Article Text

Abstract
Background CD47 is an attractive immunotherapeutic target because it is highly expressed on multiple solid tumors. However, CD47 is also expressed on T cells. Limited studies have evaluated CD47-chimeric antigen receptor (CAR) T cells, and the role of CD47 in CAR T-cell function remains largely unknown.
Methods Here, we describe the development of CD47-CAR T cells derived from a high affinity signal regulatory protein α variant CV1, which binds CD47. CV1-CAR T cells were generated from human peripheral blood mononuclear cells and evaluated in vitro and in vivo. The role of CD47 in CAR T-cell function was examined by knocking out CD47 in T cells followed by downstream functional analyses.
Results While CV1-CAR T cells are specific and exhibit potent activity in vitro they lacked antitumor activity in xenograft models. Mechanistic studies revealed CV1-CAR T cells downregulate CD47 to overcome fratricide, but CD47 loss resulted in their failure to expand and persist in vivo. This effect was not limited to CV1-CAR T cells, since CD47 knockout CAR T cells targeting another solid tumor antigen exhibited the same in vivo fate. Further, CD47 knockout T cells were sensitive to macrophage-mediated phagocytosis.
Conclusions These findings highlight that CD47 expression is critical for CAR T-cell survival in vivo and is a ‘sine qua non’ for successful adoptive T-cell therapy.
- Receptors, Chimeric Antigen
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Limited studies have evaluated the antitumor activity of CD47-chimeric antigen receptor (CAR) T cells and the role of CD47 surface expression in CAR T-cell function remains to be investigated.
WHAT THIS STUDY ADDS
Our results demonstrate CD47-CAR T cells downregulate CD47 surface expression to overcome fratricide and function in vitro. However, loss of surface CD47 renders CAR T cells ineffective in vivo and sensitizes human T cells to macrophage mediated phagocytosis.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
These data provide direct evidence that interventions or genetic modifications that decrease CAR T-cell CD47 expression will inhibit CAR T-cell survival and effector function in vivo.
Background
Chimeric antigen receptor (CAR) T-cell therapy offers a promising approach to improve outcomes and decrease toxicities for patients with relapsed or refractory solid tumors. Multiple CAR T-cell products are United States Food and Drug Administration approved to treat CD19 positive malignancies or multiple myeloma,1 highlighting success of the approach. Despite these successes, developing CAR T-cell therapies for patients with solid tumors remains a challenge, evidenced by limited objective responses across clinical trials targeting different tumor antigens.2 3 Obstacles to success against solid tumors include a limited array of targetable antigens, tumor expression of immune inhibitory molecules, and the presence of immunosuppressive cells within the tumor microenvironment (TME).4
CD47, originally called integrin associated protein, is a five-pass transmembrane protein of the immunoglobulin superfamily that binds integrins, thrombospondin, and signal regulatory protein alpha (SIRPα).5 SIRPα is primarily expressed on macrophages and other myeloid cells.6 Binding of CD47 to SIRPα inhibits macrophage phagocytosis via phosphorylation of immunoreceptor tyrosine-based inhibitory motifs and subsequent recruitment of phosphatases SHP1 and SHP2.6 7 Numerous functions of CD47 have been proposed in T-cell biology, including activation/co-stimulation, inhibition, cell death, and transendothelial migration, depending on the method of CD47 activation and other experimental conditions.8–17 The importance of CD47 in CAR T-cell function is largely yet to be explored.
CD47 is expressed at high levels on multiple solid tumors and associated with poor outcomes, making it an attractive immunotherapeutic target.18 CD47 protects solid tumors by preventing macrophage mediated phagocytosis and other immunosuppressive mechanisms.18 19 Benefits of targeting the CD47/SIRPα axis are exemplified by complete responses to CD47 antibody (αCD47) combined with rituximab for treating patients with lymphoma.20 Efficacy of combining αCD47 with pro-phagocytic, tumor-specific antibodies is also described for solid tumors.21 22 Given that CAR T cells combine the specificity of monoclonal antibodies with the tissue infiltrating and direct killing capabilities of T cells, adoptive T-cell therapy targeting CD47 offers a promising approach. However, CD47 is expressed on normal tissues and toxicity is a concern. A long-term goal is to develop CD47-specific T cells that are activated only within the TME. As a first step, here we aimed to develop and study constitutively active CD47-CAR T cells that recognize both human and murine CD47. Thus far, limited studies with CD47-CAR T cells have been performed,23 24 and none of these studies performed in vivo experiments with CD47-CAR T cells that recognize human and murine CD47, or explored the importance of CD47 expression in CAR T-cell function.
To address these gaps in our knowledge we designed a CD47-CAR that consists of CV1, a high affinity SIRPα variant that binds human and murine CD47,25 as the antigen recognition domain (CV1-CAR). We show that CV1-CAR T cells have potent antitumor activity in vitro, but surprisingly lack antitumor activity in vivo. Mechanistic studies revealed that CV1-CAR T cells downregulate CD47 surface expression to overcome fratricide post-transduction, and that CV1-CAR T cells are rapidly eliminated in vivo. Deleting CD47 in EphA2-CAR T cells also resulted in their rapid elimination in vivo, likely due to macrophage-mediated phagocytosis, highlighting the critical role of CD47 expression for CAR T-cell survival in vivo.
Materials and methods
Study design
The objectives of this study were to evaluate the effector function of CV1-CAR T cells and determine the role of CD47 expression in CAR T-cell function. Control and CAR T cells were donor-matched for all in vitro and in vivo experiments. For all experiments, the number of replicates, statistical test used, and p values are reported in the figure legends. For in vitro experiments, the reported replicates refer to biological replicates (T cells from different healthy donors). For in vivo experiments the reported replicates refer to the number of mice treated in each condition. For in vivo experiments, mice were treated with T cells from one healthy donor unless otherwise specified in the figure legend. Cages of mice were randomly assigned to experimental groups.
Cell lines
The OV10 (human ovarian cancer) and OV10-CD47 (modified to express CD47) cell lines were provided by Dr William Frazier (Washington University School of Medicine, St. Louis, Missouri, USA) in 2014. The osteosarcoma cell line LM7 was provided by Eugenie Kleinerman (The University of Texas MD Anderson Cancer Center, Houston, Texas, USA) in 2011. The generation of LM7.GFP.ffluc was previously described.26 The A549 (lung adenocarcinoma) cell line was purchased from the American Type Culture Collection (ATCC). All adherent tumor cell lines were grown in Dulbecco’s Modified Eagle Medium (DMEM; Lonza or Thermo Fisher) supplemented with 10% fetal bovine serum (FBS; Cytiva), 1% GlutaMAX (Thermo Fisher), and subcultured with 0.05% trypsin-EDTA (Thermo Fisher). The murine MH-S (alveolar macrophage) and RAW264.7 (macrophage) cell lines were purchased from ATCC. MH-S cells were cultured in high-glucose Roswell Park Memorial Institute media (RPMI; Thermo Fisher), supplemented with 10% FBS, 1% GlutaMAX, 55 nM 2-mercaptaethanol (Invitrogen), and penicillin–streptomycin (Invitrogen). RAW264.7 cells were cultured with DMEM, supplemented with 10% FBS, 1% GlutaMAX, 55 nM 2-mercaptaethanol, and penicillin–streptomycin. All cells were maintained at 37°C in 5% CO2. Cell lines were authenticated by short tandem repeat profiling using the service of the ATCC (FTA sample collection kit) and routinely checked for mycoplasma using the MycoAlert mycoplasma detection kit (Lonza).
Production of viral vectors
A codon optimized minigene encoding the CV1-CAR containing the immunoglobulin heavy-chain signal peptide, high affinity SIRPα variant CV1,25 human CD8α hinge, human CD28ζ endodomain, P2A, and ZsGreen was synthesized by GeneArt (Thermo Fisher) and subcloned via In-Fusion cloning (Takara Bio) into an SFG gamma retroviral backbone that has been previously described.27 The cloned construct was verified by sequencing (Hartwell Center, St. Jude Children’s Research Hospital (St. Jude)). The generation of SFG retroviral vectors encoding the EphA2-CAR derived from the monoclonal antibody (mAb) 4H5 with a CD28 co-stimulatory domain and the non-functional EphA2-CAR without a signaling domain (EphA2Δ-CAR) have been previously described.28 RD114-pseudotyped retroviral particles were generated by transient transfection of 293 T cells (ATCC) as previously described.29 Supernatants were collected after 48 hours, filtered, and snap-frozen for later transduction of T cells.
