous injection of E.G7-OVA cells after three times of immunization. The tumor growth was significantly inhibited in Tfam-/- mice in the prophylactic tumor model with the OVA immunization when compared with that of the control mice (figure 4I), and this group of mice showed prolonged survival (figure 4J). To investigate whether such tumor inhibition effect was due to the activation of cellular immune response, the adoptive transfer study was carried out by isolating CD8+ T lymphocytes from the immunized mouse spleen. CD8+ T lymphocytes from immunized Tfam-/- mice or control mice were injected intravenously in WT mice on day 1 before E.G7-OVA cell inoculation and on day 1 and day 3 after E.G7-OVA cell inoculation. Interestingly, the tumor growth as detected by tumor weight and tumor volume was significantly inhibited in the group that received the adoptive transfer CD8+ T cells from Tfam-/- mice when compared with that received the control CD8+ T cells (figure 4K). To further figure out the myeloid cell subset responsible for the activation of antitumor immunity, we adopted control or Tfam-/- DCs/macrophages to WT LLC tumor-bearing mice. As expected, adoption of Tfam-/- DCs instead of Tfam-/- macrophages significantly inhibited tumor metastasis and tumor growth (online supplemental figure 5). In summary, TFAM deficiency not only caused DC activation, but also led to more efficient activation of antitumor humoral and cellular immunity in vivo.
Supplemental material

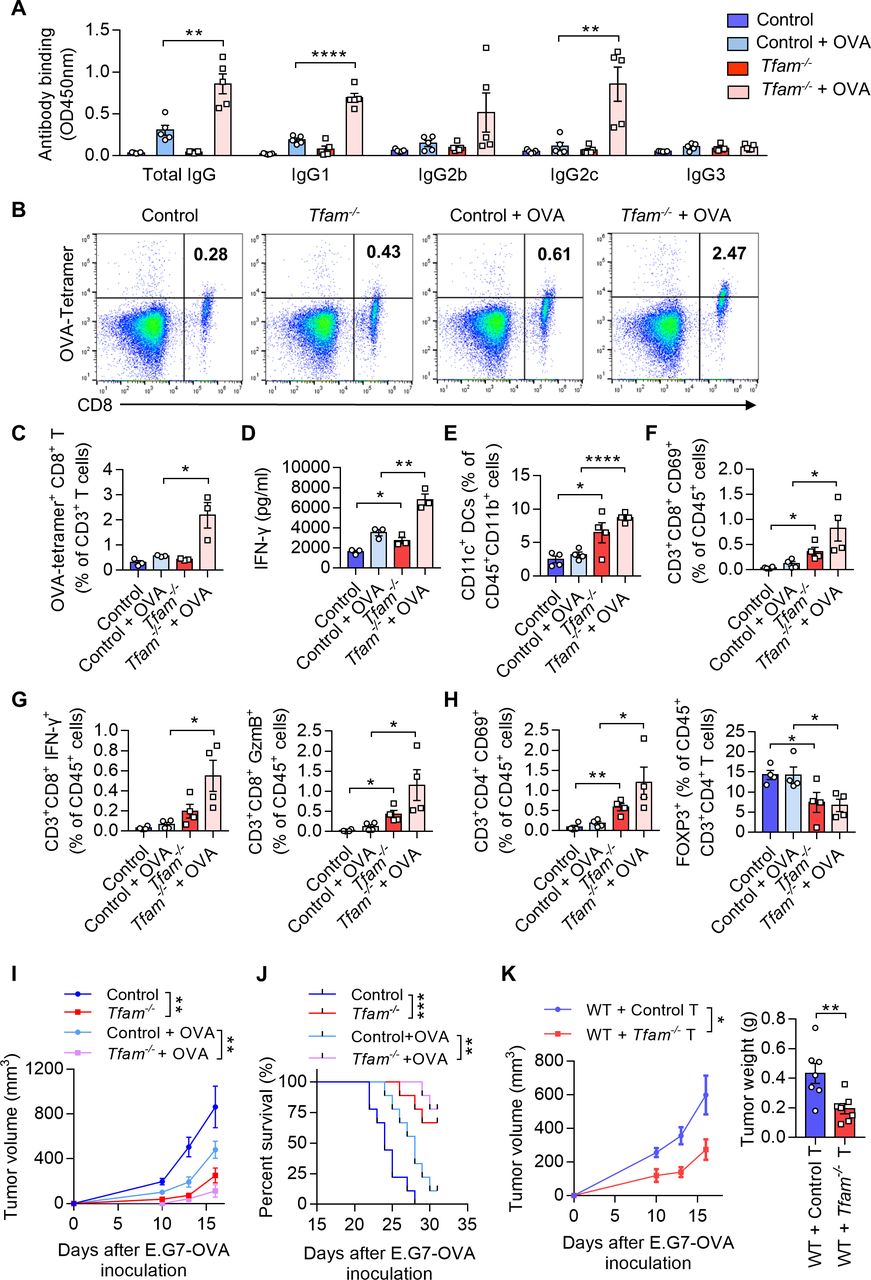
TFAM deficiency in DCs results in mitochondrial stress, cytoplasmic leak of mtDNA, and activation of cGAS-STING pathway. (A–C) Tfam deficiency induced the mitochondrial stress of DCs. (A) BMDCs from control or Tfam-/- mice were stimulated with or without LLC TS and then subjected to qPCR analysis to detect the mtDNA copy numbers (n = 3 biologically independent samples). (B) Immunofluorescence staining of CD11c (red), HSP60 (green),and DAPI (blue) in BMDCs in vitro. Scale bars represent 10 µm. (C) Representative