Article Text

Abstract
Background This phase 1 study evaluated PF-06753512, a vaccine-based immunotherapy regimen (PrCa VBIR), in two clinical states of prostate cancer (PC), metastatic castration-resistant PC (mCRPC) and biochemical recurrence (BCR).
Methods For dose escalation, patients with mCRPC received intramuscular PrCa VBIR (adenovirus vector and plasmid DNA expressing prostate-specific membrane antigen (PSMA), prostate-specific antigen (PSA), and prostate stem cell antigen (PSCA)) with or without immune checkpoint inhibitors (ICIs, tremelimumab 40 or 80 mg with or without sasanlimab 130 or 300 mg, both subcutaneous). For dose expansion, patients with mCRPC received recommended phase 2 dose (RP2D) of PrCa VBIR plus tremelimumab 80 mg and sasanlimab 300 mg; patients with BCR received PrCa VBIR plus tremelimumab 80 mg (Cohort 1B-BCR) or tremelimumab 80 mg plus sasanlimab 130 mg (Cohort 5B-BCR) without androgen deprivation therapy (ADT). The primary endpoint was safety.
Results Ninety-one patients were treated in dose escalation (mCRPC=38) and expansion (BCR=35, mCRPC=18). Overall, treatment-related and immune-related adverse events occurred in 64 (70.3%) and 39 (42.9%) patients, with fatigue (40.7%), influenza-like illness (30.8%), diarrhea (23.1%), and immune-related thyroid dysfunction (19.8%) and rash (15.4%), as the most common. In patients with mCRPC, the objective response rate (ORR, 95% CI) was 5.6% (1.2% to 15.4%) and the median radiographic progression-free survival (rPFS) was 5.6 (3.5 to not estimable) months for all; the ORR was 16.7% (3.6% to 41.4%) and 6-month rPFS rate was 45.5% (24.9% to 64.1%) for those who received RP2D with measurable disease (n=18). 7.4% of patients with mCRPC achieved a ≥50% decline in baseline PSA (PSA-50), with a median duration of 4.6 (1.2–45.2) months. In patients with BCR, 9 (25.7%) achieved PSA-50; the median duration of PSA response was 3.9 (1.9–4.2) and 10.1 (6.9–28.8) months for Cohorts 5B-BCR and 1B-BCR. Overall, antigen specific T-cell response was 88.0% to PSMA, 84.0% to PSA, and 80.0% to PSCA.
Conclusions PrCa VBIR overall demonstrated safety signals similar to other ICI combination trials; significant side effects were seen in some patients with BCR. It stimulated antigen-specific immunity across all cohorts and resulted in modest antitumor activity in patients with BCR without using ADT.
Trial registration number NCT02616185.
- vaccination
- therapies, Investigational
- prostatic neoplasms
- immunogenicity, vaccine
Data availability statement
Data are available upon reasonable request. Data may be obtained from a third party and are not publicly available. Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The use of immunotherapy in the treatment of prostate cancer (PC) has been far less successful compared with more ‘immunologically responsive’ cancers; only the vaccine sipuleucel-T has demonstrated modest survival benefit and is approved for the treatment of metastatic castration-resistant PC (mCRPC).
WHAT THIS STUDY ADDS
This phase 1 study (NCT02616185) evaluated PF-06753512, a vaccine-based immunotherapy regimen (VBIR) for PC (PrCa VBIR) that combines a vaccine prime/boost strategy with immune checkpoint inhibitors (ICIs), in two clinical states of PC, mCRPC and biochemical recurrence (BCR).
PF-06753512 overall demonstrated safety signals similar to other ICI combination trials, significant side effects were seen in some patients with BCR, a modestly prolonged radiographic progression-free survival was observed in patients with mCRPC, and a prompt and durable ≥50% decline in baseline prostate-specific antigen was observed without the use of androgen deprivation therapy in some patients with BCR.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
PrCa VBIR stimulated antigen-specific immunity in most patients, and the results are suggestive of immune-mediated antitumor activity with PrCa VBIR, particularly in BCR; therefore, further investigation of vaccine and ICI combinations in patients with PC is warranted.
Introduction
Immunotherapy with checkpoint inhibition has significantly improved the treatment of many solid tumors, especially for microsatellite instable solid tumors.1 Using immunotherapy in the treatment of prostate cancer (PC) has been far less successful compared with more ‘immunologically responsive’ cancers, such as lung cancer, melanoma, and renal cell carcinoma.2–8 At present, only the vaccine sipuleucel-T has demonstrated modest survival benefit, leading to its approval for the treatment of metastatic castration-resistant PC (mCRPC).9 Responses to checkpoint inhibitor therapy in patients with mCRPC are generally limited to those patients with microsatellite instability.10 In PC, there remains an unmet need for novel immunologic strategies to provide long-term disease control, with manageable safety profiles, in the recurrent and metastatic setting.
The natural history of recurrent PC can be viewed as a series of clinical states beginning with localized disease, followed by a state of biochemical recurrence (BCR), with progression to radiographically visible metastatic, hormone-sensitive disease; ultimately, mCRPC, an advanced form of the disease, then ensues.11–13 The use of androgen deprivation therapy (ADT) serves as the backbone of treatment for those with high-risk BCR, and all patients with hormone-sensitive and castration-resistant metastatic disease. The various clinical disease states of PC have distinct biological characteristics. These same clinical stages may harbor different immunological profiles that could influence response to immunotherapy.3 8 14–18 Therefore, there remains uncertainty surrounding the optimal patient populations in which to develop and assess vaccine-based immunotherapies.3 8 14–18
PC expresses specific tumor-associated antigens, such as prostate-specific antigen (PSA), prostate-specific membrane antigen (PSMA), and prostatic acid phosphatase (PAP), which are attractive targets for antigen-based PC vaccines.2 4 7 Apart from sipuleucel-T, other vaccination strategies, such as PROSTVAC,19 Prostate GVAX,20 21 and TG4010,22 which incorporate a variety of antigens and delivery systems, have been unsuccessful therapeutically.19 In more recent vaccine development, the incorporation of immune checkpoint inhibitors (ICIs) is proposed to induce, expand, and maintain T-cell function leading to better antitumor immune responses, perhaps by overcoming the ‘immunologically cold’ environment of PC.3 5
PF-06753512 is a vaccine-based immunotherapy regimen (VBIR) for PC (PrCa VBIR) in development for the treatment of patients in distinct clinical states of PC. PrCa VBIR combines a vaccine with ICIs, and uses novel methods of administration of these compounds, including electroporation of antigens encoded within plasmid DNA (pDNA), along with subcutaneous (SC) administration of ICIs. PrCa VBIR contains an adenovirus (AdC68) vector expressing three selected PC-specific antigens, PSA, PSMA, and prostate stem cell antigen (PSCA); pDNA (PF-06755990); and an anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) monoclonal antibody, tremelimumab (PF-06753388). Multiple overexpressed PC-specific antigens were selected to improve the probability that patients will benefit from the vaccine.23 24 AdC68 was used for priming vaccination, as antigen-specific T-cell responses have been induced by adenovirus vectors.25 26 The rationale for this combinatorial approach is to promote the induction, expansion, and long-term maintenance of T-cell derived, anti-PC immune responses at the site of metastatic disease. In preclinical models, T-cell responses were amplified at each prime and boost vaccination with PrCa VBIR.27 If effective, PrCa VBIR could potentially avoid the significant toxicity of androgen deprivation in BCR and delay the need for chemotherapy in mCRPC.
