Article Text

Abstract
Background and aims Macrophage innate immune response plays an important role in tumorigenesis. However, the role and mechanism of macrophage STING signaling in modulating tumor microenvironment to suppress tumor growth at secondary sites remains largely unclear.
Methods STING expression was assessed in liver samples from patients with colorectal cancer (CRC) liver metastasis. Global or myeloid stimulator of interferon gene (STING)-deficient mice, myeloid NOD-like receptor protein 3 (NLRP3)-deficient mice, and wild-type (WT) mice were subjected to a mouse model of CRC liver metastasis by intrasplenic injection of murine colon carcinoma cells (MC38). Liver non-parenchymal cells including macrophages and natural killer (NK) cells were isolated for flow cytometry analysis. Bone marrow-derived macrophages pretreated with MC38 were co-cultured with splenic NK cells for in vitro studies.
Results Significant activation of STING signaling were detected in adjacent and tumor tissues and intrahepatic macrophages. Global or myeloid STING-deficient mice had exacerbated CRC liver metastasis and shorten survival, with decreased intrahepatic infiltration and impaired antitumor function of NK cells. Depletion of NK cells in WT animals increased their metastatic burden, while no significant effects were observed in myeloid STING-deficient mice. STING activation contributed to the secretion of interleukin (IL)-18 and IL-1β by macrophages, which optimized antitumor activity of NK cells by promoting the expression of 4-1BBL in macrophages and 4-1BB in NK cells, respectively. Moreover, MC38 treatment activated macrophage NLRP3 signaling, which was inhibited by STING depletion. Myeloid NLRP3 deficiency increased tumor burden and suppressed activation of NK cells. NLRP3 activation by its agonist effectively suppressed CRC liver metastasis in myeloid SITNG-deficient mice.
Conclusions We demonstrated that STING signaling promoted NLRP3-mediated IL-18 and IL-1β production of macrophages to optimize the antitumor function of NK cells via the co-stimulation signaling of 4-1BBL/4-1BB.
- liver neoplasms
- macrophages
Data availability statement
Data are available in a public, open access repository.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The tumor microenvironment plays an important role in tumorigenesis and response to therapies.
Both macrophages and natural killer (NK) cells are implicated in modulating tumor microenvironment of colorectal cancer liver metastasis.
WHAT THIS STUDY ADDS
Myeloid stimulator of interferon gene (STING)-deficient mice have exacerbated colorectal cancer liver metastasis.
Macrophage STING signaling promotes NOD-like receptor protein 3 (NLRP3) activation to optimize the antitumor function of NK cells.
4-1BBL/4-1BB co-stimulation signaling is essential for the regulation of NK cell activation by macrophages.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
We have identified a novel function of macrophage STING-NLRP3 signaling in regulating the antitumor function of NK cells.
This provides a potential strategy to optimize the antitumor response of macrophages and NK cells to suppress colorectal cancer liver metastasis.
Introduction
Colorectal cancer CRC) remains one of the most common cancers with a leading cause of cancer-related deaths. Although good prognosis has been achieved in patients with early-stage CRC due to the advances in surgical and medical therapies, approximately 40% of patients with CRC progress to fatal metastasis.1 The liver is the most common anatomical site for hematogenous metastases from CRC, which has become one of the most difficult and challenging situations in the management of CRC.
Cancer immunotherapy has made a great breakthrough with the discovery of immune checkpoint inhibitors mainly targeting the adaptive immune system of T lymphocytes.2 Moreover, emerging evidence have suggested the vital role of innate immune response in tumorigenesis and antitumor therapy.3 Macrophages have been implicated in modulating tumor microenvironment (TME), which exert pleiotropic effects in inflammation and tumorigenesis. Tumor-associated macrophages (TAMs), one of the main tumor-infiltrating immune cells, exert to promote tumor development and progression.4 Different phenotypes and functions of macrophages have been found in metastatic tumors from primary tumors. The presence of macrophages or macrophage-related cytokines/chemokines in human solid tumors is associated with poor prognosis. TAMs can also regulate TME by interacting with other types of cells. Natural killer (NK) cells participate in antitumor and proinflammatory response by killing target cells and producing cytokines.5 Inhibitory molecules produced by cancer cells or TAMs impairs the function of NK cells to promote tumor escape in the immunosuppressive TME.6
The cyclic GMP-AMP synthase (cGAS) and its downstream signaling effector stimulator of interferon genes (STING) pathway is an evolutionary conserved defense mechanism against viral infections.7 The macrophage cGAS-STING pathway has emerged to participate in human inflammatory diseases and cancer by sensing self-DNA.8–11 However, both antitumorigenic and protumorigenic effects of cGAS-STING pathway have been reported, which is dependent on the specific context as well as stage of tumor progression. Pharmacologic activation of STING has been shown to restrict tumor growth and enhance immunogenicity in mouse models and clinical patients.12 Although cGAMP-STING dependent inhibition of tumor cell proliferation, viability, and invasion has been reported,13 the role of STING signaling in macrophages in regulating CRC liver metastasis remains unclear. In the present study, we found that STING signaling promoted interleukin (IL)-18 and IL-1β production of macrophages by activating NOD-like receptor protein 3 (NLRP3). Synergistic effect of IL-18-mediated 4-1BBL expression in macrophages and IL-1β-mediated 4-1BB expression in NK cells were found to be essential for the antitumor functions of NK cells regulated by macrophages via the 4-1BBL/4-1BB signaling pathway. Our study demonstrated a critical role for macrophage STING signaling in promoting antitumor response of NK cells to eliminate CRC liver metastasis.
Materials and methods
Patients and specimens
Ten patients with CRC liver metastasis undergoing liver partial resection in the First Affiliated Hospital of Nanjing Medical University were enrolled in the present study. Liver resection specimens were collected for bioassay.
Mice and cell line
Wild-type (WT) control mice (C57BL/6J background) and Global STING deficient (STINGKO) were generated by GemPharmatech. FloxP-STING (STINGFL/FL) (Shanghai Model Organisms Center) or FloxP-NLRP3 (NLRP3FL/FL) (GemPharmatech) mice were bred with myeloid-specific Cre (Lyz2-Cre) mice to create myeloid-specific STING (STINGΔMФ) or NLRP3 (NLRP3ΔMФ) knockout mice. Rag1 deficient (Rag1−/−) mice were purchased from Shanghai Model Organisms Center. All animals used throughout the study were sex-matched 8–12 weeks old mice on a C57BL/6J background and were housed and maintained under a 12 hours light/12 hours dark cycle with ad libitum access to water and standard chow with supplements under specific pathogen-free conditions. All animals received humane care.
The mouse colon cancer cells (MC38) were purchased from the Shanghai Cell Bank (Chinese Academy of Sciences, Shanghai, China) and were verified by isoenzyme testing and DNA fingerprinting. All cell lines tested negative for Mycoplasma.
Transplant tumor models and intervention in vivo
To induce the development of liver metastasis, mice were injected intrasplenically with 5×10ˆ5 MC38 cells in 100 µL of phosphate-buffered saline (PBS), followed by splenectomy 5 min after injections under isoflurane anesthesia. PBS-injected mice served as controls. Mice were sacrificed on Day 14 or 21 after the initial injection. To quantify the metastatic burden, H&E slides were scanned to calculate the metastatic burden with the ImageScope software, the area occupied by metastatic foci was divided by the total surface area.
For NK cell depletion in the transplant tumor model, mice were intraperitoneal injected with 200 µg anti-NK1.1 (Bio X Cell, BE0036)14 15 or its isotype control 3 days before and 100 µg anti-NK1.1 or isotype control on Day 3, 7 and 10 after challenge. For 4-1BBL or 4-1BB blockade in vivo, mice were injected with InVivoMAb anti-mouse 4-1BBL (CD137L) or InVivoMAb anti-mouse 4-1BB (CD137) (Bio X Cell, BE0110 or BP0296) or their isotype control 150 µg on the Day −1, 3, 7, 10.
Mice were intraperitoneal injected with 200 µg 2’3’-cGAMP (for STING activation, InvivoGen, Hong Kong, China) or 5 µM nigericin (for NLRP3 activation, InvivoGen) at Day 7 after challenge and every other day.
Cells isolation, culture and treatments
Liver non-parenchymal cells (NPCs) were isolated by dissociating MC38 metastatic liver tissue in the presence of collagenase IV (0.1% w/v, Sigma) and DNAse (0.005% w/v, Sigma) for 1 hour before centrifugation on a discontinuous Percoll gradient (GE Healthcare). Isolated NPCs were then used in various assay.
Freshly obtained mouse spleen smashed through 70 µm filter and resuspended in red blood cell lysis solution (BioLegend, San Diego, California, USA). Then splenic NK cells were purified by depletion of magnetically labeled non-NK cells with MACS (Magnetic Activated Cell Sorting) NK Cell Isolation Kit (Miltenyi Biotec, B.G, Germany) according to the manufacturer’s protocol. Purified and enriched NK cells were co-cultured with bone marrow-derived macrophages (BMDMs) in contact or transwell (1:2) in vitro for 12 hours at 37°C and then used for flow cytometry analysis.
