Article Text

Abstract
Background Tumor progression and resistance to therapy in children with neuroblastoma (NB), a common childhood cancer, are often associated with infiltration of monocytes and macrophages that produce inflammatory cytokines. However, the mechanism by which tumor-supportive inflammation is initiated and propagated remains unknown. Here, we describe a novel protumorigenic circuit between NB cells and monocytes that is triggered and sustained by tumor necrosis factor alpha (TNF-α).
Methods We used NB knockouts (KOs) of TNF-α and TNFRSF1A mRNA (TNFR1)/TNFRSF1B mRNA (TNFR2) and TNF-α protease inbitor (TAPI), a drug that modulates TNF-α isoform expression, to assess the role of each component in monocyte-associated protumorigenic inflammation. Additionally, we employed NB-monocyte cocultures and treated these with clinical-grade etanercept, an Fc-TNFR2 fusion protein, to neutralize signaling by both membrane-bound (m) and soluble (s)TNF-α isoforms. Further, we treated NOD/SCID/IL2Rγ(null) mice carrying subcutaneous NB/human monocyte xenografts with etanercept and evaluated the impact on tumor growth and angiogenesis. Gene set enrichment analysis (GSEA) was used to determine whether TNF-α signaling correlates with clinical outcomes in patients with NB.
Results We found that NB expression of TNFR2 and monocyte membrane-bound tumor necrosis factor alpha is required for monocyte activation and interleukin (IL)-6 production, while NB TNFR1 and monocyte soluble TNF-α are required for NB nuclear factor kappa B subunit 1 (NF-κB) activation. Treatment of NB-monocyte cocultures with clinical-grade etanercept completely abrogated release of IL-6, granulocyte colony-stimulating factor (G-CSF), IL-1α, and IL-1β and eliminated monocyte-induced enhancement of NB cell proliferation in vitro. Furthermore, etanercept treatment inhibited tumor growth, ablated tumor angiogenesis, and suppressed oncogenic signaling in mice with subcutaneous NB/human monocyte xenografts. Finally, GSEA revealed significant enrichment for TNF-α signaling in patients with NB that relapsed.
Conclusions We have described a novel mechanism of tumor-promoting inflammation in NB that is strongly associated with patient outcome and could be targeted with therapy.
- Neuroblastoma
- Macrophages
- Inflammation
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Tumor progression and resistance to therapy in children with neuroblastoma (NB), a common childhood cancer, are often associated with infiltration of monocytes and macrophages that produce inflammatory cytokines. However, the mechanism by which tumor-supportive inflammation is initiated and propagated remains unknown, precluding rational therapeutic interventions.
WHAT THIS STUDY ADDS
In this work, we demonstrate a novel role for NB cell TNFRSF1B mRNA and monocyte membrane tumor necrosis factor alpha (TNF-α) in the initiation of tumor-supportive inflammation in vitro and in vivo. Food and Drug Administration-approved TNF-α inhibitor etanercept disrupts this interaction between monocytes and NB cells, shutting down production of multiple tumorigenic cytokines and inhibiting tumor growth in a xenogeneic NB model.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study reveals the mechanism by which tumor-supportive inflammation is initiated and propagated, as well as how it can potentially be targeted for therapy in patients with high-risk NB.
Background
Neuroblastoma (NB) is a heterogeneous pediatric tumor of neural crest origin. It is the second most common solid tumor in children and causes 15% of all pediatric cancer deaths.1 Treatment remains a significant clinical challenge as disease course ranges from spontaneous regression to treatment-resistant progression and death.2 Representing about half of total diagnoses, high-risk NB cases present with aggressive, unfavorable histology and are often metastatic and refractory to treatment.3 Despite intensive therapies including surgery, radiation, high-dose chemotherapy with stem cell transplantation, retinoic acid, and antibody-based immunotherapy, the long-term survival for patients with high-risk disease is less than 50%.4
Approximately half of high-risk NB tumors are characterized by amplification of the MYCN and/or ALK oncogenes.5 These genetic aberrations activate transcription of genes involved in cell cycle progression and repress those that induce cell differentiation leading to highly proliferative, dedifferentiated tumors.6 7 However, the remaining half of high-risk NB tumors lack clearly defined genetic drivers of tumor progression. High-risk MYCN-non-amplified NB tumors are often infiltrated by immune cells including tumor-associated monocytes and macrophages.8–10 Moreover, patients with tumors expressing higher levels of an inflammatory gene signature associated with monocytes or macrophages (CD14, CD16, IL-6, IL-6R, and TGFB1) had worse 5-year progression-free survival than patients without this signature, highlighting the role of inflammation in promoting a tumor microenvironment (TME) favorable for NB growth.9 11
Monocytes and macrophages are major components of the TME in many types of solid tumors. In most tumor types including NB, these cells promote growth and metastasis and inhibit antitumor immunity.12 13 Therefore, targeting tumor-associated monocytes and/or macrophages is a promising strategy for cancer therapy.14 However, therapies designed to target monocytes and/or macrophages, including CSFR1 antibodies and receptor tyrosine kinase (RTK) inhibitors, have demonstrated modest therapeutic activity as single agents in early-stage clinical trials.15–17
Monocytes are an immediate developmental precursor of macrophages, and these cell types share many phenotypical markers. Recent advances in single-cell RNA sequencing (scRNAseq) and other multiparameter methods have now made it possible to clearly differentiate tumor-associated monocytes, macrophages, and their respective subsets.18 Using a combination of scRNAseq, multiplex immunohistochemistry, and in silico analysis, a recent publication demonstrated that primary NB tissues contain a heterogeneous mix of monocyte and macrophage subsets.10 The authors also showed that an inflammatory monocyte cell state is more prevalent in tumor tissues than non-malignant tissues.
To effectively neutralize proinflammatory monocytes and/or their ability to promote tumor growth, it is critical to understand how these cells interact with tumor cells. We previously demonstrated that NB cells stimulate monocytes to produce interleukin (IL)-6, which was shown to be partially responsible for promoting NB growth in mice.9 However, the mechanism by which NB cells interact with monocytes and initiate this tumor-promoting inflammatory response remains unknown.
Here, we describe a novel inflammatory feedback loop between NB cells and monocytes that is triggered and sustained by tumor necrosis factor alpha (TNF-α) signaling. We found that NB cells activate monocytes through a contact-dependent reverse signaling interaction between TNFRSF1B mRNA (TNFR2) on the NB cell surface and membrane-bound tumor necrosis factor alpha (mTNF-α) on monocytes. This interaction initiates downstream NF-κB signaling in monocytes, leading to production of protumorigenic cytokines including IL-6, G-CSF, IL-1, and soluble (s)TNF-α. These cytokines then complete the feedback loop by stimulating NB cell proliferation that results in enhanced tumor growth and angiogenesis. Importantly, we found that this proinflammatory signaling loop is completely abrogated by Food and Drug Administration (FDA)-approved etanercept, a TNF-α-neutralizing Fc-TNFR2 fusion protein, leading to reversal of monocyte-mediated NB growth promotion in vitro and in vivo.
Methods
Cell culture
Human NB cell lines SK-N-AS, SK-N-BE2, and IMR-32 were purchased from American Type Culture Collection. Human NB cell lines CHLA-136, CHLA-255, LA-N-1, LA-N-5, and LA-N-6 were established and maintained as previously described.19 Cell lines were maintained in Iscove Modified Dulbecco Medium (IMDM) supplemented with 20% heat-inactivated FBS (Gibco, Invitrogen) and 2 mM GlutaMax (Gibco, Invitrogen) without antibiotics. All cell lines were STR fingerprinted at MD Anderson Cancer Center within 1 year of use and checked for mycoplasma contamination (Lonza MycoAlert) every 2 months.
Monocyte isolation
Human peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Paque (GE Healthcare) density centrifugation from buffy coats (Gulf Coast Regional Blood Center). Monocytes were isolated by negative selection using the Pan Monocyte Isolation Kit, human (Miltneyi Biotec) according to the manufacturer’s guidelines. Purified monocytes were cultured in complete monocyte medium (IMDM with 10% heat inactivated, dialyzed FBS (Gibco, Invitrogen) with 2 mM GlutaMax) in ultralow-attachment (ULA) tissue culture plates (Corning), unless otherwise specified. Where indicated, freshly isolated monocytes were polarized to M2 macrophages by culturing with M-CSF (25 ng/mL, Peprotech) for 4 days followed by IL-4 (20 ng/mL, R&D Systems) and IL-10 (20 ng/mL, R&D Systems) for 24 hours.
