Article Text

Abstract
Background Chimeric antigen receptor (CAR) T-cell therapies have demonstrated transformational outcomes in the treatment of B-cell malignancies, but their widespread use is hindered by technical and logistical challenges associated with ex vivo cell manufacturing. To overcome these challenges, we developed VivoVec, a lentiviral vector-based platform for in vivo engineering of T cells. UB-VV100, a VivoVec clinical candidate for the treatment of B-cell malignancies, displays an anti-CD3 single-chain variable fragment (scFv) on the surface and delivers a genetic payload that encodes a second-generation CD19-targeted CAR along with a rapamycin-activated cytokine receptor (RACR) system designed to overcome the need for lymphodepleting chemotherapy in supporting successful CAR T-cell expansion and persistence. In the presence of exogenous rapamycin, non-transduced immune cells are suppressed, while the RACR system in transduced cells converts rapamycin binding to an interleukin (IL)-2/IL-15 signal to promote proliferation.
Methods UB-VV100 was administered to peripheral blood mononuclear cells (PBMCs) from healthy donors and from patients with B-cell malignancy without additional stimulation. Cultures were assessed for CAR T-cell transduction and function. Biodistribution was evaluated in CD34-humanized mice and in canines. In vivo efficacy was evaluated against normal B cells in CD34-humanized mice and against systemic tumor xenografts in PBMC-humanized mice.
Results In vitro, administration of UB-VV100 resulted in dose-dependent and anti-CD3 scFv-dependent T-cell activation and CAR T-cell transduction. The resulting CAR T cells exhibited selective expansion in rapamycin and antigen-dependent activity against malignant B-cell targets. In humanized mouse and canine studies, UB-VV100 demonstrated a favorable biodistribution profile, with transduction events limited to the immune compartment after intranodal or intraperitoneal administration. Administration of UB-VV100 to humanized mice engrafted with B-cell tumors resulted in CAR T-cell transduction, expansion, and elimination of systemic malignancy.
Conclusions These findings demonstrate that UB-VV100 generates functional CAR T cells in vivo, which could expand patient access to CAR T technology in both hematological and solid tumors without the need for ex vivo cell manufacturing.
- Immunotherapy
- Receptors, Chimeric Antigen
- Cell Engineering
- Drug Evaluation, Preclinical
- Translational Medical Research
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- Immunotherapy
- Receptors, Chimeric Antigen
- Cell Engineering
- Drug Evaluation, Preclinical
- Translational Medical Research
WHAT IS ALREADY KNOWN ON THIS TOPIC
Chimeric antigen receptor (CAR) T cells have demonstrated unprecedented success in the treatment of B-cell malignancies, but their widespread use is hindered by the logistic complexities of ex vivo manufacturing, a prolonged wait and hospital stay for the patient, efficacy challenges due to lack of in vivo expansion and persistence, and safety challenges related to lymphodepletion and cytokine release syndrome.
WHAT THIS STUDY ADDS
In this report, we demonstrate successful engineering of a lentiviral vector which transduces functional CAR T cells in vivo and demonstrates a favorable biodistribution profile. We also demonstrate the use of a rapamycin-activated cytokine receptor system to use rapamycin to selectively drive proliferation of CAR T cells.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
These findings demonstrate the feasibility of an in vivo engineering approach, a successful strategy to armour CAR T cells for enhanced proliferation, and proof-of-concept data for a clinical approach.
Introduction
Chimeric antigen receptor (CAR) T-cell therapies have demonstrated unprecedented success in the treatment of relapsed/refractory B-cell malignancies, which has generated substantial investment in engineered T cells as a therapeutic modality.1–3 Current autologous CAR T-cell manufacturing requires cell collection from the patient, genetic manipulation, expansion, and quality release before starting treatment. In addition, patients undergo preconditioning with lymphodepleting chemotherapy before receiving CAR T-cell treatment as a requirement for successful engraftment. The complexity of the manufacturing process, logistical challenges required to maintain this supply chain, and toxicity related to lymphodepleting chemotherapy and CAR T cells results in prolonged waiting periods to receive therapy and greatly limits the number of facilities where these products can be administered.4 5 Another consideration is the high cost associated with approved autologous CAR T-cell products which introduces challenges related to hospital payer and insurance coverage.
In vivo engineering to generate CAR T cells is a potential solution to the difficulties associated with ex vivo cell manufacturing as demonstrated successfully in preclinical models.6–8 Viral vector manufacturing is well established in the context of ex vivo CAR T-cell manufacturing and can be further engineered to enable direct administration to the patient. Viral vectors are ‘off-the-shelf’ products that, in contrast to off-the-shelf allogeneic cell therapies, generate autologous CAR T cells that are compatible with the patient’s immune system.
To enable in vivo T-cell engineering, we developed VivoVec, a surface-engineered third-generation self-inactivating replication-incompetent lentiviral vector (LVV) platform designed to generate CAR T cells without ex vivo cell manufacturing or lymphodepletion. UB-VV100, the first VivoVec platform-derived clinical candidate drug product, targets CD19 for the treatment of B-cell malignancies (figure 1A). UB-VV100 particles (comprising VivoVec particles packaging the UB-VV100-specific payload) are manufactured at clinical scale using a suspension process to enable in vivo administration. UB-VV100 particles are pseudotyped with the cocal fusion glycoprotein, a glycoprotein with a close structural relationship to that of VSV-G (71.5% shared amino acid sequence).9 Compared with VSV-G, cocal is resistant to serum inactivation in humans, improving in vivo persistence and enabling direct administration to patients.9 10 UB-VV100 is designed to express an anti-CD3− single-chain variable fragment (scFv) on the viral envelope to mediate T-cell binding, activation, and efficient T-cell transduction.
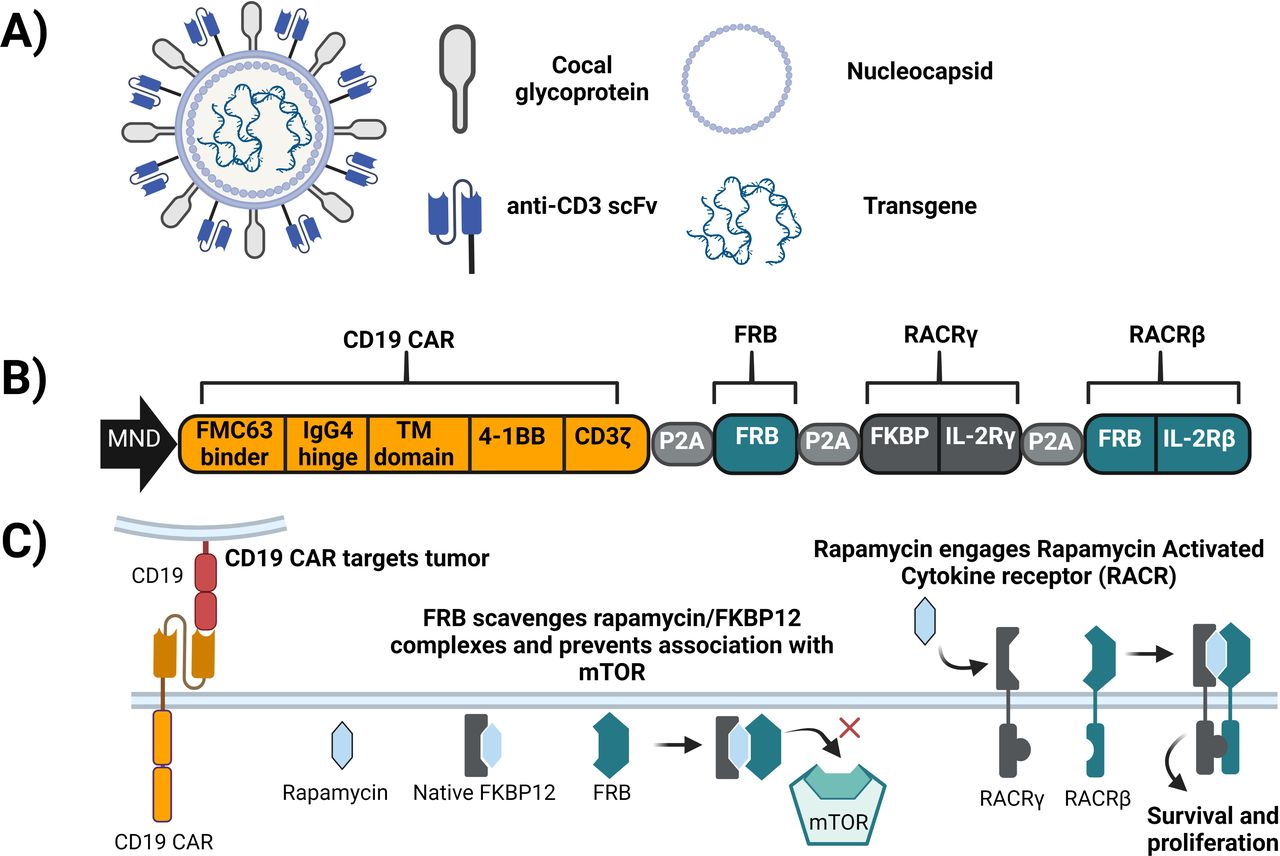
Overview of UB-VV100. (A) UB-VV100 is a third-generation replication-incompetent lentiviral vector. UB-VV100 is pseudotyped with cocal glycoprotein and displays anti-CD3 scFv moieties to mediate T-cell activation and transduction. (B) The UB-VV100 transgene encodes a polycistronic payload driven by the MND promoter and separated by P2A peptide fragments. The resulting proteins are a second-generation anti-CD19 CAR, FRB, and the RACR. RACR is a chimeric heterodimer consisting of FKBP extracellular unit attached to an IL-2Rγ signaling domain and an FRB extracellular unit attached to an IL-2Rβ signaling domain. (C) The components of UB-VV100 work together to mediate tumor killing and promote cell survival. CAR, chimeric antigen receptor; FRB, FKBP12–rapamycin-binding protein; IL, interleukin; RACR, rapamycin-activated cytokine receptor; scFv, single-chain variable fragment.
