Article Text

Abstract
Chimeric antigen receptor (CAR)-T cells have revolutionized the treatment of multiple types of hematological malignancies, but have shown limited efficacy in patients with glioblastoma (GBM) or other solid tumors. This may be largely due to the immunosuppressive tumor microenvironment (TME) that compromises CAR-T cells’ delivery and antitumor activity. We previously showed that blocking vascular endothelial growth factor (VEGF) signaling can normalize tumor vessels in murine and human tumors, including GBM, breast, liver, and rectal carcinomas. Moreover, we demonstrated that vascular normalization can improve the delivery of CD8+ T cells and the efficacy of immunotherapy in breast cancer models in mice. In fact, the US FDA (Food and drug administration) has approved seven different combinations of anti-VEGF drugs and immune checkpoint blockers for liver, kidney, lung and endometrial cancers in the past 3 years. Here, we tested the hypothesis that anti-VEGF therapy can improve the delivery and efficacy of CAR-T cells in immunocompetent mice bearing orthotopic GBM tumors. We engineered two syngeneic mouse GBM cell lines (CT2A and GSC005) to express EGFRvIII—one of the most common neoantigens in human GBM—and CAR T cells to recognize EGFRvIII. We found that treatment with the anti-mouse VEGF antibody (B20) improved CAR-T cell infiltration and distribution throughout the GBM TME, delayed tumor growth, and prolonged survival of GBM-bearing mice compared with EGFRvIII-CAR-T cell therapy alone. Our findings provide compelling data and a rationale for clinical evaluation of anti-VEGF agents with CAR T cells for GBM patients.
- tumor microenvironment
Data availability statement
Data is available upon reasonable request. The final research data underlying this publication is available, upon request, in printed or electronic form (as appropriate).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Chimeric antigen receptor (CAR)-T cell therapy, in which T cells derived from patients are genetically engineered to express synthetic molecules designed to redirect T cells to specific tumor (neo)antigen(s), has revolutionized the treatment of hematological malignancies, but has no or limited effect in solid malignancies1 2. Several designs of CAR-T cells have been tested in clinical trials for glioblastoma (GBM)3 4 and, in at least one case, mediated the regression of late-stage, multifocal, bulky disease.3 However, these responses have not been durable. For a durable response, CAR-T cells must: (1) infiltrate into and homogeneously distribute throughout the tumor microenvironment (TME) in adequate quantities, (2) enact potent and localized anti-tumor function and (3) persist and multiply in TME.5
The ideal target epitope for CAR-T cell therapy should be expressed exclusively on tumor cells while being absent in normal cells. One attractive therapeutic target for GBM is epidermal growth factor receptor variant III (EGFRvIII), although Her2 or IL-13Rα2 targeted CAR-T cells have also been evaluated in GBM. EGFRvIII is formed by the in-frame deletion of exons 2–7. EGFRvIII is present in about 30%–50% of human GBM,6 but is absent from all normal tissues. Autologous EGFRvIII-CAR-T cell therapy trials in GBM patients showed that intravenous infusion of EGFRvIII-CAR-T cells is safe, without evidence of significant toxicity, including cytokine release syndrome.4 6 However, preclinical and clinical studies have suggested that intravenously administered EGFRvIII-CAR-T cells are only capable of migrating to either the invasive edges of GBM or only in limited areas of the TME,4 which prevents CAR T cells from reaching all tumor cells. Thus, strategies that can improve CAR-T cell infiltration and overcome resistance mechanisms posed by the abnormal TME are urgently needed.
We previously showed that blocking vascular endothelial growth factor (VEGF) signaling can normalize tumor vessels in murine and human tumors, including GBM, breast, liver and rectal carcinomas, and improve the efficacy of various anticancer therapies.7 8 Moreover, blocking VEGF-signaling has been shown to increase trafficking of adoptively transferred T (ATC) cells into the tumor and improved ATC-based immunotherapy.9 Here, we hypothesized that normalizing GBM TME with an anti-VEGF agent will not only increase the infiltration of CAR-T cells in the GBM TME, but also improve the CAR-T function, resulting in increased efficacy of CAR-T cells. In this study, we targeted VEGF using an anti-VEGF antibody (B20) in two preclinical models of GBM that recapitulate different features in human GBM: CT2A (limited neoantigen load) and GSC005 (stem-cell like).10 Our results show that B20 increases both delivery and efficacy of EGFRvIII-CAR-T cells in these GBM models.