Coculture experiments and treatments
Monocytes were directly cultured with wild-type or TNF, TNFRSF1A mRNA (TNFR1), or TNFR2 knockout (KO) NB cell lines in ULA plates for 24 hours. NF-κB activation was measured by flow cytometry and cytokine production by ELISA. To evaluate contact dependence of monocyte activation by NB, monocytes were cultured in ULA plates with 7-day NB conditioned medium mixed 1:1 with complete monocyte medium for 24 hours and analyzed for IL-6 production. To evaluate the individual signaling contributions of NB and monocytes within coculture, NB cell lines were fixed in 4% paraformaldehyde (PFA) CytoFix Buffer (BD) for 15 min at 4°C and then cultured at a 4:1 ratio with freshly isolated human monocytes. Similarly, freshly isolated human monocytes were fixed using the same protocol and cocultured with NB 4:1 (NB:mon). Signaling activation was measured via downstream IL-6 production. To assess the growth advantage of NB cultured with monocytes, NB-luc cell lines and monocytes were seeded in tissue culture-treated white 96-well plates (1:1 ratio) and were cultured for 4 days. Luminescence was measured using a TECAN Spark plate reader. To evaluate changes in proliferation, NB was pulsed for 10 min in CellTrace Violet (CTV) (2 µM, Invitrogen) and seeded at 100 000 cells per well in a 12-well tissue culture-treated plate. Monocytes were added directly to the NB monolayer at a density of 100 000 per well and incubated for 4 days. CTV expression was analyzed by flow cytometry.
To evaluate phagocytosis, NB cell lines were cocultured with freshly isolated monocytes for 20 hours in ULA plates (4:1 ratio). NB cells were then isolated from cocultures using CD14+ magnetic bead isolation (Classical Monocyte Isolation Kit, Miltenyi Biotec). Purified NB was then labeled with CellTrace carboxyfluorescein succinimidyl ester (CFSE) (2 µM, Invitrogen) and cocultured with M2-polarized macrophages for 3 hours (2:1 ratio). Macrophage phagocytosis of CFSE-labeled NB was quantified by flow cytometry.
Etanercept (Amgen) and human IgG control (MP Biomedical) were used at 10 µg/mL. Small molecule inhibitors of TNF-α or NF-kB signaling were used at indicated concentrations: TAPI-1 (40 µm, Sigma) and IKK Inhibitor VII (0–20 µM, APExBio).
Clustered regularly interspaced short palindromic repeats (CRISPR)
CRISPR/Cas9-mediated KO of TNF, TNFRSF1A, and TNFRSF1B was performed as previously described.20 Briefly, sgRNAs targeting TNF, TNFRSF1A, and TNFRSF1B were designed using CRISPRscan and were selected for low predicted off-target effects and low off-target cutting frequency determination score.21 22 sgRNA guides were transcribed using the HiFi T7 Transcription Kit (NEB) from sgDNA intermediates generated by PCR from guide-specific oligo forward primers (online supplemental table 2), a universal reverse primer, and the px458 plasmid DNA template. sgRNA was concentrated using RNA Clean & Concentrate 25 (Zymo). Cas9 protein (PNABio) and purified sgRNA guides were mixed (2 µg Cas9, 1 µg sgRNA) and incubated for 20 min at RT to form ribonucleoprotein (RNP) complexes. RNPs were electroporated into NB cell lines using the Neon System (ThermoFisher) (CHLA-255 1600 V, 10 ms, three pulses; SK-N-AS and SK-N-BE2 1450 V, 20 ms, two pulses). Cells were seeded at 0.5 cells per well in 96-well tissue culture plates and expanded. Wells with single clones were genotyped; homozygous KOs were sequenced (Genewiz); and functional KO was confirmed by loss of protein expression via flow cytometry or lysate ELISA.
Supplemental material
Flow cytometry
To evaluate TNF-α expression in NB and monocytes, cells were stimulated with lipopolysaccharide (LPS) (50 ng/mL) with GolgiStop (BD) and fixed using CytoFix/Cytoperm buffer (BD). Cells were then stained with TNF-α-PE (Clone MAb11, BD). TNFR1 and TNFR2 surface expression was evaluated by staining with TNFR1-BV421 (Clone MABTNFR1-B1, BD) and TNFR2-APC (Clone hTNFR-M1, BD) in fluorescence-activated cell sorting (FACS) staining buffer containing 0.05% sodium azide to prevent receptor/antibody processing. mTNF-α surface staining was performed with anti-mTNF (Clone 6401 5 µL/test, R&D Systems) for 30 min on ice in FACS staining buffer containing 0.05% sodium azide to prevent receptor/antibody processing. To estimate changes in NF-κB expression, cells were stained with ZombieViolet (1 µL/test, BioLegend) counterstain and APC-H7 CD45 (Clone 2D1, BD) and/or FITC-CD14 (Clone M5E2, BD) to label monocytes for 20 min at room temperature. Cells were fixed and permeabilized with Phosflow Fix Buffer I (BD) and Phosflow Perm Buffer IV (BD) per manufacturer’s protocols. Fixed and permeabilized cells were then stained with PE-IκBα (Clone MAD-3, BD) for 30 min at room temperature. To evaluate changes in proliferation, CTV-pulsed NB cocultures were counterstained with APC-H7 CD14 (Clone M5E2, BD) resuspended in FACS staining buffer with 7-AAD counterstain (10 µL/test, BD). To evaluate changes in CD47 expression, cocultures were stained with CD14-APC-Cy7 (Clone M5E2, BD), GD2-BV421 (Clone 14 .G2a, BD), CD47- PE-Cy7 (Clone CC2C6, Biolegend), and Live/Dead Near IR (ThermoFisher). All flow samples were run using an LSRII 5-laser flow cytometer (Texas Children’s Hospital Cancer Center Flow Cytometry Core, Houston, TX) and Symphony A1 four-laser flow cytometer (BD) and analyzed using FlowJo Software (Treestar).
Cytokine analysis
Cytokines were detected using the Human TNF-alpha Quantikine ELISA Kit (tumor necrosis factor alpha (TNF-α), R&D Systems) and the Human IL-6 Quantikine ELISA Kit (IL-6, R&D Systems) according to manufacturer’s guidelines and read using a TECAN Spark plate reader. Global cytokine regulation was evaluated using a Luminex panel containing the following cytokines: CX3CL1, CXCL10, EGF, FGF-2, Flt3L, G-CSF, GM-CSF, IL-1α, IL-1β, IL-1Ra, IL-4, IL-6, IL-8, IL-10, IL-12 (p40), IL-12 (p70), M-CSF, MCP-1, MCP-3, MDC, MIP-1α, MIP-1β, TGF-α, TNF-α, TNF-β, and VEGF-A (Human Cytokine Panel A, Millipore-Sigma).
RNA analysis
Total RNA from cell pellets was isolated using the RNeasy Micro Plus Kit (Qiagen). One-step cDNA synthesis and quantitative reverse transcription PCR (qRT-PCR) was performed using the KAPA SYBR Fast One Step qRT-PCR kit (Roche) with the CFX96 Touch RT-PCR Detection System (BioRad). Primers were from Sigma (online supplemental table 2). Relative changes in gene expression were calculated using the ΔCt method with housekeeping gene GAPDH as control. Snap-frozen tumor specimens (30 mg) were incubated in RLT buffer (Qiagen) and homogenized using manual microtube homogenizers (BioMasher, Takara). The lysates were sheared using QiaShredder columns (Qiagen). Total RNA was isolated using the RNeasy Micro Plus Kit with gDNA elimination columns (Qiagen).
Eukaryotic mRNA sequencing with 30 M depth and 150 paired-end reads was performed by Novogene. Quality control checks on FASTQ sequencing files were performed by FastQC V.0.11.2. Reads were aligned to human (GRCh38 V.84) or mouse (GRCm38 V.81) reference genomes separately by hisat2 V.2.1.0.23 Using XenoFilteR,24 we aligned the mouse genome to the human transcript file to remove reads that mapped to both genomes, resulting in unique human reads (representing NB tumor cells) and vice versa for unique mouse reads (TME). Filtered reads were then subject to transcript assembly and quantification using stringtie V.1.3.5.25 Human and mouse transcripts were then subject to differential gene expression analysis (DESeq2 R package) using a twofold change difference in expression, and p-adj value of <0.05 as cut-off thresholds.
Raw RNA sequencing data have been deposited in the Gene Expression Omnibus at accession number GSE218915.
In vivo experiments
Animal experiments were performed according to IACUC-approved protocol AN-5194 at Baylor College of Medicine (BCM). Six-week-old NOD/SCID/IL-2Ry-null (NSG) mice were purchased from Jackson Laboratory and maintained at BCM animal care facility. At 8 weeks, mice were injected in the subcutaneous flank with either 1×106 CHLA-255-luc (or SK-N-AS-luc) or a combination of 1×106 CHLA-255-luc and 1×106 human monocytes (or SK-N-AS-luc/monocytes) embedded in growth factor-reduced Matrigel (Corning) as previously described.26 Where indicated, mice were injected intraperitoneally with 100 µg/mouse of etanercept (Amgen, 5 mg/kg/dose), or isotype human IgG control (MP Biomedical) two times a week. Tumor growth was measured by bioluminescent imaging (Small Animal Imaging Core Facility, Texas Children’s Hospital). After 3 weeks, mice were euthanized and tumors were saved for immunohistochemistry (IHC) and RNA-seq.