transmission electron microscopy (TEM) images of mitochondria in BMDCs from control or Tfam-/- mice stimulated with or without LLC TS. Scale bars represent 200 nm (upper two rows) or 500 nm (lower two rows), respectively. (D) Tfam deficiency induced the cytoplasmic leak of mtDNA of DCs. BMDCs from control or Tfam-/- mice were stimulated with or without LLC TS and then subjected to qPCR analysis to detect the quantity of mtDNA (n = 3 biologically independent samples). (E, F) Tfam deficiency in DCs results in altered mitochondrial respiratory chain or oxidative stress. BMDCs from control or Tfam-/- mice were subjected to extracellular oxygen consumption rates (OCR) (E) or ROS level (F) detection after TS stimulation at indicated times (n=3 biologically independent samples). (G) Deletion of Tfam induces the target genes of cGAS-STING pathway expression. Relative mRNA levels of indicated genes in BMDCs from control or Tfam-/- mice were measured by qPCR (n = 3 biologically independent samples). (H) Tfam deficiency activates cGAS-STING pathway of DCs. BMDCs from control or Tfam-/- mice were stimulated with LLC TS for the indicated times and cell lysates were collected for Western blot to detect the phosphorylation and protein levels of the indicated proteins. (I) Deletion of Tfam induces the expression of downstream target genes of cGAS-STING pathway. BMDCs from control or Tfam-/- mice were stimulated with or without LLC TS and then Cxcl10, Ifna4, and Ifnb1 mRNAs were measured by qPCR (n = 3 biologically independent samples). LLC, Lewis lung carcinoma. Data represent the mean±SEM. Statistical significance was determined by two-way ANOVA in (A, D, F, I) or a two-sided unpaired t-test in (G). Representative results in (A, D, E, F, G, I) and pictures in (B, C) from three independent experiments are shown. The western blot in (H) was performed twice with similar results. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance; BMDCs, bone marrow-derived DCs; DCs, dendritic cell; TS, tumor supernatant.