The purpose of this phase 1 study (NCT02616185) was to explore the safety, antitumor activity, immune response, pharmacokinetics (PK), and pharmacodynamics (PD) of escalating doses of PrCa VBIR (PF-06753512) in patients with PC.
Methods
Study design
This was a phase 1, open-label, multicenter, multiple-dose, safety, PK, PD, and immunogenicity study. The study enrolled and treated patients at 11 study centers in the USA between December 30, 2015, and February 23, 2021. The study was divided into two parts: Part 1, dose escalation enrolling patients with mCRPC, followed by Part 2, dose expansion enrolling patients with mCRPC and BCR (figure 1).

Study design. The dose for AdC68 was 6×1011 VP if not specified. Post-secondary hormone was defined as a failed androgen receptor-targeted therapy. AdC68, adenovirus vector; BCR, biochemical recurrence; mCRPC, metastatic castration-resistant prostate cancer; pDNA, plasmid DNA; RP2D, recommended phase 2 dose; TRM, tremelimumab; PF-06801591, sasanlimab; VP, viral particles.
The PrCa VBIR regimen comprised the following components: (1) a priming immunization with a replication-deficient adenovirus (AdC68) vector, derived from a chimpanzee-originated AdC68 expressing three selected PC-specific antigens (PSA, PSMA, and PSCA), administered intramuscularly; (2) DNA plasmid booster vaccinations, encoding the same antigens, administered intramuscularly with an electroporation device (TriGrid Delivery System); (3) SC administration of tremelimumab; and (4) for some cohorts, a programmed cell death protein 1 (PD-1) monoclonal antibody, sasanlimab (PF-06801591), was given SC. All intramuscular injections (AdC68 and the DNA booster vaccine) were given bilaterally in the deltoid muscles or in the vastus lateralis muscles of the quadriceps. SC injections (tremelimumab and sasanlimab) were given near the vaccinated muscles and the vaccine draining lymph nodes.
In the dose-escalation portion enrolling only patients with mCRPC, a 3+3 design was used. A cycle of PrCa VBIR included AdC68 priming dose (intramuscularly) on Day 1, followed by three pDNA boosts (5 mg fixed dose, intramuscularly) using an electroporation device at 4-week intervals for 12 weeks (16 weeks total). AdC68 dose of 4×1011 viral particles (VP) was given to Cohort 1A-mCRPC and 6×1011 VP to Cohort 2A-mCRPC. Since toxicity was determined to be acceptable in the first two cohorts, the AdC68 dose of 6×1011 VP was used for all subsequent cohorts. From Cohort 3A-mCRPC, the PrCa VBIR cycle included SC tremelimumab every 4 weeks (Q4W). For Cohorts 3A-mCRPC, 6A-mCRPC, and 7A-mCRPC, SC tremelimumab was given at 80 mg Q4W; for Cohort 9A-mCRPC, SC tremelimumab was dosed at 40 mg Q4W. Starting with Cohorts 6A-mCRPC through 9A-mCRPC, the PrCa VBIR cycle also included the anti-PD-1 antibody sasanlimab given SC Q4W, starting at 130 mg (Cohorts 6A-mCRPC and 9A-mCRPC) and escalated up to 300 mg (Cohort 7A-mCRPC).
In the dose-expansion portion, PrCa VBIR alone (AdC68 6×1011 VP, pDNA, SC tremelimumab 80 mg) and in combination with sasanlimab were evaluated in two populations: (1) patients with mCRPC who had disease progression despite abiraterone and/or enzalutamide treatment were treated with PrCa VBIR plus SC sasanlimab 300 mg (Cohort 3B-mCRPC); or (2) patients with BCR were treated with PrCa VBIR alone (Cohort 1B-BCR) or PrCa VBIR plus SC sasanlimab 130 mg (Cohort 5B-BCR). After completing two cycles of treatment (treatment duration was approximately 8 months), a maintenance treatment period with pDNA and tremelimumab administered every 2 months was provided as long as patients were deriving clinical benefit. Patients continued to receive sasanlimab Q4W where applicable. Treatment continued until disease progression, participant refusal, or unacceptable toxicity occurred.
Blood samples were collected from patients who received tremelimumab and sasanlimab for PK, PD, and immunogenicity assessments at scheduled times (details see online supplemental materials).
Supplemental material
The study was approved by either institutional review boards or an independent ethics committee at each study center. There was no patient involvement in the study design. The study was conducted in accordance with all local legal and regulatory requirements, as well as the general principles set forth in the International Ethical Guidelines for Biomedical Research Involving Human Patients, Guidelines for Good Clinical Practice, and the Declaration of Helsinki. All patients provided written informed consent.