Tumor IHC
Tumors were snap-frozen in optimum cutting temperature (OCT)Media (Tissue-Tek, VWR) and cryosectioned into 5–7 µm thin sections (Texas Children’s Hospital Histology Core, Houston, Texas, USA). Samples were fixed and permeabilized in ice-cold acetone for 10 min at −20C and blocked in Tris-buffered saline (TBS) containing 5% normal goat serum (CST) and 1% bovine serum albumin (BSA) (Sigma) for 2 hours at RT. Mouse anti-human/mouse Alpha-Smooth Muscle Actin-AF488 (Clone 1A4, ThermoFisher) and mouse anti-human/mouse CD31-PE (Clone WM59, ThermoFisher) were diluted 1 µg/mL in staining buffer (TBS, 1% NGS, 1% BSA) and incubated overnight at 4°C. Slides were counterstained with 1 µg/mL DAPI (4′,6-diamidino-2-phenylindole) (BD Biosciences) for 5 min and mounted with fluorescence mounting media (Fluoromount G, Invitrogen). Whole tumor section scans were performed using the EVOS M7000 imaging system (Thermofisher). Tumors were further imaged using a DeltaVision Live High-Resolution Deconvolution microscope (GE Healthcare) in an unbiased manner by the BCM Integrated Microscopy Core (Houston, Texas, USA). Images were processed and analyzed using FIJI/ImageJ software (NIH). To quantify CD31+ staining, images were thresholded (default, 1800 MIN, Infinity MAX), converted to binary, and analyzed using the FIJI/ImageJ ‘Analyze Particle’ function excluding regions smaller than 25 µm2.
Computational data
Analysis of human NB samples was performed within R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl). ‘Tumor Neuroblastoma Kocak-649’ was used to analyze differential gene expression pathways between stage IV MYCN-A and -NA tumors.27 ‘Tumor Neuroblastoma non-MYCN amplified Seeger-102’ was used for Kaplan Meier analysis of TNF (TNF-α), TNFRSF1A (TNFR1) and TNFRSF1B (TNFR2) gene expression and to correlate pathway gene expression with survival in MYCN-NA disease.28 Survival analysis was performed using the median value as a cut-off between low and high expression. To evaluate TNF signaling and progression, the dataset was subjected to differential gene expression analysis based on disease progression using p<0.05 and twofold expression cutoffs. Gene set enrichment analysis (over-representation test) was performed using 2×2 contingency table analysis with continuity correction. Statistical tests, as indicated, were performed within R2 and p<0.05 was considered significant.
Statistics
Statistical analysis was performed using GraphPad Prism V.9.0 software (GraphPad). Unless otherwise indicated, the distribution of data was assumed normal, and parametric analyses were performed. Comparisons between groups were performed using Student’s t-test or one-way or two-way analysis of variance, with recommended post-test correction for multiple comparisons when appropriate. A p value of <0.05 was considered statistically significant.
Results
NB activates monocytes in a cell contact-dependent, TNF-α-dependent, and NF-κB-dependent manner
We previously demonstrated that coculturing monocytes with CHLA-255 MYCN-non-amplified NB cells induces monocytes to produce IL-6.9 To determine whether this response relates to the MYCN amplification status of NB cells, we cocultured monocytes with MYCN-amplified (A) and non-amplified (NA) NB cell lines and found that all lines induced monocytes to produce IL-6 regardless of MYCN status (figure 1A). To examine whether NB cells and monocytes must be in direct contact to elicit IL-6 production, monocytes from four donors were cocultured with CHLA-255 NB cells or cultured alone in medium conditioned for 1 week with CHLA-255 cells. After 24 hours, monocytes in the conditioned medium did not produce significantly more IL-6 than those cultured in control medium. In contrast, monocytes cocultured with CHLA-255 cells strongly upregulated IL-6 production, suggesting that the effect is contact-dependent (p<0.0001) (figure 1B).

NB induces monocyte IL-6 production through a contact-dependent and TNF-α-dependent mechanism. (A) Representative panel of NB lines and freshly isolated monocytes were directly cocultured at a 4:1 (NB:mono) ratio for 24 hours. Coculture supernatants were analyzed for IL-6 levels by ELISA. Mean±SD. (B) Four monocyte donors were cultured in regular 10% IMDM medium (control), 50% 7-day CHLA-255 conditioned medium (conditioned), or directly with CHLA-255 (NB cells) at a 4:1 ratio for 24 hours, and cytokine levels were determined by IL-6 ELISA. Mean±SD, pooled from four independent experiments run in triplicate. (C) Five NB cell lines were cocultured with monocytes treated with 10 µg/mL anti-TNF-α antibody or isotype control for 24 hours. IL-6 levels were measured by ELISA and normalized to isotype-treated control. Mean normalized IL-6 secretion across five cell lines±SD. (D) Intracellular IκBα expression level within CD45+GD2- monocyte population was determined by PhosFlow flow cytometry and IκBα mean fluorescence intensity (MFI) normalized to isotype-treated control. Results are averaged across all five cell lines±SD. *P<0.05, ****P<0.0001. A, MYCN amplified; IL, interleukin; IMDM, Iscove Modified Dulbecco Medium; NA, MYCN non-amplified; NB, neuroblastoma; ns, not significant; TNF-α, tumor necrosis factor alpha.
In monocytes and macrophages, canonical NF-κB signaling is a primary route to induce IL-6 production.29 We therefore treated NB-monocyte cocultures with IKK inhibitor VII (IKKi), which blocks canonical NF-κB signaling, and observed dose-dependent abrogation of both IL-6 and soluble tumor necrosis factor alpha (sTNF-α) production from monocytes (online supplemental figure 1). Given that TNF-α is a major activator of the NF-κB pathway, we evaluated the effect of TNF-α neutralization on monocyte activation in the coculture. Treatment with an anti-TNF-α antibody reduced monocyte IL-6 production by 60% during coculture with five different NB cell lines (p<0.0001) (figure 1C) and led to a 35% increase in IκBα levels compared with isotype antibody-treated controls (p<0.05) (figure 1D). IκBα expression relates inversely to NF-κB signaling activity; sample staining is shown in online supplemental figure 2).30 Together, these data demonstrate that NB cells activate NF-κB signaling in monocytes in a contact-dependent manner that leads to downstream IL-6 production and depends in part on TNF-α.
Monocyte mTNFα mediates NF-κB activation and protumorigenic inflammation
Next, we evaluated the contributions of NB cells and monocytes to the production of TNF-α within the coculture. We found that CHLA-255 cells did not express TNF-α, even after stimulation with LPS (<0.1%); conversely, LPS-stimulated monocytes expressed TNF-α (>65% TNF-α+) with a low level of basal expression observed (2% TNF-α+) (figure 2A). Furthermore, NB cell lines showed no TNF mRNA expression by qRT-PCR (online supplemental figure 3A,B). To verify this, we generated TNF-α KO NB using CRISPR/Cas9 (online supplemental figure 3C) and validated successful TNF KO in monocytic leukemia cell line U937 before generating CHLA-255 KO single-cell clones (online supplemental figure 3D–G). Coculture of monocytes with TNF-α KO versus wild-type CHLA-255 yielded similar levels of IL-6 production, indicating that monocytes are the source of TNF-α (figure 2B).