Murine M-CSF (Macrophage Colony-Stimulating Factor, 20 ng/mL, PerproTech, New Jersey, USA) in DMEM (Dulbecco's Modified Eagle Medium) supplemented with 10% (v/v) fetal calf serum was used to generate BMDMs in vitro. BMDMs were then replated and cultured in a new 6-well plate overnight for further experiments.16
Flow cytometry
After blocking with TruStain fcX (anti-mouse CD16/32, BioLegend, San Diego, California, USA), cells were incubated with the indicated fluorescence-labeled mAbs (monoclonal antibodys) and then washed twice with PBS then acquired on CytoFLEX (Beckman Coulter, Brea, California, USA) and were analyzed using CytExpert software (V.2.4). Macrophages were detected as Live/F4/80+, and the NK cells were detected as Live/CD45+/CD3−/NK1.1+(online supplemental figure 1A). NK cell degranulation functional assays were particularly investigated by the CD107a assay and intracellular cytokines interferon (IFN)-γ, granzyme B, perforin production. CD4+ or CD8+ T cells were gated on Live/CD45+/CD3+. All flow cytometry antibodies used in the experiment were purchased from BioLegend and the details were shown in the online supplemental table 1.
Supplemental material
Supplemental material
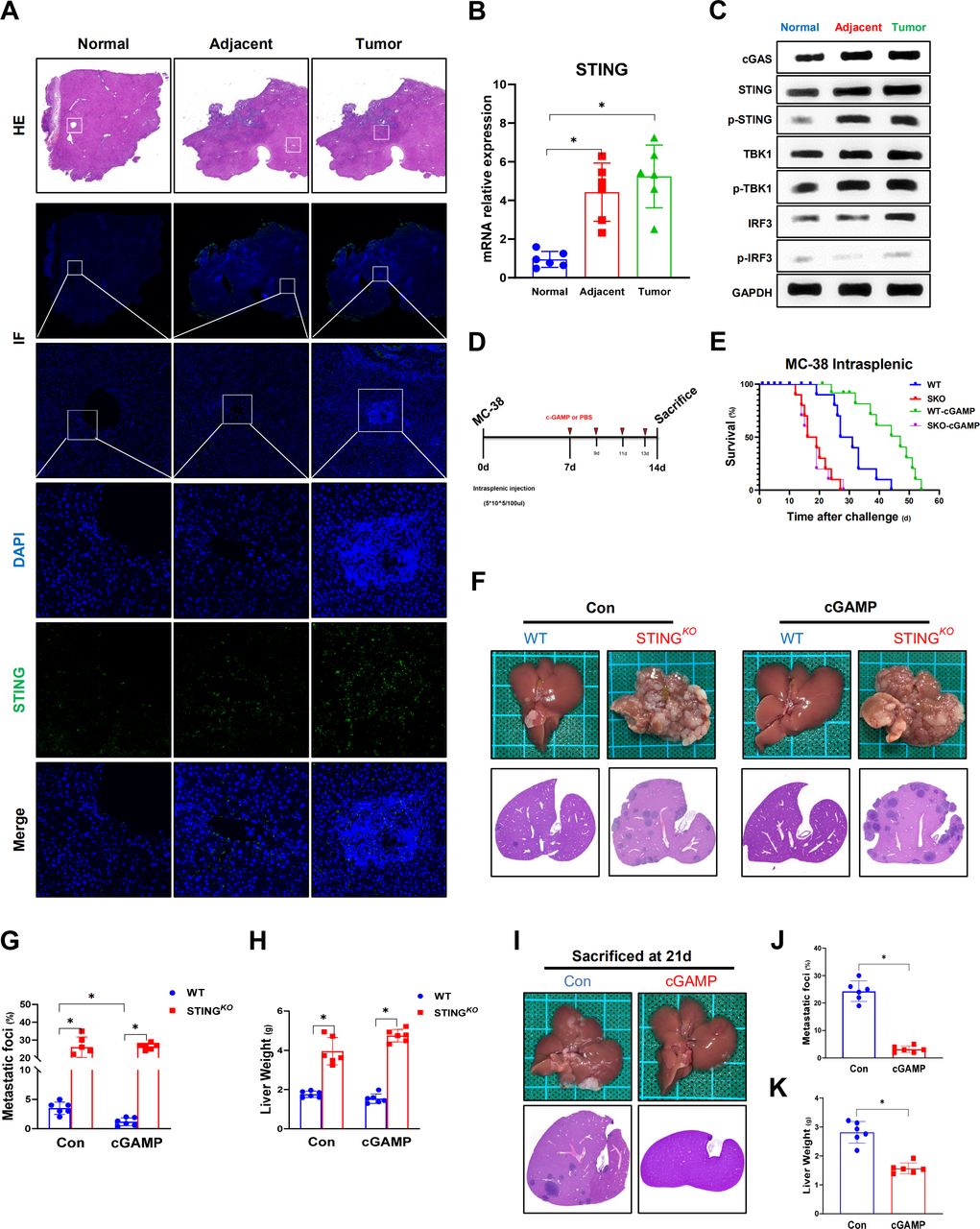
STING signaling contributes to the suppression of colorectal cancer metastatic growth in the liver. STING detected by immunoflourescent staining in normal, adjacent and tumor liver tissues section from patients with CRC liver metastasis (A). STING signaling expression measured by quantitative real-time PCR (B) and WB (C) in human colon liver metastatic tissue. Schematic representation of CRC metastatic growth in the liver model induced by intrasplenic injection of the C57BL/6 colon carcinoma MC38 cells (D). An overview of survival after MC38 intrasplenic injection (E). Representative pictures of the liver and H&E staining of liver sections harvested from WT and STINGKO mice sacrificed on Day 14 (CON vs cGAMP intraperitoneal for STING activation) or after MC38 intrasplenic injection (F). WT or STINGKO mice CRC foci coverage (G) and liver weight (H) on Day 14 after MC38 injection (n=6/group per time point per genotype). Representative pictures and H&E section (I) harvested from WT mice (CON vs cGAMP), and liver CRC foci coverage (J) and liver weight (K) on Day 21 after MC38 injection. Data are presented as mean±SEM, p<0.05*. CRC, colorectal cancer; mRNA, messenger RNA; STING, stimulator of interferon genes; WT, wild-type. WB, Western Blot; IF, immunoflourescent; DAPI, 4',6-diamidino-2-phenylindole.
H&E, immunohistochemical and IF
Livers were fixed in 10% buffered formalin overnight and paraffin-embedded subsequently, 4 µm thick sections were cut onto glass slides and processed for H&E staining. Immunohistochemical (IHC) staining was performed with Double Stain IHC Kit: Rabbit and Rat on human/mouse tissue (HRP(Horseradish Peroxidase ploymer)/Green and AP(Alkaline Phophatase polymer)/Red) (Abcam; ab183285). Specimen sections were scanned using PANNORAMIC MIDI digital scanner (3DHISTECH). For immunofluorescence, slides were permeabilized with 0.25% Triton X-100 (Thermo Fisher Scientific, Massachusetts, USA) in PBS for 20 min at room temperature and then blocked (10% fetal bovine serum, 3% bovine serum albumin (BSA)) for 30 min at 37°C and tissues were incubated with primary antibodies in PBS containing 3% BSA overnight. After that, slides were followed by a 1 hour incubation with conjugated secondary antibodies coupled with FITC (FluoresceinIsothiocyanate) or Cy3 and DAPI (4',6-diamidino-2-phenylindole). Stained sections were imaged with a confocal microscope system LSM880 (Zeiss, GER) and analyzed using ZEN software (Zeiss). All antibodies used in the experiment and western blots were shown in the online supplemental table 2.
Supplemental material
Western blotting
Cellular proteins were extracted with an ice-cold lysis buffer. Proteins (20 µg) were subjected to SDS-PAGE and transferred to polyvinylidene difluoride nitrocellulose membrane (Bio-Rad). Monoclonal rabbit anti-mouse cGAS, p-STING, TBK1, p-TBK1, IRF3, p-IRF3, NLRP3, cleaved caspase-1, GAPDH (Cell Signaling Technology, Massachusetts, USA) were used.
Quantitative real-time PCR
Following the manufacturer’s instructions, total RNA was extracted from frozen liver tissue and cells using TRIzol reagent (Invitrogen, Carlsbad, California, USA) and was reverse-transcribed into complementary DNA (cDNA) using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Indianapolis, Indy, USA). Quantitative real-time PCR was performed using SYBR green (Roche, Indianapolis, Indy, USA). The expression levels of target genes and the results were normalized against HPRT (hypoxanthine phosphoribosyltransferase) expression.
Statistical analysis
Statistical analysis data are represented as mean±SEM. Multiple group comparisons were performed by one-way analysis of variance followed by Bonferroni’s post hoc test. P values<0.05 (two-tailed) were considered statistically significant. All statistical analyses were performed using STAT software, V.11.0.