The UB-VV100 payload encodes a polycistronic sequence with an MND promoter (a synthetic promoter with constitutive expression in the hematopoietic lineage)11 driving the expression of four proteins separated by P2A sequences (figure 1B). These include a second-generation anti-CD19 CAR with the FMC63 scFv, CD3ζ, and a 4-1BB costimulatory endodomain, and the components of the rapamycin-activated cytokine receptor (RACR) system. In the presence of rapamycin, the RACR system provides prosurvival and expansion signals to CAR T cells in vivo and confers resistance to rapamycin-mediated mammalian target of rapamycin (mTOR) suppression. The RACR system is composed of
An FKBP12–rapamycin-binding protein (FRB) domain which is intended to act as a sink for intracellular rapamycin-FKBP12 complexes and prevent their inhibition of mTOR.
A synthetic chimeric cytokine receptor consisting of an FKBP-interleukin (IL)-2Rγ fusion and FRB-IL-2Rβ fusion which heterodimerize on engagement with rapamycin, resulting in intracellular signaling that mimics IL-2 and IL-15 to promote growth and proliferation (figure 1C).12
Here we demonstrate that UB-VV100 activates and transduces T cells in vitro and in vivo in a dose-dependent manner, generating CAR T cells that mediate tumor control. We demonstrate RACR-mediated expansion and enrichment in the presence of rapamycin, and characterize the biodistribution of UB-VV100 following intranodal and intraperitoneal administrations to evaluate the safety of this approach.
Methods
Vector production
Lentiviral particles were generated by polyethlenimine-mediated transient transfection of in-house suspension adapted HEK293Ts (American Type Culture Collection (ATCC), Manassas, Virginia, USA) or viral production cells (Thermo Fisher Scientific, Waltham, Massachusetts, USA) grown in a shaking flask incubator in FreeStyle or LV-Max medium (Thermo Fisher Scientific), respectively. For larger preparations of suspension culture-generated lentiviral particles, single-use bioreactors replaced shake flasks for bulk production. The lentiviral particles were further purified, concentrated, and formulated through a series of filtration steps to remove cell debris, host cell proteins, host cell DNA, and residual plasmid DNA. The final particles were then sterilized via 0.2 µM filtration and frozen. Lentiviral lots were titered on SUP-T1 cells by flow cytometry against surface CAR expression and ddPCR against lentiviral Psi integration element using the following primer sets: forward 5′-ACT TGA AAG CGA AAG GGA AAC-3′, reverse 5′-CACCCATCTCTCTCCTTCTAGCC-3, Taqman probe PSI-FAM: 5′-/56-FAM/ AGCTCTCTC/ZEN/GACGCAGGACTCGGC/3IABkFQ/-3′. Samples were analyzed on the QX200 automated droplet generator and reader system (Bio-Rad, Hercules, California, USA). Vector formulations were evaluated for particle count by p24 ELISA (ZeptoMetrix, Buffalo, New York, USA) and evaluated by western blot to confirm anti-CD3scFv incorporation (Abcam ab40844, Cambridge, UK).
Cell lines
Cryopreserved peripheral blood mononuclear cells (PBMCs) were purchased from AllCells (Alameda, California) and Bloodworks (Seattle, Washington, USA). Nalm-6 and Raji parental lines, and lines carrying mCherry and GFP::ffluc marker genes were obtained from Seattle Children’s Therapeutics. CD19 KO Nalm6 lines were gifted from Seattle Children’s Therapeutics. HEK-293T (CRL-11268) and SUP-T1 (CRL-1942) cells were obtained from the ATCC. Cryopreserved human patient samples were acquired from ProteoGenex (Inglewood, California, USA).
Assessment of UB-VV100-mediated PBMC transduction
Donor PBMCs were cultured in RPMI 1640 (Gibco) with 10% heat-inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific) and 50 IU IL-2 (R&D Systems, Cambridge, Massachusetts, USA). PBMCs were transduced at a density of 2E+06 cell/mL by adding vector directly to the well. Unless otherwise noted, PBMCs were transduced at a multiplicity of infection of 5 for 3 days for all in vitro studies. Rapamycin (1–40 nmol) was added to the indicated cultures on day 3. Activation and transduction were assessed on days 3 and 7 by flow cytometry using surface detection of FMC63 to identify CAR T cells unless otherwise noted (ACROBiosystems, Newark, Delaware, USA). Cells were washed with PBS, stained with a fixable viability dye, washed with FACS buffer (2% FBS in PBS), and then stained for 30 min in FACS buffer (online supplemental table S1). Cells were enumerated with CountBright beads for all flow cytometry studies (Thermo Fisher Scientific). Data were acquired on a Cytoflex (Beckman Coulter, Brea, California, USA).
Supplemental material
Assessment of CAR T-cell function in healthy PBMCs
Healthy donor PBMCs were transduced with UB-VV100. In indicated studies, PBMCs were activated with CD3/CD28 Dynabeads (Thermo Fisher Scientific) but not transduced. Ten days post transduction, PBMCs were plated with CD19+ or CD19KO Nalm-6 tumor cells for 24 hours at the indicated effector-to-target ratio. Nalm-6 viability was assessed by flow cytometry. Supernatant cytokines were analyzed using Meso Scale Discovery multiplex detection with the Proinflammatory Panel 1 (human) V-plex Kit (Rockville, Maryland, USA). T-cell degranulation was assessed in cultures of 1:2 CAR T:Nalm-6 for 4 hours in the presence of 1× monensin, 1× brefeldin A (BioLegend) and 1:200 diluted CD107a- PE antibody (BioLegend).
Cotransduction of Nalm-6 tumor and PBMCs
PBMCs from eight healthy donors were plated at a density of 5×105 cells per well with an equal number of Nalm-6 GFP+tumor cells. The cocultures were transduced with UB-VV100 or a vector encoding a control CAR. Transduction was assessed by intracellular detection of P2A in both T cells and Nalm-6 GFP cells. Detection of CD19 was assessed by flow cytometry (online supplemental table S1).
Transduction of B-cell malignancy PBMCs from patient samples
PBMCs from B-cell acute lymphoblastic leukemia (B-ALL) and relapsed/refractory (R/R) diffuse large B-cell lymphoma (DLBCL) patient samples were transduced with UB-VV100 (online supplemental table S2). From day 7 to day 20, UB-VV100-transduced PBMCs were expanded in 10 nM rapamycin. On day 20, rapamycin-expanded CAR T cells were assessed for cytokine release target killing in Nalm-6 cultures as previously described.
Depletion of B cells in CD34 humanized NSG mice
Studies were conducted at Fred Hutch Cancer Research Center under approval of the Institutional Animal Care and Use Committee (IACUC) protocol Proto202000003 (online supplemental table S3). Female Nod.Cg-PrkdcscidIL2rgtm1Wjl/SzJ (NSG) mice were irradiated and engrafted with CD34+ human hematopoietic stem cells at Jackson Laboratories (Bar Harbor, Maine, USA). UB-VV100 formulation was thawed on the day of treatment, diluted in PBS, and injected via intraperitoneal injection. Blood was collected via retroorbital sinus into sodium–EDTA coated microtainers and centrifuged at 4000×g for 10 min to collect plasma. Blood was lysed in 1× Pharmlyse (BD Biosciences, San Diego, California, USA) and washed in PBS (Life Technologies, Rockville, Maryland, USA). Cells were stained for viability and surface antibody (online supplemental table S1). Samples were acquired on a 4-laser Cytoflex S (Beckman Coulter) and analyzed using FlowJo V.10 (Ashland, Oregon, USA). Data were graphed in Microsoft Excel and plotted on GraphPad Prism V.9 (San Diego, California, USA).
Biodistribution of UB-VV100 in CD34-humanized NCG mice
Studies were conducted at Charles River under the approval of IACUC protocol Umoja 2021–3225. All vector formulations were thawed day of administration. Female CD34-humanized mice of NOD-Prkdcem26Cd52 Il2rgem26Cd22/NjuCrl (NCG) mice were acquired at Charles River Laboratories and were accepted for enrollment based on >45% humanization (online supplemental table S4). UB-VV100 formulation was thawed the day of treatment, diluted in PBS, and injected via intraperitoneal injection once on study day 1. Blood was prepared for flow cytometry and acquired on FACS Lyric flow cytometer (BD Biosciences). Formalin-fixed, paraffin embedded (FFPE) tissues were stained with H&E and analyzed by a board-certified pathologist at Charles River Laboratory.
Biodistribution of αCD3 cocal pseudotyped vector encoding enhanced green fluorescent (eGFP) in canines
Studies were conducted at Charles River Laboratories under the approval of IACUC protocol Umoja 2021–3232 (online supplemental table S5). Male and female beagles 6–7 months of age were sourced from Marshall Bioresources (North Rose, New York, USA). The test article evaluated (CD3-cocal-GFP) consisted of VivoVec particles containing an EGFP payload under an MND promoter. Vector was administered via ultrasound-guided inguinal lymph node injection or intraperitoneal injection once on study day 1. FFPE tissues were stained with H&E and analyzed by a board-certified pathologist at Charles River Laboratory.