Generation of CAR T cells
Human peripheral blood mononuclear cells (PBMCs) were obtained from the whole blood of healthy donors or from deidentified healthy donor pheresis products. Retroviral transduced T cells were generated as previously described.28 Briefly, on day 1 PBMCs were stimulated on non-tissue culture-treated 24-well plates, which were precoated with OKT3 (Miltenyi) and CD28 (Miltenyi) antibodies. Recombinant human interleukin (IL)-7 (10 ng/mL; PeproTech) and IL-15 (5 ng/mL; PeproTech) were added on day 2. On day 3, OKT3/CD28-stimulated T cells (2.5×105 cells/well) were transduced with RD114-pseudotyped retroviral particles on RetroNectin (Takara Bio) coated plates in the presence of IL-7 and IL-15. On day 5, transduced T cells were transferred into new 24-well tissue culture plates and expanded with IL-7 and IL-15. GFP.ffluc expressing CAR T cells were prepared similarly, except that activated T cells were simultaneously transduced with a mixture of CV1-CAR, EphA2-CAR, or EphA2Δ-CAR plus GFP.ffluc retroviral particles on day 3. Non-transduced (NT) T cells were prepared similarly, except that no retrovirus was included in the RetroNectin wells on day 3. All T cells were cultured with RPMI supplemented with 10% FBS and 1% GlutaMAX. CAR T-cell expression was determined 6–9 days post-transduction, and experiments were performed 6–14 days post-transduction.
Flow cytometry
A FACSCanto II (BD Biosciences) instrument was used to acquire flow cytometry data, which were analyzed using FlowJo V.10 (BD Biosciences). For surface staining, samples were washed with and stained in phosphate buffered saline (PBS; Lonza) with 1% FBS. For all experiments, matched isotypes or known negatives (eg, NT T cells) served as gating controls. LIVE/DEAD Aqua (Thermo Fisher) or DAPI (BD Biosciences) were used to exclude dead cells from analysis. CV1-CAR detection and ability to bind CD47 was performed using two-step staining; incubated with recombinant human or murine CD47-Fc protein (R&D Systems), washed, then stained with anti-Fc antibody (Southern Biotech goat anti-human IgG Fc or mouse anti-human IgG Fc). CV1-CAR expression was characterized by double positive expression of ZsG and Fc. EphA2-CAR detection was evaluated by staining for truncated CD20 with CD20 antibody (Clone L27 or 2H7, BD Biosciences). EphA2Δ-CAR detection was evaluated by staining for truncated CD19 with CD19 antibody (Clone J3-119 Beckman Coulter). To determine phenotype, T cells were stained with fluorochrome-conjugated antibodies using combinations of the following markers: CD4 (Clone SK3, BD Biosciences), CD8 (Clone SK1, BD Biosciences), CCR7 (Clone G043H7, BioLegend), and CD45RO (Clone UCHL1, BD Biosciences). CD47 expression for T cells and tumor cell lines was evaluated by using CD47 antibody (Clone CC2C6, BioLegend). To assess exhaustion markers, T cells were stained with fluorochrome-conjugated antibodies for programmed cell death protein-1 (PD-1) (Clone EH12.2H7, BioLegend), T-cell immunoglobulin and mucin domain-containing protein 3(TIM3; Clone F38-2E2, BioLegend), and lymphocyte activation gene 3 protein (LAG3; Clone T47-530, BD Biosciences), and expression determined based on CAR-positive T cells. Murine SIRPα was evaluated on murine macrophage cell lines by using anti-mouse CD172a (clone P84, BioLegend). Murine macrophage cell line binding to human CD47 was evaluated by incubating MH-S or RAW264.7 cells with recombinant human CD47-Fc protein (R&D Systems), followed by washing and staining with anti-Fc antibody (Southern Biotech goat anti-human IgG Fc). To evaluate intracellular CD47, cells were first stained for extracellular CD47 (CD47-APC, Clone CC2C6; BioLegend) and fixable LIVE/DEAD Aqua. Cells were then fixed and permeabilized using the Cyto-Fast Fix/Perm Solution kit (BioLegend) per the manufacturer’s protocol, followed by staining for intracellular CD47 (CD47-PE, Clone CC2C6; BioLegend).
Fratricide assay
5×105 CV1-CAR T cells were co-cultured with 5×105 NT T cells from matched donors in 2 mL of RPMI media with 10% FBS and 1% GlutaMAX supplemented with IL-7 (10 ng/mL) and IL-15 (5 ng/mL) in a 24-well tissue-culture treated plate for 24 hours at 37°C. Cells were harvested and analyzed by flow cytometry for remaining, live, ZsG-negative/CD47-positive NT T cells and ZsG-positive/CD47-negative CV1-CAR T cells.
Analysis of cytokine production
For T-cell and recombinant protein co-cultures, recombinant human or murine CD47-Fc protein was precoated overnight in PBS on a 48-well non-tissue culture treated plate at 125 ng/well, 250 ng/well, or 500 ng/well. Recombinant human B7-H3-Fc protein (R&D Systems) was precoated at 500 ng/well using the same methods. Wells were washed with media and T cells were plated at 2.5×105 cells per well without the provision of exogenous cytokines for 24 hours at 37°C. Supernatant was then collected and frozen at −80°C. Cytokines (interferon (IFN)-γ and IL-2) were measured using a quantitative ELISA per the manufacturer’s instructions (R&D Systems).
For T-cell and tumor cell line co-cultures, 1×106 T cells were co-cultured with no tumor cells or 5×105 OV10 or OV10-CD47 without the provision of exogenous cytokines for 24 hours at 37°C. Supernatant was then collected and frozen at −80°C. Cytokines (IFN-γ and IL-2) were measured using a quantitative ELISA per the manufacturer’s instructions (R&D Systems).
MTS cytotoxicity assay
A CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) was used to assess CAR T-cell cytotoxicity. In a tissue culture-treated 96-well plate, 30,000 OV10 or OV10-CD47, 20,000 LM7, or 3000 A549 cells were co-cultured with T cells at 1 to 1 ratio (OV10 and OV10-CD47) or 2 to 1 ratio (LM7 and A549) in technical triplicates. Media only and tumor cells alone served as negative controls. After 24 hours, the media and T cells were removed by gentle pipetting to avoid disrupting adherent tumor cells. CellTiter 96 AQueous One Solution Reagent ([3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS)] + phenazine ethosulfate) in RPMI with 10% FBS was added to each well and incubated at 37°C for 2 hours. The absorbance at 492 nm was measured using an Infinite 200 Pro M Plex plate reader (Tecan) to assess the number of viable cells in each well. Percentages of live tumor cells were determined by the following formula: (sample−media alone)/(tumor alone−media alone)×100.
Xenograft mouse models
All animal experiments were performed on protocols approved by the St. Jude Institutional Animal Care and Use Committee. Xenograft experiments were performed with 8–12 weeks old male or female NSG mice purchased from The Jackson Laboratory or obtained from the St. Jude NSG colony. For the intraperitoneal (i.p.) LM7 model, mice were injected i.p. with 1×106 LM7.GFP.ffluc cells, and on day 7 received a single i.p. dose of 1×106 or 5×106 EphA2Δ-CAR, EphA2-CAR or CV1-CAR T cells. For the subcutaneous (s.c.) A549 model, mice were injected s.c. with 2×106 A549 cells into the right flank, and on day 7 received a single intravenous dose of 1.5×106 or 5×106 GFP.ffluc expressing T cells. To evaluate macrophage depletion, mice were treated with 200 uL intravenous of control liposomes (Encapsome; Encapsula NanoSciences) or clodronate liposomes (Clodrosome; Encapsula NanoSciences) and bone marrow and spleens harvested 48 hours post-treatment. Tissues were processed by manual dissociation, treated with red blood cell lysing buffer (Gibco), and stained with LIVE/DEAD Aqua, F4/80 (Clone T45-2342, BD), and CD45 (Clone 30-F11, BioLegend). For the macrophage depletion model, mice were injected s.c. with 2×106 A549 cells into the right flank, followed by a single 200 uL intravenous dose of clodronate or control liposomes on day 5, and 5×106 GFP.ffluc expressing T cells on day 7 post-tumor inoculation.
Bioluminescence imaging
Mice were injected i.p. with 150 mg/kg of D-luciferin 5–10 min before imaging, anesthetized with isoflurane and imaged with a Xenogen IVIS-200 imaging system (PerkinElmer). The photons emitted from luciferase-expressing cells were quantified using Living Image software (PerkinElmer). A total body region of interest was drawn around each mouse, and radiance was recorded in units of photons/s/cm2/sr.