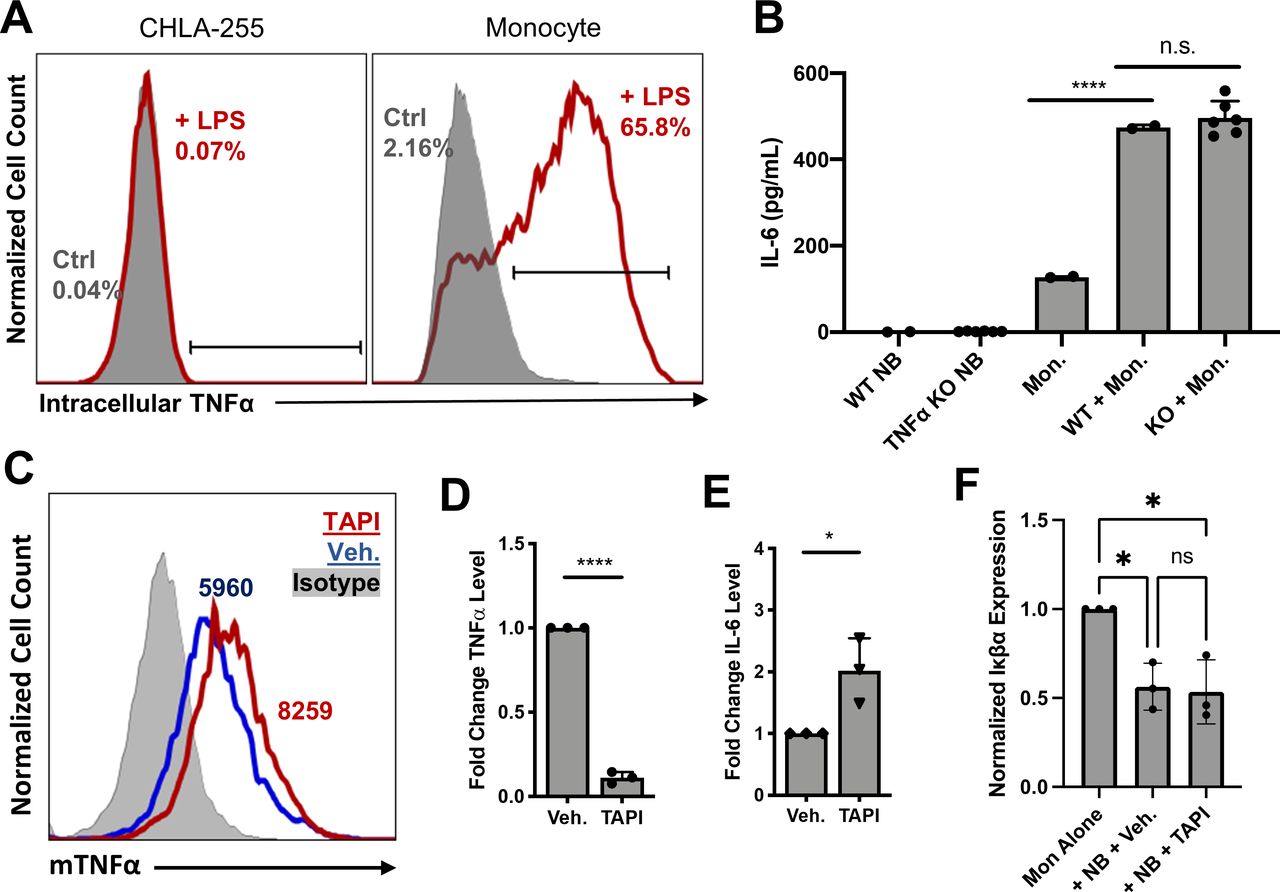
Monocyte mTNF-α, not NB TNF-α, enhances monocyte NF-κB activation. (A) CHLA-255 and monocytes were cultured alone in the presence of LPS (100 ng/mL) and GolgiStop (1.5 µL/mL) for 6 hours, and intracellular accumulation of TNF-α was measured by flow cytometry. Data are from a representative of two experiments run in duplicate. (B) WT and TNF-α KO CHLA-255 NB were cocultured with monocytes for 24 hours, and supernatant IL-6 levels were measured by IL-6 ELISA. Mean±SD of six validated KO clones. (C) NB and monocyte cocultures were treated with TNF converting enzyme inhibitor TAPI (40 µM) or Veh. (0.2% dimethyl sulfoxide (DMSO)) for 24 hours. Surface expression of mTNF-α within CD45+GD2− monocyte population was performed using flow cytometry. MFI as indicated. Data are from a representative of two experiments run in duplicate. (D) TNF-α and (E) IL-6 levels within coculture supernatants were determined by ELISA. Mean fold change in cytokine production±SD from three independent experiments run in duplicate. (F) Intracellular IκBα expression level within CD45+GD2− monocyte population was determined by PhosFlow flow cytometry and normalized to monocyte-only Ctrl. Data shown are mean normalized IκBα±SD from three independent experiments run in duplicate. *P<0.05, ****P<0.0001. Ctrl, control; IL, interleukin; KO, knockout; mTNF-α, membrane-bound tumor necrosis factor alpha; NB, neuroblastoma; ns, not significant; TNF-α, tumor necrosis factor alpha; Veh., vehicle control; WT, wild type.
TNF-α is expressed in two isoforms within cells: as a trimeric transmembrane protein (mTNF-α) and as a soluble cytokine (sTNF-α) cleaved from mTNF-α by TNF-α converting enzyme (TACE).31 32 To evaluate the contribution of each isoform to NF-κB activation, we treated cocultures with TACE inhibitor TAPI, which led to a 40% increase in surface staining for mTNF-α and a 90% reduction in sTNF-α levels in the coculture supernatant (p<0.0001) (figure 2C,D). Despite this reduction, TAPI treatment significantly increased IL-6 levels compared with a vehicle control (p<0.05) (figure 2E). Additionally, TAPI treatment did not alter NF-κB activation in monocytes during coculture compared with vehicle control (p<0.05) (figure 2F). Collectively, these findings demonstrate that monocyte-derived mTNF-α is sufficient to activate NF-κB signaling in monocytes, leading to downstream IL-6 production; however, we cannot exclude the possibility that both sTNF-α and mTNF-α are active in this process.
TNFR1 and TNFR2 play distinct roles in protumorigenic signaling loop between NB cells and monocytes
TNF-α signals through TNF-α receptors 1 and 2 (TNFR1/2), with sTNF-α mainly interacting with TNFR1 and mTNF-α with TNFR2. We evaluated the expression of TNFR1 and TNFR2 in NB cell lines and found that all lines expressed both receptors, with the highest levels in SK-N-AS and SK-N-BE2 (figure 3A). Surface expression also correlated with TNFRSF1A (encodes TNFR1) and TNFRSF1B (encodes TNFR2) mRNA expression (figure 3B,C).

NB TNFR2 expression is required for monocyte activation. (A) Representative panels of NB cell lines were analyzed for TNFR1 and TNFR2 surface expression (black line) by flow cytometry. Results are shown as percentage TNFR positive with gating threshold from isotype (gray histogram). Data are representative of two experiments run in duplicate. (B) TNFR1 expression was evaluated by qRT-PCR. C(t) values for TNFRSF1A were normalized against GAPDH housekeeping gene. Mean±SD run in duplicate. (C) TNFR2 expression was evaluated by qRT-PCR. C(t) values for TNFRSF1B were normalized against GAPDH housekeeping gene. Mean±SD run in duplicate. (D) WT, TNFR1, and TNFR2 KO SK-N-BE2 cells were cocultured with human monocytes from two different donors for 24 hours. Supernatant IL-6 levels were determined by ELISA. Mean IL-6 production normalized from monocyte background±SD, normalized data from two monocyte donors run in duplicate. (E) WT, TNFR1, and TNFR2 SK-N-BE2 KO clones were cocultured with human monocytes from two different donors for 24 hours. Intracellular IκBα expression level within the CD45-GD2+NB population was determined by PhosFlow flow cytometry and IκBα MFI normalized to NB alone control. Mean normalized IκBα MFI±SD, normalized data from two monocyte donors run in duplicate. **P<0.01,***P<0.001, ****P<0.0001. IL, interleukin; KO, knockout; NB, neuroblastoma; ns, not significant; qRT-PCR, quantitative reverse transcription PCR; TNFR1, TNFRSF1A mRNA; TNFR2, TNFRSF1B mRNA ; WT, wild type.
Next, we generated single-cell KO clones for TNFR1 (TNFRSF1A) and TNFR2 (TNFRSF1B) in SK-N-AS, CHLA-255, and SK-N-BE2 (online supplemental figure 4A,B); however, only SK-N-BE2 KO clones were evaluated further because clones of the other lines differentiated and stopped growing prior to analysis. SK-N-BE2 TNFR KO clones showed reduced surface protein expression of TNFR1 or TNFR2 without a compensatory increase in expression of the other TNF receptor (online supplemental figure 4C). To determine the role of each TNFR in TNF-α signaling within the NB-monocyte coculture, we cultured these SK-N-BE2 TNFR KO clones with freshly isolated monocytes and evaluated IL-6 levels after 24 hours. We found that only TNFR2 KO in NB altered monocyte activation, reducing coculture IL-6 levels by 80% (p<0.01) (figure 3D). These findings demonstrate that NB TNFR2 is crucial for induction of IL-6 production in monocytes, consistent with the observation that mTNF-α mediates this induction via non-canonical reverse signaling on the monocytic side.33
To determine the role of TNFR1 and TNFR2 in NB, we measured IκBα expression levels as a read-out for NF-κB activation following monocyte coculture. We found that NB NF-κB activation was only altered with TNFR1 KO as shown by a relative increase in IκBα levels following monocyte coculture compared with the wild-type control (figure 3E). To further investigate NB NF-κB signaling, which was more active in NB cell lines with the highest TNFR expression (online supplemental figure 5A), we explored whether TNF-α and/or IL-6 contribute to this activation. In NB cell lines treated with TNF-α, IL-6, IL-6 with secreted (s)IL-6Rα, or all three, TNF-α led to a drop in IκBα levels compared with untreated controls that became even more pronounced in combination with IL-6 and sIL-6Rα (online supplemental figure 5B). To determine whether this activation depends on a particular TNF-α isoform, NB-monocyte cocultures were treated with TAPI to block sTNF-α production. This resulted in a significant increase in IκBα expression versus the vehicle control group, indicating that sTNF-α partially activates NF-κB signaling in NB cells (online supplemental figure 5C). These results suggest that sTNF-α signals through TNFR1, leading to protumorigenic NF-κB activation in NB cocultured with monocytes.