Quantification of vector integration events in mouse and canine
Genomic DNA was extracted from flash-frozen whole blood using the Nucleospin Blood QuickPure kit (Macherey-Nagel) and from flash-frozen tissues using the DNeasey Blood and Tissue kit (Qiagen). DNA was quantified using the NanoDrop Lite (Thermo Fisher Scientific). The qPCR reaction was performed in triplicate using 200 ng of DNA per well on a TaqPath ProAmpTM Master Mix (Applied Biosystems) against the viral PSI element of the UB-VV100 payload as previously described. QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems) was used for acquisition. RNA in situ hybridization (ISH) against the desired cellular targets (online supplemental table S6) was performed using the RNAscope LS Multiplex Fluorescent Reagent Kit (Advanced Cell Diagnostics, Newark, California, USA). Briefly, 5 µm FFPE sections were pretreated with heat and protease prior to hybridization with the target oligo probes. Preamplifier and amplifier were hybridized sequentially, followed by tyramide signal amplification (TSA) fluorophore reaction. Specific RNA staining signal was identified as fluorescent, punctate dots. Samples were counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Fluorescent images were acquired using Panoramic SCAN II digital slide scanner (3DHistech) under ×40 magnification. Sections were manually examined to quantify and identify transduced cells (online supplemental table S7-S9).
PBMC humanized mouse model
Studies were conducted at Seattle Children’s Research Hospital under the approval of IACUC protocol ACUC00654. Female NSG mice, 6–12 weeks of age, lacking expression of MHC I and II molecules (NSG DKO, stock number 025216) were purchased from the Jackson Laboratories (online supplemental table S10). Animals were engrafted with 0.25E+06 Nalm-6 tumor cells expressing green fluorescent protein (GFP) and firefly luciferase (GFP::ffluc) via tail vein injection on study day −4. Animals were injected with 15 mg/kg of XenoLight D-Luciferin (Perkin Elmer, Waltham, Massachusetts) and evaluated after 25 min via non-invasive In Vivo Imaging System (IVIS) Spectrum instrument (Perkin Elmer). Animals were humanized with 20E+06 PBMCs via intraperitoneal injection on day −1, and then dosed intraperitoneal with vehicle (10 mM Tris, 10% sucrose, 0.1% poloxamer 188 (v/v), pH 7.1), UB-VV100 diluted in vehicle, or a cocal pseudotyped vector encoding an FMC63-RACR transgene (cocal control) on day 0. Blood was collected for flow cytometry as previously described (online supplemental table S1).
Results
UB-VV100 anti-CD3 scFv surface engineering enables targeted activation and transduction of T cells in a dose-dependent manner, generating functional CAR T cells
We first evaluated UB-VV100-mediated activation and transduction of unstimulated T cells. We incubated PBMCs from healthy human donors with UB-VV100 or a control LVV containing the same payload but lacking anti-CD3 scFv surface engineering (figure 2A). We found UB-VV100 treatment resulted in dose-dependent activation and transduction of CD4 and CD8 T cells. In contrast, the LVV lacking anti-CD3 scFv surface engineering did not activate or efficiently transduce T cells, consistent with previous observations that unstimulated T cells are inefficiently transduced by VSV-G and cocal-pseudotyped lentiviruses9 13 (figure 2B). We did not observe any transduction events in the CD3-negative population, as compared with cells treated with empty vector particles (online supplemental figure 1A,B).
Supplemental material
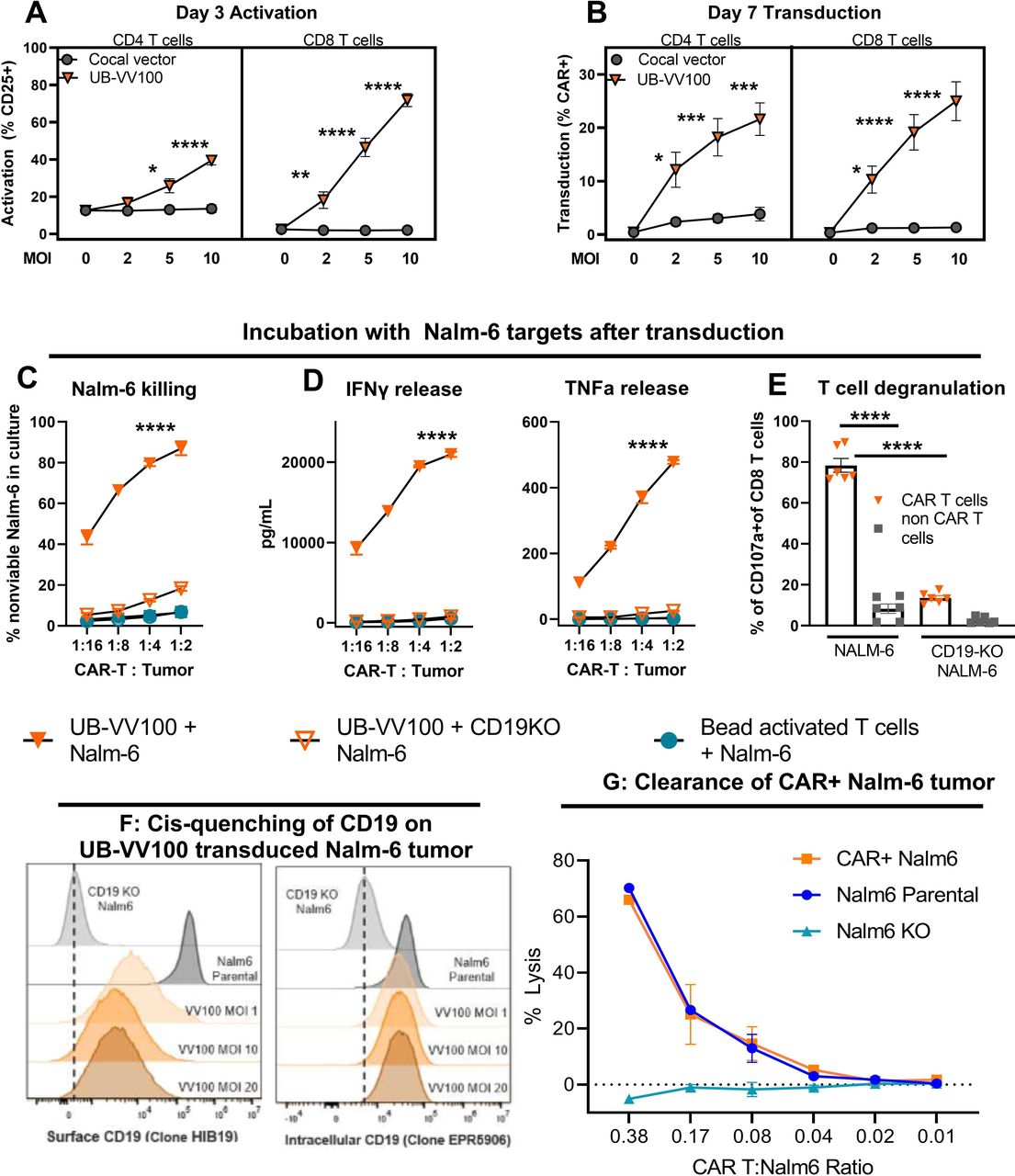
UB-VV100 activates and transduces unstimulated T cells. UB-VV100 is added directly to cultured PBMCs in the presence of IL-2 and no additional stimulation. Activation, transduction, and rapamycin-mediated expansion are evaluated in the wells. (A) T-cell activation, measured by expression of CD25 (IL-2 receptor alpha), 3 days after vector addition. (B) T-cell transduction frequency, measured by surface FMC63 CAR expression, 7 days after vector addition at the indicated MOI. Data points represent mean±1 SEM. Data represent pooled results of n=5 for three unique donors analyzed in two independent experiments. The symbol * indicates significance using two-way ANOVA full interaction model with Tukey’s tests for multiple comparison on the indicate time point. (C) PBMCs transduced with UB-VV100 were incubated with CD19 KO Nalm-6 or parental Nalm-6 at varying E:T cell ratios as indicated. PBMCs activated with CD3/CD28 beads and incubated with parental Nalm-6 were used as an additional negative control. Cocultures were assessed for detection of dead Nalm-6 cells by flow cytometry and (D) release of IFN-γ and TNF-α into the culture supernatant. The symbol * denotes values indicating significance for ordinary two-way ANOVA, comparison of UB-VV100+Nalm-6 compared with the other groups at the indicated time point by Tukey’s postcomparison test. Partial results are related for simplicity. n=3 unique donors per group, combined the result of two independent experiments originally performed in technical duplicate. (E) CAR T cells were cultured with Nalm6 cells at an E:T of 1:2 for 4 hours, and degranulation was measured in P2A+CAR+ CD8+ T cells or P2A−negative non-CAR CD8+ T cells by flow cytometric detection of extracellular CD107a. The symbol * indicates significance for one-way ANOVA. Data represent two donors pooled from two independent experiments in technical duplicates. Nalm-6 cells were transduced with UB-VV100 at MOIs 1, 10, and 20. On day 10, CAR+Nalm-6 cells were stained with an anti-CD19 antibody (clone HIB19) to assess (F) surface CD19 expression levels and an anti-CD19 antibody that binds to an intracellular CD19 epitope (clone EPR5906) to determine the total CD19 protein level. (G) CAR T cells (VV100, MOI 5) from three healthy donors were cocultured with CAR+Nalm6 cells (VV100, MOI 10), non-transduced Nalm6 parental cells, or CD19 KO Nalm6 cells (Nalm6 KO) at multiple CAR T to Nalm6 ratios. After 24 hours of coculture, CAR+Nalm6 cells were identified based on P2A transgene expression, and the frequency of dead Nalm6 cells was determined by viability dye staining via flow cytometry. The percentage of lysis was calculated as the frequency of dead CAR+Nalm6 cells normalized to lysis of Nalm6 cells cocultured with mock transduced PBMCs. Data points represent mean±SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 for all data panels for the indicated analysis. ANOVA, analysis of variance; CAR, chimeric antigen receptor; E:T, effector-to-target; IFN-γ, interferon gamma; KO, knockout; MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cell; TNF-α, tumor necrosis factor alpha.