TFAM deficiency in DCs results in mitochondrial stress, mtDNA leak, and cGAS-STING pathway activation
We next investigated whether the activation of DCs by TFAM depletion relates to the alteration in mitochondrial function. It is reported that TFAM plays a critical role in maintaining the structure of mtDNA, by wrapping mtDNA to form a nucleoid structure and protecting it against ROS.14 15 We first characterized the quantity of mtDNA and the relative mtDNA copy numbers from control and Tfam-/- BMDCs. The results showed that TFAM deficiency led to lower mtDNA copy number and the same tendency was also observed under TS stimulation condition (figure 5A).HSP60 is important for the protein folding and protein transportation into mitochondria, which can also act as a danger signal for stress or damaged cells.25 It is interesting to notice that HSP60, a major mitochondrial chaperonin, exhibited decreased granule formation, more diffused positive staining, and the changes of localization in the cultured Tfam-/- BMDCs (figure 5B). The exhibited immunofluorescence changes in HSP60 after TFAM depletion in BMDCs also suggested mitochondrial stress. Electron microscope images also revealed the sharply altered mitochondrial morphology in BMDCs from Tfam-/- mice in vitro (figure 5C). Mitochondria swelling, hyperfusion, and mitochondria cristae disappearance were observed in Tfam-/- group when compared with the control. The similar results were also observed when Tfam-/- BMDCs were stimulated with TS when compared with the normal control cells (figure 5C). Considering TFAM contributes to maintaining the stability of mtDNA, it is hypothesized that TFAM deficiency might result in mtDNA leakage. As we characterized the concentration of mtDNA in the cytoplasm, it is found that much higher concentrations of mtDNA were detected in Tfam-/- BMDCs with or without TS stimulation when compared with that of control BMDCs (figure 5D).
As TFAM is closely related to mitochondrial metabolism, it is not surprising that the levels of numerous genes related with oxygen consumption rates metabolic function were decreased while genes associated with glycolysis were increased in Tfam-/-BMDCs (online supplemental figure 6A). In addition, the depletion of TFAM in BMDCs also led to lower oxygen consumption rates (figure 5E), as well as more production of ROS (figure 5F) . We dissected the pathways related with immune-stimulation in the further study. Through RNA-Seq, we found that immune-active or inflammatory-related genes ranked top in the heat map of representative enriched genes analysis for Tfam-/- BMDCs. Among them, type I interferon-activated genes were notably enriched, including the expression of genes related with cytoplasmic RNA and DNA sensors, such as Ddx58, Irf3, Irf7, Stat1, and Tmem173 (online supplemental figure 6B). The elevated transcript levels of mRNA of the cGAS-STING pathway were further evidenced in Tfam-/-BMDCs (figure 5G). It is reported that the leakage of mtDNA and ROS production could synergistically contribute to the activation of cGAS-STING pathway.26 As the two considerations have already been observed in the Tfam-/- BMDCs in the current study, we further investigated the activation of cGAS-STING pathway in Tfam-/-BMDCs . The expression levels of pTBK1, pIRF3, and STING, the makers associated with cGAS-STING pathway, were significantly activated in Tfam-/- group, which directly implicated the activation of the cGAS-STING pathway (figure 5H). In addition, the significantly elevated levels of Cxcl10, Ifnb1, and Ifna4 were detected in Tfam-/- BMDCs compared with the control group before and after TS stimulation, indicating the activation of cGAS-STING signaling and the enhanced type I interferon responses (figure 5I). Notably, the Tfam-/- STING-activated DCs did not undergo apoptosis either with or without TS stimulation when compared with the control DCs (online supplemental figure 7). These data suggested that TFAM deficiency in DCs induced mitochondrial stress and metabolic alterations, leading to mtDNA leakage and ROS production, thus triggering the cGAS-STING pathway, which might be the reason for an enhanced activation of Tfam-/- BMDCs and its efficiently provoked immune responses.