Methods and materials
Reagents
We obtained B20 (a monoclonal antibody against murine VEGF) from Genentech, and purchased the following reagents: RPMI1640 Medium, Dulbecco’s Modified Eagle Medium (DMEM), Fetal Bovine Serum (FBS), Non-Essential Amino Acids (NEAA), Sodium pyruvate, Insulin-Transferrin-Selenium, 2-mercaptoethanol, polyethylenimine (PEI), Retronectin (Takara Bio), CellTracker Red CMTPX Dye (ThermoFisher), Myc-Tag Alexa Fluor 647 (Cell Signaling, clone: 9B11). Murine EGFRvIII-CAR-expressing construct was a gift from Dr. Darrell Irvine’s lab at MIT.11 It contains antigen-specific scFv (clone 139 scFv for EGFRvIII CAR), mouse CD8a hinge and transmembrane domain, CD28 costimulatory domain, CD3z intracellular domain, and a Myc tag for CAR detection by flow cytometry (online supplemental figure 1A).
Supplemental material
Mice
C57BL/6 mice were obtained from the Cox-7 animal facility operated by the Edwin L. Steele Laboratories, Department of Radiation Oncology at the MGH. All animal experiments were performed in the Cox-7 defined flora animal facility, accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Both female and male mice were used. All animal studies are fully approved by the Institutional Animal Care and Use Committee of MGH prior to the initiation of the Project. MGH adheres to the Public Health Service Policy on Humane Care of Laboratory Animals. The OPRR Animal Welfare Assurance number is A3596-01, 9/17/97. The animal protocol we used in this study is 2004N000050.
Isolation of murine primary T cells and generation of EGFRvIII-CAR-T cells
Murine T cells were isolated from mouse spleens using a T cell isolation kit (STEMCELL Technologies), and cultured in T cell culture medium (RPMI+10% FBS + 1 x NEAA+1 x Sodium pyruvate+1 x Insulin-Transferrin-Selenium + 1 x 2-mercaptoethanol).11 To activate T cells, a T-75 flask was precoated with 10 mL of PBS with anti-CD3 (0.5 µg/mL) and anti-CD28 (5 µg/mL) at 4°C overnight. T cells were then cultured at 37°C for 48 hours without disturbance. For retrovirus production, 293 T cells were cultured in DMEM supplemented with 10% FBS. One hour before transfection, each well was replenished with prewarmed medium. For transfection, 3 µg of plasmid (2.25 µg of CAR plasmid plus 0.75 µg of Eco packaging plasmid) was added to 50 µL of PBS. Then, 9 µg of PEI (1 µg/µL) was added into 50 µL PBS. Next, plasmids and PEI were mixed by vortexing for 10 s. After a 15 min incubation at room temperature, the transfection mixture was gently added to 293 T cells. 24 hours later, old medium was removed and replenished with prewarmed medium without disturbing the cells. Virus-containing supernatant was collected another 48 hours later and passed through a 0.45 µm filter to remove cell debris.
On the transduction day, activated T cells were collected, counted and resuspended at 2×106 cells/mL in T cell culture medium supplemented with 40 IU/mL of murine IL-2. Then, 1 mL of virus supernatant was first added into each well of the Retronectin-coated six-well plates, and 1 mL of the above cell suspension was added and mixed well by gentle shaking. Spin infection was carried out at 2000 ×g for 120 min at 32°C. Plates were then carefully transferred to an incubator and maintained overnight. 24 hours later, old medium was removed and replenished with prewarmed T cell culture medium with 20 IU/mL of murine IL-2. Transduction efficiency was evaluated by surface staining of Myc tag using an anti-Myc antibody (Cell Signaling Technology) 72 hours after transduction. For the following experiments, CAR-T cells were collected and used 4 days after transduction.