UB-VV100-transduced CAR T cells were incubated with CD19+ and CD19-deficient (CD19KO) Nalm-6 B-ALL target cells. CAR T cells lysed Nalm-6 cells and secreted effector cytokines in an antigen-specific and dose-dependent manner (figure 2C,D). CAR T-cell degranulation, measured as CD107α positivity, was readily observed when CAR T cells were cocultured with CD19+ but not with CD19KO Nalm-6 target cells (figure 2E). Together these results demonstrate the expected functionality and specificity of UB-VV100-transduced CAR T cells.
Although UB-VV100 is intended to activate and transduce T cells, transduction of malignant B cells in vivo poses theoretical safety and efficacy risks to patients due to the potential for CD19 epitope masking. This phenomenon occurred in a pediatric B-ALL patient treated with ex vivo manufactured anti-CD19 CAR T-cell product, CTL019, which induced resistance to CTL019 therapy.14 To evaluate the risk of epitope masking by UB-VV100, CAR+Nalm-6 cells were generated with UB-VV100 and characterized for surface CD19 epitope detection and resistance to CAR T cytotoxicity.
CAR+Nalm-6 cells demonstrated reduced, but not absent, surface detection of CD19 as measured by median fluorescence intensity (MFI), while total CD19 levels were similar to that of non-transduced Nalm-6 cells (figure 2G). This data suggests that the surface CD19 epitope of CAR+Nalm-6 cells is only partially masked. Despite diminished surface detection of CD19 on CAR+Nalm6 cells, anti-CD19 CAR T cells were able to kill CAR+Nalm6 cells in an antigen-dependent manner. Importantly, the percentage of lysis was similar between CAR+Nalm6 cells and non-transduced Nalm6 cells with normal surface CD19 expression (figure 2G). These results were corroborated in a separate in vitro study in which CAR T cells completely eliminated CAR+Nalm-6 cells when a 1:1 mixture of PBMCs and Nalm6 cells were transduced with UB-VV100 (online supplemental figure S2). The CD19-CAR encoded by UB-VV100, encodes a CD19-CAR with a short IgG4 hinge (14 amino acids) that would be predicted to inefficiently interact with the membrane-distal CD19 epitope on the cell surface, in contrast to CTL019, which encodes a longer CD8a hinge (45 amino acids) and cis-masks CD19 from CTL019 recognition.15 16
Supplemental material
Rapamycin-induced RACR activation drives UB-VV100-transduced T-cell expansion and enrichment
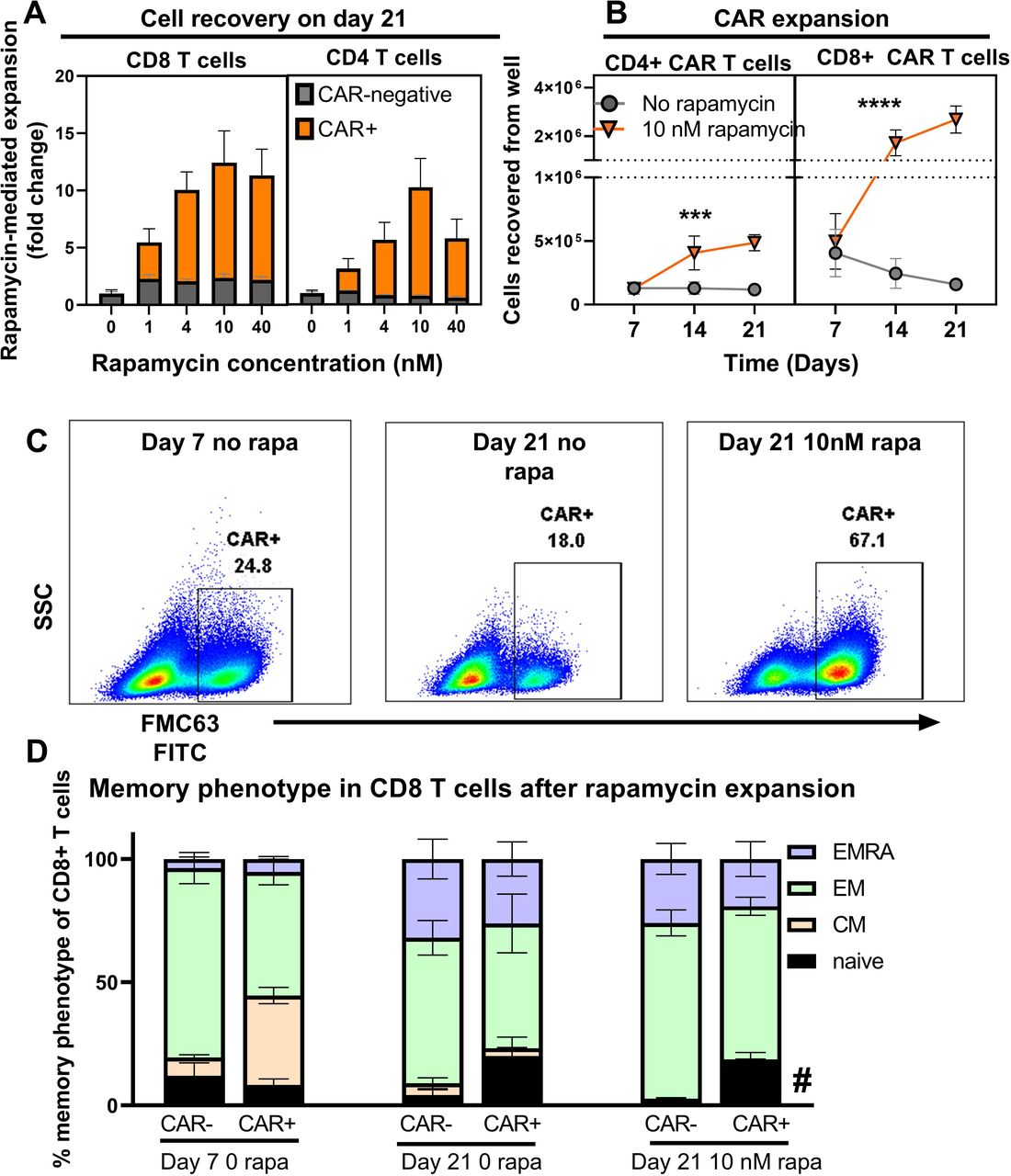
To evaluate CAR T-cell expansion driven by the RACR system, we treated UB-VV100-transduced PBMCs with 0, 1, 4, 10, and 40 nM of rapamycin. Treatment with 1–40 nM of rapamycin resulted in a concentration-dependent expansion of CAR+CD4 and CD8 T cells compared with PBMC cultures without rapamycin, and compared with CAR-negative cells within the same well. Peak expansion was evident at 10 nM for most, but not all, individual donors (figure 3A). Together, the data demonstrate effective engagement of the RACR system along a range of rapamycin concentrations that are consistent with clinical rapamycin dosing troughs (5–20 ng/mL), depending on indication.17 Notably, we observed that T cells were the only cell type to reliably survive in culture under all tested conditions.