Results
STING signaling contributes to the suppression of CRC metastatic growth in the liver
To investigate the role of STING signaling in liver CRC metastasis, we first analyzed STING expression in normal, adjacent and tumor liver tissues from patients with CRC liver metastasis (figure 1A). Compared with normal tissues, significantly increased protein levels of cGAS/STING/TBK1/IRF3 signaling and gene induction of STING were found in adjacent and tumor tissues (figure 1B,C). Next, we evaluated the metastatic burden of STING-deficient mice compared with that of WT mice in a mouse model of CRC liver metastasis by intrasplenic injection of murine colon carcinoma cells (MC38). Meanwhile, cGAMP, an STING agonist, was administered on Day 7 post injection (figure 1D). Significantly shortened survival was observed in STING-deficient mice compared with WT mice (figure 1E). STING-deficient mice had many more metastatic lesions (figure 1F) with larger hepatic MC38 foci coverage (figure 1G), and heavier livers (figure 1H) were observed on Day 14 post MC38 injection. In contrast, STING activation by cGAMP resulted in prolonged survival and less liver metastasis in WT mice, while no significant effects were observed in STING-deficient mice (figure 1E–H). Interestingly, cGAMP-treated WT mice showed no obvious tumor lesions on Day 14 and only a few tumor lesions on Day 21 post MC38 injection (figure 1I–K), indicating that STING activation by exogenous cGAMP treatment effectively delayed liver metastatic tumor development. Together, these results suggest that the activation of STING signaling suppresses liver CRC metastasis.
Myeloid STING-deficient mice are highly susceptible to CRC liver metastatic growth
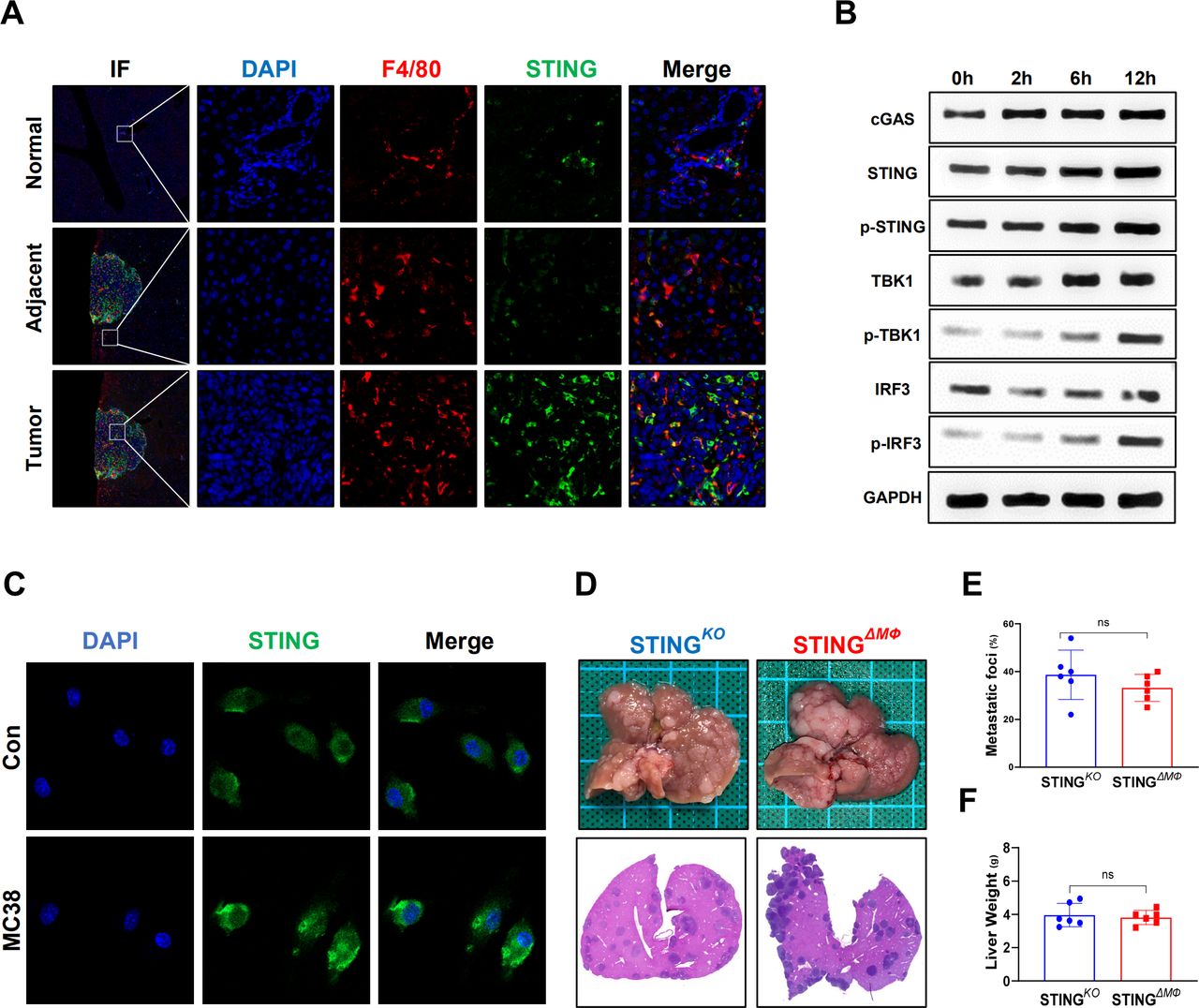
TAMs play an important role in tumor development and progression by modulating the TME.17 Macrophage STING signaling has also been implicated in antitumor immunity.8 As shown in figure 2A, significantly enhanced double staining of F4/80 and STING was detected inside the tumor compared with that of adjacent and normal liver tissues from mice with CRC liver metastasis. Furthermore, in vitro MC38 cell stimulation triggered STING signaling activation in macrophages, as evidenced by enhanced immunostaining of the cGAS/STING/TBK1/IRF3 signaling pathway (figure 2B,C). Therefore, myeloid-specific STING KO mice were used to further determine whether the activation of macrophage STING signaling contributes to the above antitumor activity. Indeed, compared with global STING KO mice, myeloid STING KO mice exhibited similar MC38 growth in the liver, as shown by analysis of metastatic lesions, hepatic MC38 foci coverage, and liver weight (figure 2D–F). These results indicate that STING activation in macrophages is required to suppress CRC metastatic growth in the liver.
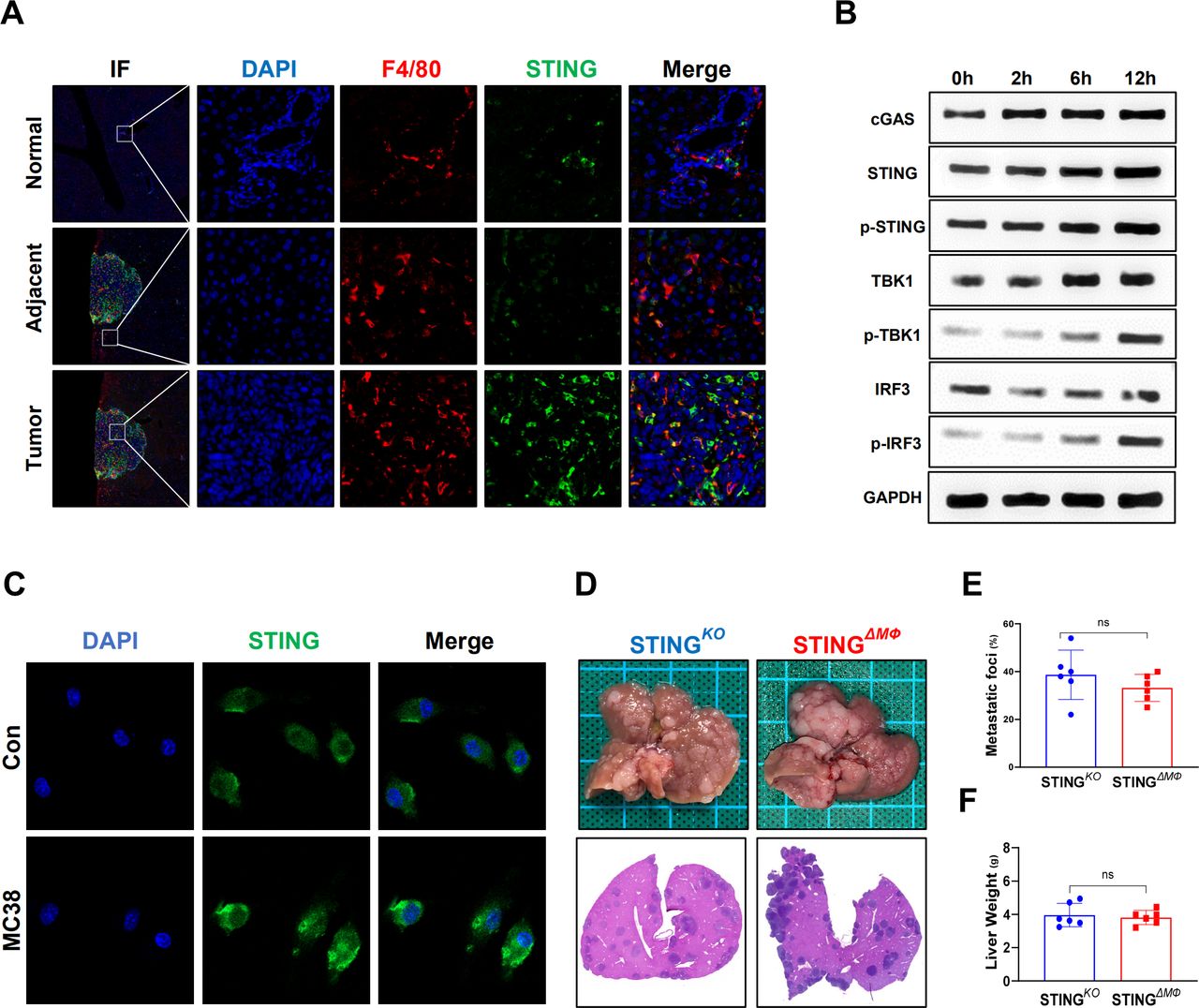
Myeloid STING-deficient mice are highly susceptible to colorectal cancer liver metastatic growth. STING in F4/80+macrophages in normal, adjacent and tumor liver tissues (selected from the same liver tissue section) from wild-type mice sacrificed on Day 14 after MC38 intrasplenic injection were detected by immunohistochemical (A). STING signaling of BMDMs co-cultured with MC38 at different time point were measured by WB (B). STING detected by IF in BMDMs co-cultured with MC38 for 12 hours or control (C). Representative pictures of the liver and H&E of liver sections (D), liver colorectal cancer foci coverage (E) and liver weight (F) of STINGKO and STINGΔMФ mice sacrificed on Day 14 after MC38 intrasplenic injection (n=6/group per genotype). Data are presented as mean±SEM, p<0.05*. BMDMs, bone marrow-derived macrophages; STING, stimulator of interferon genes. WB, Western Blot; IF, Immunoflourescent; DAPI, 4',6-diamidino-2-phenylindole.