Cell culture and generation of EGFRvIII-expressing GBM cells
The murine CT2A was obtained from Dr. Thomas N. Seyfried’s laboratory at Boston College12 and cultured in DMEM supplemented with 10% FBS. GSC005 cells expressing GFP were obtained from Dr. Samuel D. Rabkin’s laboratory at MGH and cultured as neurospheres.13 The CT2A cell lines expressing GFP and secretable Gaussia luciferase (GLUC) were generated by transducing cells with a lentiviral vector coexpressing GFP and GLUC,14 provided by the MGH vector core, followed by sorting. EGFRvIII-expressing CT2A cells and GSC005 cells were generated by lentiviral transduction of GFP-GLUC-CT2A cells or GFP-GSC005 cells with a murine EGFRvIII construct, followed by sorting. All cell lines were grown in a humidified atmosphere of 5% CO2 and 95% air at 37°C and repeatedly tested and were negative for mycoplasma using the Mycoalert Plus Mycoplasma Detection Kit (Lonza).
In vitro co-culture assays
EGFRvIII-CT2A tumor cells (or parent CT2A) were seeded in 96-well plate as 1×105 cells in 100 µL tumor cell culture medium. Then 1×105 of EGFRvIII-CAR-T cells (or naïve T cells) 100 µL T cell culture medium was added to the plate. Six hours and 24 hours later,11 supernatant was collected, and cytokines were measured using ELISA.
Tumor models
C57BL/6 mice were injected with tumor cells (1×105 EGFRvIII-CT2A-GFP-GLUC or 5×104 EGFRvIII-GSC005-GFP) orthotopically using a stereotactic device.10 All brain tumor cells were implanted in the forebrain. Tumor size was measured either by high frequency ultrasound or blood GLUC.10 15 We also found good correlation (R2=0.78) between Gluc value and tumor volume measured by ultrasound in EGFRvIII-GFP-Gluc-CT2A tumor bearing mice (online supplemental figure 2). Tumors were size-matched and randomized to treatment groups.
For survival studies, C57BL/6 mice bearing EGFRvIII-CT2A-GFP-GLUC or EGFRvIII-GSC005-GFP-GLUC were treated with: (A) PBS, (B) EGFRvIII-CAR-T cells (2×106 cells per mouse, intravenous injection), (C) B20 (2.5 mg/kg of body weight, every 3 days for four doses), (D) isotype control IgG (2.5 mg/kg of body weight, every 3 days for four doses) + EGFRvIII-CAR-T cells or (E) B20 (2.5 mg of body weight/kg, every 3 days for four doses) + EGFRvIII-CAR-T cells. Tumor size was measured every 3 days until animals showed the first clinical sign of morbidity (including serious movement problems, hunch-back, and/or weight loss beyond 15%), at which time the mice were euthanized.
Cranial window and multiphoton intravital imaging
To implant the transparent cranial window, a 6 mm circle was drilled on the skull bilaterally using a highspeed drill.16 The brain was cleaned, and the window was sealed with a 7 mm circular cover slip (glass or plastic for ultrasound), glued to the bone with histocompatible cyanoacrylate glue. For imaging experiment, EGFRvIII-CAR-T cells were fluorescently labeled with CellTracker Red CMTPX Dye.17 Mice were imaged 24 hours after the retro-orbital injection of CAR-T cells. The cranial window-bearing animals were anaesthetized and fixed with a bilateral plastic holder. Power for the multiphoton microscopy was estimated to be 2.5 mW. The emission band-pass filters used were 610±75 nm for fluorescently labeled CAR-T cells and 535±43 nm for GFP-GSC005. The imaging was performed and visualized with high-resolution z stacks (2.5 µm per step, total step ~250 µm).
Flow cytometry
For the immunological studies, mice were sacrificed 3 days after the fourth treatment. Tumors were harvested and single-cell suspension was conducted for flow cytometry experiment. Single-cell suspensions were prepared from tumor tissues. Cells were washed and stained with different antibodies with suggested concentrations. Surface staining was performed in FACS buffer (2% BSA in PBS) on ice for 20 min. For cytokine expression analysis, cells were placed in RPMI containing 1× Brefeldin A and 10% FBS for 4.5 hours. After surface staining, cells were fixed in Fix/Perm buffer for 20 min, washed in permeabilization buffer, and stained for intracellular factors in permeabilization buffer for 20 min on ice. Flow cytometry was performed on BD LSRFortessa-X20, and data analysis was performed on FlowJo.
