Article Text

Abstract
Background The wider application of T cells targeting viral tumor-antigens via their native receptors is hampered by the failure to expand potent tumor-specific T cells from patients. Here, we examine reasons for and solutions to this failure, taking as our model the preparation of Epstein-Barr virus (EBV)-specific T cells (EBVSTs) for the treatment of EBV-positive lymphoma. EBVSTs could not be manufactured from almost one-third of patients, either because they failed to expand, or they expanded, but lacked EBV specificity. We identified an underlying cause of this problem and established a clinically feasible approach to overcome it.
Methods CD45RO+CD45RA− memory compartment residing antigen-specific T cells were enriched by depleting CD45RA positive (+) peripheral blood mononuclear cells (PBMCs) that include naïve T cells, among other subsets, prior to EBV antigen stimulation. We then compared the phenotype, specificity, function and T-cell receptor (TCR) Vβ repertoire of EBVSTs expanded from unfractionated whole (W)-PBMCs and CD45RA-depleted (RAD)-PBMCs on day 16. To identify the CD45RA component that inhibited EBVST outgrowth, isolated CD45RA+ subsets were added back to RAD-PBMCs followed by expansion and characterization. The in vivo potency of W-EBVSTs and RAD-EBVSTs was compared in a murine xenograft model of autologous EBV+ lymphoma.
Results Depletion of CD45RA+ PBMCs before antigen stimulation increased EBVST expansion, antigen-specificity and potency in vitro and in vivo. TCR sequencing revealed a selective outgrowth in RAD-EBVSTs of clonotypes that expanded poorly in W-EBVSTs. Inhibition of antigen-stimulated T cells by CD45RA+ PBMCs could be reproduced only by the naïve T-cell fraction, while CD45RA+ regulatory T cells, natural killer cells, stem cell memory and effector memory subsets lacked inhibitory activity. Crucially, CD45RA depletion of PBMCs from patients with lymphoma enabled the outgrowth of EBVSTs that failed to expand from W-PBMCs. This enhanced specificity extended to T cells specific for other viruses.
Conclusion Our findings suggest that naïve T cells inhibit the outgrowth of antigen-stimulated memory T cells, highlighting the profound effects of intra-T-cell subset interactions. Having overcome our inability to generate EBVSTs from many patients with lymphoma, we have introduced CD45RA depletion into three clinical trials: NCT01555892 and NCT04288726 using autologous and allogeneic EBVSTs to treat lymphoma and NCT04013802 using multivirus-specific T cells to treat viral infections after hematopoietic stem cell transplantation.
- Immunotherapy
- Immunotherapy, Adoptive
- T-Lymphocytes
- Adaptive Immunity
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are included in the article or uploaded as supplementary information. Any other relevant data is available upon reasonable request to crooney@bcm.edu and sandhyas@bcm.edu. Any specific data analysis codes used for TCR sequencing data analysis can be available upon reasonable request to u235274@bcm.edu.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
T cells targeting tumor-associated antigens via their native receptors can be effective as therapy for various malignancies, but most T-cell products have low antigen specificity and so complete responses are rare. It has been assumed that tumor antigen-specific T cells are anergized at the tumor site, hence circulate with low frequency. As a result, many investigators have used transgenic T-cell receptor-modified T cells as therapy. However, these target only a single epitope in a single antigen, leading to the outgrowth of escape mutants. By contrast, T cells activated from blood by stimulation with whole antigens recognize multiple epitopes in multiple tumor antigens and so effort to reactivate and expand such T-cell products are warranted.
WHAT THIS STUDY ADDS
Our study suggests that viral tumor antigen-specific T cells do circulate in patients but are refractory to reactivation and expansion. By removing CD45RA expressing peripheral blood mononuclear cell subsets prior to antigen stimulation, we were able to relieve this suppression, producing virus-specific T cells with higher antigen specificity from both patients and healthy donors. This holds true for T cells specific to other viruses and provides investigators with a strategy to increase the potency of their therapeutic T-cell products. Although we could not pinpoint the exact mechanism underlying this clonal inhibition, we speculate that it may represent a physiological mechanism to prevent the oligoclonal outgrowth of cross-reactive T cells with poor protective capacity at the expense of a broad repertoire of naïve T cells with protective capacity in response to infection with new pathogens.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The availability of a clinically feasible strategy to increase the antigen specificity and potency of viral antigen-specific T cells from patients and healthy donors will enable clinical investigators and biotech to improve their cell therapy products. Our studies also open up a new avenue of research into the mechanisms by which naïve T cells can affect antigen-stimulated memory T cells.
Introduction
While autologous T-cell immunotherapies are changing the face of cancer treatment, broad accessibility to these treatments remains problematic, in part because of difficulties in manufacturing products with the desired potency. T cells may be rendered tumor-specific by selection of T cells with appropriate native T-cell receptor (TCR) specificities,1–3 or by expression of transgenic TCRs,4–7 or chimeric antigen receptors (CARs).8–10 Although all three approaches may be clinically effective, transgenic TCRs and CARs each target individual epitopes on single antigens, increasing the risk of immune evasion.