STING signaling in macrophages suppresses hepatic metastasis by promoting the antitumor properties of NK cells
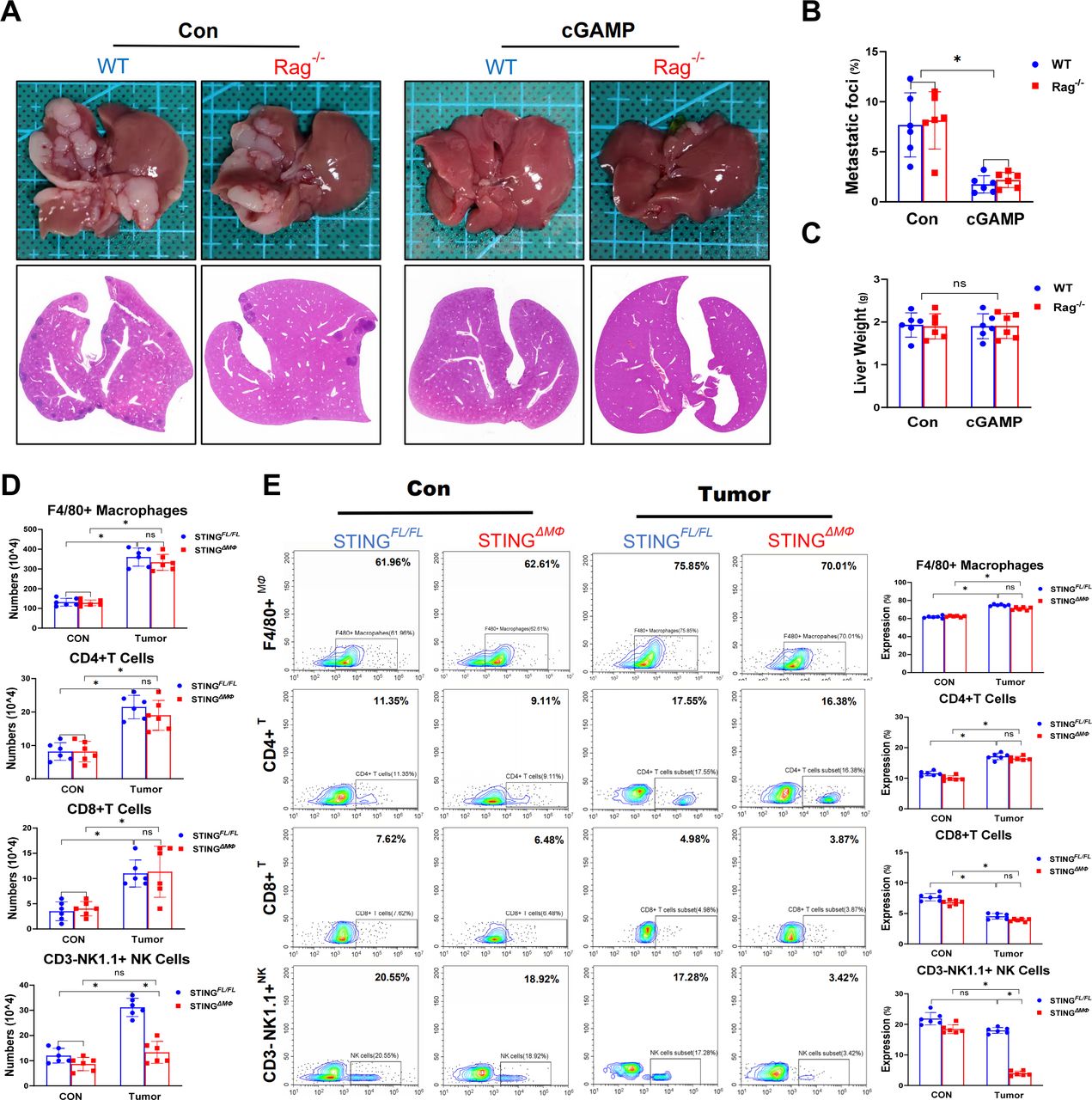
Next, we determined the cell type of immune cells contributed to the control of MC38 hepatic metastasis. Interestingly, while the Rag1–/– mice exhibited similar metastatic burden as compared with WT mice, STING activation by cGAMP significantly suppressed intrahepatic MC38 growth in the Rag1–/– mice (figure 3A–C). These results indicated that the adaptive immune cells, such as T, B and natural killer T (NKT) cells, were not required for the suppression of MC38 liver metastatic growth by macrophage STING activation. Moreover, the intrahepatic numbers (figure 3D) and proportions (figure 3E) of immune cells were compared in FloxP-STING and myeloid STING-deficient mice with CRC liver metastasis. Compared with STINGFL/FL mice, STINGΔMФ mice showed similar numbers and proportions of F4/80+ macrophages, CD4+ and CD8+ T cells but significantly decreased numbers and proportions of NK cells.

Control of hepatic metastasis by macrophage STING signaling occurs independently of the adaptive immune system WT and Rag–/– mice were intrasplenic injected with MC38. Representative pictures of the liver and H&E staining of liver sections harvested from WT and Rag–/– mice sacrificed on Day 14 (CON vs cGAMP intraperitonealy for STING activation) after MC38 intrasplenic injection (A). And liver colorectal cancer foci coverage (B) and liver weight (C) on Day 14 after MC38 injection. STINGΔMФ and STINGFL/FL control mice were intrasplenic injected with MC38. Livers and intrahepatic NPCs were collected on Day 14. Numbers (D) and proportions (E) of F4/80+ macrophages, CD4+ or CD8+ T cells and NK1.1+ NK cells in liver NPCs. Data are presented as mean±SEM, p<0.05*. NK, natural killer; NPCs, non-parenchymal cells; STING, stimulator of interferon genes; WT, wild-type.
Immunostaining of NKG2D in liver tissues further confirmed that myeloid STING KO mice had significantly less infiltration of NKG2D-positive NK cells (figure 4A). NK cells have been implicated in antitumor immunity by killing tumor cells and producing cytokines, while their function may be modified by other cells in the immunosuppressive TME.5 The antitumor properties of NK cells in the CRC liver metastasis model in STINGFL/FL and STINGΔMФ mice were further analyzed. FACS (Fluorescence Activated Cell Sorter) staining for CD69 and KLRG1, markers of NK cell cytotoxic activity, also revealed decreased frequencies of CD69hiKLRG1lo hepatic NK cells in myeloid STING KO mice compared with FloxP-STING animals (figure 4B). NK cells from STINGΔMФ mice also produced less perforin but similar levels of IFN-γ and granzyme B (figure 4C). To confirm the critical role of macrophage STING signaling in tumor immunosurveillance by promoting NK cell antitumor activity, we administered an NK cell-depleting anti-NK1.1 antibody to STINGΔMФ mice and STINGFL/FL controls and determined their response to MC38 liver metastatic growth. Depletion of NK cells in FloxP-STING animals increased their metastatic burden compared with PBS-treated controls, while no significant effects were observed in myeloid STING KO mice (figure 4D–F). Overall, this suggests that the activation of STING signaling in macrophages suppresses hepatic metastasis by promoting the antitumor properties of NK cells.