Supplemental material
Supplemental material

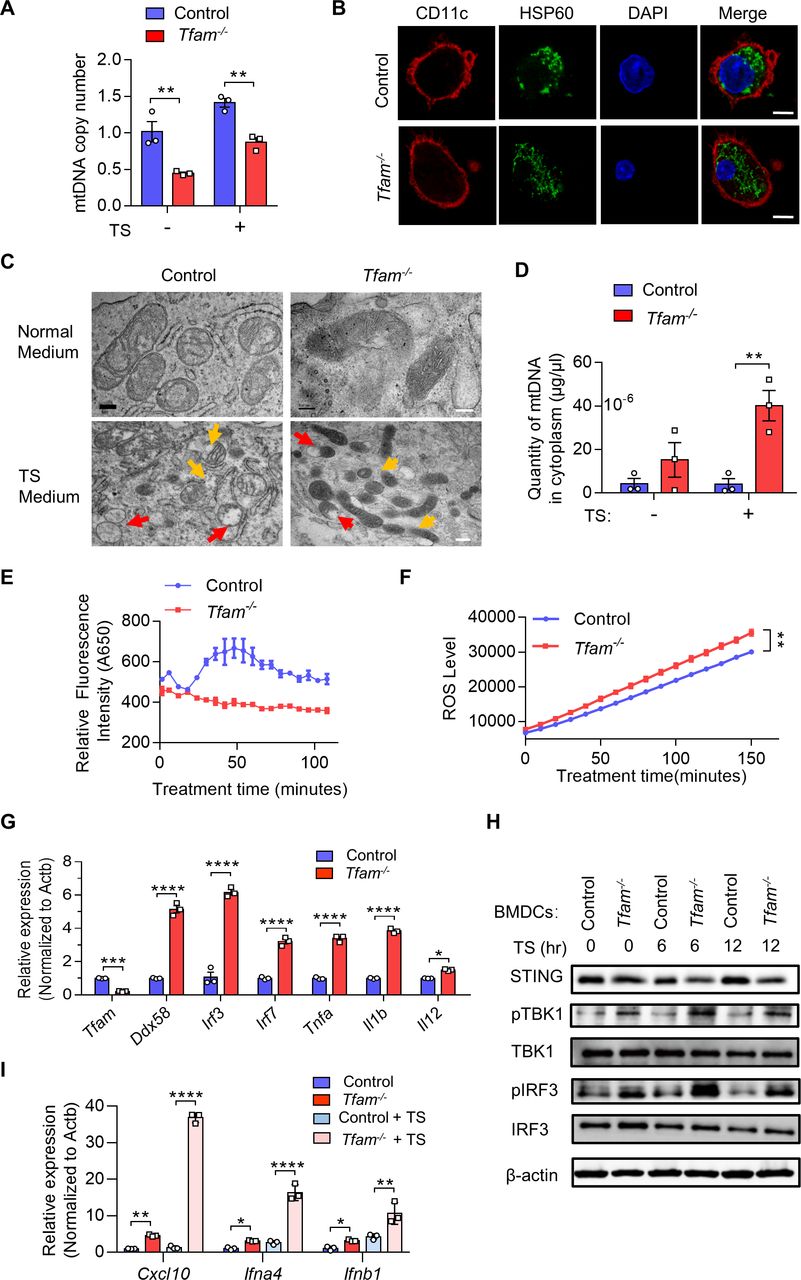
STING antagonism restricts DC maturation and potentiates tumor progression in Tfam-/- mice. (A) Blockade of STING strongly repressed the maturation of DCs induced by Tfam deficiency. BMDCs from Tfam-/- mice were stimulated with or without TS or H-151 for 24 hours and then subjected to flow cytometry analysis to detect the expression of costimulatory molecules. The left panel shows the representative histograms of the gated CD11c+ Tfam-/- DCs. The quantitative data of flow cytometry results are shown in the right panel (n=3 biologically independent samples). (B) Blockade of STING noticeably reduced the secretion of inflammatory cytokines of DCs induced by Tfam deficiency. BMDCs from Tfam-/- mice were similarly treated as in (A), and then levels of TNF-α and IL-12 p40 in the supernatant were detected by ELISA (n = 3 biologically independent samples). The original levels of cytokines in TS were subtracted. (C) Blockade of STING significantly abolished the migration and maturation of Tfam-/-DCs in vivo. Single-cell suspension of lymph nodes from Tfam-/- mice treated with C-176 (13.4 mg/kg) or Veh (solvent) were subjected to flow cytometry analysis. The percentages of CD197+, MHCII+, CD40+, or CD80+ DCs are quantified from CD11c+ gated subpopulation (n=4 mice). (D–F) Blockade of STING re-accelerates tumor progression in Tfam-/- mice. LLC cells (5×105) were intravenously injected into control or Tfam-/- mice to establish