Given the extensive heterogeneity of human tumors,11 12 it may be beneficial to manufacture therapeutic products with a broader array of antigen/epitope specificities. Such tumor antigen-specific T cells expanded from tumor biopsies (tumor infiltrating lymphocytes (TILs)) have produced tumor responses in patients with melanoma,13 14 and cervical cancer3 15 while T cells selectively expanded from patient peripheral blood have been effective for melanoma,16 lymphoma,17 18 and virus-associated malignancies.19–21 However, for cell therapies to be both accessible and optimally effective, it is essential to consistently manufacture potent products from patients whose T-cell function may have been impaired by the immunosuppressive effects of their disease and its treatment. While TILs offer one solution, most tumors lack accessible TILs and their manufacture is complex and invasive,22–24 the expansion of tumor antigen-specific T cells from patient peripheral blood frequently fails at the manufacturing stage.18 25 26
To examine the difficulties in selectively expanding tumor antigen-specific T cells for therapy, we made use of our clinical studies of T cells targeting the four Epstein-Barr virus (EBV) antigens expressed in EBV+ lymphoma. Although EBV-specific T cells (EBVSTs) can produce complete and sustained remissions,19 we were unable to generate EBVSTs from almost one third of patients with refractory/relapsed EBV+ lymphoma because patient T cells either failed to expand or lacked sufficient antigen-specificity to meet the potency release criteria.
T cells targeting autologous tumor antigens via their native TCRs should have been exposed to antigen and reside mostly in the CD45RO+ CD45RA− memory compartment.27–30 Therefore, in an attempt to enrich EBV tumor antigen-specific T cells, we removed the CD45RA+ fraction of peripheral blood mononuclear cells (PBMCs) that includes naïve T cells (TNs) as well as terminally differentiated effector memory (TEMRA) and stem cell memory (TSCM) T cells prior to antigen stimulation.27 30–32 This manipulation enabled the expansion of low-frequency T-cell clonotypes that did not expand from unfractionated PBMCs, leading to increased antigen specificity even from patient PBMCs that otherwise failed to respond. The inhibitory activity of CD45RA+ PBMCs could be attributed to the TN fraction since other CD45RA+ cells were not inhibitory. This improvement extended to T cells specific for other viruses including oncogenic human papillomavirus (HPV), allowing the generation of more potent virus-specific T cells (VSTs) with a more focused TCR repertoire. Hence our strategy has wide application to the improved implementation of native TCR-based therapeutics.
Materials and methods
Blood donors and cell lines
Blood samples were obtained from EBV seropositive healthy volunteers and patients with EBV-positive lymphoma with informed consent using Baylor College of Medicine, Institutional Review Board-approved protocols. Buffy coats were purchased from the Gulf Coast Regional Blood Center, Houston, Texas, USA. PBMCs isolated on Lymphoprep gradients (Axis Shield, Oslo, Norway) were used to generate VSTs and activated T cells (ATCs) for use as antigen presenting cells (APCs). The universal (U) EBV-transformed lymphoblastoid cell line (LCL) and K562cs used as co-stimulatory cell lines are described in the online supplemental materials and methods.
Supplemental material
CD3 and CD28 ATCs
PBMCs were stimulated with CD3 (from the OKT3 hybridoma cell line ATCC# CRL 8001, Manassas, Virginia, USA) and CD28 (Becton Dickinson BD, Franklin Lakes, New Jersey, USA) antibodies and expanded in interleukin (IL)-2 (National Institutes of Health, Bethesda, Maryland, USA) as previously described.33 ATCs were restimulated with CD3/CD28 antibodies, pulsed with a mastermix of EBV T2 antigen pepmixes, irradiated at 30 Gy using an RS2000 X-ray irradiator (RadSource, Suwanee, Georgia, USA) and used as autologous APCs.
Pepmixes
Overlapping peptide libraries—(15 mers overlapping by 11 amino acids) spanning the protein sequences of EBV type-2 latency (T2) antigens (EBNA-1, LMP-1 and LMP-2, BARF-1), were purchased from JPT technologies (Berlin, Germany).17 33
PBMC subset depletion
CD45RA+ PBMCs CD56+ natural killer (NK) cells or CD25+ regulatory T cells (Tregs) were depleted or selected from PBMCs using CD45RA antibody-coated beads and MACS MicroBead Technology (Miltenyi Biotec, Bergisch Gladbach, Germany) as per the manufacturer’s instructions. The eluted CD45RA negative fraction was used to generate RAD-VSTs. In some experiments, the CD45RA+ cells were plunged from the columns for VST generation. The isolation purity was determined by flow cytometry.
Immunophenotyping
T cells were phenotyped using CD3, CD4, CD8, CD56, CD45RA, CD45RO, CCR7, CD62L, TIM-3, PD-1, LAG-3, KLRG-1, CD25 surface antibodies (BioLegend, San Diego, California and BD Biosciences, Franklin Lakes, New Jersey, USA) as previously described.17 33 The cells were acquired using the Gallios Flow Cytometer or BD FACSCanto II, and results were analyzed using Kaluza software (Beckman Coulter) or FlowJo analysis software (FlowJo, Ashland, Oregon, USA)
Separation of CD45RA subsets
Magnetic bead selected CD45RA+ PBMCs were sorted into CD3+CD45RA+, CD3–CD45RA+, TEMRAs(CD45RA+, CCR7–), TSCMs(CD45RA+, CCR7+, CD95+) and TNAIVE (CD45RA+, CCR7+, CD95−) subsets using SH800S Sony cell sorter using antibodies from BioLegend (San Diego, California, USA) or BD Biosciences. Details are in the online supplemental materials.