Generation of knockout T cells
CD47 knockout (KO) or control AAVS1 KO T cells were generated using CRISPR-Cas9 technology. On day 1 PBMCs were stimulated on non-tissue culture-treated 24-well plates that were precoated with OKT3 and CD28 antibodies. Recombinant human IL-7 (10 ng/mL) and IL-15 (5 ng/mL) were added on day 2. On day 3, CD3/CD28 activated T cells were electroporated with precomplexed ribonucleoproteins (RNPs) using the Lonza Nucleofection system (4D-Nucleofector X unit; program EH-115; Lonza). If applicable, electroporated T cells were transduced on day 4 with RD114-pseudotyped retroviral particles on RetroNectin coated plates in the presence of IL-7 and IL-15. sgRNAs were designed by the St. Jude Center for Advanced Genome Engineering to target CD47 (5’-AGUGATGCUGUCUCACACAC-3’) or AAVS1 (5’-GGGGCCACUAGGGACAGGAU-3’). RNPs were precomplexed at an sgRNA/Cas9 ratio of 2:1, prepared by adding 2.7 µL of 60 µM sgRNA (Synthego) to 2 µL 40 µM Cas9 (MacroLab, University of California, Berkeley) and frozen prior to use. A total of 1×106 T cells were resuspended in 15 µL of P3 Primary Human kit solution (Lonza) with 5 µL RNPs and electroporated. Cells were plated on a recovery 48-well tissue-culture treated plate in RPMI with 20% FBS, 1% GlutaMAX, and supplemented with IL-7 (10 ng/mL) and IL-15 (5 ng/mL). After 24–72 hours recovery, the medium was switched to RPMI with 10% FBS and 1% GlutaMAX, and the cells expanded with IL-7 and IL-15 added every 2–3 days. CD47 KO efficiency was evaluated by flow cytometry using CD47 antibody (Clone CC2C6, BioLegend).
Histology and immunohistochemistry
Tissues were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned at 4 µm, mounted on positive charged glass slides (Superfrost Plus; 12-550-15, Thermo Fisher) that were dried at 60°C for 20 min, and stained with H&E. The following immunohistochemistry protocol was used for the detection of human CD3: Anti-CD3 antibody (Clone EP41, Biocare Medical, CME324B), 60 min incubation, heat-induced epitope retrieval with cell conditioning media 1 (950-500; Ventana Medical Systems), 92 min; Visualization with DISCOVERY OmniMap anti-Rb HRP (760–4311; Ventana Medical Systems), DISCOVERY ChromoMap DAB kit (760-159; Ventana Medical Systems), Hematoxylin II (790–2208; Ventana Medical Systems), and Bluing reagent (790–2037; Ventana Medical Systems).
Phagocytosis assay
MH-S (murine alveolar macrophage) and RAW24.6 (murine macrophage) cell lines were stained with CellBrite Red (Biotium) for 20 min at 37°C. They were then washed and plated at 1.25×105, 6.25×104, or 3.13×104 cells per well in a 48-well tissue culture treated plate and incubated at 37°C for 1–2 hours. NT, control KO (AAVS1-KO), or CD47-KO T cells were then stained with CellBrite Green (Biotium) for 20 min at 37°C. They were then washed and plated on macrophages at a 1 to 1 effector to target ratio. The co-culture was then imaged on a Sartorius Incucyte S3 machine every 20 min for 4 hours. Phagocytosis was determined by analyzing the merge of green and red. All data were normalized to hour 0. To compare across donors, all data were then normalized to NT T cells (of the respective donor). Area under the curve analysis was then performed on the normalized merge data.
Statistical analyses
For all experiments, the number of biological replicates and statistical analysis used are described in the figure legends. Statistical analyses were performed using GraphPad Prism (GraphPad Software) and presented as means±SEM or min to max. Paired t-tests, unpaired t-tests, simple linear regression, two-way analysis of variance with Sidak’s multiple comparisons test, and Wilcoxon matched-pairs signed rank test were used in data analysis. Two-tailed t-tests were performed in all instances. P value<0.05 was considered statistically significant. ns, non-significant: p>0.05, *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Results
CV1-CAR T cells overcome fratricide by downregulating CD47 surface expression
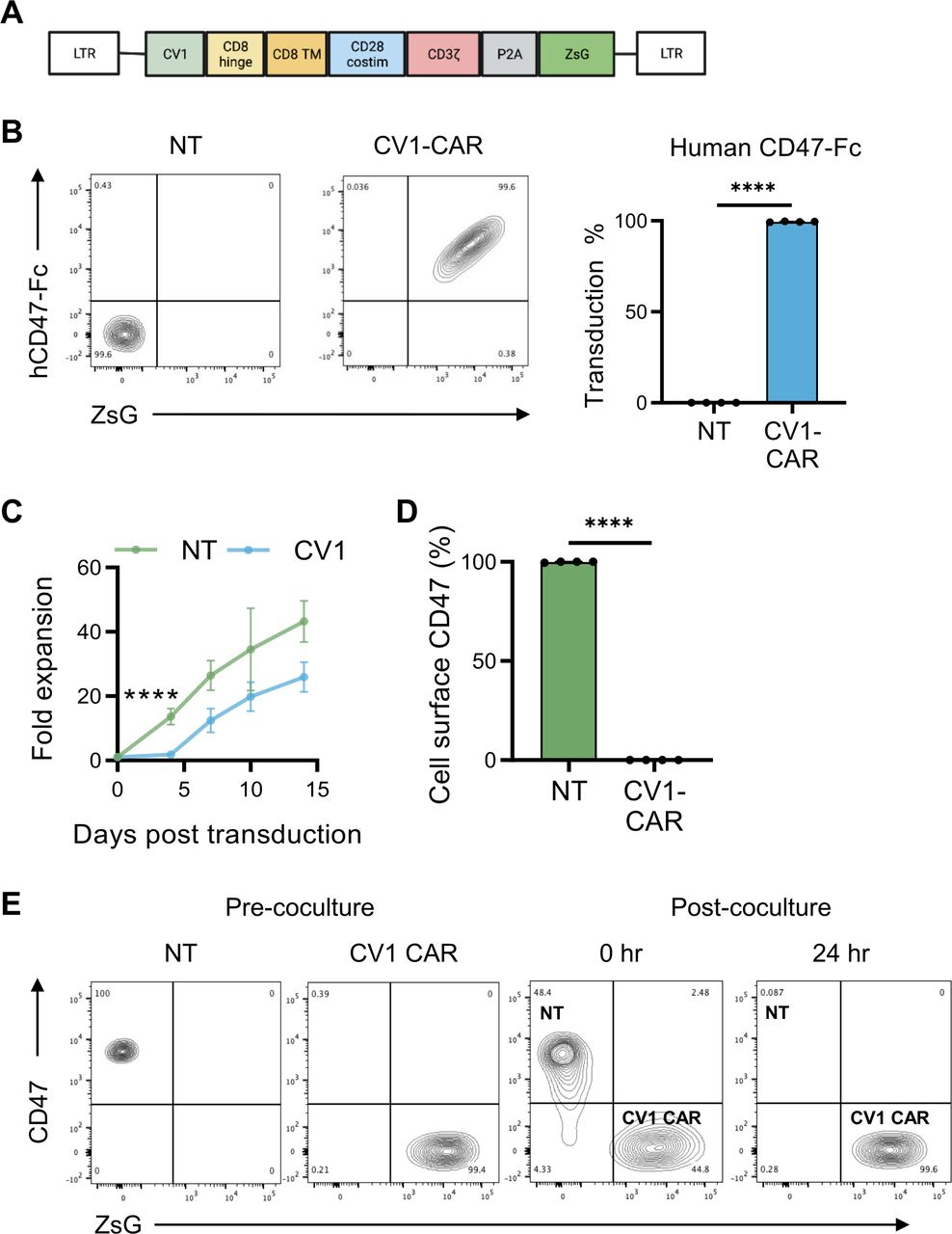
To preclinically evaluate the effector function of CD47-CAR T cells, we constructed a second generation CD47-CAR consisting of an antigen recognition domain derived from the high affinity SIRP⍺ variant CV1,25 which recognizes both human and murine CD47, a CD8⍺ hinge/transmembrane domain, and a CD28ζ endodomain. The CV1-CAR retroviral vector also encoded a P2A ribosomal skip sequence and ZsGreen (ZsG) as a detection marker (figure 1A and online supplemental figure 1). To confirm CV1-CAR expression and its ability to bind human and murine CD47, we incubated NT or CV1-CAR T cells with recombinant human or murine CD47-Fc protein, and measured Fc by flow cytometry. T-cell transduction was also confirmed by flow cytometry for ZsG. Post-transduction, CV1-CAR T cells bound both human and murine CD47, and close to 100% of T cells were CAR positive (figure 1B and online supplemental figure 2A). CV1-CAR T cells did not expand for the first 4 days post-transduction, but thereafter expanded at a similar rate to NT T cells (figure 1C). At the end of expansion, NT and CV1-CAR T cells had a similar CD4+/CD8+ ratio and a predominate effector memory phenotype (CCR7−, CD45RO+) (online supplemental figure 2B,C). Intriguingly, NT T cells expressed high levels of CD47, while CV1-CAR T cells were CD47-negative (figure 1D), suggesting that CV1-CAR T cells downregulate CD47 and kill CD47-positive NT T cells, explaining limited early expansion and the near 100% CAR positive product. Indeed, co-culture assays revealed CV1-CAR T cells killed CD47-positive NT T cells within 24 hours, demonstrating T-cell fratricide (figure 1E and online supplemental figure 2D).