experimental pulmonary metastasis models (n=8 mice). Mice simultaneously received daily intraperitoneal injections of C-176 (13.4 mg/kg) or Veh (solvent) and sacrificed on day 24. The gross appearance (D) and H&E staining (E) of the lungs were also examined and the lung metastasis nodules and metastatic area (n=3 mice’s lungs were paraffin embedded) were evaluated (F). Scale bars represent 2 mm. LLC, Lewis lung carcinoma. Data represent the mean±SEM. Statistical significance was determined by a two-sided unpaired t-test in (AB,C, F). Representative results in (A, B) and picture in (A) from three independent experiments are shown. Representative results in (C–F) and pictures in (E) from two independent experiments are shown. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. BMDCs, bone marrow-derived DCs; DC, dendritic cell; MHC, major histocompatibility complex; TS, tumor supernatant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
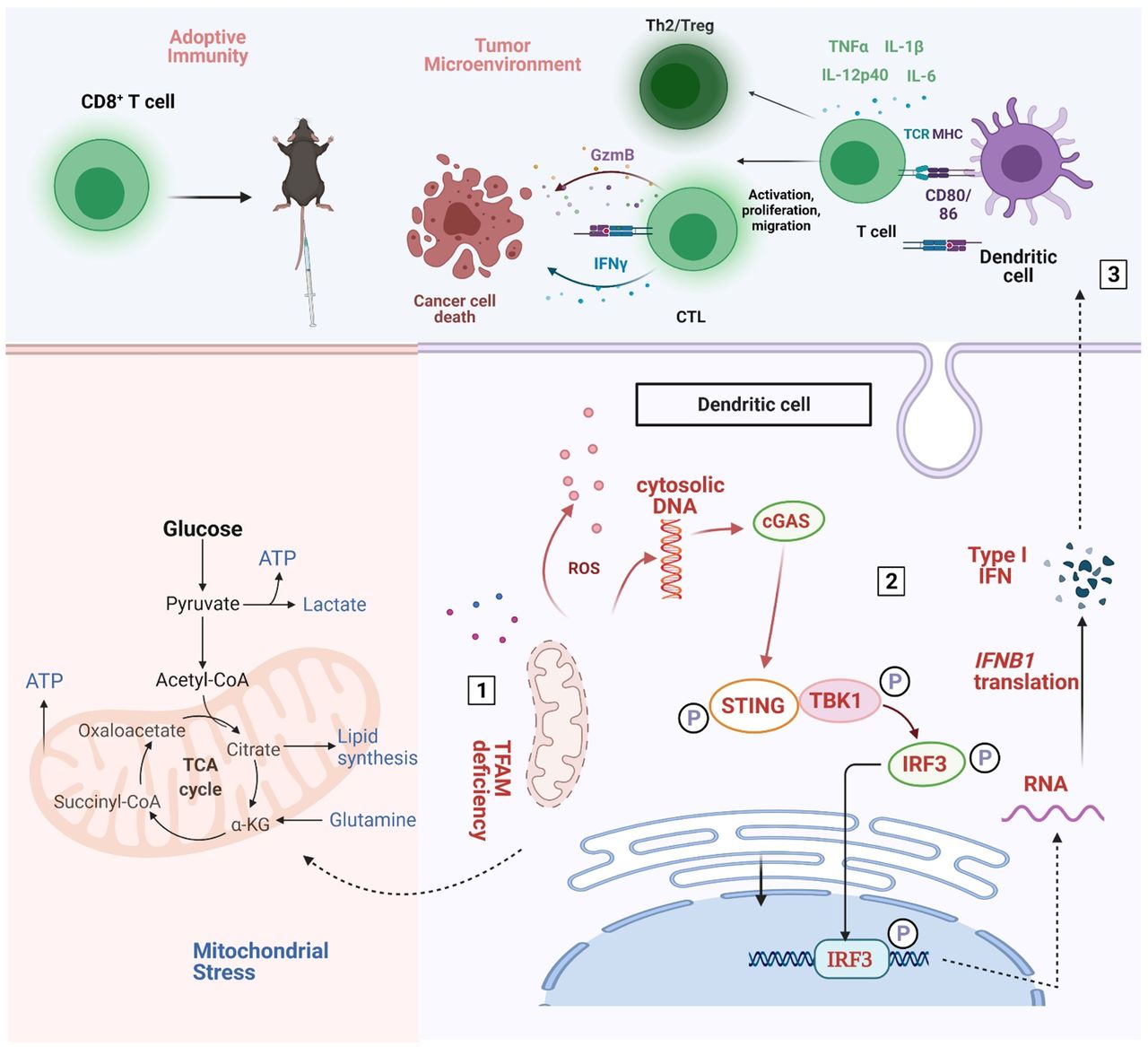{kind=link}
Schematic diagram of the mechanistic findings. Tfam deficiency in DCs resulted in mitochondrial metabolism abnormalities (number 1), then the cGAS-STING pathway actived (number 2), subsequently enhanced the immunity of DCs and T cells and resultantly inhibiting lung metastases of the tumor by remodeling the tumor microenvironment (number 3). Furthermore, spleen T lymphocytes of immunized Tfam-/- mice displayed stronger antitumor immunity when adoptively transferred into tumor-bearing wild type mice. DCs, dendritic cells.