TNF-α neutralization reduces tumor-promoting inflammatory reaction in vitro
The results presented thus far show that NB TNFR2 activates reverse signaling of mTNF-α on the monocyte surface, leading to activation of NF-κB and production of protumorigenic cytokines. To explore the clinical relevance of this mechanism, we used FDA-approved etanercept, an Fc-TNFR2 fusion protein that neutralizes signaling by both TNF-α isoforms.34 Treatment of NB-monocyte cocultures with etanercept blocked nearly all sTNF-α and IL-6 production and led to a complete and partial reversal of IκBα degradation in monocytes and NB, respectively, compared with an IgG control (figure 4A–D). To determine whether NB cell activation by monocyte mTNF-α is required for induction of IL-6 production by monocytes, we cocultured monocytes with fresh or fixed NB cells (online supplemental figure 6). We found that etanercept does not activate IL-6 production in monocytes at baseline and that fixed NB cell lines are still able to induce IL-6 production in monocytes in an etanercept/TNF-α-dependent fashion. These results reinforce the idea that surface expression of NB TNFR receptors is critical for monocyte activation and that NB cell signaling is not required for TNF-α-dependent induction of IL-6 production in monocytes.

Etan reduces protumorigenic signaling in vitro. (A–E) CHLA-255 was cultured with freshly isolated human monocytes for 24 hours in the presence of 10 µg/mL Etan or IgG in a vehicle Ctrl. (A) TNF-α level within coculture supernatant was determined by TNF-α ELISA. Mean TNF-α concentration±SD run in triplicate. (B) Supernatant IL-6 levels were determined by ELISA. Mean IL-6 concentration±SD run in triplicate (C) Intracellular IκBα expression level within CD14+GD2− monocyte population was determined by flow cytometry, and IκBα MFI was normalized to monocyte-only Ctrl. Mean normalized IκBα MFI±SD pooled from three independent experiments run in triplicate. (D) Intracellular IκBα expression level within the CD14-GD2+NB population was determined by flow cytometry, and IκBα MFI was normalized to NB-only Ctrl. Mean normalized IκBα MFI±SD pooled from three independent experiments run in triplicate. (E) Multiplex Luminex cytokine assay was performed on coculture supernatants. Heatmap shows mean log10 transformation of cytokine concentration in picogram per millilitre. Red arrows indicate cytokines significantly upregulated during coculture and ablated by Etan treatment across two monocyte donors. Luminex assay performed in duplicate on conditions run in triplicate. P values for relevant comparisons shown in online supplemental table 1. (F) CHLA-255-luc was cultured with freshly isolated human monocytes treated with 10 µg/mL Etan or Ctrl for 72 hours. NB luminescence was used to evaluate relative viability of cell line. Results are normalized mean luminescence±SD pooled from three independent experiments with six replicates. (G) CHLA-255 was cultured with freshly isolated human monocytes for 24 hours in the presence of 10 µg/mL Etan or Ctrl. The percentage of cells undergoing early apoptosis was identified by annexin-V(+), 7-AAD(−). Data expressed as mean percent early apoptosis±SD pooled from three monocyte donors run in triplicate. (H) CHLA-255 was pulsed for 10 min with CTV then cocultured with monocytes in the presence of 10 µg/m L Etan or Ctrl for 4 days. Shown is relative proliferation (normalized to the inverse MFI of NB CTV staining)±SD of three monocyte donors run in triplicate. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Ctrl, control; CTV, CellTrace Violet; Etan, etanercept; IL, interleukin; NB, neuroblastoma; ns, not significant; TNF-α, tumor necrosis factor alpha.
Next, we performed a multiplex Luminex cytokine array to evaluate global changes in cytokine production in the coculture following etanercept treatment. We found that 12 cytokines/chemokines (G-CSF, GM-CSF, IL-1α, IL-1β, IL-6, IL-10, IL-12-p40, IP-10, MCP-3, MIP-1α, MIP-1β, and TNF-α) were upregulated in CHLA-255-monocyte cocultures compared with NB or monocytes alone and subsequently ablated following treatment with etanercept (figure 4E) (online supplemental table 1). These results were consistent across multiple monocyte donors and NB cell lines (online supplemental figure 7).
Next, we evaluated the effect of etanercept on NB growth and viability in vitro by coculturing firefly luciferase-labeled CHLA-255 (CHLA-255-luc) with monocytes and treating with etanercept. Coculture with monocytes and control IgG resulted in a significant increase in NB luminescence intensity compared with NB cells cultured alone (p<0.01), and etanercept treatment completely abrogated the effect of monocytes on NB cells (p<0.001) (figure 4F). To evaluate the impact of etanercept on NB apoptosis, CHLA-255 was cultured with monocytes and etanercept or IgG control for 24 hours, and the percentage of NB cells undergoing early apoptosis was identified as annexin-V(+) 7-AAD(−) (online supplemental figure 7A). Coculture with monocytes led to a 50% reduction in NB early apoptosis (p<0.001), while etanercept induced a 44% increase in NB undergoing apoptosis compared with the control (p<0.05) (figure 4G). To examine changes in NB proliferation, CHLA-255 cells were pulsed with CTV and cocultured with monocytes in the presence of etanercept or control IgG (online supplemental figure 7B,C). Monocytes induced a 32% increase in NB proliferation (range 24%–42% across donors, p<0.01), which etanercept treatment then reduced by 72% (range 57%–78% across donors, p<0.01, data normalized to inverse MFI of NB CTV staining) (figure 4H). To determine the effect of etanercept treatment on phagocytosis, we evaluated NB CD47 expression and direct phagocytosis (online supplemental figure 9A,B). Coculture of CHLA-255 and SK-N-AS NB cells and monocytes led to an increase in CD47 expression in both NB cell lines, and this upregulation was ablated following etanercept treatment (p<0.0001) (online supplemental figure 9C). However, when these same NB cells were cultured with M2-polarized macrophages, we did not detect a significant increase in CFSE-pulsed NB cells within the macrophages, indicating that there is not a functional difference in phagocytosis following etanercept treatment (online supplemental figure 9D).
Together, these results indicate that etanercept effectively disrupts the protumorigenic mTNF-α/TNFR2 signaling axis between NB cells and monocytes, preventing NF-κB activation in both cell types. This reduces the production of protumorigenic cytokines by monocytes, ultimately resulting in increased apoptosis and decreased proliferation of NB cells in vitro.