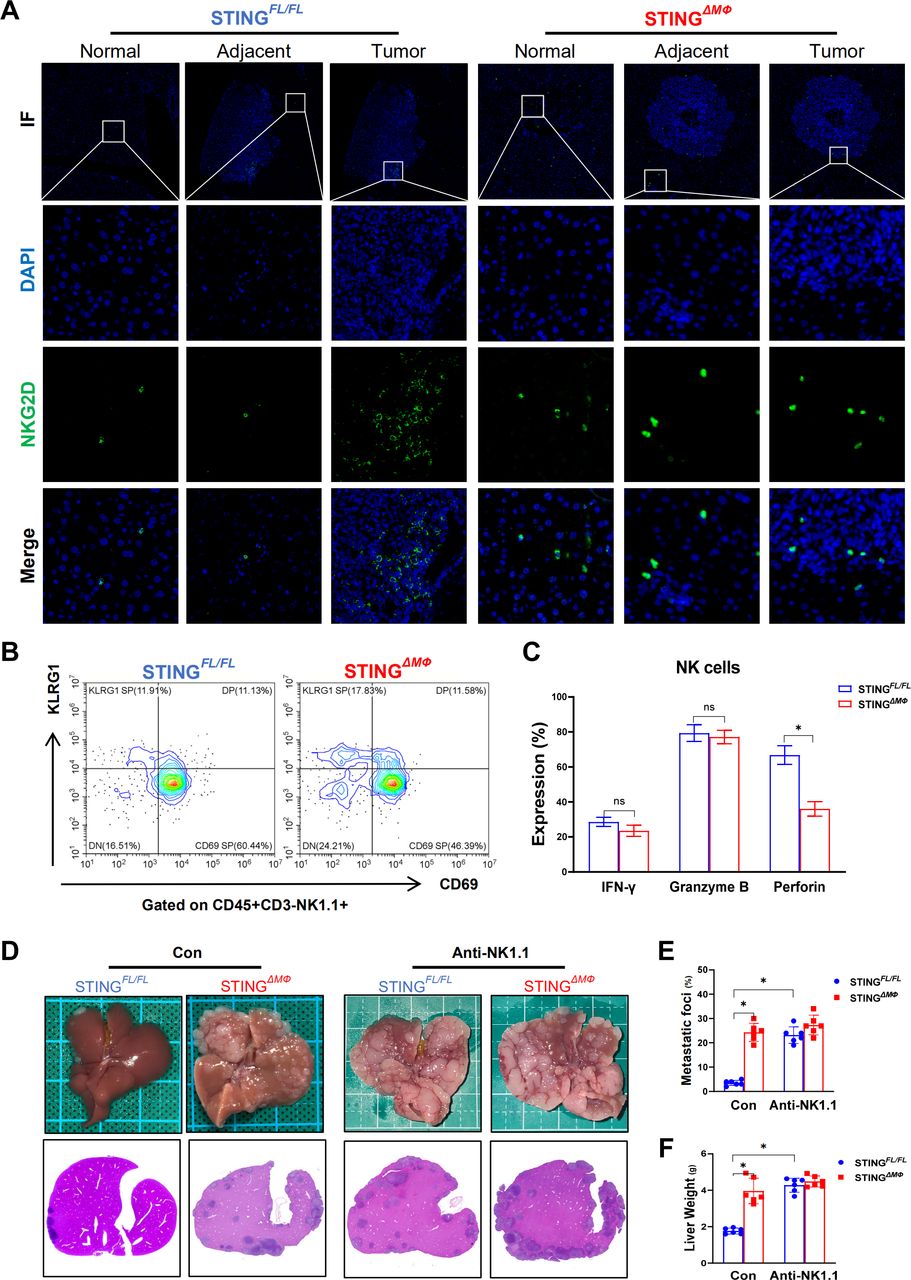
STING signaling in macrophages suppresses hepatic metastasis by promoting the antitumor properties of NK cells. NKG2D+ NK cells in normal, adjacent and tumor liver tissues (selected from the same liver tissue section) were detected by immunohistochemical (A). Representative FACS plots of CD69 and KLRG1 (B) and mean fluorescence intensity expressions of IFN-γ, granzyme B, perforin (C) of NK cells in hepatic NPCs. Anti-NK1.1 antibody were used to deplete NK cells in STINGFL/FL and STINGΔMФ mice with MC38 intrasplenic injection. Representative pictures of the liver and H&E sections (D), liver colorectal cancer foci coverage (E) and liver weight (F). (n=6/group per genotype). Data are presented as mean±SEM, p<0.05*. IFN, interferon; NK, natural killer; NPCs, non-parenchymal cells; STING, stimulator of interferon genes. FACS, Fluorescence Activated Cell Sorter; IF,Immunoflourescent; DAPI, 4',6-diamidino-2-phenylindole.
Co-stimulation signaling of 4-1BBL/4-1BB is required for NK cells to control hepatic metastasis
To dissect the precise role of macrophage STING signaling in regulating NK cell antitumor activity, BMDMs were stimulated with or without MC38 cells and then co-cultured with NK cells (figure 5A). BMDMs without MC38 pretreatment failed to activate NK cells, as indicated by the lack of significant changes in CD107a levels (online supplemental figure 1B). Cell-to-cell uncontacted co-culture with MC38-primed WT BMDMs promoted the antitumor properties of NK cells, as shown by the increased levels of CD107a, IFN-γ, granzyme B, and perforin, which were further enhanced by cell-to-cell contact. STING deficiency in BMDMs significantly inhibited the expression of CD107a and perforin in both cell-to-cell contact and uncontacted co-cultured NK cells (figure 5B,C). These results demonstrated that cell-to-cell contact promoted NK cell activation, indicating an important role of co-stimulatory factors in the activation of NK cells by macrophages. In vivo co-staining of macrophage and NK cells also showed that most NK cells were adjacent to the macrophages in both STINGFL/FL and STINGΔMФ mice (online supplemental figure 3A). Therefore, we analyzed the expression of co-stimulatory factors and found that MC38 treatment significantly induced 4-1BBL expression in BMDMs, which was restricted by macrophage STING depletion (figure 5D,E). Myeloid STING-deficient mice with CRC liver metastasis also showed significantly lower levels of 4-1BBL in macrophages and lower levels of 4-1BB in NK cells (figure 5F,G). While 4-1BBL blockade suppressed the antitumor activity of NK cells primed by BMDMs, supplementation with exogenous 4-1BBL reversed the role of macrophage STING deficiency in impairing NK cell function (figure 5H & online supplemental figure S1). In vivo, blockade of 4-1BBL or 4-1BB significantly increased MC38 liver metastatic growth in STINGFL/FL but not in STINGΔMФ mice (figure 5I–K). The NK cells also showed decreased expression of perforin, but not IFN-γ and granzyme B, in FloxP-STING animals post 4-1BBL or 4-1BB blockade (online supplemental figure 1D). Collectively, these results demonstrate that 4-1BBL/4-1BB signaling is required for macrophage STING activation to promote the antitumor function of NK cells.