Patients
Eligible patients were men aged ≥18 years with a histological or cytological diagnosis of adenocarcinoma of the prostate accompanied by adequate bone marrow, renal, and liver function. Patients were excluded if they met any of the following conditions: a diagnosis of neuroendocrine PC or a neuroendocrine component; metastases to the brain or liver; Eastern Cooperative Oncology Group performance status ≥2; prior malignancy other than PC <3 years, excluding successfully-treated basal cell carcinoma or squamous cell carcinoma of the skin; systemic anticancer therapy <4 weeks or 5 half-lives; chemotherapy in mCRPC; sipuleucel-T and PROSTVAC <12 months; or investigational agents, immunosuppressive dose of corticosteroids, or other immunosuppressive medication <30 days.
For patients with mCRPC, those in Cohorts 1A-mCRPC, 2A-mCRPC, and 3A-mCRPC had progressed on ADT, but were not required to have prior novel androgen receptor-directed therapy (eg, enzalutamide or abiraterone); those in Cohorts 7A-mCRPC through 9A-mCRPC and 3B-mCRPC were required to have prior therapy with enzalutamide and/or abiraterone, testosterone <50 ng/dL, disease progression per modified Prostate Cancer Working Group 3 (PCWG3) criteria, and currently being treated with a gonadotrophin-releasing hormone agonist or antagonist (unless surgically castrated). Measurable disease based on the Response Evaluation Criteria in Solid Tumors (RECIST) criteria was not required for patients with mCRPC; however, for patients with bone-only metastatic disease, this was required to be evaluable using PCWG3 criteria.
Patients with BCR (Cohorts 1B-BCR and 5B-BCR) were required to meet the following criteria: definitive local therapy for primary diagnosis; rising PSA with a minimum PSA of 0.2 ng/mL; PSA doubling time (PSADT) <10 months, testosterone >150 ng/dL; and no metastatic disease on conventional imaging (CT/MRI or bone scintigraphy).
Endpoints
Dose-escalation portion
The primary endpoint was incidence and grade of treatment-emergent adverse events (TEAEs), including dose-limiting toxicities (DLTs), as graded by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) V.4.03. Secondary endpoints included: immune response such as T cells specific to the three selected PC tumor-antigens, laboratory abnormalities, tremelimumab and sasanlimab single-dose PK parameters, and the incidence and titers of anti-drug antibodies (ADA) against tremelimumab and sasanlimab (sampling time for PK and ADA assessments see online supplemental materials). Exploratory endpoints were: objective response rate (ORR) per RECIST V.1.1; antitumor response and tumor control duration based on total measurable tumor burden per immune-related RECIST V.1.1 (irRECIST); bone metastatic disease as evaluated per PCWG3 criteria; radiographic progression-free survival (rPFS) as assessed by RECIST V.1.1 and PCWG3; and baseline and changes from baseline for PSA and PSADT.
Dose-expansion portion
The primary endpoints were: rates of TEAEs, including DLTs, and laboratory abnormalities. Secondary endpoints included: PSA-50 response rate (defined as the proportion of patients whose on-study locally measured PSA declined from baseline by ≥50% at two consecutive measurements ≥3 weeks apart prior to other systematic anticancer therapy) and duration of response (DOR); baseline and changes from baseline for PSA and PSADT; and the incidence and titers of ADA against tremelimumab and sasanlimab. Immune response, including T cells specific to the three selected PC tumor-antigens, was assessed as an exploratory endpoint.
Dose-escalation and expansion portions
For patients with mCRPC in Cohort 3B-mCRPC and 7A-mCRPC who were treated at the same dose (AdC68+DNA plasmid booster vaccine+tremelimumab 80 mg + sasanlimab 300 mg), additional secondary endpoints were: ORR and DOR per RECIST; antitumor response and tumor control duration based on total measurable tumor burden per irRECIST; and rPFS by RECIST, irRECIST, and PCWG3 criteria.
Specific T-cell response analysis was conducted on Cohort 5B-BCR, and on Cohort 7A-mCRPC and 3B-mCRPC.
Statistical analyses
Details on sampling, sample size, and analysis populations are in online supplemental materials. The results are presented as overall, all patients with mCRPC, and patients with BCR.
Safety was assessed with adverse events (AEs) and serious AEs (SAEs) described by Medical Dictionary for Regulatory Activities (V.23.1) preferred term and graded by NCI CTCAE (V.4.03). Immune-related AEs (irAEs) were defined as immune-related, based on the need for immunosuppressive therapy such as steroids, or endocrine replacement therapy. The definition of irAE also required that no alternative clinical explanation for the AE could be found, other than immune-related. In the first 28 days following the first AdC68 vaccination, the occurrence of specific hematologic and non-hematologic AEs (not considered related to disease or disease progression) potentially attributable to the drug combination was classified as a DLT (definitions see online supplemental materials).
Antitumor activity was assessed through radiologic tumor assessments, including bone scintigraphy and CT or MRI conducted at baseline, during treatment, at suspected disease progression, and at the time of withdrawal from treatment. Assessment of tumor response was per RECIST and irRECIST, and bone metastatic disease assessed per PCWG3. Progression-free survival was estimated using the Kaplan-Meier method.
Blood samples for PK analyses were assayed for all analytes using validated analytical methods. Plasma PK parameters were estimated using non-compartmental analysis, and PK parameters were summarized descriptively.
Analyses of immunogenicity and PD endpoints were summarized descriptively. Peripheral blood mononuclear cell (PBMC) samples were assayed for antigen-specific T-cell response against PSMA, PSCA, and PSA antigens by applying a modified enzyme-linked immunospot (ELISpot) assay including an initial in vitro expansion. Numbers of interferon (IFN)-γ secreting spot-forming cells were counted and scored as a positive T-cell response if there was a ≥2-fold count at any time point post vaccination compared with baseline.
Software used was SAS V.9.4 (SAS Institute, North Carolina, USA). T-cell response analysis was calculated using R V.3.6.1.