Supplemental material
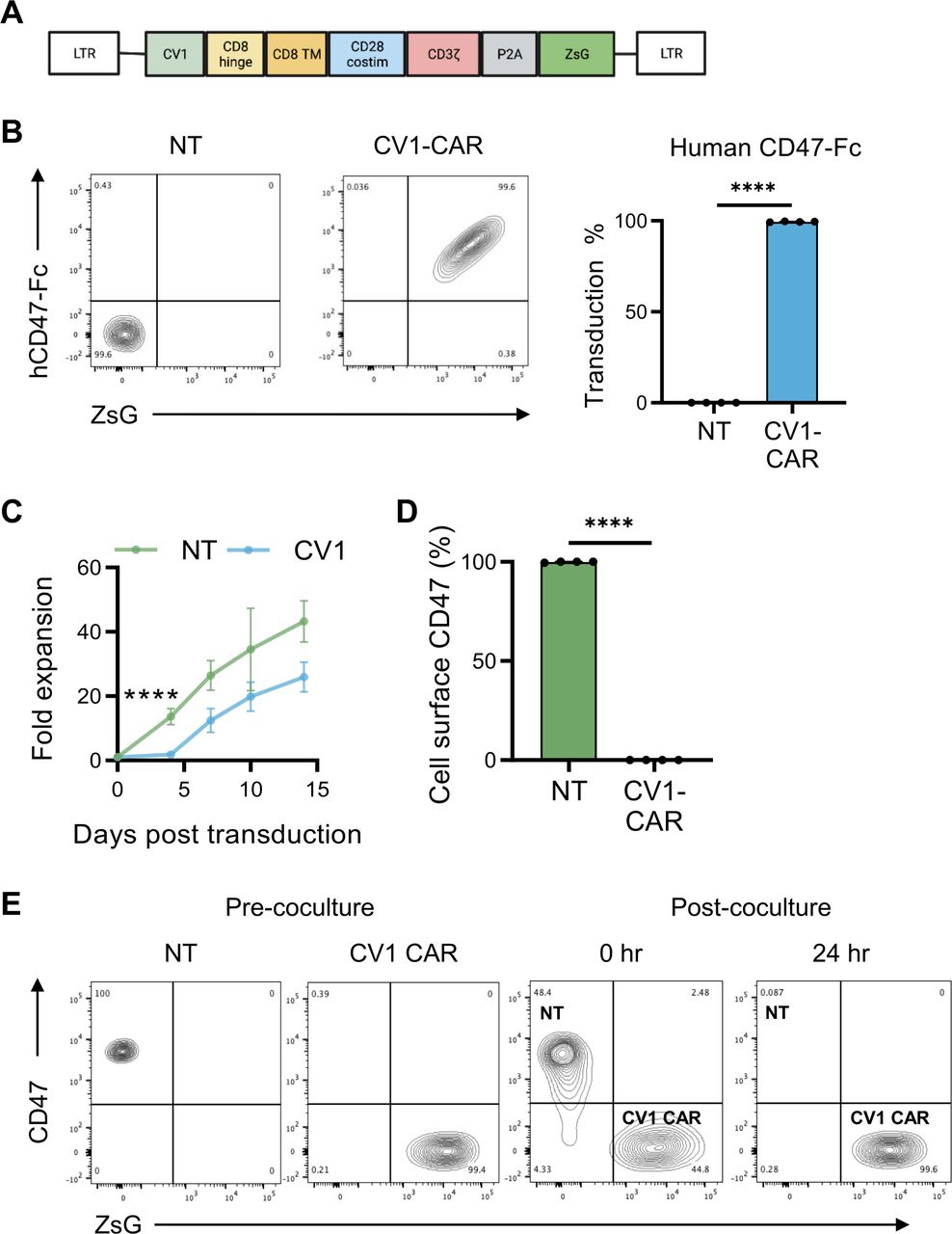
CV1-CAR T cells overcome fratricide by downregulating CD47 surface expression. Activated T cells were transduced with gamma retroviral particles encoding the CV1-CAR and surface expression was measured by flow cytometry. (A) Schematic representation of the retroviral vector encoding the CV1-CAR and ZsGreen (ZsG) as a detection marker. (B) Representative flow plots of non-transduced (NT) or CV1-CAR T cells binding recombinant human CD47-Fc protein (left panel), and summary data (right panel; n=4). (C) Expansion of NT (n=3) and CV1-CAR T cells (n=6). (D) Surface CD47 expression on NT and CV1-CAR T cells 7 days post-transduction (n=4). (E) CV1-CAR T-cell killing of CD47-positive/ZsG-negative NT T cells was determined by flow cytometry. NT and CV1-CAR T cells were harvested immediately (0 hours) and 24 hours post co-culture. Data represents mean±SEM (B, C, and D). ****p<0.0001 by paired t-test (B and D) or simple linear regression (C). CAR, chimeric antigen receptor.
To determine if loss of CD47 in CV1-CAR T cells was due to outgrowth of CD47-negative T cells versus internalization or downregulation of CD47, we asked if CD47-negative T cells are present prior to transduction (day 0), and if intracellular CD47 is detected in CV1-CAR T cells post-transduction. Pre-transduction (day 0), all CD3-positive T cells expressed high levels of CD47 (online supplemental figure 3A), suggesting CD47-negative cells are not available for potential outgrowth. Additionally, while NT T cells had high intracellular CD47 expression (online supplemental figure 3B,C), limited intracellular CD47 was detected in CV1-CAR T cells (online supplemental figure 3B,C). Together these findings indicate CV1-CAR T cells downregulated CD47 post-transduction to overcome fratricide.
CV1-CAR T cells are cytotoxic in vitro but lack antitumor activity in vivo
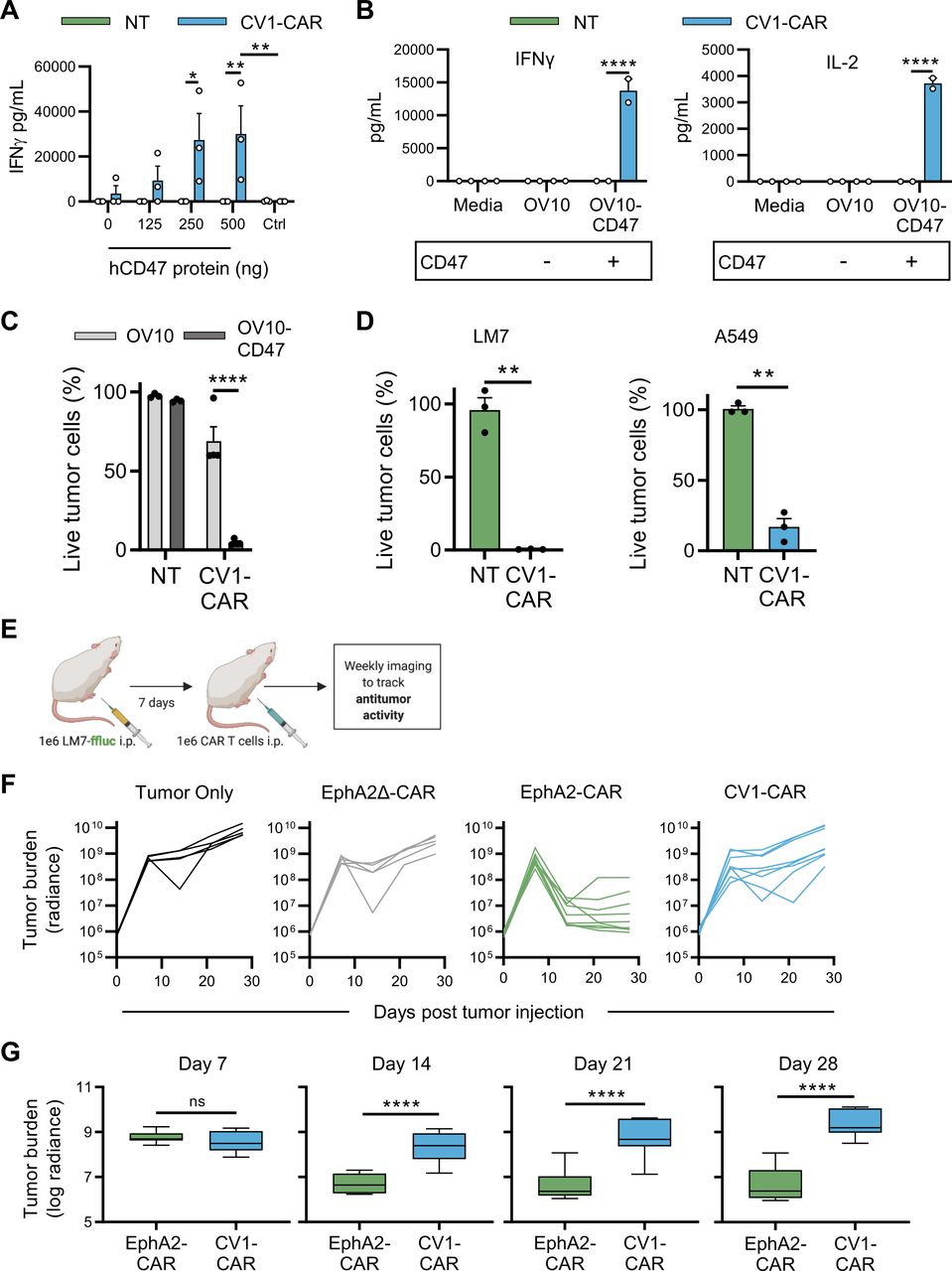
To evaluate CV1-CAR T-cell specificity, we first cultured CV1-CAR or NT T cells on plates coated with human CD47, murine CD47 or control protein (human B7-H3). CV1-CAR T cells secreted significantly greater levels of IFN-γ when cultured with human or murine CD47 compared with control protein (figure 2A and online supplemental figure 4A). Additionally, IFN-γ production increased when cultured with higher quantities of CD47, indicating that CV1-CAR T cells are activated in a dose-dependent manner by human or murine CD47. To evaluate specificity and cytotoxicity against cancer cells we used CD47-negative OV10 cells or OV10 modified to express human CD47 (OV10-CD47; online supplemental figure 4B). CV1-CAR T cells produced high levels of cytokines (IFN-γ and IL-2) and were cytotoxic against OV10-CD47, further demonstrating antigen specificity (figure 2B,C). They were also cytotoxic against other CD47-positive cancer cell lines including LM7 (osteosarcoma) and A549 (lung adenocarcinoma) (figure 2D and online supplemental figure 4B), which were used in downstream in vivo experiments. These data confirmed that CV1-CAR T cells recognize and kill targets in a CD47-specific fashion.