Supplemental material
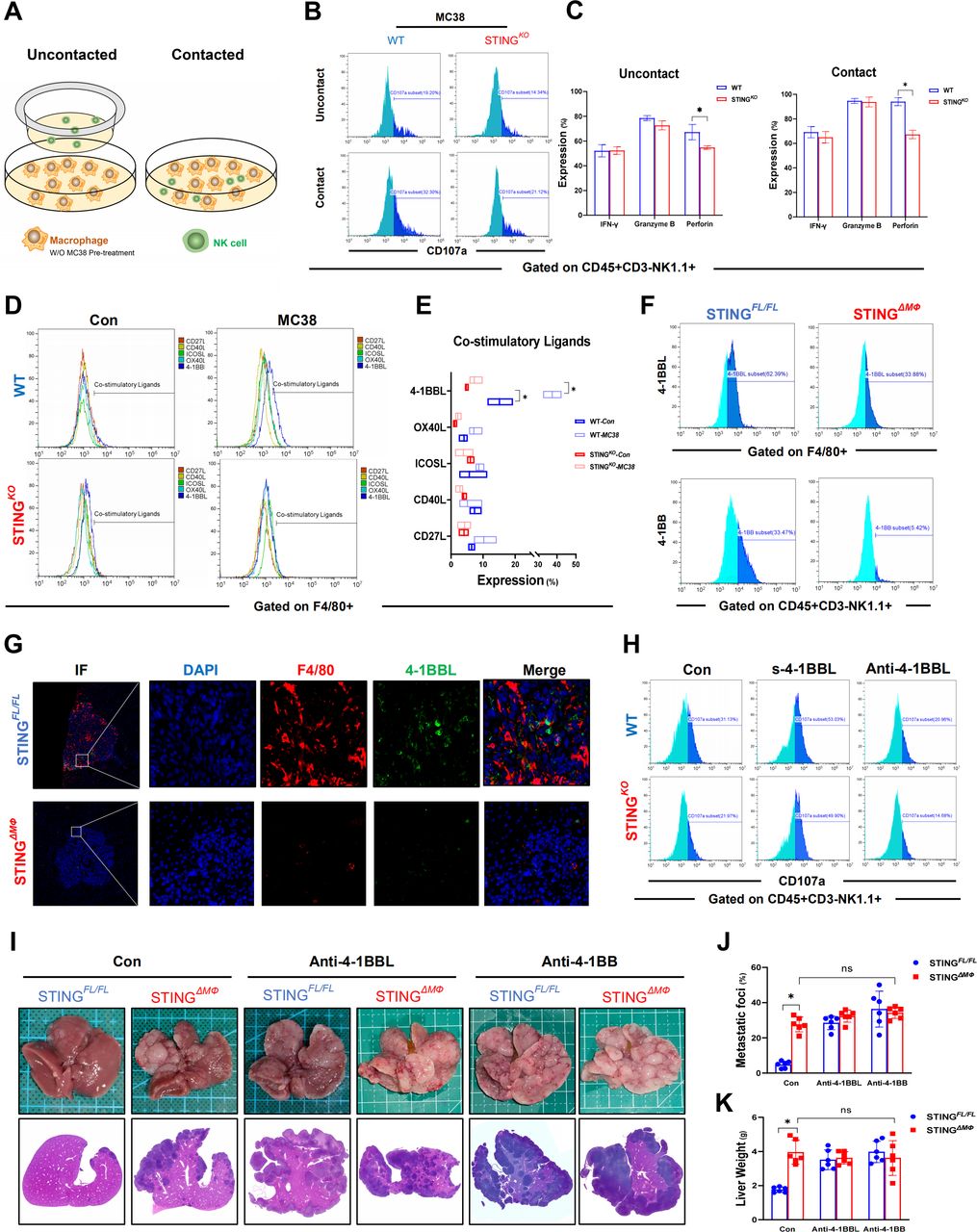
Co-stimulation signaling of 4-1BBL/4-1BB is required for NK cells to control hepatic metastasis. Schematic representation of macrophages and NK cells co-culture model (A). Representative MFI expression of CD107a on NK cells co-cultured with BMDMs or which pretreated with MC38 (B) in contact or uncontact methods. MFI expressions of IFN-γ, granzyme B, perforin represented secretory function of NK cells co-cultured with BMDMs (C). MFI expressions of Co-stimulatory ligands on BMDMs (WT mice) treated with MC38 (D and E). 4-1BBL on macrophages and 4-1BB on NK cells in non-parenchymal cells isolated from STINGFL/FL or STINGΔMФ mice on Day 14 after MC38 intrasplenic injection (F). And 4-1BBL on F4/80+ macrophages in liver were detected by immunohistochemical (G). Expressions of CD107a of NK cells undergone 4-1BBL/4-1BB co-stimulation interference when co-cultured with BMDMs pretreated with MC38 (H). STINGFL/FL and STINGΔMФ mice were intrasplenic injected with MC38. Representative pictures of the liver and H&E staining of liver sections harvested from STINGFL/FL and STINGΔMФ mice sacrificed on Day 14 (Anti-4-1BBL/4-1BB antibody for blockage) after MC38 intrasplenic injection (I). And liver colorectal cancer foci coverage (J) and liver weight (K) on Day 14 after MC38 injection. Data are presented as mean±SEM, p<0.05*. BMDMs, bone marrow-derived macrophages; IFN, interferon; MFI, mean fluorescence intensity; NK, natural killer; STING, stimulator of interferon genes; WT, wild-type. IF,Immunoflourescent; DAPI, 4',6-diamidino-2-phenylindole.
Synergistic effect of macrophage-derived IL-18 and IL-1b signaling on 4-1BBL/4-1BB signaling-mediated optimal antitumor activity of NK cells
To further determine the molecular mechanism of macrophage STING signaling in regulating 4-1BBL/4-1BB activation and the subsequent antitumor activity of NK cells, we analyzed the cytokine expression profiles of WT and STING-deficient BMDMs in response to MC38 treatment. Compared with WT BMDMs, STING-deficient BMDMs produced significantly decreased levels of IL-18 and IL-1β, with similar levels of tumor necrosis factor (TNF)-α, IL-6, IL-8, IL-10 IL-12, and IL-23 (figure 6A). We also analyzed other cytokines of IL-1 family in BMDMs. Interestingly, compared with WT BMDMs, STING-deficient BMDMs expressed higher level of IL-33 with lower levels of IL-1a, IL-37 and IL-38 (online supplemental figure 3B). Besides, we also analyzed the expression of gene induction of some chemokines in macrophages. STING-deficient BMDMs showed decreased levels of CCL3, CCL21, CXCL9 and CXCL10 in response to MC38 treatment (online supplemental figure 3C). Then, neutralizing antibodies were used to confirm whether IL-18 and IL-1β are essential for modulating 4-1BBL/4-1BB signaling and subsequent NK cell activation. Indeed, while IL-18 neutralization reduced 4-1BBL expression in macrophages, IL-1β neutralization decreased 4-1BB expression in NK cells (figure 6B). Both IL-18 and IL-1β neutralization effectively suppressed NK activation, as shown by the reduced CD107a expression and decreased production of perforin (figure 6C). Therefore, we further investigated whether IL-18/4-1BBL and IL-1β/4-1BB were essential for macrophage STING signaling in promoting NK cell activation. Exogenous supplementation with IL-18 or IL-1β restored 4-1BBL expression in STING-deficient BMDMs and 4-1BB expression in NK cells, respectively, and both promoted NK cell activation, as indicated by increased CD107a expression. The combination of IL-18 and IL-1β treatment significantly improved NK cell activation (figure 6D,E), indicating a synergistic effect of macrophage-derived IL-18 and IL-1β signaling on the 4-1BBL/4-1BB signaling-mediated optimal antitumor activity of NK cells.
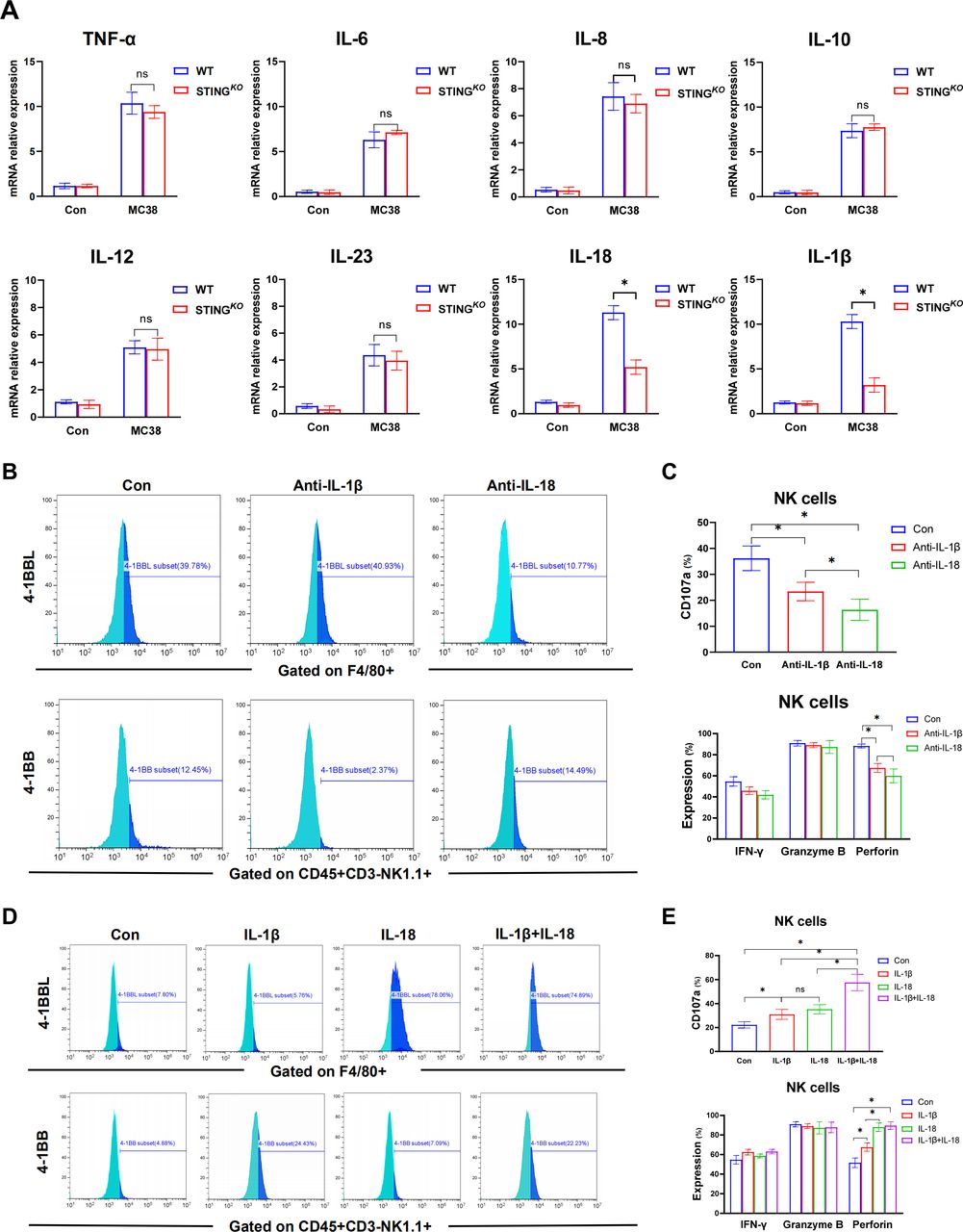
Synergistic effect of macrophage-derived IL-18 and IL-1β signaling on 4-1BBL/4-1BB signaling-mediated optimal antitumor activity of NK cells. The synthesis of multiple cytokines in BMDMs detected by real-time PCR (A). MFI of the expression of 4-1BBL on BMDMs isolated from WT mice undergone the blockage of IL-18 and IL-1β in the presentence of MC38 (B). MFI of CD107a and secretory function of NK cells undergone co-cultured with BMDMs pretreated with MC38 under the synergistic interference with IL-18 and IL-1β (C). MFI of the expression of 4-1BBL on BMDMs extracted from STINGKO mice undergone the stimulation of IL-18 and IL-1β in the presence of MC38 pretreatment (D). MFI of CD107a and secretory function of NK cells undergone co-cultured with BMDMs prestimulated though MC38 under the synergistic stimulation with IL-18 and IL-1β (E). Data are presented as mean±SEM, p<0.05*. BMDMs, bone marrow-derived macrophages; IL, interleukin; MFI, mean fluorescence intensity; mRNA, messenger RNA; NK, natural killer; STING, stimulator of interferon genes; TNF, tumor necrosis factor; WT, wild-type.