STING antagonism restricts DC maturation and potentiates tumor progression in Tfam-/- mice
To investigate the critical role of STING activation induced by TFAM depletion in DCs, we pretreated the primary BMDCs isolated from Tfam-/- mice with a STING antagonist, H-151 in vitro. We found that the expression of costimulatory molecules including CD40, CD86, and MHCII in the BMDCs from Tfam-/- mice were significantly decreased by the treatment of H-151, and the similar results were recorded in the presence of TS (figure 6A, left panel). However, there is no such trend in the BMDCs from control mice (figure 6A). In accordance with this, the secretion of the cytokines by the Tfam-/- BMDCs, such as TNF-α and IL-12 p40, was also significantly blocked by H-151 either with or without the stimulation of TS (figure 6B).
In the next set of experiment, we examined how STING inhibition in Tfam-/- mice affects their DC behavior and immune activation. We used C-176 which is a commonly used STING antagonist for animal experiments in vivo. After the treatment with C-176, the migration and activation of DCs in lymph nodes in Tfam-/- mice were assessed. The results showed that the STING inhibitor C-176 significantly inhibited the expression of CD197, CD40, MHCII, and CD80 in DCs from the lymph nodes of Tfam -/- mice (figure 6C). We also studied the effect of C-176 in the LLC lung metastasis model. The results showed that STING inhibition by C-176 notably reversed the suppressive effects on tumor growth in Tfam -/- mice and promoted tumor progression and metastasis (figure 6D–F). In summary, these results illustrated a strong correlation between the cGAS-STING pathway, DC activation, and tumor suppressive effects in Tfam-/- mice. The activation of cGAS-STING pathway in Tfam-/- DCs plays a key role in provoking the enhanced antitumor immune response.
Discussion
Several observations have been made concerning mitochondria, TFAM, and anti-cancer immunity. In the present study, we identified that the depletion of Tfam gene in mitochondria of DCs led to the suppression of tumor growth and enhancement of immunity through the cGAS-STING pathway, which is supported by the following results: first, the deletion of TFAM in tumor-infiltrating myeloid immune cells inhibits tumor progression; second, targeted reduction of Tfam gene expression in DCs results in mitochondrial abnormalities and metabolic changes; third, mtDNA escapes to the cytoplasm and activates the cGAS-STING pathway; last but not the least, Tfam deletion could also promote maturation, migration, antigen presentation and inflammatory cytokine secretion of DCs, further activating T cells to exert antitumor effects. In summary, our results elucidate how mitochondrial abnormalities caused by Tfam deletion enhance downstream immune responses through activation of DCs, providing new insights with respect to anti-cancer therapy targeting immunometabolism.