CV1-CAR T cells are cytotoxic in vitro but lack antitumor activity in vivo. CV1-CAR T-cell effector function was evaluated in vitro using co-culture (cytokine production) and cytotoxicity assays. (A) IFN-γ production in culture with recombinant human CD47 (hCD47) protein (n=3). (B) IFN-γ (left panel) and IL-2 production (right panel) post co-culture with CD47-positive or CD47-negative tumor cells, or media alone. Media was collected after 24 hours and cytokines determined by ELISA (n=2). (C) Cytotoxicity assay using OV10 or OV10-CD47 as targets and NT (n=3) or CV1-CAR (n=4) T cells as effectors at a 1 to 1 effector to target cell ratio. (D) CV1-CAR T-cell cytotoxicity at 2 to 1 T-cell to target cell ratio against LM7 osteosarcoma (left panel; n=3) and A549 lung adenocarcinoma (right panel; n=3). CV1-CAR T-cell effector function was evaluated in vivo using the LM7.GFP.ffluc intraperitoneal (i.p.) osteosarcoma model. (E) Diagram of the in vivo study. Mice were injected with 1×106 LM7.GFP.ffluc cells i.p. on day 0, followed by no treatment (tumor only; n=5) or 1×106 EphA2Δ-CAR (negative control; n=5), EphA2-CAR (positive control; n=9) or CV1-CAR (n=8) T cells i.p. on day 7. In vivo data was pooled from two independent experiments using T cells from two separate donors. (F) Individual and (G) summary bioluminescence measurements (radiance=photons/s/cm2/sr) over time. Data represents mean±SEM (A, B, C and D) or min to max (G). *p<0.05, **p<0.01, ****p<0.0001, ns, non-significant by two-way analysis of variance with Sidak’s multiple comparisons test (A, B, and C), paired t-test (D) or unpaired t-test (G). CAR, chimeric antigen receptor; IFN, interferon; IL, interleukin; NT, non-transduced.
Given the specificity and cytolytic activity of CV1-CAR T cells in vitro, we hypothesized they would exhibit robust antitumor activity in vivo. We therefore tested CV1-CAR T-cell antitumor activity in an established i.p. LM7.GFP.ffluc osteosarcoma model, where multiple CAR T-cell products have potent antitumor activity.29 30 CV1-CAR or control T cells were injected i.p. and tumor growth monitored by serial bioluminescence imaging (figure 2E). EphA2-CAR T cells, which have robust antitumor activity in this model,29 served as a positive CAR T-cell control, while T cells expressing an EphA2-CAR that lacked intracellular signaling domains (EphA2Δ-CAR) served as a negative control (online supplemental figure 5A). As expected, EphA2-CAR T cells exhibited rapid antitumor activity, while EphA2Δ-CAR T cells had no antitumor activity in comparison to untreated (tumor only) animals (figure 2F). To our surprise however, tumors rapidly progressed post-treatment with CV1-CAR T cells (figure 2F,G). To confirm these findings were not dose-specific, we repeated the experiment at a fivefold higher CAR T-cell dose and found similar results (online supplemental figure 5B-D). Thus, while CV1-CAR T cells recognized and killed CD47-positive cancer cells in vitro, they lacked antitumor activity in vivo.
CV1-CAR T cells have limited persistence in vivo
The lack of in vivo antitumor activity suggested that CV1-CAR T cells either failed to persist or were otherwise incapable of killing CD47-positive tumors despite appropriate persistence post injection. Given that CD47 surface expression allows cells to evade macrophage mediated phagocytosis,31 we hypothesized that limited T-cell persistence resulted in the failure of CV1-CAR T cells to eradicate tumors in mice. To evaluate CV1-CAR T-cell persistence, we injected NSG mice with A549 tumor cells s.c. into the right flank, followed by intravenous injection of GFP.ffluc-expressing CV1-CAR or EphA2-CAR T cells 7 days later (figure 3A). As expected, EphA2-CAR T cells trafficked to tumors, expanded and persisted with high bioluminescence signals over flank tumors by day 14 post-treatment, while CV1-CAR T cells had limited detection 1 day post-infusion (figure 3B–D). To confirm that CV1-CAR T cells could be detected in vivo post-infusion, we repeated the study and measured T-cell bioluminescence starting 4 hours post-treatment (online supplemental figure 6A). While CV1-CAR T cells were detected 4–8 hours post T-cell injection, their signal rapidly disappeared thereafter (online supplemental figure 6B-D). Evaluation of cell surface exhaustion markers showed similar expression of PD-1, TIM3, and LAG3 between EphA2-CAR and CV1-CAR T cells (online supplemental figure 7A-C). Together, these data demonstrated that CV1-CAR T cells have limited persistence in vivo, which was unlikely due to an exhausted phenotype pre-infusion.

CV1-CAR T cells do not expand or persist in vivo. CV1-CAR T-cell expansion and persistence were evaluated in vivo using the A549 subcutaneous (s.c.) model. (A) Diagram of the in vivo study. Mice were injected with 2×106 A549 cells s.c. on day 0, followed by 1.5×106 GFP.ffluc expressing EphA2-CAR (positive control; n=5) or CV1-CAR (n=5) T cells given intravenously (i.v.) on day 7. (B) T-cell bioluminescence images for indicated days. Images for EphA2-CAR T-cell treated mice are spliced due to the location of two mice in a separate cage. (C) Individual and (D) summary bioluminescence measurements (radiance=photons/s/cm2/sr) over time. Data represents min to max (D). **p<0.01, ***p<0.001, ****p<0.0001 by unpaired t-test (D). CAR, chimeric antigen receptor.