NLRP3-mediated IL-18 and IL-1β activation is responsible for macrophage STING signaling to promote 4-1BBL/4-1BB-dependent NK cell antitumor properties
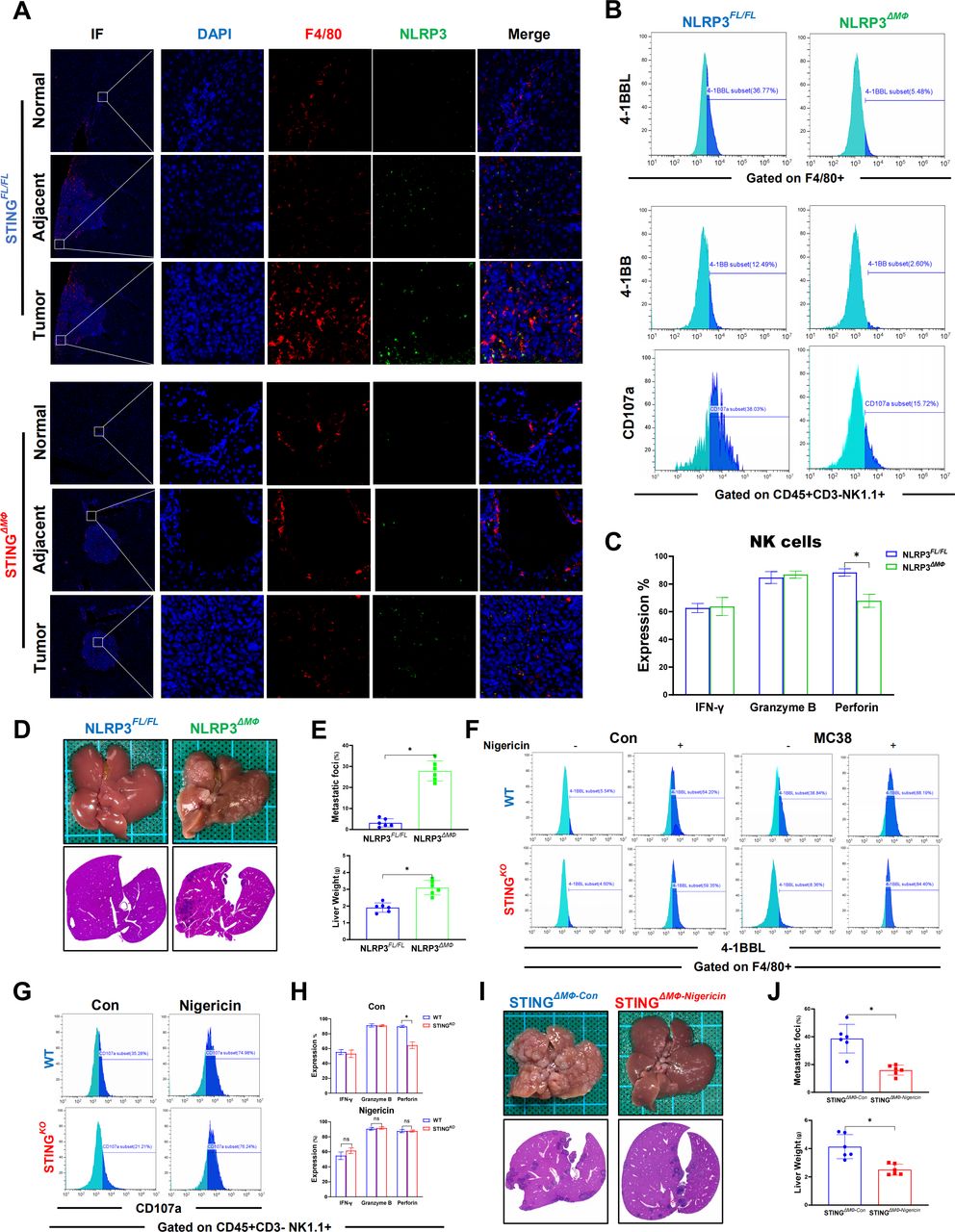
IL-18 and IL-1β are both important downstream signaling molecules of NLRP3, and we previously found that STING activation promoted NLRP3 activation in macrophages.18 Therefore, we investigated whether NLRP3 was essential for macrophage STING to promote NK cell activation via IL-18 and IL-1β signaling. Significantly enhanced double staining of F4/80 and NLRP3 was observed inside the tumor compared with that of adjacent and normal liver tissues (figure 7A). In vitro MC38 treatment induced activation of macrophage NLRP3 signaling, which was suppressed by STING depletion (online supplemental figure 2A,B). Similar to the above findings in STING-deficient macrophages, NLRP3 deficiency significantly inhibited the expression of 4-1BBL in macrophages and 4-1BB in NK cells. Macrophage NLRP3 depletion also suppressed the activation of NK cells, as indicated by lower levels of CD107a and perforin (figure 7B,C). Interestingly, myeloid NLRP3-deficient mice also exhibited a worse tumor burden than FloxP-NLRP3 control animals (figure 7D,E). Moreover, NLRP3 activation by its agonist nigericin was able to induce 4-1BBL expression in both WT and STINGKO macrophages in the absence or presence of MC38 treatment and abrogated the role of macrophage STING deficiency in suppressing 4-1BBL expression in macrophages (figure 7F). NLRP3 activation by nigericin also reversed the effects of macrophage STING deficiency on inhibiting NK cell activation, as indicated by the increases in 4-1BB and CD107a expression and perforin production (figure 7G,H).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Macrophage STING signaling promotes antitumor properties of NK cells by activating NLRP3. NLRP3 in F4/80+macrophages in normal, adjacent and tumor liver tissues (selected from the same liver tissue section) from STINGFL/FL or STINGΔMФ mice sacrificed on Day 14 after MC38 intrasplenic injection were detected by immunohistochemical (A). MFI expression of 4-1BBL on BMDMs extracted from NLRP3FL/FL and NLRP3ΔMФ mice and MFI of 4-1BB or CD107a on NK cells co-cultured with BMDMs in the presentence of MC38 (B). Secretory function of NK cells (C). Representative pictures of the liver and H&E of liver sections from NLRP3FL/FL or NLRP3ΔMФ (n=6/group per genotype) sacrificed on Day 14 after MC38 intrasplenic injection (D). NLRP3FL/FL and NLRP3ΔMФ mice liver CRC foci coverage and liver weight on Day 14 after MC38 injection (E). Expressions of 4-1BBL on BMDMs extracted from WT and STINGKO mice treated with nigericin in the present of MC38 or not (F). MFI expression of CD107a (G) and secretory function (H) of NK cells co-cultured with BMDMs from WT or STINGKO mice after nigericin treatment. STINGΔMФ mice were treated with nigericin or vehicle control on Day 7 and every other day after MC38 intrasplenic injection. Representative pictures of the liver and H&E of liver sections (I) and liver CRC foci coverage and liver weight (J). Data are presented as mean±SEM, p<0.05*. CRC, colorectal cancer; BMDMs, bone marrow-derived macrophages; MFI, mean fluorescence intensity; NK, natural killer; NLRP3, NOD-like receptor protein 3; STING, stimulator of interferon genes; WT, wild-type.