TNF-α neutralization reduces tumor growth and alters the TME
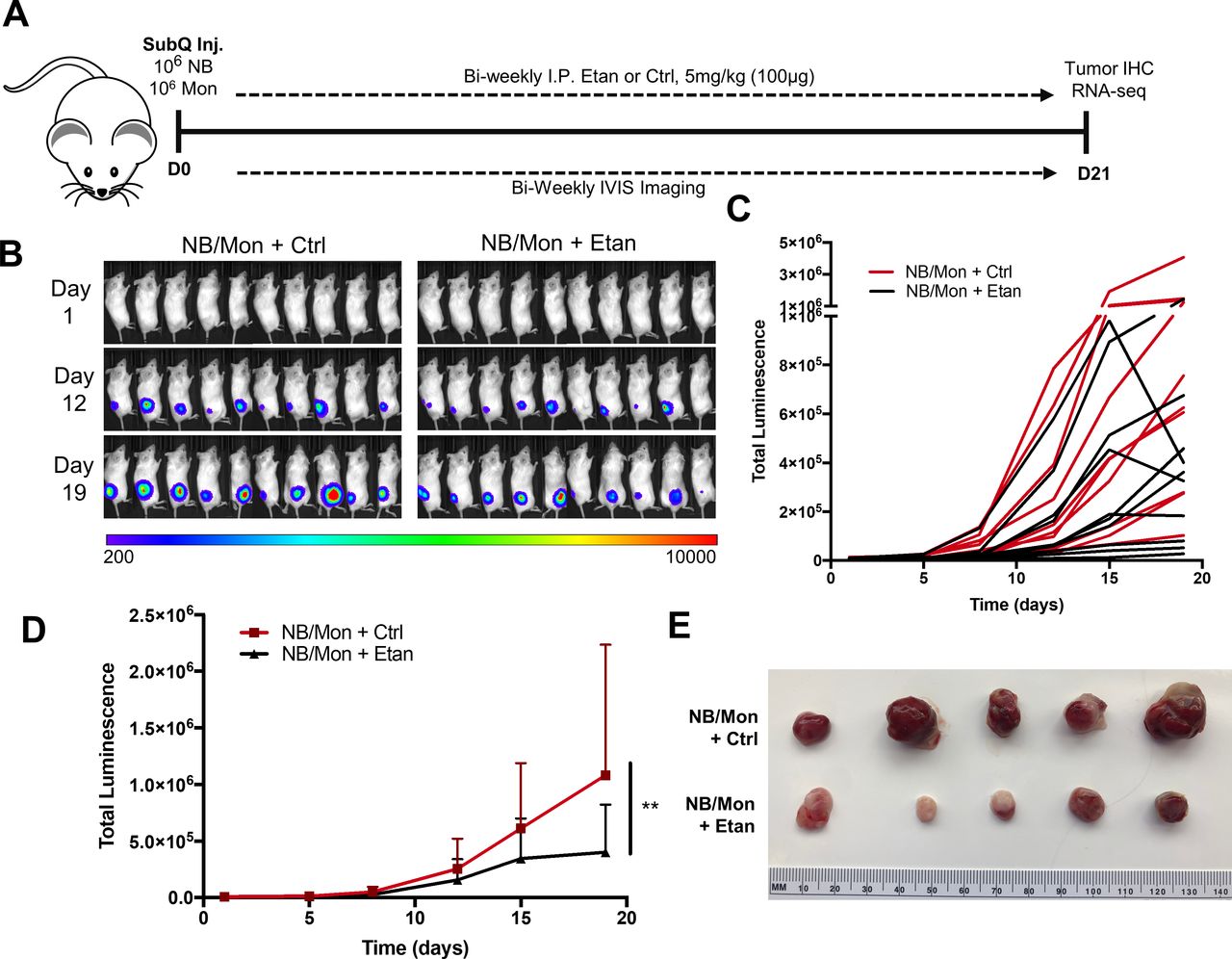
To evaluate the impact of etanercept in vivo, we engrafted Matrigel-embedded CHLA-255-luc and monocytes into the subcutaneous flanks of NSG mice and treated biweekly with etanercept or IgG control (figure 5A). Using luminescence as a readout for tumor growth (figure 5B,C), we found that etanercept treatment reduced tumor growth by 65% compared with control (p<0.01) (figure 5D,E). We observed similar results in mice engrafted with SK-N-AS-luc/monocyte xenografts, where etanercept treatment reduced tumor growth by 30% (p<0.05) (online supplemental figure 10). Importantly, treated tumors appeared to lack blood vessel development and were confined to the subcutaneous tissue of the flank, while control tumors appeared highly angiogenic and invaded underlying muscles.
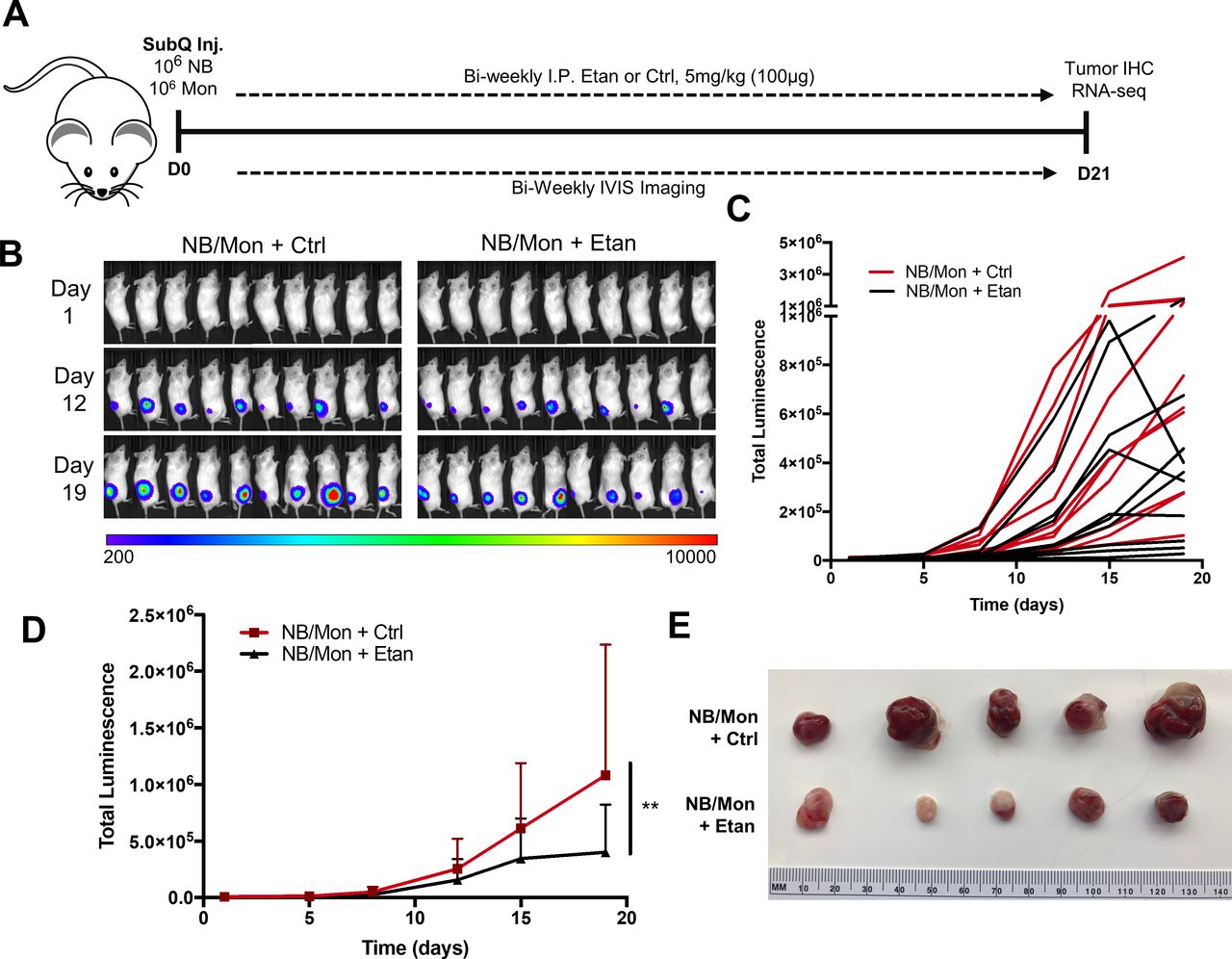
Etan treatment inhibits tumor growth in an NB/monocyte xenogeneic mouse model. (A) In vivo subcutaneous xenograft model. CHLA-255-luc and freshly isolated human monocytes were embedded in Matrigel and injected into the subcutaneous right flank of 8-week-old female NSG mice. Mice were injected intraperitoneally biweekly with 5 mg/kg (100 µg) Etan or IgG in a vehicle Ctrl, and tumor growth was tracked indirectly through luminescence imaging using IVIS imaging system. (B) Luminescence readings for each mouse (n=10/group) measured in radiance (photons/second/cm2/steradian). (C) Graphical representation of total luminescence per tumor from (B) over time. (D) Mean tumor luminescence over time, two-way analysis of variance with Bonferroni multiple comparisons test (n=10 per group). (E) Representative tumors for each group following sacrifice at day 21. **P<0.01. Ctrl, control; Etan, etanercept; NB, neuroblastoma.
To assess angiogenesis, we evaluated expression of murine endothelial marker CD31 and pericyte/fibroblast marker alpha smooth muscle actin (α-SMA) within the tumor parenchyma. Immunofluorescent staining for CD31 and α-SMA in tumor tissue showed the formation of complex vasculature in control tumors, while etanercept-treated tumors failed to form distinct vessels despite the presence of colocalized CD31+ and α-SMA+ cells (figure 6A and online supplemental figure 11). Quantification of ×20-magnified images of CD31 staining revealed a similar number of CD31+ regions per field of view (FOV) (figure 6B). However, in etanercept-treated tumors, the average area of CD31+ regions per FOV was significantly smaller (p<0.01), and vessel length as estimated by the maximum linear distance across CD31+ regions was significantly shorter than in control tumors (p<0.05) (figure 6C,D). Overall, these results validate the macroscopic observation that etanercept treatment reduces tumor angiogenesis.