Results
Patient disposition and demography
Overall, 91 patients were treated in the dose escalation (mCRPC: n=38) and dose expansion (n=53; BCR: n=35, mCRPC: n=18). Most patients were white (86.8%), with a median (range) age of 70.0 (52–88) years and a median Gleason score of 7.0 (table 1). The median (range) duration of treatment, in months, was 3.94 (0.03–47.11) for all patients, 3.22 (0.03–46.49) for patients with mCRPC, 22.06 (0.95–47.11) for Cohort 1B-BCR (patients with BCR), and 2.79 (0.03–17.48) for Cohort 5B-BCR (patients with BCR). Median number of cycles was two for all patients (including patients with mCRPC), three for Cohort 1B-BCR, and one for Cohort 5B-BCR; the corresponding treatment duration (4 months per cycle) was 8, 12, and 4 months. The median (range) duration of follow-up, in months, was 9.8 (1–51) for all patients, 8.5 (1–51) for patients with mCRPC, and 12.9 (2–48) for patients with BCR.
Baseline demographics and characteristics
Safety
Overall, one patient in Cohort 6A-mCRPC (mCRPC) had a DLT of Grade 4 myositis and Grade 3 myasthenia gravis. Both events were attributable to AdC68, tremelimumab, and sasanlimab, which resulted in permanent discontinuation of study treatment.
mCRPC
In patients with mCRPC, all-causality AEs were reported in 55 (98.2%) and SAEs in 16 (28.6%) patients. The most common all-causality AEs were anemia (46.4%), fatigue (41.1%), and arthralgia (39.3%). Treatment-related AEs (TRAEs) were reported in 51 (91.1%) and serious TRAEs in 11 (19.6%) patients. The most common TRAEs were fatigue (35.7%), nausea (21.4%), and diarrhea (21.4%). Grade 3 or 4 TRAEs developed in 23 (41.1%) patients, and 1 patient had a Grade 5 TRAE of pulmonary embolism as a complication of myasthenia gravis (table 2).
Summary of TRAEs occurring in ≥20% of patients in Cohort 1B-BCR, Cohort 5B-BCR, and all patients with mCRPC
Permanent discontinuations due to all-causality AEs occurred in 16 (28.6%) patients, among whom, 13 (23.2%) were due to TRAEs. The non-serious AEs leading to discontinuation were Grade 3 rash maculopapular, amylase increased, hyperglycemia, rash papular, and weight decreased; and Grade 4 lipase increased. The SAEs leading to discontinuation were Grade 2 myositis; Grade 3 colitis and diarrhea; Grade 4 myasthenia gravis (DLT), and Grade 5 pulmonary embolism.
The irAEs occurred in 23 (41.1%) patients, 12 (21.4%) had Grade 3 irAEs (colitis, rash, hepatitis, and myositis), 1 (1.8%) had Grade 4 irAEs of myasthenia gravis and myositis, and none had Grade 5 irAEs. The most common irAEs were thyroid dysfunction (23.2%), hypothyroidism (12.5%), hyperthyroidism (10.7%), and diarrhea (10.7%).
BCR
In patients with BCR (Cohorts 1B-BCR and 5B-BCR), all-causality AEs were reported in 20 (100%) and 15 (100%) patients, and SAEs were reported in 2 (10.0%) and 6 (40.0%) patients. The most common all-causality AEs were fatigue (60.0%), influenza-like illness (60.0%), and arthralgia (40.0%) in Cohort 1B-BCR, and diarrhea (53.3%), fatigue (53.3%), and increased aspartate aminotransferase (AST; 46.7%) in Cohort 5B-BCR. TRAEs and serious TRAEs were reported in 19 (95.0%) and zero patients in Cohort 1B-BCR, and 15 (100%) and 6 (40.0%) patients in Cohort 5B-BCR. The most common TRAEs were influenza-like illness (60.0%), fatigue (45.0%), and injection site pain (30.0%) in Cohort 1B-BCR; and fatigue (53.3%), increased AST (40.0%), and diarrhea (40.0%) in Cohort 5B-BCR. In Cohorts 1B-BCR and 5B-BCR, Grade 3 or 4 TRAEs developed in 6 (30.0%) and 9 (60.0%) patients, and Grade 5 TRAE in 0 and 1 (immune-mediated myocarditis) patient (table 2).
In Cohort 1B-BCR, 1 (5.0%) patient discontinued due to a Grade 4 TRAE of uveitis. In Cohort 5B-BCR, of the 10 (66.7%) patients who permanently discontinued due to AEs, 9 (60.0%) patients had TRAEs. The non-serious TRAEs leading to discontinuation were Grade 2 muscular weakness, diarrhea, and hyperthyroidism; and Grade 3 alanine aminotransferase increased, fatigue, influenza-like illness, and a non-TRAE of right kidney clear cell carcinoma. The treatment-related SAEs leading to discontinuation were Grade 3 hypothyroidism and syncope, Grade 4 colitis and immune-related myocarditis, and Grade 5 immune-related myocarditis.
The irAEs occurred in 4 (20.0%) patients in Cohort 1B-BCR, with 1 patient having Grade 4 immune-related uveitis and no other Grade ≥3 irAEs. In Cohort 5B-BCR, 12 (80.0%) patients had irAEs, with immune-related thyroid dysfunction as the most common (8, 53.3%); of the 12 patients, 4 had Grade 3 irAEs (rash, colitis, adrenal insufficiency, pituitary dysfunction, thyroid dysfunction, and hepatitis), 2 had Grade 4 irAEs (colitis, hepatitis, myocarditis, and myositis), and 1 had Grade 5 immune-mediated myocarditis.
Death
Overall, 6 (6.6%) patients died; 2 (2.2%, immune-mediated myocarditis and pulmonary embolism) were considered treatment-related. The six TEAEs with fatal outcome included neoplasm progression in two patients (one patient with mCRPC in Cohort 3B-mCRPC and one patient with mCRPC in Cohort 7A-mCRPC), sepsis in one patient with mCRPC in Cohort 3B-mCRPC, immune-mediated myocarditis in one patient with BCR in Cohort 5B-BCR, and pulmonary embolism in one patient with mCRPC in Cohort 6A-mCRPC.