Statistical analysis
Statistics were performed using Prism V.9. Student’s t-test or one-way analysis of variance with Tukey’s post-hoc test was used as indicated in the figure legends. The Kaplan-Meier method and log-rank test was used for survival studies as indicated in the figure legends. Student’s t-test was used for two arm studies as indicated in the figure legends. N represents the number of mice used in the experiment, with the number of individual experiments listed in the legend. Graphs show individual or in case of survival studies combined experiments/samples. Results are presented as mean with or without error bars showing the SE of the mean. Differences with p<0.05 were considered statistically significant.
Results
Anti-VEGF treatment improves the efficacy of EGFRvIII-CAR-T cells in murine GBM tumor models
We first generated the murine EGFRvIII-CAR-T cells using RetroNectin-based transduction.11 We found the transduction efficiency to be higher than 75%, as evaluated by surface staining of c-Myc tag with an anti-Myc antibody 72 hours after transduction (online supplemental figure 1B). As murine GBM cells do not spontaneously express EGFRvIII, we stably expressed EGFRvIII in CT2A and GSC005 GBM cell lines to (online supplemental figure 1C). EGFRvIII expression in both cell lines was greater than 90%. Next, we co-incubated EGFRvIII-CAR-T cells and EGFRvIII-GBM cells for 6 and 24 hours, and found elevated levels of IFN-γ in culture medium (online supplemental figure 1D), indicating the killing activity of EGFRvIII-CAR-T cells.
Next, we tested the killing ability of EGFRvIII-CAR-T cells in vivo, but only observed minor difference between CAR-T cells and vehicle control (figure 1C,E). To test if VEGF-blockade can potentiate the therapeutic response of EGFRvIII-CAR-T cells, we treated mice bearing size-matched GSC005 and CT2A tumors (volume of 1.5 mm3) with (A) PBS, (B) EGFRvIII-CAR-T cells (2×106 cells per mouse, intravenous injection), (C) B20 (2.5 mg/kg, every 3 days, four times), (D) isotype control IgG (2.5 mg/kg, every 3 days, our times) + EGFRvIII-CAR-T cells or (E) B20 (2.5 mg/kg, every 3 days, four times+EGFRvIII-CAR-T cells. While B20 or EGFRvIII-CAR-T cell monotherapy showed only minor improvement in the animal survival, the combination of B20 with EGFRvIII-CAR-T cells significantly improved the survival benefit compared with monotherapy with B20 or CAR-T cells (figure 1C,E).