Etanercept treatment alters NB TME. (A) Tumors recovered from mice in figure 5 were snap frozen in OTC media, thin sectioned, and stained for murine CD31 expression (red) and DAPI counterstain (blue). Results shown are representative images from five FOVs per tumor, with five tumors per group. Scale bar 50 µm. (B) Total number of CD31+ regions per FOV with box and whisker plots showing quartiles±range (Ctrl n=15, Etan n=25). (C) Size of each CD31+area quantified per FOV; shown is median CD31+ area per FOV with box and whisker plots showing quartiles±range (Ctrl n=15, Etan n=25) (D) Longest distance across each CD31+ area (max Feret distance) per FOV; shown is median distance per FOV with box and whisker plots showing quartiles±range (Ctrl n=15, Etan n=25). (E) Eukaryotic mRNA-seq was performed on tumor samples, and mouse transcripts were excluded from analysis by the XenoFilterR program. Differentially regulated genes were identified using twofold expression difference and adjusted p value of <0.05 as cut-offs. (F) Eukaryotic mRNA-seq was performed on murine component of tumor samples, and human transcripts were excluded from analysis by the XenoFilterR program. Differentially regulated genes were identified using twofold expression difference and adjusted p value of <0.05 as cut-offs. Over-representation GSEA revealed pathways differentially regulated within NB or the murine TME by Etan treatment compared with IgG Ctrl and are presented in table 1. Ctrl, control; Etan, etanercept; FOV, field of view; GSEA, gene set enrichment analysis; NB, neuroblastoma; ns, not significant; TME, tumor microenvironment.
Next, we performed eukaryotic mRNA-seq on representative etanercept-treated and control tumors shown in figure 5E. On average, 93% of obtained reads mapped to the human genome 6% mapped to the mouse genome, and 1% were unmappable and subsequently excluded. Within NB transcripts (uniquely human), we found 138 upregulated genes and 117 downregulated genes in etanercept-treated tumors compared with control tumors (figure 6E). Over-representation gene set analysis revealed a down-regulation of genes associated with angiogenesis, hypoxia and stress responses; inflammatory signaling including mitogen-activated protein kinase (MAPK) and IL-1 signaling, and neurogenesis in etanercept-treated tumors versus control (table 1). Over-representation gene set analysis on the murine component of etanercept-treated tumors showed reduced erythrocytes, platelets, and neutrophils (figure 6F and table 1). Notwithstanding the limitations of the immunodeficient NSG mouse model, these results are consistent with the observation that etanercept mediates potent antitumor activity in vivo by blocking tumor angiogenesis and inhibiting inflammatory and prosurvival signaling in NB cells.
Human and murine signaling pathways downregulated by etanercept treatment in mouse model of NB
TNF signaling is elevated in MYCN-NA NB and correlates with poor outcome
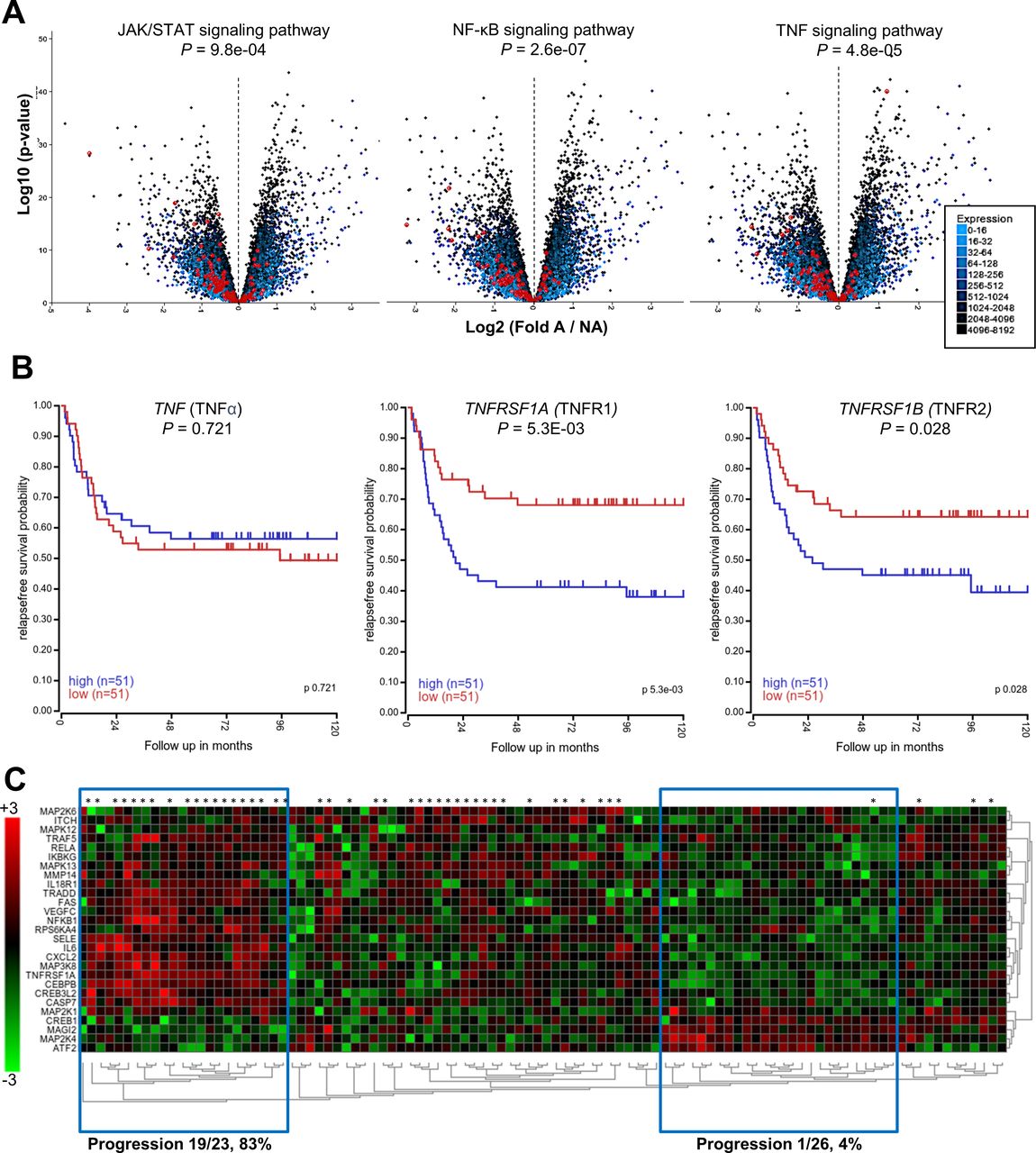
Given the importance of TNF-α in the protumorigenic interaction of NB and monocytes, we asked whether the expression of genes that mediate TNF-α signaling correlates with clinical outcome. Using datasets from the R2 Genomics Analysis and Visualization platform, we evaluated the differential expression of inflammatory signaling pathways in MYCN-A versus MYCN-NA stage IV NB tumors. We found that JAK/STAT, NF-κB, and TNF-α signaling pathways were all highly enriched in MYCN-NA NB (p<0.0001) (figure 7A). Using Kaplan-Meier analysis, we found that elevated expression of TNFRSF1A (TNFR1) and TNFRSF1B (TNFR2) correlated with reduced relapse-free survival (p=5.3×10−3 and p=2.8×10−2, n=102) in patients with stage IV MYCN-NA NB (figure 7B). We then used unsupervised clustering of gene expression to evaluate differential expression of genes in the Kyoto Encyclopedia of Genes and Genomes (KEGG) ‘TNF_Signaling_Pathway’, comparing patients with MYCN-NA NB that progressed with those who did not. We identified two distinct gene expression clusters that correlated significantly with progression-free survival (p=8.0×10−3): patients with elevated NF-κB signaling (TNFRSF1A, NFKB1, downstream cytokines, etc) had high frequency of progression (19/23, 83%), and patients with low NF-κB signaling had low frequency of progression (1/26, 4%) (figure 7C). Overall, these results reinforce the clinical relevance of TNF-α signaling, particularly for patients with MYCN-NA NB.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MYCN-non-amplified NB tumors are enriched in TNF-α signaling pathways. (A) Kocak neuroblastoma dataset of stage IV tumors only (n=211) was subjected to differential gene expression analysis based on the presence or absence of MYCN amplification (A, NA). Volcano plots depicting differential gene expression are overlayed with genes from the indicated pathways in red. Genes that are differentially expressed in NA tumors are left of the dashed axis line, and p values for pathway enrichment are indicated. A, n=65; NA, n=146. (B) Seeger NA dataset was subjected to Kaplan-Meier survival analysis based on gene expression using median expression as a cut-off for high (n=51) or low (n=51) expression. Shown are relapse free survival curves for TNF-α (TNF), TNFR1 (TNFRSF1A), and TNFR2 (TNFRSF1B). (C) Seeger NA neuroblastoma dataset was subjected to differential gene analysis between patients that relapsed (*) or had no progression. Unsupervised clustering of KEGG ‘TNF_Signaling_Pathway’ is displayed as a heatmap of differentially expressed genes; blue boxes indicate clusters of patients with distinct gene expression patterns that correlate highly with progression. Overall pathway association with progression free survival p=8.0×10-3. A, MYCN amplified; NA, MYCN non-amplified; NB, neuroblastoma TNF-α, tumor necrosis factor alpha; TNFR1, TNFRSF1A mRNA; TNFR2, TNFRSF1B mRNA.