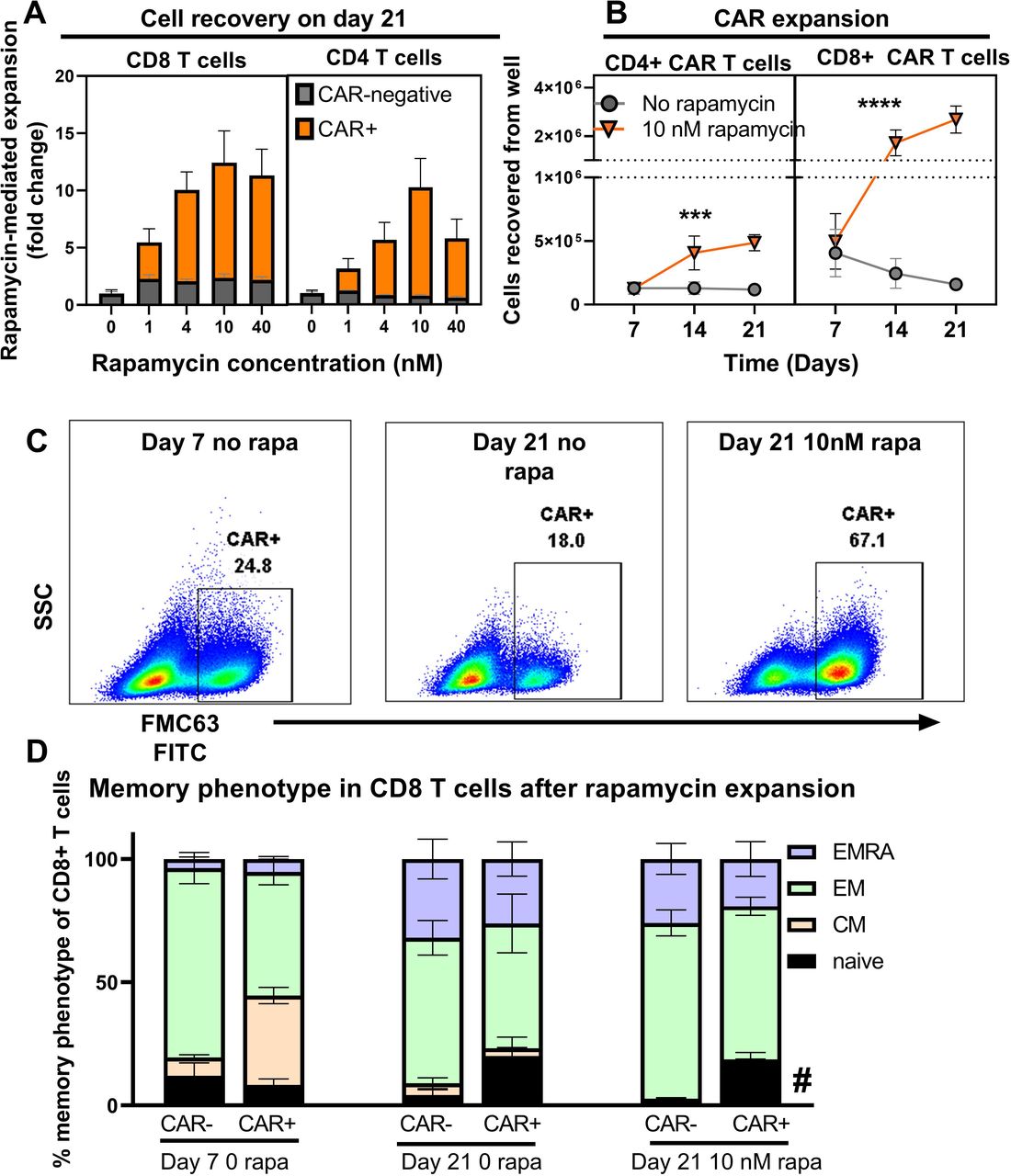
Rapamycin promotes selective expansion of CAR T cells. (A) PBMCs were transduced with UB-VV100 at an MOI of 5 for 3 days before vector removal. Cells were transferred to wells containing the indicated concentration of rapamycin and cultured for an additional 18 days. On study day 21, CAR+ and CAR-negative T-cell expansion was enumerated by flow cytometry. n=5, pooled results from two independent experiments using three separate PBMC donors. Data points indicate mean±1 SEM. (B) PBMCs treated as described (A) at the 10 nM rapamycin concentration were analyzed by flow cytometry to determine the absolute CD4 and CD8 CAR T-cell counts. n=6 per group per time point, pooled results of two independent experiments using three PBMC donors. Axis is split to allow separation of CD4 and CD8 T cells to be seen on the same scale. Symbols (*) indicate statistical significance for two-way ANOVA, main column analysis of effect of rapamycin treatment on cell expansion or enrichment over time series. Data points indicate mean±1 SEM; some error bars not visible due to eclipse by data symbol. PBMCs were transduced with MOI=5 with UB-VV100 and expanded for 18 days with rapamycin (study day 21 after transduction). T cells were assessed by flow cytometry for (C) representative flow staining of surface FMC63 detection and (D) memory phenotype. Representative plots show a single donor. CAR+ T cells are overlaid in red over total T-cell population. Memory phenotype was quantified for CD8+ T cells for day 7 no rapamycin, day 21 no rapamycin, day 21+10 nM rapamycin, in both the CAR+ and CAR-negative fractions. Naïve (CCR7+CD45RA+). Symbol (*) indicates p value of < 0.05 for two-way ANOVA, Tukey’s multiple comparison’s test for comparing the ‘naïve’ population of the CAR+ fraction to the naïve population of the CAR-negative fraction on study day 21+10 nM rapamycin. n=6 from replicate measurements of three unique PBMC donors per condition, results from one experiment. ANOVA, analysis of variance; CAR, chimeric antigen receptor; CM, central memory (CCR7+CD45RA−); EM, effector memory (CCR7−CD45RA+); EMRA, effector memory re-expressing CD45RA (CCR7−CD45RA+); MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cell.
To characterize the kinetics of RACR-driven expansion and enrichment, CAR T-cell frequency and total number were measured over time with and without 10 nM rapamycin treatment. Rapamycin treatment led to absolute expansion of both CD4+ and CD8+ CAR T cells between days 7 to 21 (figure 3B). These findings demonstrate that RACR system engagement selectively expands transduced T cells at clinically relevant concentrations of rapamycin.
To determine the impact of prolonged RACR signaling on T-cell memory differentiation state, we assessed the memory phenotype (CD45RA and CCR7) of the CAR+ and CAR-negative T cells from wells treated with 10 nM rapamycin for 18 days. On day 21, the CAR+ T cells displayed increased frequencies of phenotypically naïve T cells (CD45RA+CCR7+) than the CAR-negative fraction, but we did not find evidence that adding rapamycin for RACR engagement drove terminal differentiation of CAR T cells in culture (figure 3D).
UB-VV100 generates functional CAR T cells from PBMCs collected from a heavily pretreated patient population
Compared with healthy donors, patients with B-cell malignancies can have a heightened inflammatory state due to disease condition and prior cancer therapies. This can lead to increased heterogeneity in the overall cellular composition and the quality of patient T cells (eg, CD4:CD8 ratio, differentiation state, and T-cell subtypes), which could impact T-cell permissiveness to transduction, potency, and persistence properties of CAR T cells.18–20 To evaluate the function of UB-VV100 and transduced T cells with patient material, PBMCs from B-ALL (n=3) and R/R DLBCL (n=5) patients were transduced with UB-VV100 (day 0). In both B-ALL and R/R DLBCL patient samples, UB-VV100 mediated T-cell activation and generated CAR T cells (figure 4A). To determine if rapamycin treatment can enrich patient-derived CAR T cells, cells were further cultured in the presence or absence of 10 nM rapamycin starting on day 7. Treatment with rapamycin led to a higher frequency of CAR T cells by day 20 in all patient samples compared with the no rapamycin control group (figure 4B). Rapamycin-enriched patient CAR T cells were functional as demonstrated by in vitro cytokine secretion and killing of Nalm-6 cells (figure 4C,D). Taken together, these data demonstrate that UB-VV100 can generate functional CAR T cells from patient PBMCs from relevant disease indications.
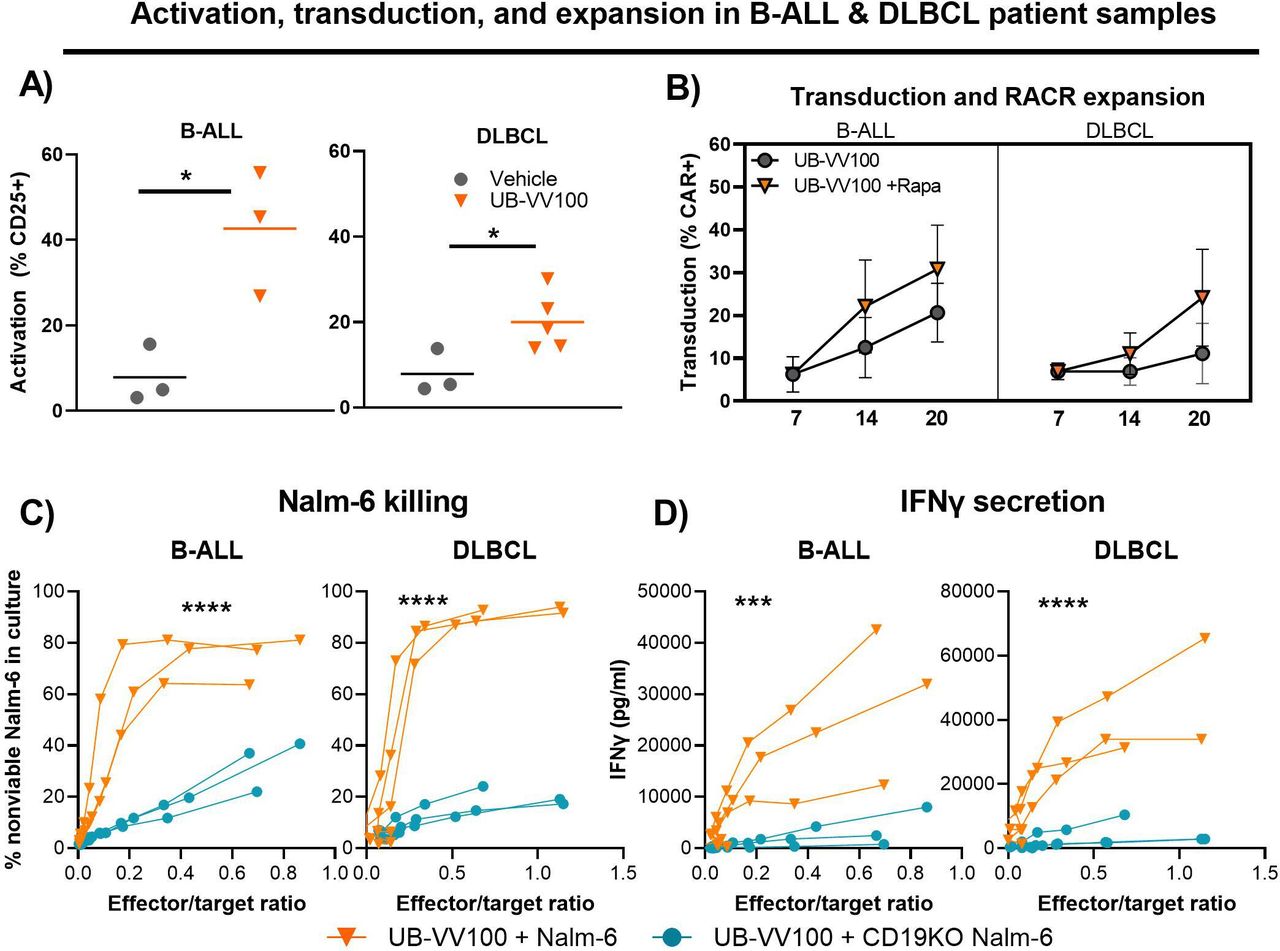
UB-VV100 transduces functional CAR T cells from patient samples. In two separate experiments, PBMCs from patients with B-ALL and DLBCL were transduced with UB-VV100 at MOI of 5. (A) On day 3, the frequencies of T cells expressing CD25 were assessed in B-ALL and DLBCL samples. On day 7 post UB-VV100 transduction, cells were further cultured in the absence or presence of 10 nM rapamycin. *P<0.05, Student’s t-test. Bars indicate mean. (B) The frequencies of CAR T cells on days 7, 14, and 20 were determined by flow cytometry. On day 20, rapamycin-expanded CAR T cells from B-ALL and DLBCL patient samples were cocultured with Nalm-6 or CD19KO Nalm-6 tumor cells. (C) After 24 hours of coculture, the frequency of non-viable Nalm-6 tumor targets was determined by viability dye staining via flow cytometry. ****P<0.001, two-way ANOVA, main effect analysis for Nalm-6 identity. (D) The concentration of IFN-γ in the coculture supernatant was also measured in both B-ALL and DLBCL samples. Data points indicate mean±1 SEM. ***P<0.001, ****P<0.0001, two-way ANOVA, main effect analysis for Nalm-6 identity. ANOVA, analysis of variance; CAR, chimeric antigen receptor; IFN-γ, interferon gamma; MOI, multiplicity of infection.