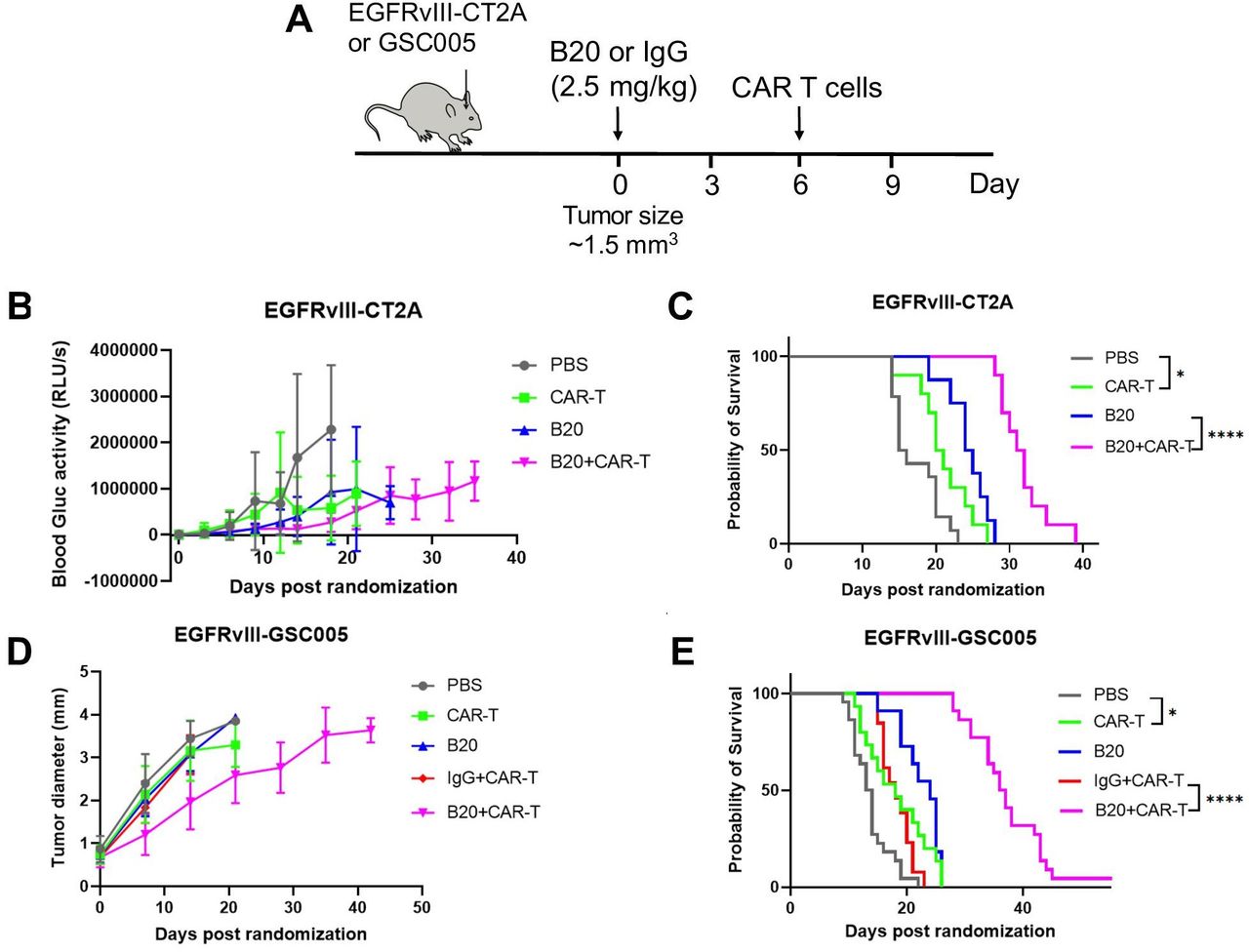
Anti-VEGF treatment improves the efficacy of EGFRvIII-CAR-T cells in murine GBM tumor models. (A) Schematic representation of experimental setup to evaluate the effect of PBS, CAR-T, B20, IgG+CAR T and B20+CAR T on the survival of GSC005 and C2TA GBM-bearing mice. (B) and (C) Tumor growth kinetics and median survival for CT2A tumors (PBS (n=22, 15.5 days), CAR-T (n=14, 20.5 days), B20 (n=8, 24.5 days), B20+CAR T (n=19, 32 days)). (D) and (E) Tumor growth kinetics and median survival for GSC005 tumors (PBS (n=12, 13.5 days), CAR-T (n=17, 18.5 days), B20 (n=10, 24 days), IgG+CAR T (n=13, 18 days), B20+CAR T (n=21, 37 days)). Error bars show median±SEM. Statistical analysis was performed using Student’s t-test or one-way ANOVA test. *p<0.05, ****p<0.0001. ANOVA, analysis of variance; CAR, chimeric antigen receptor; GBM, glioblastoma; VEGF, vascular endothelial growth factor. PBS, phophate buffered saline.
Anti-VEGF treatment improves the infiltration of EGFRvIII-CAR-T cells
To test whether the improved survival resulted from the increased number of EGFRvIII-CAR-T cells in the GBM TME, we first used intravital microscopy to observe the infiltration of CAR-T cells into GFP-GSC005 TME with or without B20 treatment (figure 2A). EGFRvIII-CAR-T cells were fluorescently labeled with CellTracker Red CMTPX Dye.17 Twenty-four hours after CAR-T cell injection, we found that the number of infiltrating CAR-T cells significantly increased after B20 treatment compared with control (PBS+EGFRvIII-CAR-T cells) (figure 2A, online supplemental movies SM1 and SM2). In addition to the intratumoral distribution and number of CAR-T cells, we also measured the distribution of CAR-T cells in brains using immunostaining. We collected tumors from GFP-GSC005 bearing mice after treatment with (1) PBS+EGFRvIII-CAR-T cells and (2) B20+EGFRvIII-CAR-T cells. Treatment with PBS did not show CAR-T cell infiltration, whereas treatment with B20 did (figure 2B). Lastly, we performed flow cytometry to quantify the CAR-T cell numbers in tumor (figure 2C). We found that B20 increased CAR-T accumulation from 3.4% to 14.9%. Collectively, these data suggest that anti-VEGF therapy improves the infiltration of EGFRvIII-CAR-T cells in GSC005 TME.