Finally, we tested the in vivo role of NLRP3 activation by nigericin. Treatment of nigericin effectively restored activation of NLRP3 signaling (online supplemental figure 2C) and significantly increased the expression of IL-18 and IL-1β (online supplemental figure 2D) in livers from myeloid STING-deficient mice. Consistent with the findings of in vitro studies, restoration of NLRP3 activation by nigericin suppressed CRC liver metastasis in myeloid STING-deficient mice (figure 7I,J). Moreover, nigericin treatment also increased the number (online supplemental figure 2E) and perforin production (online supplemental figure 2F) of intrahepatic infiltrating NK cells. Together, these results indicated that macrophage STING-mediated NLRP3 activation promoted 4-1BBL/4-1BB-dependent NK cell antitumor activity via IL-18 and IL-1β signaling.
Discussion
Liver metastasis remains a major cause of CRC mortality. Although NK cells limiting liver metastasis has been reported,19 the role of macrophage STING signaling in regulating NK cell activation in CRC liver metastasis remains unclear. Using an intrasplenic model of CRC metastasis to the liver, we investigated the regulatory role and mechanism of macrophage STING signaling in promoting antitumor functions of NK cells. Macrophage STING signaling-mediated NLRP3 activation and subsequent IL-18 and IL-1β expression were found to be essential for NK cell activation. IL-18 and IL-1β induced the expression of 4-1BBL and 4-1BB in macrophages and NK cells, respectively, which facilitated macrophage STING signaling to promote the antitumor function of NK cells via 4-1BBL/4-1BB pathway.
The host immune system and inflammation shapes the fate of tumor progression at all stages.20 21 Interplay between tumor cells and immune cells play an important role in forming specific TME to regulate the engraftment, survival and progression of CRC metastasis in the liver. Multiomics integrative analysis of CRC reveals significant heterogeneity between primary colorectal tumors and their liver metastases.22 With a unique composition of parenchymal and immune cells that regulate innate and adaptive immunity, the liver is one of the most immune tolerogenic organ to prevent unwanted immune responses.23 The liver harbors the largest proportion of macrophages among all solid organs in the body.24 Liver macrophages serve as a gatekeeper for initiating or suppressing immune response by sensing pathogen-associated molecular patterns of bacteria and microbial products from the intestine via the portal vein, or danger-associated molecular patterns.24 We previously showed that macrophage-related inflammation regulated ischemic liver injury.25 Infiltration of macrophages in both primary and metastatic solid tumors is associated with poor prognosis, making TAM-targeting strategies as promising therapeutic approaches in cancer.26
cGAS-STING signaling has demonstrated an expanding role in microbial infections, cellular senescence, autoimmune and inflammatory diseases and antitumor immunity.7 27 DNA or cGAMP from tumors activates cGAS-STING signaling to initiate antitumor immunity.28 Blockade of MERTK enhanced the transport of tumor-derived cGAMP into TAM and subsequent STING activation, which increased tumor immunogenicity and antitumor immunity.29 Microbiota-derived STING agonists induced type I IFN production to reprogrammed TME, and the absence of microbiota skewed the TME toward protumorigenic macrophages.30 IFN-independent activities of STING were also found in macrophages and T cells, and tumor induced STING-mediated cell death of T cells contributed to tumor immune evasion.31 However, persistent inflammation mediated by chronic activation of cGAS-STING signaling can also have tumor and metastasis-promoting functions by inducing an immune-suppressive TME.8 cGAMP generated in cancer cells induced STING activation and production of inflammatory cytokines in astrocytes, which thereby supporting tumor growth and chemoresistance.32
NK cells are cytotoxic lymphocytes which participate in viral infection and cancer immunity by killing target cells and producing cytokines.33 However, in the immunosuppressive TME, the function of tumor-infiltrating NK cells may be impaired on exposure to inhibitory molecules produced by cancer cells, leading to tumor escape.5 Therefore, NK cell-based immunotherapies include adoptive transfer therapies, direct stimulation, recruitment of NK cells into the TME, and modulation of the TME to enhance antitumor NK cell function.34 35 High amounts of circulating or tumor-infiltrating NK cells were found to be negatively correlated with the presence of metastasis in various types of solid tumors.36 The innate lymphoid cells (ILCs) are lymphocytes with unique functional properties in immune defense and tissue homeostasis but lack rearranged antigen receptors.37 ILCs are composed of NK cells, ILC1, ILC2 and ILC3 according to their developmental and effector program.38 Despite distinct developmental origins, ILC1 and conventional NK cells share many characteristics and are therefore commonly referred to as type I ILCs. Similar to NK cells, ILC1s promote type 1 immune responses against intracellular pathogens via the production of IFN-γ and TNF. Comparable numbers of ILC1s and NK cells are found in the liver and are both essential to control the liver metastasis.39 40 However, recent discovery of functional characteristics and transcriptional programs shared by ILC1s and NK cells has made it difficult to distinguish them unequivocally.41 In addition, conventional NK cells have been found to acquire ILC1 characteristics under certain conditions such as in a cancer setting, which may complicate the identification of NK cells and ILC1s.42 43 In the present study, the potential role of ILC1s were not excluded and analyzed precisely. Since anti-NK1.1 administration may potentially deplete ILC1 and NKT cells, mice with selective loss of NK cells would be employed to better define the specific role of NK cells in CRC development. Of note, while lack of perforin expression by ILC1s has reported,44 we found that NK cells from STINGΔMФ mice produced less perforin but similar levels of IFN-γ and granzyme B, indicating a critical role of NK cells but not ILC1s regulated by macrophage STING signaling.
Interplay between macrophages and other immune cells plays an important role in shaping the TME and antitumor immunity.45 TIM-4 (T cell immunoglobulin and mucin domain-containing protein 4) cavity-resident macrophages engage with phosphatidylserine-expressing cytotoxic T lymphocytes, which inhibited antitumor activity of CD8 T cells even during programmed cell death protein-1 blockade.46 Regulatory T cells (Tregs) recruitment was crucial for TAM-mediated anti-programmed death ligand-1 resistance and immunosuppression in hypoxic TME.47 Inhibition of STAT3 in Tregs enhanced the antitumor activity of T cells and M1 macrophages.48 AXL inhibition in macrophages stimulated anti-leukemic immunity of NK cells and T cells.49 Dectin-1 mediated activation of macrophages promoted the antitumor function of NK cell in metastasis control.50 NLRP3-IL-18 signaling of macrophages was critical for the maturation and tumoricidal activity of NK cells in CRC metastatic growth in the liver.51
However, there are some limitations in current study. Since both MC38 and CT26 cell lines are used in CRC liver metastasis model,52 experiments with a second cell line would strengthen our findings. In addition, the effects of in vivo suppression of IL-18 and IL-1β by neutralizing antibody or transgenic mice were not evaluated.
Conclusions
Taken together, we demonstrated that STING signaling promoted NLRP3-mediated IL-18 and IL-1β production of macrophages and subsequent the antitumor response of NK cells via the co-stimulation signaling of 4-1BBL/4-1BB. Our results identified that the STING/NLRP3 axis is critical for macrophages to promote the antitumor function of NK cells in preclinical models, now poised for clinical evaluation.
Data availability statement
Data are available in a public, open access repository.
Ethics statements
Patient consent for publication
Ethics approval
The study protocol was approved by the Institutional Review Board of the First Affiliated Hospital of Nanjing Medical University. Ethics Committee of the First Affiliated Hospital of Nanjing Medical University exempted consent for participation in the study (No.2022-SR-308). All animal procedures met the relevant legal and ethical requirements according to the protocols approved by the Institutional Animal Care and Use Committee (IACUC)of Nanjing Medical University (No. IACUC-2009050).
Acknowledgments
We would like to thank the Core Facility of the First Affiliated Hospital of Nanjing Medical University for its help in the experiment.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
YS, HH and ZL contributed equally.
Contributors HZ developed the study concept. LK, HZ and ZR directed experimental design and interpreted data. YS, HH and ZL performed experiments and computational analysis. HH, ZL, JX, YG, XZ, SZ, WZ, DW and PW conducted experiments and additional data analysis. ZR, LK and HZ contributed reagents/analytic tools and/or grant support. YS, ZR and HZ wrote the manuscript. All authors read, edited, and approved the manuscript.
Funding This study was supported by the National Natural Science Foundation of China (No. 82071798, 81871260, 81901628 and 81600450), CAMS Innovation Fund for Medical Sciences (No. 2019-I2M-5-035), Jiangsu Provincial Medical Innovation Center (CXZX202203), Jiangsu Provincial Medical Key Laboratory (ZDXYS202201), the Natural Science Foundation of Jiangsu Province BK20191490, the Six Talent Peaks Project in Jiangsu Province (No. 2018-WSN-011), the Jiangsu Science and Technology Association Young Science and Technology Talents Lifting Project (No. DG000D4007) and A Project Funded by the PAPD.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.