Discussion
This study defines a novel positive feedback loop by which NB cells activate monocytes, initiating a self-sustaining inflammatory reaction that supports tumor growth. Central to this mechanism, we demonstrate an unexpectedly fundamental role for both secreted and membrane-bound TNF-α signaling in the crosstalk between NB and monocytic cells. In detail, NB TNFR2 reverse signals through mTNF-α expressed on monocytes, causing monocytes to produce sTNF-α that in turn binds to TNFR1 on NB cells and activates prosurvival NF-κB signaling. In addition to sTNF-α, mTNF-α-activated monocytes produce cytokines including IL-6 and G-CSF, which have been shown to promote NB growth via STAT3 activation. We also found that expression of TNF-α receptors and other genes associated with the TNF-α pathway are highly predictive of poor outcome in patients with NB. Importantly, targeting TNF-α with FDA-approved etanercept blocks monocyte activation by NB cells in vitro and inhibits angiogenesis and tumor growth in vivo.
We provide definitive evidence that cell–cell contact between NB cells and monocytes is required for activation of NF-κB in monocytes and production of downstream cytokines, and that this cascade begins with interaction of mTNF-α on monocytes and TNFR2 on NB cells. Interestingly, while both NB cells35 36 and monocytes37 38 have been reported to express mTNF-α, none of the eight NB cell lines that we tested expressed levels of TNF mRNA that could be detected by qRT-PCR. To explore this inconsistency, we tested a commonly used anti-mTNF-α monoclonal antibody (clone 6401) in validated TNF-α KO NB cells and detected positive staining that indicated non-specific activity of this antibody. Further, TNF-α KO NB clones activated monocytes to the same extent as wild-type NB, suggesting that monocytes are the relevant source of mTNF-α in the NB-monocyte axis. The results of our experiments treating monocytes with a TACE inhibitor further support the critical role of monocytic mTNF-α in the activation of monocytes by NB cells. The mTNF-α isoform has been reported to function as both a ligand (forward signaling) and a receptor (reverse signaling) in various cell types, including myeloid cells and macrophages.33 Our findings are consistent with the reverse signaling mode of mTNF-α action in monocytes following engagement by NB cell TNFR2.
The essential role of TNF-α in the NB-monocyte interaction is evidenced by experiments with etanercept, an Fc-TNFR2 fusion protein that mimics and therefore competes with NB cell TNFR2 for binding to monocytic mTNF-α. We found that etanercept blocks physical aggregation of NB cells and monocytes, further supporting the notion that TNFR2 initiates interaction between NB cells and monocytes. Strikingly, etanercept shuts down production of the full network of inflammatory cytokines, including IL-6, G-CSF, sTNF-α, and IL-1β that have been shown individually to promote tumor growth in numerous preclinical NB models and clinical studies.39–44
In particular, the role of IL-6 in supporting NB growth, metastasis, and resistance to therapy has been investigated extensively.9 39 40 For example, we previously demonstrated that tumor-infiltrating macrophages and monocytes are a major source of IL-6 in primary NB tumors and bone marrow metastases,9 and others have shown that IL-6 promotes NB cell proliferation and resistance to apoptosis in a manner that depends on STAT3 and ERK1/2 signaling.39 40 Clinically, elevated levels of IL-6 in the serum and bone marrow of patients with NB have been associated with lower event-free survival,45 and expression of IL-6R in MYCN-non-amplified NB tumors has been shown to correlate inversely with event-free survival.11 Future studies should aim to delineate the relative contributions of TNF-α and IL-6 to tumor progression and compare the therapeutic efficacy of inhibiting IL-6 and TNF-α individually and in combination.
Other STAT3-activating cytokines have also been shown to contribute to NB growth; for example, G-CSF drives tumor growth and metastasis in mouse xenograft NB models by supporting NB cells with cancer stem cell properties.42 43 Another recent study showed that NB cells, including primary tumor cells, induce monocytes to produce sTNF-α and IL-1β.44 These cytokines in turn induce NB cells to express ARG2, which catabolizes arginine and generates metabolic changes that favor NB growth and contribute to the immunosuppressive TME. We have shown here that interaction of NB TNFR2 with monocytic mTNF-α triggers an inflammatory reaction resulting in production of multiple tumor-promoting cytokines and that this reaction can be terminated by treatment with etanercept. In addition to its proinflammatory effects, TNF-α production in the TME has been shown to directly induce the epithelial-to-mesenchymal transition (EMT) and dedifferentiation of tumor cells.46 Therefore, we cannot exclude the possibility that EMT inhibition contributes to the overall therapeutic activity of etanercept against NB; this should be considered in further mechanistic studies beyond the scope of this work.
Our proof-of-concept therapeutic experiments demonstrate that etanercept also impacts tumor growth in vivo—NSG mice engrafted with human NB cells and primary human monocytes had smaller, less vascularized tumors when they were treated with etanercept compared with tumors from control mice. This disruption of the TME is consistent with the role of tumor-associated monocytes, monocyte-derived macrophages, and their respective cytokines in tumor angiogenesis, invasion, and metastasis in many types of cancer.47 48 Additional studies beyond the scope of this investigation may be needed to evaluate the effect of etanercept on adaptive immunity, which cannot be addressed in a xenogeneic model. Another limitation of the model employed in our in vivo studies is the fact that human monocytes do not survive long-term in NSG mice; specifically, we did not detect monocytes or macrophages of human origin in tumor tissues at 3 weeks post injection. To compensate for this limited window of persistence, we used a high monocyte-to-NB cell ratio during tumor implantation as in previous studies using the same model.9 26 Future studies will require the development of tumor models that allow endogenous generation and differentiation of human monocytes and macrophages—for example, NSG-SGM3 mice reconstituted with human stem cells.49
Gene expression analysis of NB xenografts revealed that etanercept-treated tumors had decreased ability to respond to stress and hypoxia, as well as decreased expression of genes associated with the prosurvival MAPK pathway activated during the innate immune response.50 The RAS–MAPK pathway is also commonly impacted by gain-of-function mutations in many types of cancer51; indeed, mutations in this pathway were recently shown to occur frequently in relapsed NB and to correlate with poor survival.52 This suggests that tumor-induced inflammation not only creates a TME that is conducive to tumor progression and activates prosurvival NF-κB and STAT3 signaling pathways but can also enhance key cell-intrinsic oncogenic signaling in NB cells. Therefore, combining etanercept with small-molecule MEK inhibitors could be a promising therapeutic strategy for recurrent/resistant NB. Overall, elucidation of this novel molecular feedback loop that underlies a tumor-initiated inflammation cascade will inform the design of new pathophysiology-driven therapeutic strategies for high-risk NB and other types of cancer.
Supplemental material
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
The authors are grateful for the excellent technical assistance provided by the staff of the Flow Cytometry Core Laboratory of the Texas Children’s Cancer and Hematology Center, Small Animal Imaging Core Facility at Texas Children’s Hospital, and the Integrated Microscopy Core at Baylor College of Medicine.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors The study was designed by JAT and LSM. LSM is guarantor for this work. Monocyte isolation, neuroblastoma coculture, and functional testing were performed by JAT, ANC, KHD, and SW. Design, isolation, and functional testing of CRISPR knockouts was performed by JAT. In vivo experiments were performed by JAT, XX, LG, JB, GAB, and GT. RNA-seq analysis was performed by CZ. Data analysis including statistical analysis was performed by JAT, ANC, CZ, and LSM. The manuscript was written by JAT and LSM and edited by EJDP.
Funding This work was supported by grants from the National Institutes of Health (RO1 CA116548 and RO1 CA262250 to LSM), Cancer Prevention and Research Institute of Texas (Baylor College of Medicine Comprehensive Cancer Center Training Program, RP160283, to JAT), and Robert and Janice McNair Foundation (McNair Medical Institute at Baylor College of Medicine, to JAT). Clinical grade etanercept was supplied by Amgen free of charge.
Competing interests JAT and LSM are coinventors on a pending patent application that relates to targeting protumorigenic inflammation for cancer therapy. Other authors have declared that no conflict of interest exists.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.