Clinical response
mCRPC
Of 54 patients with mCRPC in the modified intention-to-treat population inclusive of those without RECIST measurable disease (n=30, 55.6%) (table 1), at a median follow-up of 8.5 (range 1–51) months, 1 patient in Cohort 3B-mCRPC had a confirmed complete response (CR, the CR was a 1.5-cm para-aortic lymph node in this patient who withdrew from study therapy early after Grade 3 diarrhea), 1 patient each in Cohorts 7A-mCRPC and 3B-mCRPC had a confirmed partial response (PR), and 8 (4 in Cohort 3B-mCRPC, 1 each in Cohorts 2A-mCRPC, 3A-mCRPC, 6A-mCRPC, and 7A-mCRPC) had stable disease (SD) (figure 2, online supplemental table S1). ORR was 5.6% (n/N=3/54, 95% CI: 1.2% to 15.4%) and disease control (confirmed and unconfirmed CR+PR+SD+non-CR/non-progressive disease) rate was 29.6%. Of the 24 (44.4%) patients with mCPRC who had RECIST measurable disease at baseline (table 1), the ORR was 12.5% (3/24). The median (range) duration of SD was 5.4 (1.4–13.3) months, the median (range) DOR (CR+PR) was 5.6 (3.9–7.4) months, and the median (95% CI) rPFS (defined as time from start date of treatment to the date of first documented progression of disease by PCWG3-defined bone progression or RECIST, or death due to any cause, whichever came first) was 5.6 (3.5 to not estimable) months in patients with mCRPC (online supplemental figure S1). Three patients had progression in bone per PCWG3 criteria. Overall, the PSA-50 response rate was 7.4% (4/54, based on PSA assessments performed at the study center’s local laboratory) (table 3). The best response for PSA from baseline is shown in online supplemental figure S2.

Swimmer plot of duration of treatment and tumor response over time per RECIST V.1.1 in patients with mCRPC. (A) Patients with mCRPC excluding Cohorts 7A-mCRPC and 3B-mCRPC. (B) Patients with mCRPC in Cohorts 7A-mCRPC and 3B-mCRPC. mITT population. Treatment of each cohort see the footnote of table 2. Measurable disease at baseline was defined as showing new or progressive metastatic lymph node and/or local recurrence or visceral metastatic disease (with the exception of metastases to the liver) on CT or MRI scans. Duration of treatment was defined as (last dose date−first dose date+1). One patient from Cohort 3B-mCRPC achieved a confirmed CR (CR was 1.5-cm para-aortic lymph node, patient withdrew from study therapy after early Grade 3 diarrhea). CR, complete response; mCRPC, metastatic castration-resistant prostate cancer; mITT, modified intention-to-treat; NE, not evaluated; PD, progressive disease; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease.
Summary of PSA parameters
Patients with mCRPC treated at the recommended phase 2 dose (RP2D, that is, Cohorts 7A-mCRPC and 3B-mCRPC) had an ORR of 9.4% (n/N=3/32, 95% CI: 2.0% to 25.0%). The median DOR was 5.6 (3.9–7.4) months, the median rPFS was 5.6 (95% CI: 2.0 to NE) months, and the rPFS rate was 45.5% (95% CI: 24.9% to 64.1%) at 6 months and 30.4% (95% CI: 7.6% to 57.6%) at 12 months. The PSA-50 response rate was 6.3% (2/32, local laboratory assessments). Measurable disease at baseline was confirmed in 18 (56.3%) out of the 32 patients with mCRPC in these two cohorts. If only including these 18 patients, ORR was 16.7% (n/N=3/18, 95% CI: 3.6% to 41.4%).
BCR
Of patients with BCR, the PSA-50 response rate was 25.0% (5/20) in Cohort 1B-BCR and 26.7% (4/15) in Cohort 5B-BCR, and the median (range) duration of PSA-50 response was 10.1 (6.9–28.8) months and 3.9 (1.9–4.2) months, respectively (table 3). Minimum percent change from baseline for PSA assessed at local laboratory are shown in online supplemental figure S1.
PK
Serum PK parameters determined included the maximum concentration (Cmax), time to maximum concentration (Tmax), area under the concentration versus time curve (AUC) from time 0 to 672 hours (AUC0–672h), and observed accumulation ratio for trough concentrations after multiple dosing (Rac, Ctrough). AUC0–672h, Cmax, and accumulation ratio Rac, Ctrough were presented as geometric mean (geometric % coefficient of variation), and Tmax was presented as median (range).
For tremelimumab administered at 80 mg SC (n=6), on Cycle 1 Day 1, AUC0–672h was 2,389,000 (32) ng×hour/mL; Cmax was 4360 (31) ng/mL; and Tmax was 307 (165–477) hours; on Cycle 2 Day 1 (n=33), Rac, Ctrough was 1.454 (73). For sasanlimab administered at 300 mg SC, on Cycle 1 Day 1, AUC0–627h (n=13) was 10,250,000 (44) ng×hour/mL; Cmax (n=14) was 19,960 (40) ng/mL; and Tmax (n=14) was 188 (67.3–378) hours; on Cycle 2 Day 1, Rac, Ctrough(n=15) was 0.7794 (220). For sasanlimab administered at 130 mg SC (n=11), on Cycle 1 Day 1, AUC0–627h was 4,289,000 (44) ng×hour/mL; Cmax was 8612 (44) ng/mL; and Tmax was 165 (71.0–380) hours; on Cycle 2 Day 1, Rac, Ctrough (n=8) was 0.2734 (253) (table 4).
Serum tremelimumab and sasanlimab pharmacokinetics parameters
Immunogenicity and PD
A total of 84 patients were evaluable for ADA against tremelimumab. Of these 84 patients, 17 (20.2%) were tremelimumab-induced ADA-positive, with a median onset at Day 84 (sampling time for ADA assessments see online supplemental materials). Of the 17 patients who were ADA-positive and the 67 patients who were ADA-negative, the numbers of patients who experienced Type I hypersensitivity reaction were 2 (11.8%) and 9 (13.4%), respectively. A Type I hypersensitivity reaction was defined as any event occurring on the same day of drug injection, including headache, nausea, fever, chills, dizziness, pruritus, musculoskeletal chest pain, non-cardiac chest pain, and back pain. Injection site reactions were experienced in 4 (23.5%) and 5 (7.5%) patients, respectively.