CD47 is required for CAR T-cell expansion and persistence in vivo
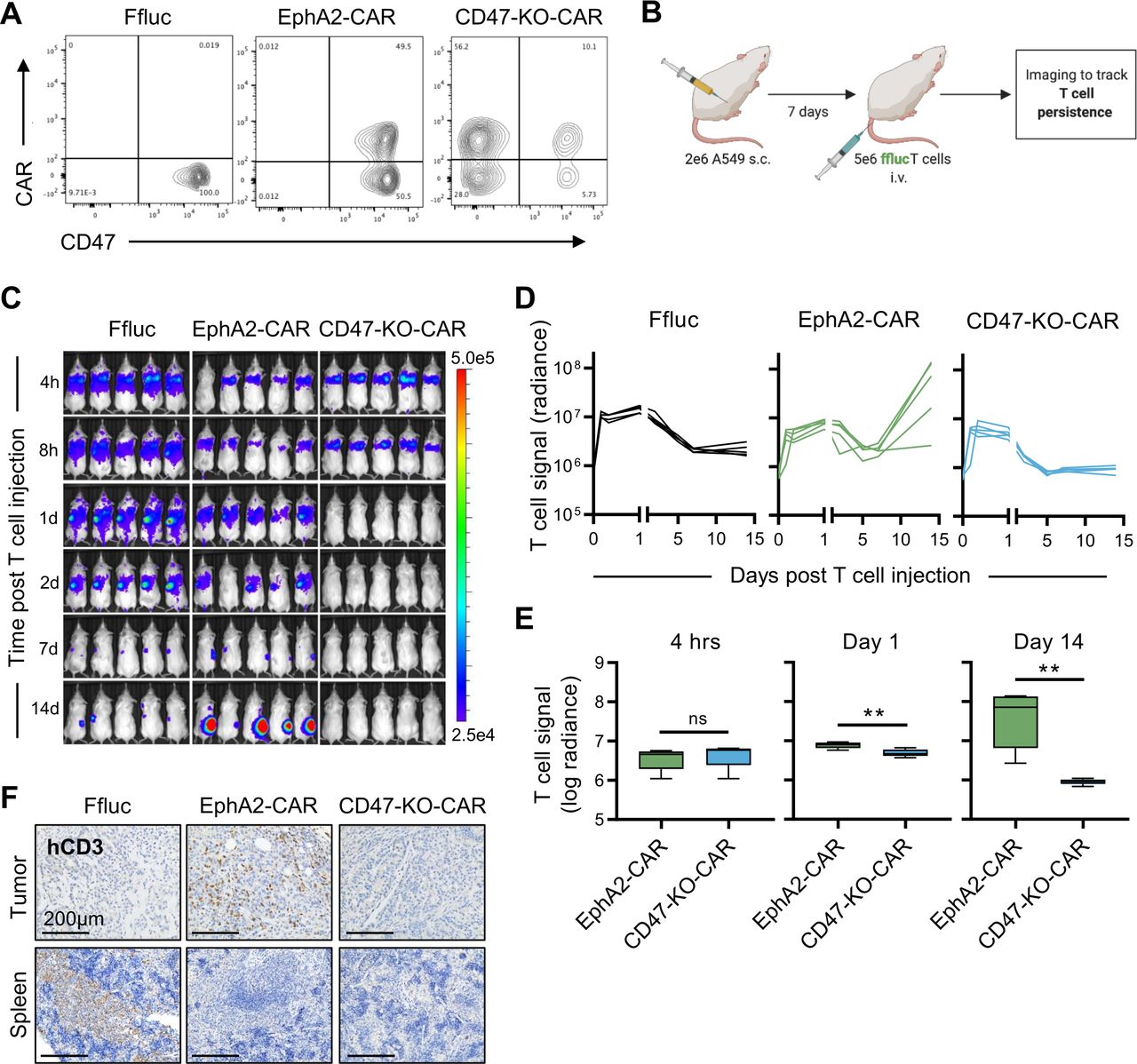
These findings led us to ask an important question, is CD47 surface expression required for CAR T-cell persistence in vivo, or are these findings limited to CV1-CAR T cells? To answer this, we knocked out CD47 in EphA2-CAR T cells (CD47-KO-CAR) (figure 4A), and compared their in vivo fate to CD47-positive EphA2-CAR T cells in our s.c. A549 model in which tumors were injected into the right flank of NSG mice followed by intravenous injection of GFP.ffluc-expressing EphA2-CAR or CD47-KO-CAR T cells on day 7 (figure 4B). While CD47-positive EphA2-CAR T cells expanded and persisted at flank tumor sites, CD47-KO-CAR T cells rapidly disappeared as judged by bioluminescence imaging (figure 4C–E). Mice were euthanized and the imaging findings were confirmed by immunohistochemistry for CD3-positive human T cells (figure 4F). To confirm these findings were not due to the knockout procedure, we repeated the study using AAVS1-KO EphA2-CAR T cells and obtained similar results (online supplemental figure 8A-E). These data indicated that CD47 is critical for CAR T-cell expansion and persistence in vivo.

CD47 knockout CAR T cells fail to persist in vivo. CD47-negative CAR T-cell persistence was evaluated in vivo using the A549 subcutaneous (s.c.) model and GFP.ffluc expressing CD47 knockout EphA2-CAR (CD47-KO-CAR) or control T cells. (A) CD47 surface expression for EphA2-CAR (positive control), CD47-KO-CAR, or otherwise unmodified (Ffluc; negative control) T cells. (B) Schematic of the in vivo study. Mice were injected with 2×106 A549 cells s.c. on day 0, followed by 5×106 EphA2-CAR (n=5), CD47-KO-CAR (n=5), or Ffluc (n=5) T cells intravenously (i.v.) on day 7. (C) Bioluminescence images for indicated time points (h=hour, d=day). (D) Individual and (E) summary bioluminescence measurements over time. (F) Immunohistochemistry staining for human CD3 in tumors and spleens extracted 16 days post T-cell injection. Images represent one mouse per treatment condition. Scale bars=200 µm. Radiance=photons/s/cm2/sr. Data represents min to max (E). **p<0.01, ns, non-significant by unpaired t-test (E). CAR, chimeric antigen receptor.
CD47-negative T cells are targeted by murine macrophage cell lines
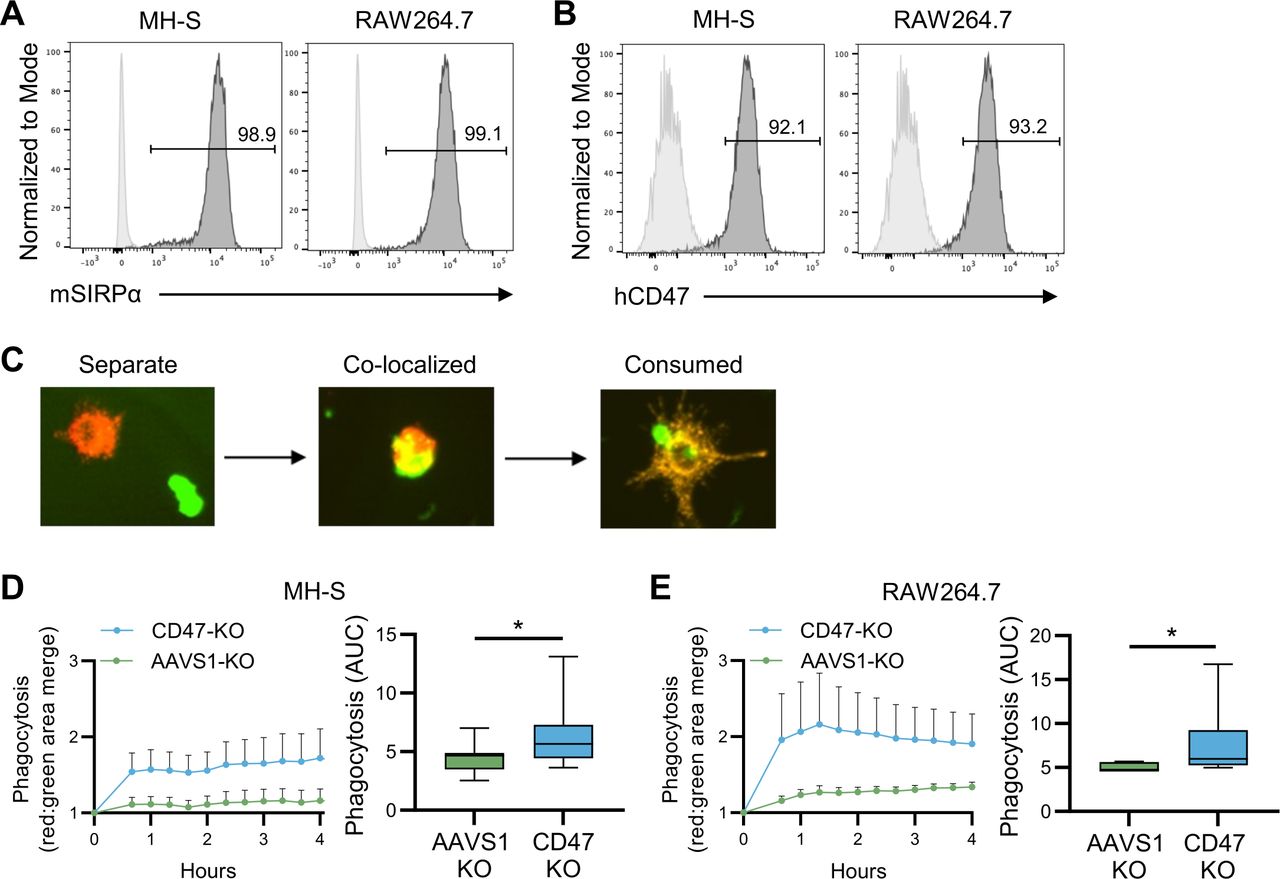
CD47 is a known macrophage anti-phagocytic signal,31 so we queried whether limited persistence of CD47-negative human CAR T cells could be due to clearance by murine macrophages.32 To determine if murine macrophages could phagocytose CD47-negative human T cells, we co-cultured NT, CD47-KO, or AAVS1-KO human T cells at a 1 to 1 ratio with MH-S or RAW264.7 murine macrophages, both of which express murine SIRPα and bind human CD47 (figure 5A,B). T cells were labeled with CellBrite Green, macrophages with CellBrite Red, and T-cell phagocytosis by macrophages was quantified by measuring the merged green/red signal detected by Incucyte live cell imaging (figure 5C). Imaging revealed murine macrophages phagocytosed CD47-KO T cells significantly greater than CD47-positive AAVS1-KO T cells (figure 5D,E), indicating that macrophage mediated phagocytosis was potentially responsible for the elimination of CD47-negative CAR T cells in vivo.