Mitochondria are complex organelles that affect the occurrence, growth, survival, and metastasis of cancer. Therefore, the dysregulations of mtDNA can also affect the development of tumors. Hence, it is very important to understand the mechanisms by which the dysregulation of mtDNA influences the process of tumorigenesis.27 Reportedly, the depletion of Tfam impairs the tumor growth in Kras-driven mouse models of lung cancer.28 However, the role of TFAM in tumor-associated immune cells has not been clarified. In particular, TFAM protein has been shown to play a dual role. First, the role of TFAM in mitochondria is similar to the role of histones in nucleosome. TFAM wraps mtDNA entirely to form a nucleoid structure that may prevent mtDNA from being disrupted by ROS. Second, TFAM regulates the replication of mtDNA and maintains the copy number of mtDNA, which is closely associated with the metabolic activity of mitochondria. Furthermore, it was revealed that even a slight variation of the ratio of TFAM to mtDNA might significantly affect the transcription and replication of mtDNA,29 which makes it important to keep this ratio in a narrow range for maintaining the normal function of mitochondria. Therefore, we hypothesize that TFAM could be a critical regulator of mitochondrial metabolism and tumor immunity. To test the hypothesis, we established the experimental pulmonary metastasis models using bone marrow-specific Tfam knockout mice. Consequently, it was observed that Tfam loss in myeloid cells resulted in the inhibition of tumor growth and the reshaping of the TIME. In Tfam-/- mice with LLC pulmonary metastasis model, the TIME was observed to be immune-promotive.
In addition, myeloid cells have the ability to infiltrate into immunological ‘cold’ tumors, while T cells are usually excluded or confined peripherally.30 During cancer progression, a microenvironment rich in myeloid cells and lacking in T cells is formed in the metastatic site before metastasis.31 In our study, the infiltration of DC in TME has been shown to be linked with the expression of Tfam. We revealed that TFAM deficiency promoted DC maturation in vivo and in vitro, boosted the proliferation of T cells, and facilitated the secretion of inflammatory cytokines. DCs are essential for T cell-mediated tumor immunity since they transport tumor antigens to lymph nodes and activate cytotoxic T lymphocytes through presentation to initiate the antitumor response. Besides, T cell activation requires interactions between the T cell receptor (TCR) and the CD80, CD86, and MHC molecular antigen peptide presented by DCs. Additionally, the increased levels of proinflammatory cytokines, including IL-6, IL-1β, and TNF-α, can further assist DCs activation and help T cells fight against tumors.2 Furthermore, we also found that Tfam deficient DCs could enhance humoral and cellular immune responses in vivo. It was shown that the IgG level in serum was significantly increased in immunized Tfam-/- mice, corresponding to the raised MHCII expression on Tfam deficient DCs in vivo and in vitro.32 It’s worth noting that the populations of activated DCs from the spleens of Tfam-/- mice in both unimmunized and immunized groups were increased. Moreover, a more intense cellular immune response mediated through DCs activation was observed. Considering that cellular immunity is the predominant antitumor immunity, activation, and homing of DCs in Tfam-/- mice already activated the specific T cells response in the spleen prior to immunization.33 To our surprise, the OVA-stimulated BMDCs from Tfam-/- mice can not only significantly induce the quick expansion of antigen-specific CD8+ T lymphocytes of OT-I mouse in vitro, but also elicit the proliferation of the cytotoxic T cells, which can continue to play strong antitumor effects after cell adoption. Studies suggest that Batf3-dependent DC is essential for effector T cell priming and transporting to the tumor environment in adoptive therapy,34 which may partly explain why the adoptive cell transfer also exerted a strong antitumor effect. Therefore, the activation of DCs dramatically enhances the activity of T cells and potently modulates the effectiveness of adoptive immunotherapy.
Notably, previous study reported that, Lyzm-Cre targets mainly lung macrophages, blood monocytes, and neutrophils, and only marginally DCs.35 However, our results found that TFAM expression was obviously decreased in DCs both in vivo and in vitro (online supplemental figures 2B, 3D), and the efficiency of TFAM recombination in GM-DCs was better than that in GM-Macs (online supplemental figure 3D). Additionally, Gao et al36 showed that TFAM deficiency did not affect DCs. We think that this inconsistency is caused by the different experimental models and designs, that is, the influenza virus infection model was used by Gao et al, whereas tumor lung metastasis model and preventive vaccine model were used in our study. Although we could not completely rule out the possible role of other myeloid cell subsets in the antitumor effect, this research is focused on DC-related mechanisms. Our FCM analysis of TME illustrated that both recruitment and activation of DCs were enhanced in Tfam-/- mice in vivo, while the percentages of AMs, M1 TAMs, and M2 TAMs were rarely altered (figure 2A–C). The most direct evidence for the contribution of DCs is yielded from the adoptive experiments. Adoption of neither WT nor Tfam-/- macrophages inhibited tumor growth and metastasis in LLC metastasis models, while adoption of Tfam-/- DCs did (online supplemental figure 5). Thus, it is unreasonable to deny the important role of Tfam-/- DCs in provoking antigen presentation and blocking tumor metastasis.