Of 58 patients evaluable for ADA against sasanlimab, 13 (22.4%) patients were sasanlimab-induced/boosted ADA-positive, with a median onset at Day 142. Of these 13 patients who were ADA-positive and the 45 patients who were ADA-negative, patients who experienced Type I hypersensitivity reaction were 3 (23.1%) and 13 (28.9%), respectively; injection site reactions were experienced in 0 and 6 (13.3%) patients, respectively. Three patients had ADA for both tremelimumab and sasanlimab. There was no apparent effect of ADA on exposure to tremelimumab or sasanlimab.
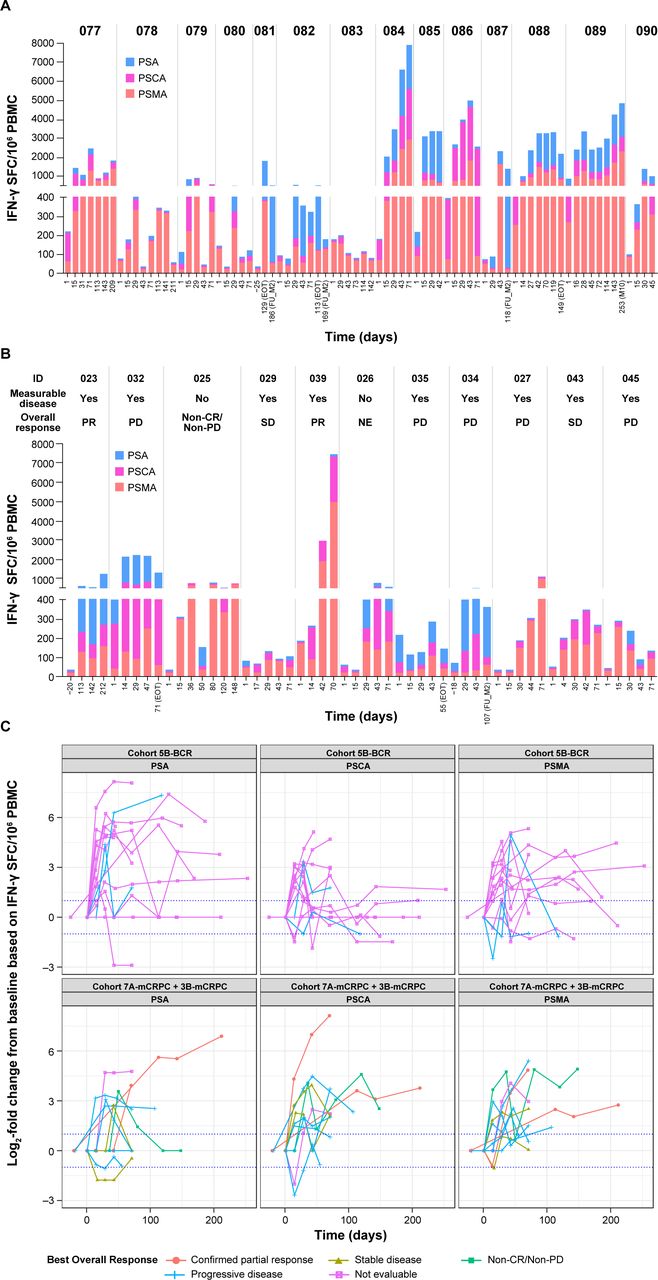
T-cell responses to ≥1 antigen (PSMA, PSCA, or PSA), based on ELISpot measurements of IFN-γ release, were seen in all patients across cohorts assessed in BCR and mCRPC. The range of positive T-cell responses for each antigen were: PSMA 85.7% to 90.9%, PSCA 71.4% to 90.9%, and PSA 72.7% to 92.9% (online supplemental table S2). T-cell response in patients with mCRPC (Cohorts 7A-mCRPC, 3B-mCRPC) and BCR (Cohort 5B-BCR) treated at the RP2D are shown in figure 3 and online supplemental figures S3 and S4.

{kind=link}
{kind=link}
{kind=link}
T-cell immune response. (A) Patients with BCR in Cohort 5B. (B) Patients with mCRPC in Cohorts 7A and 3B. (C) Fold changes from baseline of each antigen. Patients with mCRPC in Cohorts 7A-mCRPC and 3B-mCRPC were treated at the RP2D, which was AdC68 6×1011 VP, the DNA booster vaccine, tremelimumab 80 mg, and sasanlimab 300 mg. For patients with BCR in Cohort 5B-BCR, the treatment was AdC68 6×1011 VP, the DNA booster vaccine, tremelimumab 80 mg, and sasanlimab 130 mg.BCR, biochemical recurrence; CR, complete response; EOT, end of treatment; IFN, interferon; mCRPC, metastatic castration-resistant prostate cancer; NE, not evaluated; PBMC, peripheral blood mononuclear cell; PD, progressive disease; PR, partial response; PSA, prostate-specific antigen; PSCA, prostate stem cell antigen; PSMA, prostate-specific membrane antigen; RP2D, recommended phase 2 dose; SD, stable disease; SFC, spot-forming cells; VP, viral particle.
Discussion
This phase 1 open-label study evaluated the safety, antitumor activity, PK, and PD of increasing dose levels of three PrCa VBIR components alone and in combination with increasing doses of sasanlimab. An RP2D was then evaluated in patients with mCRPC, who continued on ADT, and in high-risk BCR, where patients were not treated with ADT. These seemingly disparate populations were selected to evaluate what the optimal recurrent PC population for cancer vaccination should be. It has been theorized that earlier clinical states of PC, such as BCR (rising PSA but no metastatic disease detected on conventional imaging) may harbor a less immune-suppressed microenvironment, which might allow an antitumor response to vaccination. In the current study, there was no meaningful tumor regression (based on RECIST) in patients with mCRPC; however, an encouraging signal of activity was observed in patients with BCR. The observed one death related to myocarditis in patients with BCR was concerning given that this population is at low risk for cancer-associated death. However, increasing dose levels of the study treatment were generally tolerated, with overall toxicities comparable to other ICI combination trials in patients with PC.28 29
Suggestive evidence for increased immuno-responsiveness in early-stage PC can be seen in neoadjuvant trials. In the preoperative setting, studies have demonstrated that ADT rapidly changes the tumor microenvironment with an influx of T regulatory cells.30 By comparison, a pilot neoadjuvant trial of PROSTVAC (vaccine using poxviral-based vectors with transgenes for PSA and three costimulatory molecules for T cells) without ADT demonstrated a decrease in tumor-infiltrating lymphocytes T regulatory cells and increase in T effector helper cells (CD4+FOXP3−).31 Similarly, neoadjuvant sipuleucel-T (an autologous dendritic cell vaccine using PBMCs activated ex vivo with PAP and fused with granulocyte-monocyte colony stimulating factor) without ADT before prostatectomy led to a threefold increase in CD4+FOXP3 T-cell infiltrate.32 In a trial of patients with BCR, T-cell responses were generally greater when vaccination with sipuleucel-T was administered before ADT compared with the reverse.33 Collectively, the data suggest that there may be advantages to vaccination in the absence of ADT, which is a potential approach in the BCR population. In this setting, both ADT and monitoring alone are standards of care, unlike in mCRPC, where obtaining castrate levels of testosterone (ie, ADT) is an essential component of therapy.