Murine macrophages phagocytose CD47-negative human T cells. Murine macrophage cell line (MH-S and RAW264.7) ability to phagocytose CD47-negative human T cells was evaluated in vitro. (A) MH-S and RAW264.7 expression of murine SIRPα (mSIRPα; dark gray=antibody, light gray=isotype control). (B) Post Fc block, MH-S and RAW264.7 cells were incubated with human CD47-Fc protein and anti-Fc antibody to determine human CD47 (hCD47) binding (dark gray=antibody, light gray=isotype control). (C) Representative Incucyte live cell images of separate human T cells (green) and murine macrophages (red) (left; separate), with T-cell phagocytosis (yellow) (middle; co-localized), and post-phagocytosis (right; consumed). (D) MH-S phagocytosis of CD47-negative (CD47-KO; n=8) compared with CD47-positive (AAVS1-KO; n=8) human T cells (E) RAW264.7 phagocytosis of CD47-KO (n=8) compared with AAVS1-KO (n=8) human T cells. Data represents mean±SEM (D left panel and E left panel) or min to max (D right panel and E right panel). *p<0.05 by Wilcoxon matched-pairs signed rank test (D and E). SIRPα, signal regulatory protein alpha.
Macrophage depletion enhances CD47-negative CAR T-cell persistence in vivo
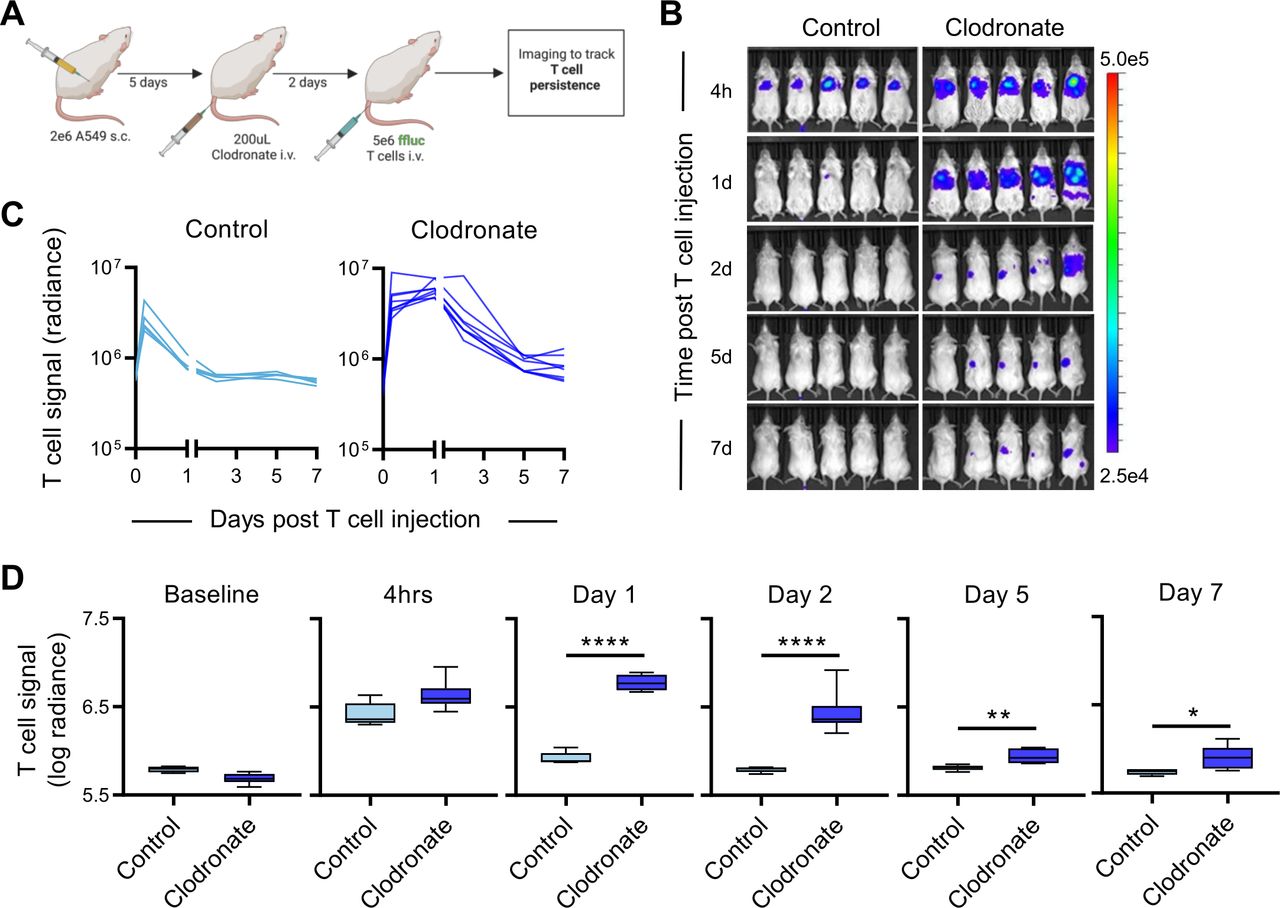
To evaluate if macrophages are involved in the rapid elimination of CD47-negative CAR T cells in vivo, we asked if depleting macrophages with clodronate liposomes33 enables CD47-negative CAR T cells to persist post-infusion. As expected, control liposomes did not alter the percent of macrophages detected in the spleen or bone marrow of NSG mice (online supplemental figure 9A,B), while clodronate liposomes reduced macrophages in both of these tissues within 48 hours (online supplemental figure 9A,B). Mice engrafted with A549 tumors s.c. were injected with a single 200 uL intravenous dose of clodronate or control liposomes 48 hours prior to administration of GFP.ffluc expressing CD47-KO-CAR T cells (figure 6A and online supplemental figure 9C). While CD47-KO-CAR T cells were rapidly eliminated in control treated mice, the same T cells were detected at high levels for 24 hours post-infusion and persisted above baseline for approximately 7 days in macrophage depleted animals (figure 6B–D). These findings indicate that macrophage depletion enhanced CD47-negative CAR T-cell persistence, and macrophages are likely responsible for the elimination of CD47-negative CAR T cells in vivo.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CD47-negative CAR T cells exhibit enhanced persistence in macrophage depleted mice. CD47-negative CAR T-cell persistence was compared +/− macrophage depletion in vivo using the A549 subcutaneous (s.c.) model. (A) Schematic of the in vivo study. Mice were injected with 2×106 A549 cells s.c. on day 0, followed by a single 200 uL intravenous (i.v.) dose of control liposomes (n=5) or clodronate liposomes (n=8) on day 5, and 5×106 GFP.ffluc expressing CD47 knockout EphA2-CAR T cells i.v. on day 7. (B) Bioluminescence images for indicated time points (h=hour, d=day; representative images for 5/8 clodronate treated mice are shown). (C) Individual and (D) summary bioluminescence measurements over time. Radiance=photons/s/cm2/sr. Data represents min to max (D). *p<0.05, **p<0.01, ****p<0.0001 by unpaired t-test (D). CAR, chimeric antigen receptor.
Discussion
Here we describe the importance of CD47 surface expression for CAR T-cell effector function in vivo. While loss of CD47 expression enables CV1-CAR T cells to overcome fratricide and retain their effector function in vitro, they fail to expand, persist, or exhibit antitumor activity in vivo. This finding was not specific for CV1-CAR T cells, since CD47-KO EphA2-CAR T cells had the same in vivo fate. Finally, we demonstrate that CD47-negative T cells are preferentially phagocytosed by macrophages in vitro, and macrophage depletion enhanced CD47-negative CAR T-cell persistence in vivo. Thus, our findings highlight that CD47 expression is critical for CAR T-cell survival in vivo and is a ‘sine qua non’ for successful adoptive T-cell therapy.