Supplementary video
Supplementary video

Anti-VEGF treatment improves the infiltration of EGFRvIII-CAR-T cells. (A) Intratumoral distribution of EGFRvIII-CAR-T cells inside of GFP-GSC005 GBM tumors imaged by multiphoton microscopy. Images were taken 24 hours after the injection of CAR-T cells (fluorescently labeled as red). Scale bars=50 µm. (B) Measurement of CAR-T cell (pink) number in GFP-GSC005 tumors (green) treated with PBS or B20 (2.5 mg/kg). scale bar=50 µm. (C) Percentage of EGFRvIII-CAR-T cells inside of GSC005 GBM tumors measuring by flow cytometry. Error bars show ±SEM. Statistical analysis was performed using Student’s t-test. *p<0.05, **p<0.01. CAR, chimeric antigen receptor; GBM, glioblastoma. PBS, phosphate buffered saline.
Anti-VEGF treatment enhances antitumor function of CAR-T cells and host T cells in GBM
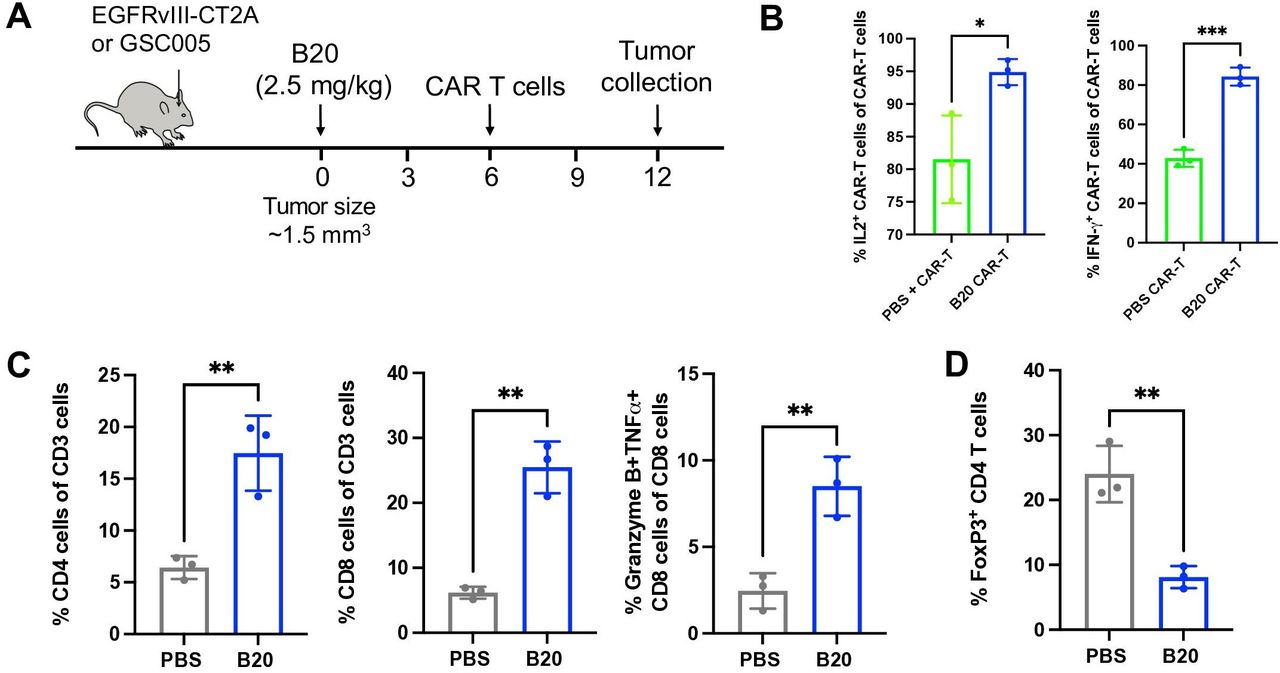
To examine the effects of anti-VEGF therapy on the function of CAR-T cells and other immune cells in the GBM TME, we performed flow cytometry of GSC005 tumors (figure 3A). Both the total CAR-T cells as well as IL-2+ and IFN-γ + CAR T cells increased after anti-VEGF therapy compared with control or CAR-T therapy alone (figure 3B). Moreover, Granzyme B+TNF-α+ CD8 T cells increased significantly during combined B20 treatment, as did the overall number of CD4+ and CD8+ in the TME (figure 3C). We also checked the myeloid population in the GBM TME (online supplemental figure 3A). We observed no changes in the percentage of CD45high/CD11b+/F4/80+/GR1- macrophages or Ly6Clow monocytes in tumors after B20 treatment (online supplemental figure 3B). However, we did observe a slight increase of neutrophils in tumors after B20 treatment.18 We did not observe any changes in CD45+TCR-CD8a+dendritic cell population after B20 treatment (online supplemental figure 3B). Taken together, the flow cytometry data suggest that anti-VEGF therapy can not only improve CAR-T cell function, but also reprogram the GBM immune compartment to a more immunostimulatory milieu.