EBVST generation
Whole, or subset depleted PBMCs were pulsed with the T2-EBV pepmix cocktail (EBNA1, LMP1, and LMP2), (BARF-1 was added only for patient samples) and expanded with IL-15 (R&D Systems, Minneapolis, Minnesota, USA) at the indicated concentrations and IL-7 (10 ng/mL) (PeproTech, Rocky Hill, New Jersey, USA). On day 9, the cells received a second stimulation (S2)—with irradiated T2 pepmix-pulsed autologous ATCs and irradiated K562cs or human leukocyte antigen (HLA)-negative LCLs (ULCLs), at an EBVST: ATCs: K562cs/ULCL ratio of 1:1:5 with IL-7 and IL-15.33 Expanding cells were split as required and analyzed for phenotype, specificity and function on day 16 of culture, unless otherwise indicated. Although EBVST lines were not 100% EBV specific, and without CD45RA depletion may have contained CD3−CD56+NK cells, for convenience we refer to them as EBVST.
Intracellular cytokine assay
VSTs were stimulated with 100 ng of EBV pepmix or control pepmix (tumor antigen PRAME). After 1-hour post stimulation, brefeldin-A and monensin were added (BioLegend, San Diego, California, USA). After 16 hours, cells were surface stained, then permeabilized with fixable live dead stain and interferon (IFN)-γ and tumor necrosis factor (TNF)-α antibodies (Becton Dickinson). Further details are in online supplemental material.
Enzyme-linked immunospot assay
The frequency of T2 antigen-specific T cells within the EBVST population was measured in IFN-γ enzyme-linked immunospot (ELISpot) assays as previously described.33 34 EBVSTs were plated at 1×105 per well in duplicate and stimulated with 100 ng pepmixes. The number of IFN-γ spot-forming cells (SFCs)/105 cells in response to viral antigen pepmixes after subtracting negative control values was used as a measure of antigen specificity.
Cytotoxicity assay
The cytolytic specificity of the VSTs was evaluated in a standard 4-hour 51Cr chromium release assay.33 34 Autologous ATCs alone or pulsed with pepmixes were labeled with 51Cr sodium chromate for 1 hour at 37°C, washed and plated at effector:target ratios of 40:1, 20:1, 10:1, and 5:1. After 4 hours of co-culture, supernatant was harvested and 51chromium release measured using a gamma counter to calculate percent specific-lysis.
DNA isolation and TCR sequencing
Genomic DNA was isolated from 3×106 whole, CD45RA+ or CD45RA depleted PBMCs and their derived expanded EBVSTs and sequenced by Adaptive Biotechnologies (Seattle, Washington, USA) using survey resolution immunoSEQ technology that uses a multiplex PCR system to amplify all possible TCR-β CDR3 sequences.35 Productive clonotype frequency was computed using the immunoSEQ Analyzer track frequency tool and imported into R for analysis.
In vivo experiments
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG), 4–6 weeks old, female mice (Jackson Laboratory, Bar Harbor, Maine, USA) were engrafted subcutaneously with EBV-LCLs suspended in Matrigel matrix (Corning, Tewksbury, Massachusetts, USA). Ten days later, EBVSTs were injected intravenously. Tumor volume, measured using an external caliper was calculated using the formula: tumor volume = ½ (length) × (width)2. Either T cells or tumor cells were modified with retroviral vectors expressing Firefly luciferase (FF-luc). Bioluminescence was measured using an IVIS Imaging system (Caliper Life Sciences, Hopkinton, Massachusetts, USA) after injecting mice with D-Luciferin (Sigma-Aldrich, St Louis, Missouri, USA). Living Image software (PerkinElmer, Waltham, Massachusetts, USA) was used to visualize and calculate total luminescence covering the region of interest drawn covering the tumor area. All procedures complied with the Institutional Animal Care and Usage Committee at Baylor College of Medicine approved protocol #AN5551.
Statistical analysis
We used GraphPad Prism V.7 (GraphPad Software, La Jolla, California, USA) for statistical analysis using paired Student’s t-test and/or as indicated in figure legends. Data are plotted as mean±SEM unless otherwise indicated. Significance is denoted by p<0.05, (0.12 (ns, non-significant), 0.033(*), 0.002(**), <0.001(***)) unless otherwise indicated.