CAR structure, including the antigen recognition domain, hinge and transmembrane domains can substantially alter CAR T-cell transduction and function.30 34 Here CD47-CAR T cells derived from CV1, a high affinity SIRPα variant,25 with CD8α hinge and transmembrane domains exhibited ~100% transduction, loss of CD47 surface expression and impaired early expansion. CD47-CAR T cells with different binding and structural domains have been described with discrepant results. Similar to our findings, those derived from the mAb Hu5F9 with CD28 hinge and transmembrane domains lost CD47 surface expression and had impaired expansion compared with NT T cells.24 In the same study B6H12-CAR T cells with CD28 hinge and transmembrane domains also had impaired expansion. In a separate study however, B6H12-CAR T cells with a CD8α hinge and CD28 transmembrane had low CAR surface expression and normal expansion.23 Therefore, even with the same scFv, CD47-CAR T cells exhibit different properties based on CAR structural domains. Thus, delineating the precise effect of hinge and transmembrane domains on CD47-CAR T-cell function requires additional studies and careful testing of different CD47 antigen binding domains with multiple iterations of hinge and transmembrane domains.
T-cell fratricide can occur when CAR targeted antigens are expressed on the T-cell surface. This phenomenon is described for multiple targets and differentially affects CAR T-cell products based on multiple factors including the capacity for antigen downregulation.35 We and others have shown that human T cells express CD47,36 and now demonstrate that CV1-CAR T cells commit fratricide by rapidly killing CD47-positive T cells. They overcome fratricide and expand post-transduction by downregulating CD47 cell surface expression. CAR T-cell fratricide is, not surprisingly, well documented for T-cell malignancy associated antigens such as CD5 and CD7. Similar to CV1-CAR T cells, CD5-CAR T cells have impaired early expansion and downregulate CD5 expression to overcome fratricide.37 In contrast, CD7 is not downregulated post CD7-CAR transduction, which leads to substantial fratricide, limited expansion, and poor function.38 39 Several methods have been established to overcome CAR T-cell fratricide including genetic deletion of the target antigen in T cells,38 40 inhibiting CAR expression with a Tet-Off system prior to in vivo infusion,41 blocking antigen or CAR recognition domains with peptides or antibodies,42 sequestering CARs in the endoplasmic reticulum during expansion,39 or transducing antigen negative T-cell populations.43 While these approaches could be explored to overcome CD47-CAR mediated fratricide, CD47-negative CAR T cells lack in vivo antitumor activity. Thus, approaches need to be developed in which, for example, a CD47 molecule is expressed in CAR T cells that still functions as a ‘don’t eat me signal’, but is not recognized by the CD47-CAR to avoid fratricide.
CD47 has important functions in the lymphohematopoietic system. It is long known to be a marker of self on red blood cells44 and important for neutrophil transendothelial migration during inflammation.45 We show CD47 is critical for T-cell therapy because it is required for CAR T-cell expansion and persistence in vivo. In T cells, CD47 is needed for persistence in some allogeneic murine models, as CD47-negative T cells are eliminated by splenic dendritic cells and macrophages within 1 day post-infusion into major histocompatibility complex disparate animals.46 CD47 also plays a role in T-cell transendothelial migration,16 17 activation/co-stimulation,8–11 inhibition,12 13 and cell death14 15 dependent on experimental conditions and the method of CD47 activation. Since we found potent CAR T-cell function in vitro, rapid CAR T-cell elimination in vivo, macrophage phagocytosis of CD47-negative T cells in vitro, and enhanced CD47-negative CAR T-cell persistence in the presence of macrophage depletion in vivo, macrophage mediated elimination of CD47-negative CAR T cells is most likely responsible for their dismal performance in vivo. While less likely, other functions of CD47 beyond the ‘don’t eat me signal’ might have also contributed to their failure in vivo, including disordered transendothelial migration, and future studies are needed to provide additional insights.
Multiple methods have been designed to target the CD47-SIRP⍺ pathway for cancer immunotherapy. CD47 antibodies combined with tumor targeting antibodies produce high rates of complete responses in patients with lymphoma,20 demonstrating clear utility of targeting the CD47-SIRPα pathway for treatment of human cancers. CD47 antibody combined with GD2 or HER2 antibodies further demonstrates the utility of this therapeutic approach against solid tumors.21 22 Given the intrinsic infiltrative and proliferative properties of T cells combined with an ability to genetically enhance effector functions, adoptive cell therapy targeting CD47 continues to hold promise despite the challenges described herein. Future approaches such as generating inducible CD47-CAR T cells for activation only within the tumor environment could potentially move the approach forward.47 Given the presence of CD47 expression in normal tissues, the development of next-generation CD47-specific cell therapy approaches will require careful toxicity evaluation. Toxicity studies could not reliably be performed for CV1-CAR T cells in this study because CV1-CAR T cells lacked effector function and persistence in vivo. Some novel methods to target the CD47/SIRPα axis using adoptive cell therapy have been described and warrant further investigation. For instance, CAR T cells secreting a SIRPα-Fc fusion molecule or an anti-CD47 single domain antibody fragment to disrupt CD47/SIRPα binding have greater antitumor activity than CAR T cells alone.48 49 There is also growing evidence supporting a role for overexpressing CD47 to enhance the survival of adoptively transferred immune cells.50 Our data supports the notion that overexpressing CD47 in immune cells could be protective in the context of cell therapy for cancer.
In conclusion, while downregulation of CD47 in CV1-CAR T cells renders them functional in vitro, CD47 surface expression was required for CAR T-cell expansion and persistence in vivo. These findings underscore the importance of CD47 as a critical protective signal for CAR T cells in vivo.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Human peripheral blood mononuclear cells (PBMCs) were obtained from the whole blood of healthy donors after written informed consent was obtained in accordance with the tenets of the Declaration of Helsinki. The protocol was approved by the St. Jude Children’s Research Hospital Institutional Review Board. The protocol ID is pro00008053. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank Deanna Langfitt, Abbas Karouni, Sagar Patil, and Cecilia Western from the St. Jude Bone Marrow Transplantation and Cellular Therapy Flow Cytometry Core Lab for their support and advice for flow cytometry experiments. We thank Chandra Savage, Clemothy Bell, Amanda George, Tanya Khan, Jennifer McCommon, and Krista Millican from the St. Jude Animal Resource Center for their expertise in care of study animals. We thank Walter Akers, Melissa Johnson, Carmen Coleman, Caroline Danehy, and Rebecca Thorne from the St. Jude Center for In Vivo Imaging and Therapeutics for their expertise in small animal imaging. We thank Diane Woods for expert management of the St. Jude Department of Bone Marrow Transplantation and Cellular Therapy Translational Research Laboratory operations. We thank Amy Prior, Shalonda Campbell, and Molly Martin from the St. Jude Department of Bone Marrow Transplantation and Cellular Therapy for their administrative support. Graphics were created with BioRender (BioRender.com), for which we have a license.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conceptualization: ANB, SG, CD. Formal analysis: ANB, PC, SG, CD. Funding acquisition: SG, CD. Investigation: ANB, PC, PN, PV, HS. Methodology: ANB, PC, SMP-M, PV, HS, GK, SG, CD. Project administration: ANB, CD. Supervision: SG, CD. Validation: ANB, CD. Visualization: ANB, CD. Writing—original draft: ANB, CD. Writing—review and editing: ANB, PC, SMP-M, PN, PV, HS, GK, SG, CD. Responsible for the overall content as guarantor: CD.
Funding Preclinical imaging was performed by the Center for In Vivo Imaging and Therapeutics, which is supported in part by grants from the US Department of Health and Human Services, National Institutes of Health, National Cancer Institute (NCI) (P01CA096832 and R50CA211481). The St. Jude Hartwell Center that performed DNA sequencing of plasmids is supported in part by a grant from the NCI (P30CA021765). Guide RNA design and validation were performed by the St. Jude Center for Advanced Genome Engineering, which is supported in part by a grant from the NCI (P30CA021765). This work was supported by the Assisi Foundation of Memphis to CD, the American Lebanese Syrian Associated Charities (ALSAC) to CD and SG, and the St. Jude Comprehensive Cancer Center Developmental Biology and Solid Tumor Program, which is funded in part by a grant from the NCI P30CA021765, to CD.
Competing interests PC, PN, GK, SG, and CD are co-inventors on patent applications in the fields of T-cell or gene therapy for cancer. SG is a consultant of TESSA Therapeutics, a Data and Safety Monitoring Board (DSMB) member of Immatics, and has received honoraria from Tidal, Catamaran Bio, Sanofi, and Novartis within the last 2 years.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.