{kind=link}
{kind=link}
{kind=link}
Anti-VEGF treatment enhances function of CAR-T cells and effector T cells in GBM. (A) GSC005 tumors were treated as shown in the schematic. (B) Percentage of IL2+and IFN-γ+ CAR T cells inside the GSC005 GBM tumors. (C) Percentage of CD4+T cells, CD8+T cells, and the cytotoxic Granzyme B+TNF-α+ CD8+ T cells in the GSC005 tumors. Error bars show ±SEM. Statistical analysis was performed using Student’s t-test. *p<0.05, **p<0.01, ***p<0.001. CAR, chimeric antigen receptor; VEGF, vascular endothelial growth factor.
Discussion and conclusion
GBM is one of the most challenging tumors to treat.19 Despite advances in the standard of care with surgery and chemoradiation, antiangiogenic therapy, and tumor-treating fields, the median survival for patients with GBM is less than 2 years. More effective therapeutic strategies for GBM are urgently needed. CAR-T cell therapy represents one such approach and has shown some efficacy in GBM; however, robust responses have not been achieved.3 GBM blood vessels are abnormal and leaky, characterized by heterogeneous permeability and poor perfusion.20 Poor perfusion impairs the delivery of endogenous CD8+T cells and CAR-T cells,8 20 and the resulting hypoxia and low pH increases immunosuppression.8 20 Thus, the abnormal TME confers resistance to CAR-T cell therapy in GBM by limiting their infiltration and efficacy. Previous studies from our laboratory and others have shown that anti-VEGF therapies can normalize the tumor vasculature and improve the outcome of various anti-cancer agents (chemotherapies, radiationtherapies, nanotherapies, molecularly targeted therapies and immunotherapies).7 20 21 Therefore, we investigated if anti-VEGF therapy could increase CAR-T cell infiltration and improve the efficacy of CAR-T cells in GBM models.
Our study has several advantages over other CAR-T studies previously performed using human GBM cell lines in immunodeficient hosts. First, we used two murine GBM models that sufficiently recapitulate different features found in human GBMs. The CT2A GBM model has a low mutational burden (specifically without IDH1-R132H mutation) and exhibits immunological features similar to human GBM, including reduced anti-PD-L1 antibody sensitivity and hypofunctional tumor infiltrating lymphocytes.22 The GSC005 model, which is derived from a genetically engineered mouse model and recapitulates the stem-like features of human GBM, has a low mutational burden.23 Second, with the full immune compartment present in C57BL/6 mice, we not only evaluated endogenous immune activity after anti-VEGF+CAR T therapy, but also used B20 to target VEGF in both murine cancer and stromal cells, which may better represent the treatment setting in clinic.