The subpopulation of patients with BCR in this trial had PSA reductions of >50% with durable response (those patients with BCR who received one ICI and VBIR had a median duration of 6.9 months; those patients with BCR who received two ICIs and VBIR had a median duration of 4.1 months). The PSA-50 rates were similar in those who received one versus two ICIs as part of PrCa VBIR, although the time to which PSA-50 was achieved was faster for those patients treated with two ICIs than one ICI. In those patients with mCRPC treated at the same expansion dose (Cohorts 7A-mCRPC and 3B-mCRPC, n=32), the ORR was 9.4% (16.7% for the 18 patients with measurable disease at baseline) and the estimated median rPFS was 5.6 months.
In mCRPC, a disease that is dominated by bone metastases that are not measurable by RECIST, outcomes such as ORR underestimate the effectiveness of therapy. In comparison, measurement of rPFS, which incorporates bone assessments per PCWG3 criteria, is a more accurate reflection of disease control in PC. While comparison to historic databases has significant limitations, previously reported rPFS rates for patients with mCRPC and progression on an androgen receptor signaling inhibitor, subsequently treated with chemotherapy, have ranged from 3.7 months34 to 5.2 months.35 The CARD trial showed that in patients with mCRPC who were previously treated with docetaxel and abiraterone or enzalutamide, the median rPFS of the cabazitaxel group was 8.0 months and the comparator (an androgen signaling-targeted inhibitor) group was 3.7 months.36 As such, the 5.6-month rPFS in the current study appears somewhat favorable. In general, the biologic/therapeutic effects of cancer vaccines may take longer to be observed, and may carry durable benefit going forward, and thus require longer follow-up time periods than typically expected with conventional treatment. For patients with BCR, metastasis-free survival would take many years of follow-up, which is an obvious limitation of efficacy analyses in an early-phase study.
PrCa VBIR uses multiple antigens as part of the prime/boost strategy to improve clinical efficacy. All patients demonstrated post-treatment T-cell responses to ≥1 antigen (PSMA, PSCA, or PSA; figure 3), suggestive of immune activation by PrCa VBIR. The ICIs were administered concurrently with vaccination priming and boosting in this trial, which may account for enhanced T-cell activation. Nevertheless, a correlation between T-cell responses and drug regimen or tumor control (rPFS) was not observed, possibly due to limited data. T-cell response against tumor-associated antigens alone may not be sufficient for conferring antitumor activity or clinical benefit, as shown in the phase 3 PROSTVAC trial19; potential underlying reasons include tumor heterogeneity rendering some areas not accessible by the administered vaccine and immune escape due to exhaustion of T effector cells or upregulation of additional inhibitory molecules such as lymphocyte-activation gene-3 and T-cell immunoglobulin and mucin domain-containing protein-3, which are not covered by the treatment regimens.37 The novel delivery by electroporation of pDNA encoding selected antigens depends on the induction of a transient state of cell membrane permeability. Delivery by electroporation has been demonstrated to increase DNA vaccine potency both preclinically and clinically.38–42
The safety profile for PrCa VBIR+/− anti-PD-1 reflected common non-immunological TRAEs as well as irAEs. Approximately 40% of patients with mCRPC experienced an irAE, most commonly thyroid dysfunction, colitis, and rash. In patients with BCR, one (Cohort 1B-BCR, n=20) versus two (Cohort 5B-BCR, n=15) ICIs were evaluated with vaccination, and while the numbers are small, there were more irAEs in the cohort receiving dual ICIs (20% one ICI vs 80% two ICIs). The irAEs also led to higher discontinuation rates in the dual-ICI cohorts relative to single ICI (5% vs 60%).
Although more irAEs may be anticipated when administering two as compared with a single ICI, the magnitude of the difference (two-fold) was not expected. The high percentage of irAEs with dual checkpoint inhibition plus vaccination in the patient with BCR population could limit the use of dual ICIs in combination with a vaccine. The selection of an anti-CTLA-4 rather than anti-PD-1 for the single-agent ICI cohorts was based on preclinical data demonstrating amplified T-cell responses with tremelimumab.27 Administration of ICIs SC was intended to amplify nodal antigen presenting cell and T-cell activity to draining lymph nodes, although nodal immune cell activity was not studied. In addition to enhancing potential activation at the site of draining lymph nodes with SC administration, it had been postulated that there may be less systemic toxicity. With the caveats of a small study, however, there was no clear signal of improved safety or preferred PK with SC ICI administration compared with historic intravenous administration.28
In summary, in this phase 1, open-label study, PrCa VBIR overall demonstrated safety signals similar to other ICI combination trials in mCRPC, with a modestly prolonged rPFS that did not warrant further study; patients with BCR (who did not receive concurrent ADT) treated with dual ICI had more irAEs than those with mCRPC, but some had prompt and durable PSA-50 responses without the use of ADT, which is noteworthy, as many patients seek an ADT-sparing approach. Future trials using immunotherapy combinations with a dual checkpoint inhibitor backbone in BCR should be carried out with careful oversight in this population with a long natural history of disease, as the potential risks must be carefully balanced with potential benefit. The sponsor will not develop PrCa VBIR further for strategic reasons; however, this study provides evidence for proof-of-principle that immunotherapy using vaccine strategies should be further studied in PC.