UB-VV100 biodistribution in humanized mice following intraperitoneal administration indicates selective transduction of immune cells
We evaluated the activity, biodistribution, and safety profile of UB-VV100 in a CD34+ hematopoietic stem cell humanized mouse model, using endogenous B-cell depletion as a surrogate indicator of UB-VV100-transduced CAR T-cell activity.21 For delivery of UB-VV100 in the clinic, an intranodal injection could potentially maximize UB-VV100 exposure in a tissue enriched with T cells while minimizing systemic distribution. While NSG mice lack developed lymph nodes, injection through the intraperitoneal route results in drainage through abdominal lymphatics22; hence, this route was chosen for murine efficacy and safety studies. CD34-humanized mice were treated with doses of 0.4E+06, 2E+06, and 10E+06 transducing units (TUs). Animals developed a UB-VV100 dose-dependent reduction in B cells, with animals treated with 10E+06 TU reaching levels consistent with B-cell aplasia. In contrast, animals treated with vehicle or 10E+06 TU of a cocal-pseudotyped vector displaying an anti-CD3 scFv but encoding a CAR of irrelevant specificity exhibited B-cell levels comparable to the untreated control animals, demonstrating antigen-specific activity (figure 5A and online supplemental table S3). Vehicle-treated animals exhibited a slight reduction in B cells over the study period, consistent with past reports that circulating B-cell levels gradually reduce over time in CD34-humanized mice.23 Circulating CAR T cells were detected in a UB-VV100 dose-dependent manner but not in mice treated with the control CAR encoding vector, suggesting antigen-dependent expansion of CAR T cells. The detection of B-cell depletion (day 11) prior to the detection of circulating CAR T cells in the peripheral blood (day 18) may be due to B-cell killing in sites other than the peripheral blood or may reflect occluded or reduced detection of surface CAR after engagement with cognate antigen.

UB-VV100 mediates B-cell depletion in CD34-humanized mice with a favorable biodistribution profile. (A) CD34-humanized NSG mice were injected with 0.4, 2.0, or 10.0 E+06 TUs of UB-VV100. Control animals were injected with vehicle only or 10E+06 TU of cocal pseudotyped vector displaying an αCD3 scFv and encoding an irrelevant (control) CAR. Circulating B cells and circulating T cells were enumerated by flow cytometry once a week for 25 days. **P<0.01, two-way ANOVA, Tukey’s multiple comparison’s test for main effect of vector dose. (B) CD34-humanized NCG mice were treated with either 36E+06 or 360E+06 TU UB-VV100 and were administered 1 mg/kg rapamycin every 48 hours beginning on day 5. Biodistribution of transduced cells was evaluated on day 28 by detection of the viral element PSI in genomic DNA tissue samples using qPCR. (C) Liver and spleen of mice treated with 360E+06 TU UB-VV100 were analyzed by RNA ISH to characterize the identity of cells expressing UB-VV100 RNA transcripts using colocalization analysis. Representative images depict human T cells (CD3+) and mouse macrophages (CD68+) transduced by UB-VV100. White indicates DAPI nuclear stain; green denotes human CD3; yellow denotes RACR sequence of transduced cells; red denotes murine CD68; blue denotes murine CD45. ANOVA, analysis of variance CAR, chimeric antigen receptor; ISH, in situ hybridization; RACR, rapamycin-activated cytokine receptor; scFv, single-chain variable fragment; TU, transducing unit.
We evaluated biodistribution and acute toxicity of UB-VV100 in CD34-humanized mice injected intraperitoneal with 36E+06 or 360E+06 TU of UB-VV100. Animals were treated with 1 mg/kg of rapamycin via intraperitoneal injection every other day beginning study day 5. There was a UB-VV100 dose-dependent reduction in B cells as highlighted earlier (online supplemental figure 3). Animals tolerated both dose levels of UB-VV100 without acute adverse events. On final study termination and necropsy, organs from all study animals were evaluated for histopathology. There were no UB-VV100-related clinical signs or effects on body weights, food consumption or local injection site irritation, and no postmortem macroscopic, microscopic, or organ weight differences that could be attributed to UB-VV100 treated as compared with vehicle control animals. On study day 28, organs were evaluated for presence of transduced cells by qPCR. Integration events were detected in a dose-dependent manner; the highest level of integrated vector genomes was detected in the spleen and liver, followed by lower levels in the bone marrow, injection site, and lung (figure 5B). Organs from animals treated with UB-VV100 were further evaluated by ISH against the UB-VV100 transgene to evaluate the identity of transduced cells. Semiquantitative colocalization scoring of integration events in spleen and liver characterized transduced cells as either human T cells (0–10% human CD3+ of transgene+cells) or murine macrophages (91%–100% murine CD68+ of transgene+cells) (figure 5C, full tissue section quantified in online supplemental table S7 and S8).
Supplemental material
Biodistribution in canines following intranodal or intraperitoneal administration indicates that UB-VV100 primarily transduces immune cells/tissues
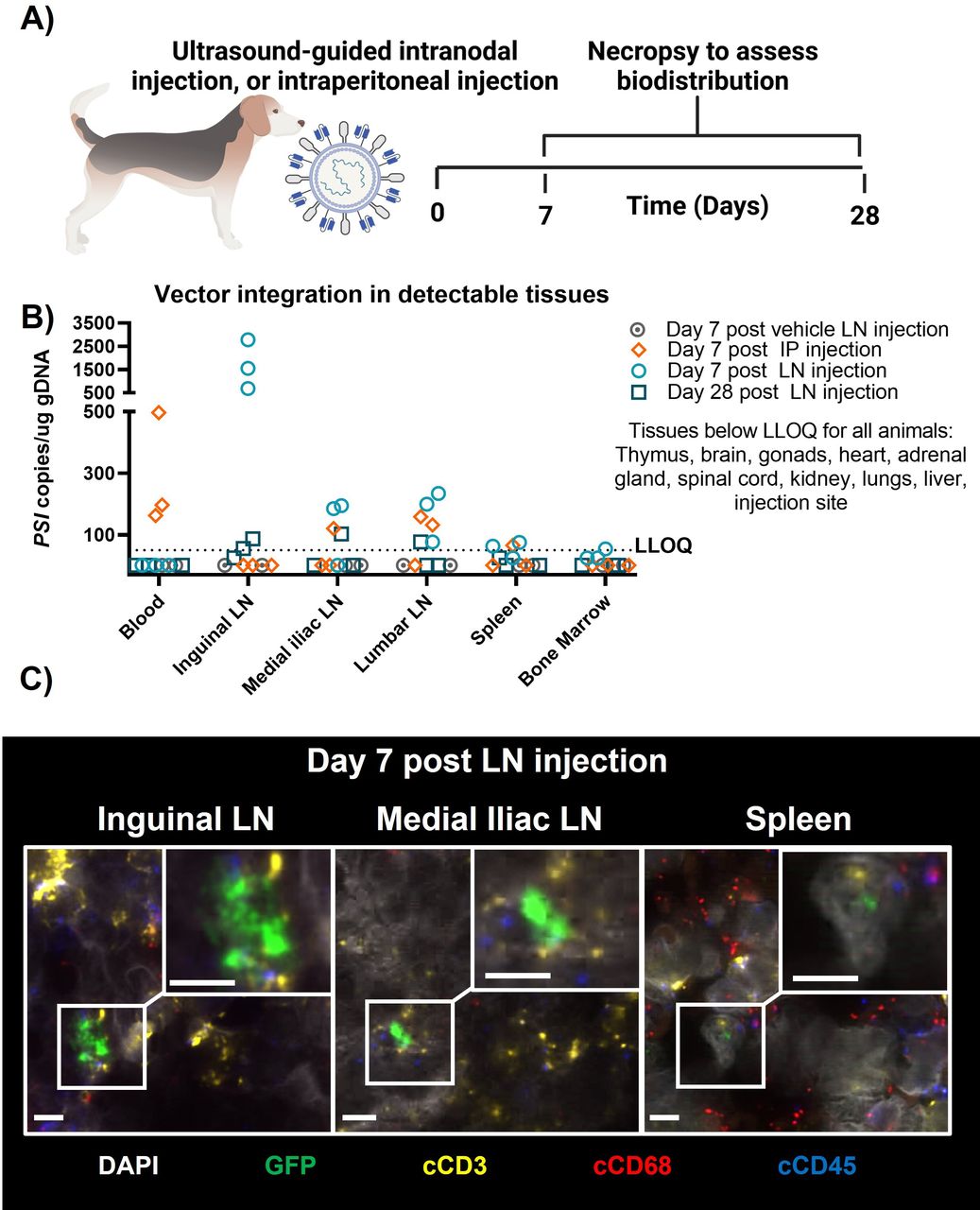
As lymph node injections are not feasible in immunocompromised NSG/NCG mouse models, biodistribution was also evaluated in canines, which can adequately assess cocal-mediated transduction,9 10 though no targeted activation of T cells is expected due to a lack of cross-reactivity with the antihuman CD3 scFv surface engineering. UB-VV100 surface engineered viral particles containing an EGFP payload (CD3-cocal-GFP) were administered to healthy beagles by bilateral, ultrasound-guided inguinal lymph node injection of 4E+08 TU CD3-cocal-GFP or intraperitoneal injection of 40E+08 TU CD3-cocal-GFP (figure 6A). CD3-cocal-GFP was well tolerated. Intranodal injection resulted in detection of integrated vector genomes primarily in the injected inguinal lymph nodes, with a 10-fold lower frequency of transduced cells detected in downstream draining medial iliac and lumbar lymph nodes. Intraperitoneal injection resulted in detection of integrated vector genomes primarily in the blood and medial iliac and lumbar lymph nodes through which interstitial fluid from the intraperitoneal cavity drains (figure 6B). There was no quantifiable detection of integrated vector genomes in any non-lymphoid tissues with either route of administration (ROA). ISH analysis identified the large majority of transduction events from both ROAs (ranging from >70% to >99% across lymph node samples) as immune cells (transgene+ and canine CD45+), including macrophages (transgene+ and canine CD68+) and T cells (transgene+ and canine CD3+). Despite a lack of canine T-cell targeting with particle surface engineering, transduction of canine T cells was observed in lymphoid tissues from intranodal injected animals, indicating the intranodal ROA is effective at delivering lentivirus to the target T-cell population (figure 6C, quantified in online supplemental table S9).