Results
EBVSTs could not be manufactured from one-third of patients with EBV+ lymphoma
In our clinical trial of autologous EBVSTs to treat EBV+ lymphoma (NCT01555892), we manufactured EBVSTs by stimulating patient PBMCs with autologous mature dendritic cells, pulsed with EBV type-2 latency (T2) antigen pepmixes, LMP1, LMP2, BARF1, and EBNA1. T2 antigen specificity in the final product was evaluated by measuring the number cells that secreted IFN-γ (SFCs) per 105 cells in response to pepmix stimulation using ELISpot assays.33 Although the ELISpot assay underestimates the true frequency of antigen-specific T cells by at least one log36 37, it is HLA agnostic and highly specific. Most patient EBVSTs contained a low frequency of T2 antigen-specific T cells (figure 1A) and 22.2% (16 of 72) failed the release criterion of greater than 20 IFN-γ SFCs/105 (figure 1B), while five additional EBVST lines failed to grow, bringing the manufacturing failures to 27% (figure 1C). Of the 56 eligible EBVSTs, 26 (46%) had less than 500 SFC/105, with 14 (25%) having less than 100 SFC (figure 1C). These results showed that a substantial number of cells lacking EBV specificity expanded in our ‘EBVST’ products. NK cells accounted for a fraction of these ‘bystander cells’ in some lines, since 25 of 68 EBVSTs contained over 20% CD3–CD56+ NK cells (median frequency 11.4%, range 0.03%–94.2%) (figure 1D). To prevent manufacturing failures, we sought to reduce the outgrowth of bystander cells lacking EBV specificity.
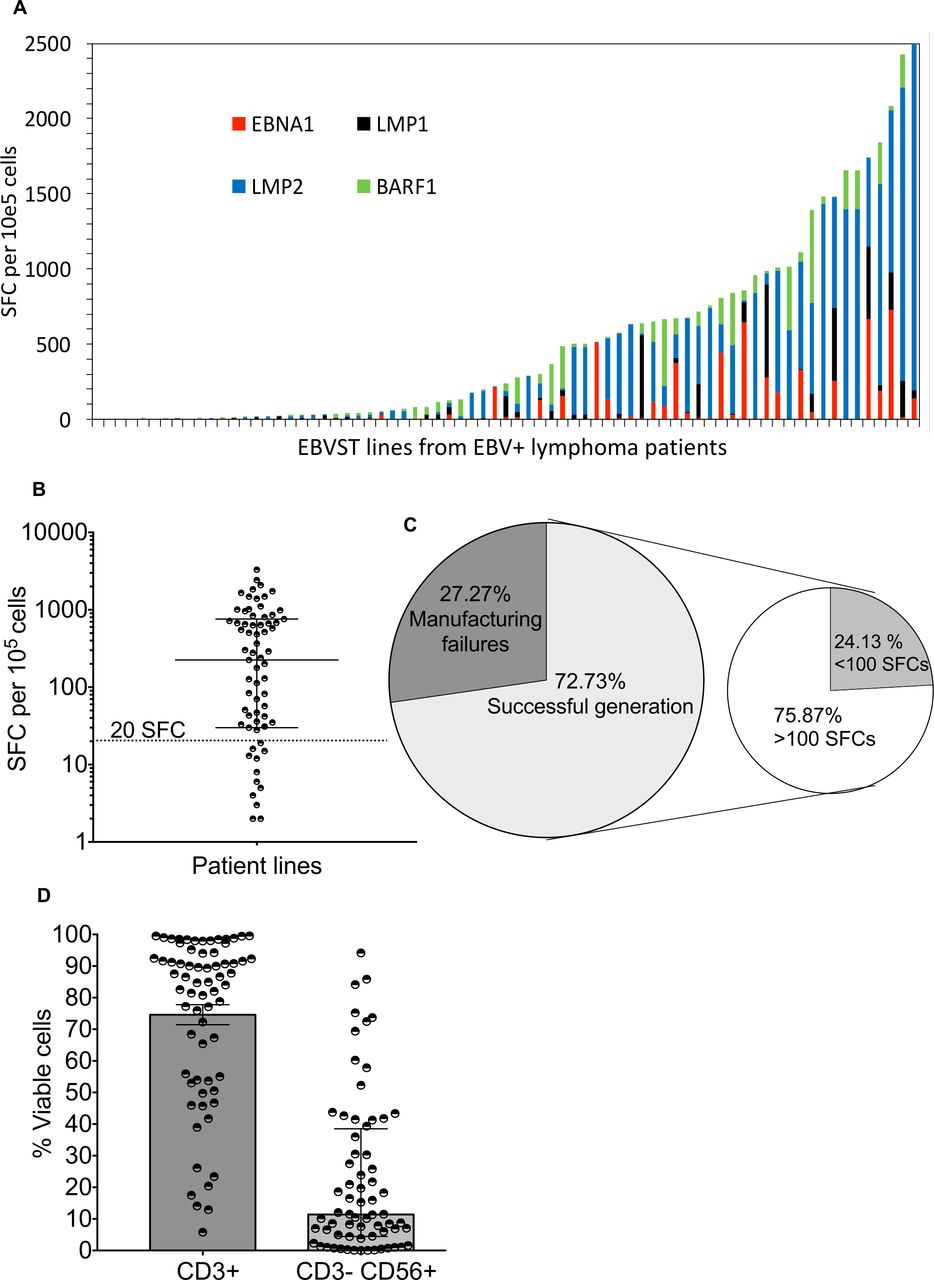
Antigen specificity and phenotype of patient EBVSTs manufactured for a clinical trial (NTC01555892). (A) Antigen specificity from 72 EBVSTs generated from 57 patients by stimulation of peripheral blood mononuclear cells with 4 EBV T2-pepmixes comprizing 15 mer peptides overlapping by 11 amino acids, spanning the entire protein sequences of EBV type-2 latency antigens (EBNA-1, LMP-1, LMP-2 and BARF-1). The Y-axis shows the number of cells per 105 EBVSTs that produced interferon-γ in response to stimulation with each EBV T2 antigen for all patient lines as measured in enzyme-linked immunospot assays. The X-axis indicates each patient line generated. EBNA1, LMP1, and LMP2 were termed T2 antigens. (B) Dot plot representation of total antigen specificity in patient EBVSTs. Each circle represents one patient line. The dotted horizontal line mark divides the 56 EBVSTs with >20 SFCs per 105 EBVSTs from the 16 EBVSTs with <20 (4 had 0 detected SFCs). (C) Pie chart illustrating the frequency of manufacturing failures including EBVSTs that failed to expand and/or produced less than 20 IFN-γ SFCs per 105 cells (27.27%) from all patients (21 of 77 EBVSTs from 62 patients, including two or more attempts from 15 patients). The secondary pie chart divides EBVSTs with antigen specificity of greater or less than 100 SFCs per 105 cells that passed the specificity criteria. (D) The frequency of CD3+ T cells and CD3−CD56+ natural killer cells gated on viable lymphocytes (n=68). Graph B is plotted with median and IQR. EBV, Epstein-Barr virus; EBVSTs, EBV-specific T cells; SFC, spot forming cells.
CD45RA depletion of PBMC prior to EBVST initiation reduces NK cells and increases viral-antigen specificity
To enrich memory T cells and remove TN and NK cells, we depleted CD45RA+ cells from the PBMCs of 14 healthy donors prior to stimulation. CD45RA+ cells were reduced from a mean of 55%±4% in whole PBMCs (W-PBMCs) to 1.6%±0.5% in CD45RA-depleted PBMCs (RAD-PBMCs) while NK cells were reduced from a mean of 16%±2% in W-PBMCs to 3%±2% in RAD-PBMCs (online supplemental figures 1A–D). The frequency of CD4+ T cells in RAD-PBMCs was also significantly increased (p=0.004) (online supplemental figures 1E).
Supplemental material
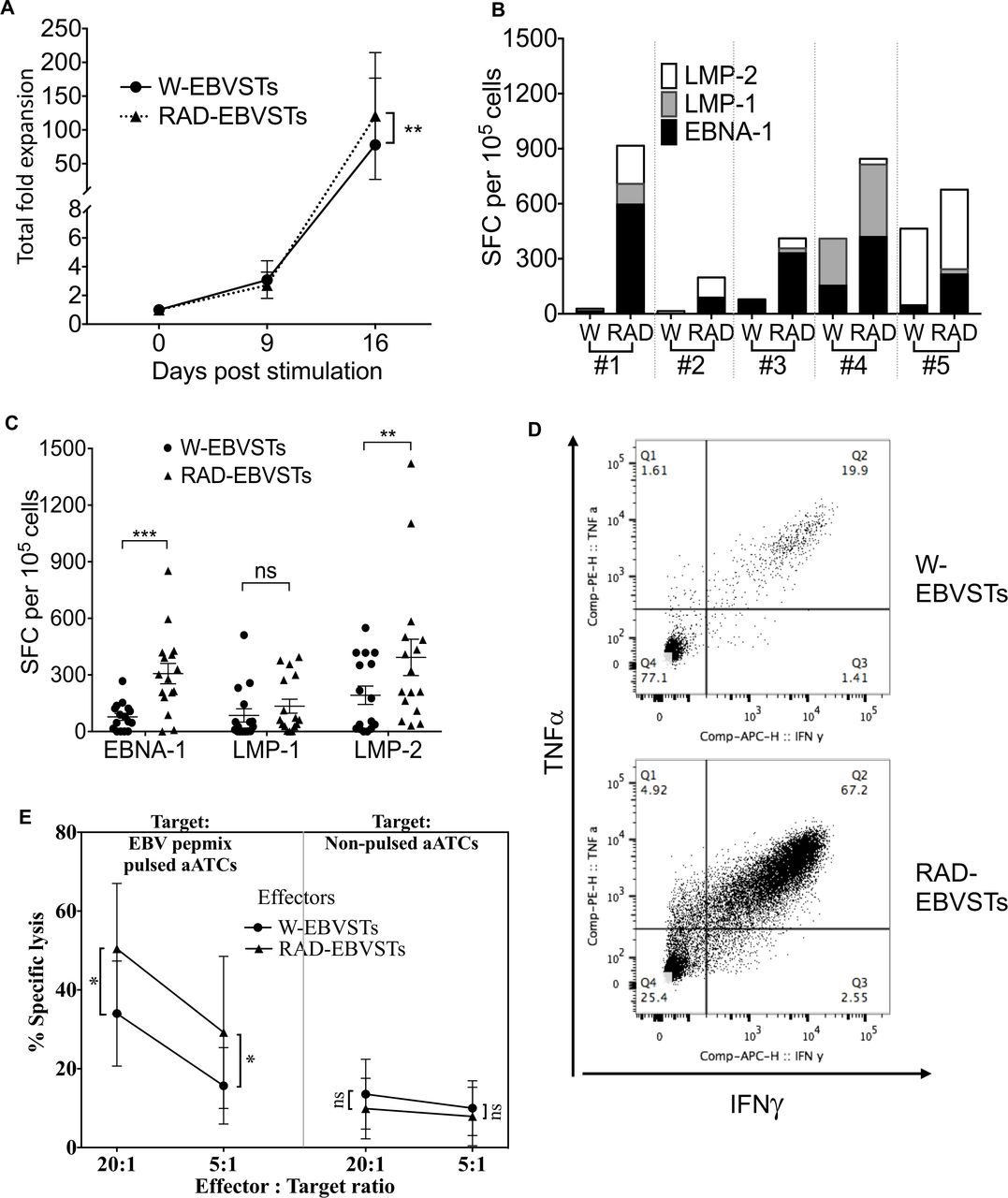
CD45RA depletion increased both the expansion and the T2 antigen specificity of EBVSTs over the 16 days of culture (121±23-fold expansion vs 78±25-fold for W-EBVSTs (p=0.009) (figure 2AB and online supplemental figure 2A p=0.002). The mean frequency of EBV antigen-specific T cells in RAD-EBVSTs was 835 (range 51–2335) IFN-γ SFCs per 105 cells compared with 354 (range 15−1113) in W-EBVSTs (figure 2C). EBV antigen-stimulated RA+ PBMCs expanded relatively poorly and showed lower EBV antigen specificity than W-EBVSTs or RAD-EBVSTs (online supplemental figures 3A,B).
Supplemental material
Supplemental material

Depletion of CD45RA-positive PBMCs increases the antigen specificity of derived EBVSTs. EBVSTs were generated from the W-PBMCs and RAD-PBMCs of 16 healthy EBV seropositive donors using EBV T2 antigen pepmixes in the presence of IL-7 and IL-15. Since monocytes are mostly CD45RA-negative, they provided antigen-presenting cells in both W-PBMCs and RAD-PBMCs. On day 9, the cells were restimulated with an antigen-presenting complex comprizing irradiated, T2 pepmix-pulsed autologous activated T cells (ATCs) as antigen-presenting