As a potent innate immune mediator stimulated by DNA, the cGAS-STING pathway has been shown to cross-talk with the anti-cancer immune response.37 Previous studies combined with our results indicated that the DNA-activated STING pathway is essential for type I IFN signaling in DCs, which is required for antitumor effects, especially in radiotherapy.34 38 Meanwhile, mitochondrial disorder and evocative oxidative stress are able to crosstalk with the cGAS-STING pathway. Mitochondrial disorders could lead to a high level of ROS production in cells, which can directly damage mtDNA.39 Next, the release of mtDNA would activate the cGAS-STING pathway and facilitate the production of type I interferon, which plays a critical role in modulating the anti-cancer immunity. Some approaches might be used to induce mitochondrial stress to potentiate the STING-dependent antitumor immunity in the TME. Radiotherapy,40 chemotherapy40 and immunotherapy,41 mtDNA mutation, ROS accumulation, or mitophagy42 could result in dysregulation of mitochondrial homeostasis and mitochondrial stress. The released mtDNA is delivered to DCs as STING-activator to modulate TME.43 Studies have shown that reduced NRF2 in PERK-deficient MDSCs would prime STING-dependent expression of antitumor type I interferon via cytosolic mtDNA.44 Here, our study highlighted a vital relationship between STING and Tfam, in which STING is a pivotal downstream regulator of metabolic defects and mitochondrial disorder in BMDCs of Tfam-/- mice. Additionally, we have also shown that blocking STING pathway inhibits the maturation of BMDCs and the secretion of inflammatory factors.
TFAM deletion in mouse embryonic fibroblasts has been previously reported to increase antiviral innate immune responses.45 However, little has been done to explore how manipulating TFAM is related with metabolism in antigen presenting cells and antitumor immune response. In this study, we uncovered that the TFAM knockout in myeloid cells was significantly associated with the suppression of tumor growth and metastasis, which was accompanied by reshaping the TME in Tfam-/- mice. Meanwhile, our findings demonstrate that the deficiency of TFAM in DCs would result in the mtDNA escape and trigger the cGAS-STING pathway, thus promoting DC maturation, migration, antigen presentation, and the secretion of inflammatory factors to enhance the activation of downstream T cells. It was further confirmed that inhibiting cGAS-STING pathway through inhibitors would revert the effect of suppressing tumor growth (figure 7). In conclusion, our results elucidate the crucial role of TFAM in DCs in activating antitumor immune response, and complementing the regulatory mechanisms of the TME from the perspective of mtDNA. Hence, strategies that allow specific regulation of mitochondrial function in selected cell populations can be designed for tumor therapy, though a great deal of works need to be done before achieving this goal.
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as online supplemental information. Data are available on reasonable request. The RNA-seq data are available in the Gene Expression Omnibus (GEO) dataset with the accession number GSE223950. All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
All animal studies carried out were approved by the Animal Care and Use Committee of Sichuan University (Chengdu, Sichuan, China).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
TL and ZZ contributed equally.
Contributors XW conceived the project, designed the experiments and revised the manuscript. ML supervised the research and designed the experiments. TL performed experiments, analyzed data, and wrote the manuscript. ZZ, ZB, FM, YL, JY, SC, XH, WH, RP and WR performed the experiments. ZZ, ZB and HZ analyzed data. YW gave technical support and conceptual advice. TL, ZZ and XT revised the manuscript. All authors commented on the manuscript. XW and ML are acting as guarantors.
Funding This work was supported by National Science Fund for Excellent Young Scholars (No. 32122052), National Natural Science Foundation Regional Innovation and Development (No. U19A2003), National Natural Science Foundation of China (82273297).
Competing interests No, there are no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.