Prior studies have employed CAR-T cells that target VEGF receptors 1, 2 and 3 (VEGFR1, VEGFR2 and VEGFR3) on the endothelial cells. However, a phase I/II clinical trial was terminated as no responses were observed using VEGFR2-CAR-T therapy.24 A recent study by Lanitis et al demonstrated that the upregulation of VEGF-A in cancer cells competed for VEGFR2 binding with VEGFR2-targeting CAR-T cells, thus diminishing the effect of this CAR-T therapy.25 Combining anti-VEGF/R2 antibodies with VEGFR2-CAR-T therapy may solve this problem. Indeed, coadministration of anti-VEGF-A antibody in vivo promoted CAR-T cell persistence and tumor control in B16 melanoma. Lanitis et al attributed this effect to restored CAR-T cell vascular adhesion. In line with this study, we found that anti-VEGF therapy improved antitumor efficacy of EGFRvIII-directed CAR-T cells, which does not rely on VEGFR2 on the endothelial cells for adhesion. Additionally, Bocca et al showed improved infiltration and efficacy of GD2-CAR T cells in a human neuroblastoma xenograft model in immunodeficient mice using bevacizumab.26 Since bevacizumab does not neutralize VEGF produced by murine cells, the role of host-derived VEGF in infiltration and efficacy of CAR-T cells in their study is not known. By using syngeneic GBM models, our study provides evidence for improved delivery and antitumor efficacy of CAR T cells by anti-VEGF therapy in a setting closer to the clinic.
In summary, our study provides a strategy to overcome major challenges in CAR-T cell therapy in GBM by: (1) increasing the CAR-T cell infiltration, intratumoral distribution, and activation in murine GBM models and (2) reprograming TME by increasing the number and activation of endogenous effector T cells, resulting in improved antitumor efficacy of CAR-T therapy in two GBM mouse models. Given that anti-VEGF therapies have been approved for a number of solid tumors, including GBM,21 our study provides mechanistic insights and compelling preclinical data in support of testing the combination of vascular normalizing agents and CAR-T therapies in GBM patients. Furthermore, this approach may also improve CAR-T therapy of other solid tumors that share similar TME features as well as for other cell-based therapies using autologous or allogenic immune cells (eg, NK cells, macrophages).27
Data availability statement
Data is available upon reasonable request. The final research data underlying this publication is available, upon request, in printed or electronic form (as appropriate).
Ethics statements
Patient consent for publication
Acknowledgments
We would like to thank Dr. Darrell Irvine of MIT for providing the murine EGFRvIII-CAR and EGFRvIII constructs, Dr. Thomas N. Seyfried of Boston University for providing CT2A cells, and Dr. Samuel D. Rabkin of Massachusetts General Hospital for providing GSC005 cells. We also thank Drs. Heena Kumra, Hye-Jung Kim, Igor dos Santos and Sampurna Chatterjee for their helpful input on this manuscript; and Anna Khachatryan and Carolyn Smith for their technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @MGHSteeleLabs
XD and JR contributed equally.
Contributors XD, JR and RKJ contributed to conception and design; XD, JR, ZA, MaD, SR and MeD contributed to methodology; XD, JR, ZA, SL, SR, MaD contributed to acquisition of data; XD, ZA contributed to analysis and interpretation of data; XD contributed to writing of the manuscript; XD, JR, ZA, SL, MeD, DF and RKJ contributed to review and revision of the manuscript. All authors approved the final version of the manuscript, including the authorship list.
Funding This work was supported by grants from the National Foundation for Cancer Research (RKJ); the Ludwig Center at Harvard Medical School (RKJ); the Jane’s Trust Foundation (RKJ); the Nile Albright Research Foundation (RKJ); and the NIH grants R35-CA197743, R01-CA269672, R01CA259253, U01CA261842 and U01-CA224348 (RKJ), R01-CA208205 (DF and RKJ), and R01NS118929 (DF). XD was supported by the Fund for Medical Discovery Fundamental Research 21-2 from Massachusetts General Hospital. JR was a Cancer Research Institute (CRI)-Dr. Keith Landesman Memorial Fellow, and was supported by the CRI Irvington Postdoctoral Fellowship and the Fund for Medical Discovery Fellowship from Massachusetts General Hospital. ZA was supported by the Aid for Cancer Research Award, and the Tosteson Fellowship and Cancer Center Excellence Award from Massachusetts General Hospital. MeD was supported by NIH/NCI K22-CA258410 and the American Association for Cancer Research-Loxo Oncology 19-40-50-DATT.
Competing interests RKJ received consultant fees from Bristol Myers Squibb, Elpis, Innocoll, SPARC, and SynDevRx; owns equity in Accurius, Enlight, and SynDevRx; is on the Board of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla Healthcare Opportunities Fund, and Tekla World Healthcare Fund; and received a research grant from Boehringer Ingelheim. JR owns stock options in Dynamic Cell Therapies.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.