cells and irradiated co-stimulatory cells, and harvested on day 16 for characterization. (A) shows proliferation data and error bars are shown as mean±SEM for all donors whereas statistical comparisons were determined using paired two-tailed Student’s t-test. p<0.05, (0.12 (ns, non-significant), 0.033(*), 0.002(**), <0.001(***). (B) Shows the EBV antigen specificity of EBVSTs from five representative donors. (C) Shows the specificity of all 16 donors for each T2 antigen. (D) Frequency of IFN-γ and TNF-α secreting CD3+T cells after stimulation with EBV latent antigens (showing one representative of five). (E) Killing of EBV pepmix pulsed and non-pulsed autologous activated T cells by W-EBVSTs and RAD-EBVSTs from nine donors in a 4-hour chromium-release assay. Less than 10% specific 51Cr release from non-pulsed autologous ATCs, was considered negative. Statistical comparisons were determined using paired two-tailed Student’s t-test. p<0.05, (0.12 (ns, non-significant), 0.033(*), 0.002(**), <0.001(***) data shown are plotted as mean±SEM. EBV, Epstein-Barr virus; EBVSTs, EBV-specific T cells; IFN, interferon; IL, interleukin; PBMCs, peripheral blood mononuclear cells; TNF, tumor necrosis factor.
Enhanced antigen-specificity in RAD-EBVSTs was mirrored by an increased frequency of IFN-γ and TNF-α producing polyfunctional T cells as measured in intracellular cytokine assays (figure 2D and online supplemental figure 2B, p=0.013). RAD-EBVSTs also demonstrated a broader target antigen repertoire, as shown for five representative donors (figure 2B). For example, donor 1 RAD-EBVSTs acquired LMP1 and LMP2 specificity in addition to EBNA1; donor 2 acquired LMP2 specificity; and donor 3 acquired LMP1 and LMP2 specificity (figure 2B). RAD-EBVSTs produced greater killing of EBV pepmix pulsed autologous ATCs than W-EBVSTs in 4-hour 51Cr release assays (p=0.011, figure 2E), and contained fewer CD3−CD56+ NK cells (p=0.002, online supplemental figure 2B).
CD45RA depletion of PBMCs also increased the antigen specificity of T cells activated with antigens from other viruses, including adenovirus (AdV) (423±114 SFCs in RAD-AdVSTs compared with 221±58 SFCs in W-AdVSTs; p=0.042), varicella zoster-virus (VZV) (770±370 SFCs in RAD-VZVSTs compared with 335±231 SFCs in W-VZVSTs; p=0.033) (online supplemental figure 2D), CMV and BK virus (not shown). These observations support the use of CD45RA depletion as a general approach to enhance antigen specificity of VST platforms.