Data availability statement
Data are available upon reasonable request. Data may be obtained from a third party and are not publicly available. Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the Memorial Sloan Kettering Cancer Center Institutional Review Board (15-325A), the Yale University Institutional Review Board (1512016984), the Western Institutional Review Board (20151318), the Duke University Health System Institutional Review Board (00067872), the University of Pittsburgh IRB (15-118), the National Cancer Institute IRB (16C0079), the Thomas Jefferson University Office of Human Research (16C.471), the Atlantic Health System IRB (1295262), and the Biomedical Research Alliance of New York (18-11-398-03).
Acknowledgments
This work was sponsored by Pfizer. Medical writing support was provided by Anne Marie McGonigal, PhD, and Shuang Li, PhD, of Engage Scientific Solutions and funded by Pfizer.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors All authors were involved in the trial conception/design, or the acquisition, analysis, or interpretation of data. All authors contributed to the drafting of the manuscript and approved the final version. The authors were thankful for the statistical programming support provided by Fang Yuan at Pfizer Global Biometrics and Data Management. This paper is dedicated to the memory of Dr Nicholas Vogelzang, who died in September 2022 during its revision. Dr Vogelzang was strongly committed to the improvement of cancer patients’ lives through his untiring work in the field. His enthusiasm and contributions will be greatly missed.
Competing interests This work was sponsored by Pfizer. KAA: PI/research funding to institution: Amgen, AstraZeneca, CytomX, Eli Lilly and Company, GlaxoSmithKline, Parker Institute for Cancer Immunotherapy, Pfizer, Trishula. CSH: consulting fees: Astellas, AstraZeneca, Bayer, Blue Earth Diagnostics, Carrick Therapeutics, Clovis Oncology, Ferring Pharmaceuticals, Genentech, Hinova, Janssen, Menarini, Merck Sharp & Dohme, Myovant Sciences, Novartis, Pfizer; contracted research support: Aragon Pharmaceuticals, Astellas, AstraZeneca, Bayer, Clovis Oncology, eFFECTOR Therapeutics, Emergent BioSolutions, Ferring Pharmaceuticals, Medivation, Roche; ownership interest: CTI BioPharma; honoraria: Astellas. LN and X-HZ: nothing to disclose. LJA: consulting or advisory role: AADi; research funding (institution as recipient): Amgen, Astellas, Aveo, Bayer, BioNTech, Bristol Myers Squibb, Calithera Biosciences, Eisai, Eli Lilly and Company, Epizyme, Exelixis, Genentech/Roche, Inovio, Merck, Novartis, Peloton Therapeutics, Pfizer, Seattle Genetics, Surface Oncology; other relationship: Pfizer; uncompensated relationships: Exelixis. TZ: PI/research funding: AbbVie/StemCentrx, Acerta, Astellas, AstraZeneca, Eli Lilly and Company, Janssen, Merck, Merrimack, Mirati Therapeutics, Novartis, OmniSeq, Personal Genome Diagnostics, Pfizer, Regeneron; speaker: Genomic Health (end 2020), Sanofi-Aventis (end 2020); advisory board/consultant: Amgen, Aptitude Health, AstraZeneca, Aveo, Bayer, Bristol Myers Squibb, Calithera Biosciences, Clinical Care Options, Dendreon, Eisai, Eli Lilly and Company, Exelixis, Janssen, Merck, MJH Associates, Peerview, Pfizer, Pharmacyclics, QED Therapeutics, Sanofi-Aventis, Seattle Genetics, Vaniam; employee/stockholder: Archimmune Therapeutics (spouse), Capio Biosciences (spouse), Nanorobotics (spouse). HB: consultancy: Caris, Idera, Myovant, Novocure, Tracon; speaker’s bureau: Guardant360. NJV: PI/research funding: none; advisory boards: Caris, Clovis, Eisai, Fujifilm, Gilead Sciences, Merck, OnQuality Pharmaceuticals, Pfizer-Myovant, Sanofi-Aventis, Seagen; speaker’s bureau: Aveo, Bayer, Caris, Janssen, PER, Pfizer-Myovant, Sanofi, Seagen. SMP: PI funding: Janssen, Merck, Pfizer, UroGen Pharma. MTS: paid consultant and/or received honoraria: AstraZeneca, PharmaIn, Resverlogix, Sanofi; research funding to institution: Ambrx, AstraZeneca, Bristol Myers Squibb, F. Hoffman-La Roche, Immunomedics, Janssen, Madison Vaccines, Merck, Pfizer, SignalOne Bio, Tmunity, Zenith Epigenetics. RAM: research funding: Bayer. SB, NC, OB, RL, MK, I-MW, JZ, S-YT, and KAK: employees of Pfizer with stock and/or stock options. KC, HC, and RH: employees of Pfizer at the time of the study. DPP: stock and other ownership interests: Bellicum Pharmaceuticals, TYME Technologies; consulting or advisory role: Advanced Accelerator Applications, Amgen, Astellas, AstraZeneca, Bayer, Bicycle Therapeutics, Boehringer Ingelheim, Bristol Myers Squibb, Clovis Oncology, Eli Lilly and Company, Exelixis, Gilead Sciences, Incyte, Ipsen, Janssen, Mirati Therapeutics, Monopteros Therapeutics, Pfizer, Pharmacyclics, Regeneron, Roche, Seattle Genetics, UroGen Pharma; research funding: Advanced Accelerator Applications (Inst), Agensys (Inst), Astellas Medivation (Inst), AstraZeneca (Inst), Bayer (Inst), BioXcel Therapeutics (Inst), Bristol Myers Squibb (Inst), Clovis Oncology (Inst), Eisai (Inst), Endocyte (Inst), Eli Lilly and Company (Inst), Genentech (Inst), Gilead Sciences (Inst), Innocrin Pharma (Inst), MedImmune (Inst), Medivation (Inst), Merck (Inst), Mirati Therapeutics (Inst), Novartis (Inst), Pfizer (Inst), Progenics (Inst), Replimune (Inst), Roche (Inst), Sanofi (Inst), Seattle Genetics (Inst); expert testimony: Celgene, Sanofi.
Provenance and peer review Not commissioned; externally peer reviewed.
Author note Authors HB, KC, HC, and RH affiliations when study was carried out.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.