Biodistribution of surface-engineered lentiviral vectors in a canine. (A) Canines were treated with either 4E+08 TU CD3-cocal-GFP via ultrasound-guided bilateral inguinal lymph node injection or 4E+9 TU CD3-cocal-GFP via intraperitoneal injection. Necropsy was performed after either 1 or 4 weeks to assess lentiviral integration biodistribution profiles. (B) Biodistribution of transduced cells was evaluated by detection of the viral element PSI in genomic DNA blood and tissue samples using qPCR. Only organs with transduction events detected over the LLOQ are shown. (C) RNA ISH was performed to characterize the identity of cells expressing EGFP RNA transcripts using colocalization analysis. Representative images depict canine T cells transduced by CD3-cocal-GFP in the inguinal lymph node, medial iliac lymph node, and spleen 1 week after intranodal injection. Original magnification ×40; scale bars indicate 5 µM. White denotes DAPI nuclear stain; green denotes eGFP of transduced cells; yellow denotes canine CD3; red denotes canine CD68; blue denotes canine CD45. eGFP, enhanced green fluorescent; ISH, in situ hybridization; IP, intraperitoneal; LLOQ, lower limit of quantification; LN, lymph node;TU, transducing unit.
UB-VV100 controls systemic leukemia in a Nalm-6 xenograft mouse model
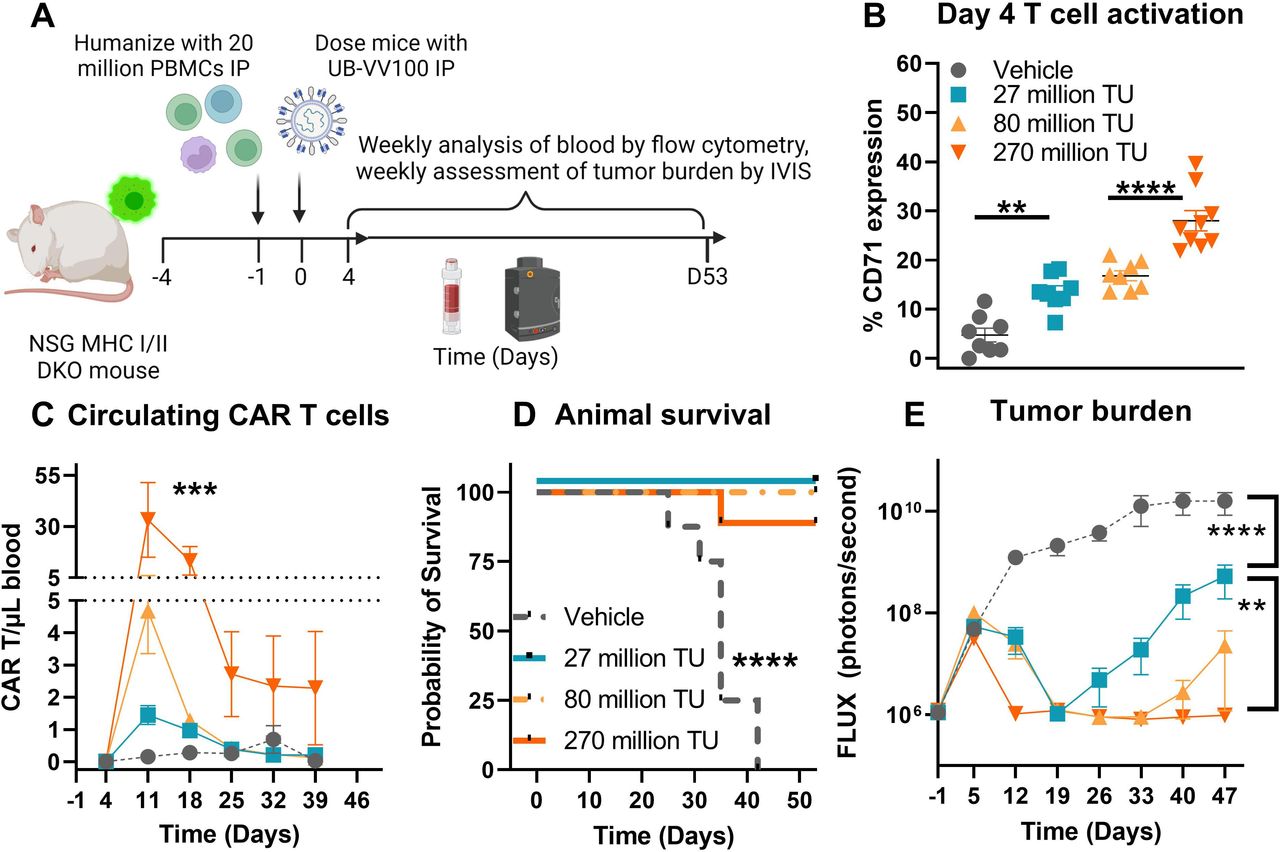
We evaluated the efficacy of in vivo generated CAR T cells in an aggressive tumor model. To do this, we developed a systemic Nalm-6 tumor model in PBMC-humanized NSG mice in which the mouse MHC class I and II genes have been knocked out to reduce graft-versus-host disease, and minimized transduction caused by activation driven by xenoreactivity.24 Mice were implanted with Nalm-6 cells via tail vein injection day −4, humanized with PBMCs via intraperitoneal injection day −1, and treated with vector intraperitoneal day 0 (figure 7A). Administration of 27, 80, and 270 million TU of UB-VV100 resulted in a dose-dependent activation of human T cells as measured by CD71 expression 4 days after administration (figure 7B). By day 11, circulating CAR T cells were detectable in all UB-VV100-treated mice in a dose-dependent manner (figure 7C). By study day 12, animals treated with UB-VV100 displayed significant tumor reduction compared with vehicle treated animals, and animals treated with the highest dose showed complete tumor elimination (figure 7D). Animals treated with all doses of UB-VV100 tolerated the treatment without signs of toxicity. All doses of UB-VV100 significantly improved animal survival as compared with vehicle-treated animals (figure 7E). Transduction of circulating murine CD45+ cells was not detected at any time point, and transduction of Nalm-6 tumor cells could not be evaluated due to absence of detectable tumor cells by flow cytometry in UB-VV100 treated mice during serial bleeds or at terminal euthanasia in the represented study (online supplemental figure 4). In an independent replicate study, Nalm-6 tumor cells were detectable in a few mice euthanized 42 and 85 days after UB-VV100 treatment. The recovered Nalm-6 tumor cells exhibited bright CD19 expression, an absence of FMC63 expression, and an absence of intracellular P2A expression as compared with Nalm-6 tumor recovered from vehicle treated animals (online supplemental figure 5). While the possibility of Nalm-6 transduction cannot be definitively disproven, we did not observe this phenomenon in this study series.
Supplemental material
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
UB-VV100 treatment results in in vivo transduction of CAR T cells and clearance of Nalm-6 tumor. (A) Animals were engrafted with Nalm-6 tumor on study day −4 via tail vein injection, humanized with 20E+06 PBMCs on day −1 via intraperitoneal injection, and treated with 27 million, 80 million or 270 million TU of UB-VV100 on study day 0 via intraperitoneal injection. (B) T-cell activation was assessed on day 4 by flow cytometry. **P<0.01, ****P<0.0001, one-way ANOVA, Tukey’s multiple comparison test between the indicated groups. (C) Circulating CAR T cells were enumerated by flow cytometry surface staining against FMC63. ***P<0.001, one-way ANOVA for day 11. n=7–9 per group per time point. (D) Animal survival was evaluated during the study. (E) Average tumor burden as assessed by bioluminescent imaging. (F) Heatmap of bioluminescent imaging data overlayed with mouse images. The last observed in-life photon flux data were reported for deceased animals at the indicated time points. **P<0.01, ****P<0.0001, two-way ANOVA, multiple comparisons between the indicated UB-VV100 dose level across the entire observation period. n=7–9 per group, ****P<0.0001, Mantel-Cox test. ANOVA, analysis of variance; CAR, chimeric antigen receptor; IVIS, In Vivo Imaging System; PBMC, peripheral blood mononuclear cell; TU, transducing unit.
During model development, we observed that treatment of PBMC-humanized mice with rapamycin inhibited humanization, both in the context of healthy animals and Nalm-6 tumor bearing animals (online supplemental figure 6A,B). Based on literature on the use of rapamycin in humanized mouse models, this is likely due to inhibition of allogeneic interactions between human PBMCs and Nalm-6 tumor cells or xenogeneic interactions with mouse antigens which are required for sustained survival of human T cells in the mouse environment.25–28 To decouple the need for PBMC humanization from the mechanism of action of the RACR system, we employed ex vivo manufactured CAR T cells in a Raji tumor-bearing NSG model. We found that administration of 0.05 or 0.5 mg/kg of rapamycin 5× a week promoted dose-dependent relative enrichment and total expansion of ex vivo manufactured CAR T cells (containing the same payload construct as UB-VV100) infused at a dose of 1 million CAR+ cells per mouse. Treatment with 0.5 mg/kg rapamycin, but not lower doses of rapamycin, was associated with complete tumor clearance (online supplemental figure 7 and online supplemental table S11). We also evaluated in our in vivo disease model the use of a rapamycin analog in which mTOR binding is greatly reduced (rapalog, AP21967). This rapalog can still engage the RACR heterodimeric cytokine receptor to provide a positive IL-2/IL-15 signal, but with substantially reduced immunosuppressive effects on all immune cells. Using 1, 5, or 10 mg/kg rapalog in combination with UB-VV100 in our systemic Nalm-6 tumor model, we observed rapalog dose-dependent enhancement of CAR T-cell expansion and improved survival (online supplemental figure 8 and online supplemental table S12).
Supplemental material
Supplemental material
Supplemental material
Discussion
We found that surface engineering of lentiviral particles with the cocal glycoprotein and anti-CD3 scFv enabled engagement, activation, and transduction of CD3+T cells to express an anti-CD19 CAR and the RACR system. The resulting CAR T cells exhibited antigen-specific tumor engagement and preferential expansion compared with non-transduced cells in vitro in the presence of rapamycin. In vivo, treatment with UB-VV100 resulted in T cell activation, CAR T-cell transduction, and elimination of endogenous and malignant B cell targets. UB-VV100 biodistribution in humanized mice and canines was limited primarily to immune cells and was not associated with tissue pathology.
Unstimulated T cells exist in the G0 phase of the cell cycle where they are not readily transduced by LVVs; they require external stimulation to enter the G2/M phase and permit gene transfer.6 29–31 Buchholz and colleagues have published several studies on their efforts to develop LVVs to overcome this.6–8 Pseudotyping LVVs with a modified paramyxovirus envelope to target CD3 has demonstrated in vivo transduction of CAR T cells and depletion of malignant and endogenous B-cell targets, particularly in combination with exogenous human IL-7 and bead activation.6 7 In vivo CAR T-cell manufacturing has also been accomplished by repeated doses of T cell-targeted nanoparticles bearing a CAR-encoding DNA or mRNA payload.32 33 In our hands, administration of a single dose of UB-VV100 without any stimulants resulted in robust in vivo T-cell activation, producing sufficient CAR T cells to eradicate a systemic B-cell malignancy. Additional vector surface engineering could further refine the quality of T-cell engagement and tissue tropism to enhance in vivo transduction.
In vivo administration of LVVs carries the risk of off-target transduction, and VivoVec particles could exhibit wide tissue tropism via cocal binding its cognate receptors (including the LDL-R family).6 34 Although we observed widespread anatomical distribution of VivoVec-transduced cells by qPCR, analysis by ISH revealed transduced cells were predominantly of immune origin (T cells and macrophages, which traffic to nearly all tissues), even in the context of high-volume intraperitoneal delivery, and even in the absence of anti-CD3 scFv targeting. Our observations represent an important extension of previous work with VSV-G pseudotyped LVVs, which reported biodistribution to the liver, spleen, and bone marrow, but did not identify transduced cell types.35 36 Although transduction of macrophages is inefficient in vitro due to their quiescent state, other groups have reported uptake of LVVs by murine Kupffer cells (a type of macrophage found in the liver), resulting in sequestration of particles from surrounding hepatocytes and transduction of the Kupffer cells.37 38 Macrophage transduction in our models may therefore be a result of macrophage scavenging behavior, followed by pH-dependent viral entry into the cytosol mediated by cocal.39 It is not clear if human macrophages, which exhibit robust postentry restriction of lentiviral transduction via SAMHD1, would exhibit the same susceptibility to transduction in vivo.40 Nonetheless, generation of CAR+ macrophages in the clinical setting could promote clearance of CAR-targeted cells and contribute to the holistic drug mechanism of action.41
The use of UB-VV100 in the clinical setting poses efficacy and safety challenges that may not replicate in preclinical models using healthy donor derived materials. Despite potential differences in the quality of T cells expected in human patient populations, UB-VV100 generated functional CAR T cells from B-ALL and R/R DLBCL-derived PBMCs, suggesting in vivo manufacturing is still effective in this patient population. An added risk with the in vivo approach is the potential transduction of CD19+ malignant B cells with a CD19-binding CAR leading to epitope masking and subsequent antigen escape.14 UB-VV100 was intentionally designed to encode a CD19 CAR comprising an FMC63 scFv positioned on a short hinge in order to minimize cis engagement with the CD19 target epitope, which is located in a membrane distal position.15 16 Indeed, Nalm-6 cells transduced with UB-VV100 retained sensitivity to CAR T-cell killing, and no antigen escape via this mechanism was observed in murine systemic tumor models.
Due to the high complexity and sensitive timeline of the systemic Nalm-6 tumor model in PBMC humanized mice, we have to date been unable to develop a study protocol in which rapamycin administration does not interfere with either humanization or tumor growth. Data from in vitro studies, in vivo studies using ex vivo manufactured CAR T cells, and in vivo studies using rapalog support that the rapamycin–RACR axis may be of clinical benefit to patients. The holistic approach of in vivo transduction paired with RACR-mediated expansion has the potential to mitigate efficacy and safety concerns of existing CAR T-cell therapies. On the efficacy side, CAR T cells that expand in their natural context may have a more favorable phenotype and enhanced activity compared with those expanded in prolonged ex vivo culture, which has been associated with undesirable differentiation and exhaustion.42–45 On the safety side, the kinetics of in vivo CAR T generation may reduce the severity of cytokine release syndrome (CRS). In hematological malignancies, CRS has been correlated with lymphodepletion regimens, the initial infused dose of CAR T cells, and tumor burden at the time of treatment.46 47 In vivo CAR T-cell generation and subsequent expansion is anticipated to result in more gradual kinetics of CAR T cells engaging tumor, possibly blunting CRS.
The removal of lymphodepletion has many potential benefits for patients. Lymphodepletion is currently required to promote full CAR T-cell efficacy but also increases the risk of serious infection in a vulnerable patient population.48 UB-VV100 is designed to be used with rapamycin in the absence of lymphodepletion, whereby rapamycin provides protection against immune-mediated deletion of nascent CAR T cells via mTOR inhibition of non-transduced cells while simultaneously supporting CAR T-cell proliferation via RACR.49–51 Rapamycin may also dampen some forms of immune toxicity without interfering with CAR T-cell function.52 53
Safe and effective engineering of CAR T cells in vivo has the potential to expand access to this class of life-saving therapies in both solid and liquid tumors by reducing the cost for therapy, reducing the wait time for treatment and avoiding the need for lymphodepleting chemotherapy, as well as providing potential safety and efficacy advantages over ex vivo manufactured CAR T cells.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
The authors thank Seattle Children’s Therapeutics for gifting of engineered Nalm-6 cell lines; Seattle Children’s Research hospital, Fred Hutch Cancer Research Center, and Charles River Laboratories for conducting in vivo studies; Advanced Cell Diagnostics for performing in situ hybridization analysis; and Biorender.com for creation of infographics used in the figures.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Scientific conception and design: AHB, SG, SAH, RPL, KRM, CJN, BYR, AS, AMSc, AMSu, and H-AT. Vector design and scale-up: JF, SG, MM, CJN, MDP, BYR, AS, and SS. Execution and generation of data in final figures: AHB, TG, SAH, BI, KRM, DP, AMP, AMSc, AMSu, and H-AT. Preparation of manuscript in current form: AHB, SG, KRM, AMSc, AMSu, and H-AT. Interpretation of data and review of final review/revision of manuscript: all authors. KRM is guarantor of scientific content and AMSc is guarantor of decision to publish.
Funding All studies were sponsored by Umoja Biopharma without use of any public funding.
Competing interests All authors hold equity with Umoja Biopharma.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.