Removal of CD45RA+ subsets from PBMCs increases the clonality of derived EBVSTs
To determine if the enhanced antigen specificity of RAD-EBVSTs was reflected in their clonal composition, we compared the clonal repertoires of EBVSTs derived from W-PBMCs, RAD-PBMCs and RA+ PBMCs with each other, and with the PBMC subsets from which they were derived, by sequencing their TCR-Vβ CDR regions, as outlined in online supplemental figure 4A using two healthy donors. As expected, the clonal diversity of EBVSTs was reduced compared with that of PBMCs but remained surprisingly broad despite 16 days of EBV antigen-driven expansion, although the clonal diversity of RAD-EBVSTs was lower than that of W-EBVSTs (figure 3A,B for donor 1, and online supplemental figures 4B, C for donor 2). We observed unexpected differences in the 50 most abundant clonotypes of W-EBVSTs and RAD-EBVSTs, with only 9 shared clonotypes in donor 1, and 17 in donor 2 (figure 3C) a more specific clonal outgrowth in RAD-EBVSTs. Indeed, the top 50 clonotypes in W-EBVST had greater identity with those in W-PBMCs, illusrating a less selective clonal outgrowth than in RAD-EBVSTs that shared few top 50 clonotypes with RAD-PBMCs (6 shared clonotypes compared with 20 in W-EBVSTs) (figure 3D and online supplemental figure 4D). Notably, W-EBVSTs also shared a greater clonal identity with RA+ EBVST (online supplemental figure 4E) that showed little to no EBV specificity as shown in online supplemental figures 3A,B. The top RAD-EBVSTs clonotypes were larger than those in W-EBVSTs (figure 3 and online supplemental table 1), which combined with reduced diversity, produced a trend to greater clonality as quantified in the Simpson clonality plot (online supplemental figure 4F, with a calculated Shannon diversity of 8.6 and 8.7 for W-EBVSTs and 6.9 and 8.3 for RAD-EBVSTs, donor 1 and 2, respectively). This figure also illustrates the surprizing finding that W-EBVSTs have lower clonality than W-PBMCs, due in part to the higher-frequency clonotypes in W-PBMCs (figure 3D and online supplemental figure 4F).
Supplemental material
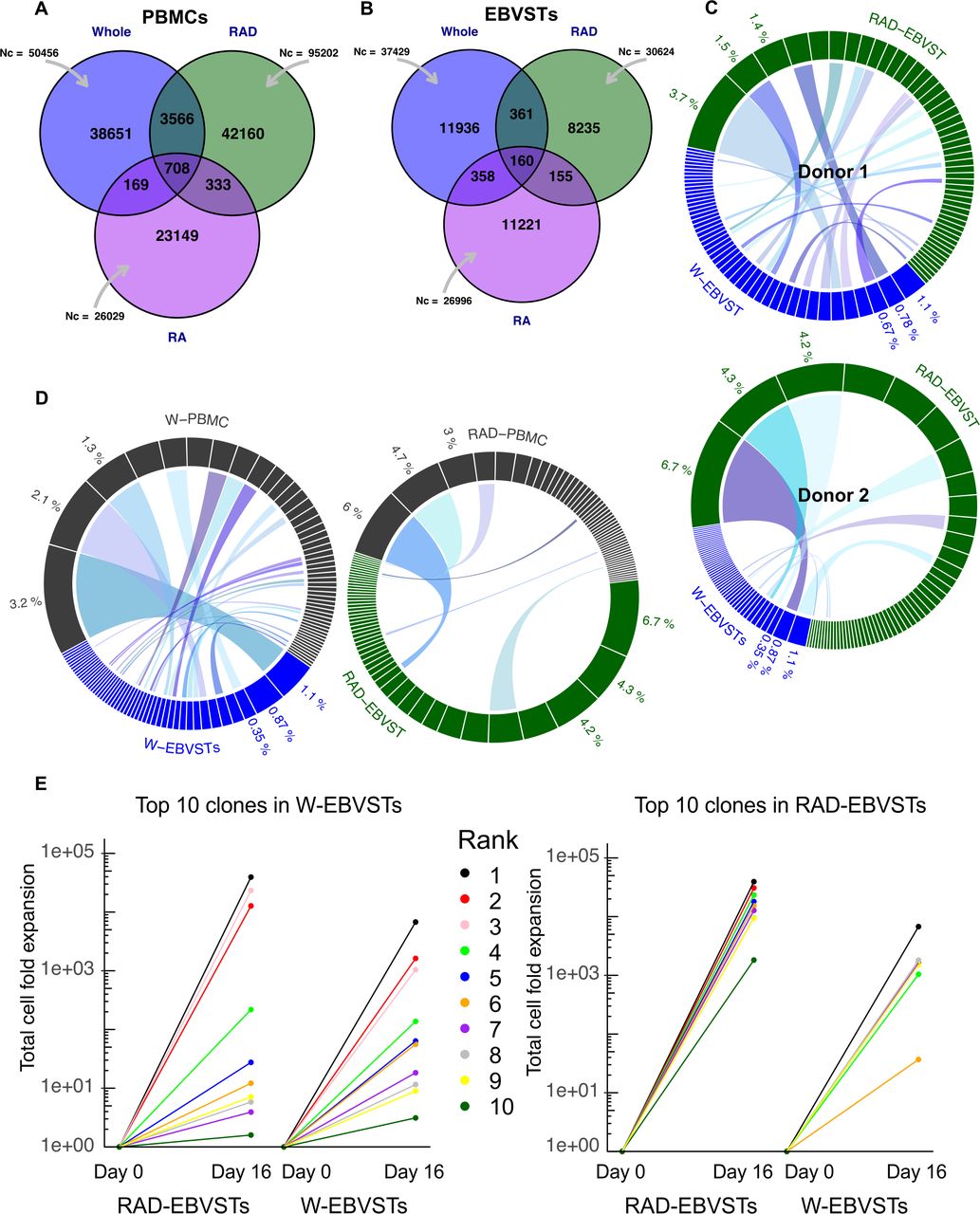
Increased clonality of EBVSTs derived from CD45RA depleted PBMCs. TCR Vβ sequencing was performed on W-PBMCs, RAD-PBMCs and RA+ PBMCs, and their derived EBVSTs. (A, B) Venn diagram showing the total number of functional templates sequenced (Nc), and the number of unique and shared TCR sequences between W-EBVSTs, RAD-EBVSTs and RA+ EBVSTs for donor 1. (C) Circos plot comparing the 50 highest frequency clonotypes of W- and RAD-EBVSTs from donors 1 and 2. Ribbons connect clonotypes with identical TCR Vβ sequences. (D) Compares the 50 most abundant clonotypes in W- and RAD-PBMCS with their derived EBVSTs (W-PBMCs (gray), W-EBVSTs (blue), RAD-PBMCs (gray), and RAD-EBVSTs (green) subsets. Ribbons identify shared TCR Vβ sequences. (E) Shows the total fold expansion of the 10 most abundant clonotypes in W-EBVSTs in both W- and RAD-EBVSTs for donor 1, calculated by multiplying the % clonal frequency in PBMCs with that in EBVSTs multiplied by the total fold expansion of EBVSTs. Each color represents a single clonotype, and those shared between W and RAD-EBVSTs are represented by the same color. (F) Shows the fold expansion of the top 10 RAD-EBVST clonotypes in W- and RAD-EBVSTs. EBV, Epstein-Barr virus; EBVSTs, EBV-specific T cells; IFN, interferon; IL, interleukin; PBMCs, peripheral blood mononuclear cells; TCR, T-cell receptor.
The larger size of RAD-EBVST clonotypes compared with W-EBVST clonotypes (figure 3C and online supplemental table 1) was explained by their greater expansion after stimulation as illustrated in figure 3E, which shows the total fold expansion of the top 10 clonotypes in RAD-EBVSTs and W-EBVSTs (mean and median of 18,428-fold and 16,716-fold in RAD-EBVSTs compared with 973-fold and 59-fold in W-EBVSTs). The top 10 RAD-EBVST clonotypes expanded relatively poorly, or not at all in W-EBVST (figure 3C,E). This could not be explained by their absence from W-PBMCs, since 8 of the top 10 RAD-EBVST clonotypes were detected in W-PBMCs (online supplemental table 1). The top 10 W-EBVST clonotypes were all detected in RAD-EBVSTs, though 6 of these expanded less in RAD-EBVSTs than in W-EBVSTs (figure 3E and online supplemental table 1).
We then compared the relative changes in frequency from PBMCs to EBVSTs of the top 10 clonotypes in each EBVST subset to their change in frequency in each other subset (online supplemental figures 5A,B). These violin plots illustrate the far greater expansion of the top RAD-EBVST clonotypes compared with the top clonotypes in W- and RA+ subsets, and show that a majority of the top W-EBVSTs and RA+ EBVSTs clonotypes decreased in frequency in RAD-EBVST, while showing remarkably little change in frequency from that in their respective PBMCs, (online supplemental figure 5A (b-c) and B (e-f). Our clonal analysis is limited by the process of TCR detection, which is highly stochastic and prone to variation, and because we have analyzed only two healthy donors. However, data from both donors illustrate the greater and more selective expansion of clonotypes in RAD-EBVSTs compared with W-EBVSTs, indicating that CD45RA+ cells in W-PBMCs inhibit the clonal outgrowth of antigen-stimulated memory T cells.
Supplemental material
TNs are the source of inhibition of EBVSTs within the CD45RA+ PBMC fraction
To identify the source of inhibition of clonal outgrowth within the CD45RA+ fraction of PBMCs, we examined the inhibitory activity of CD45RA+ subsets. Two potentially immunosuppressive CD45RA+ subsets are natural T-regulatory cells (CD4+ CD25hi) and NK cells (CD3– CD56+). However, depletion of these prior to EBVST generation did not improve EBVST specificity, suggesting that neither was the source of suppression (online supplemental file 6A). To determine if the suppressive components resided in CD3+ or CD3− subsets of the CD45RA+ fraction, we added them separately to RAD-PBMCs prior to EBVST stimulation. Only the CD45RA+ CD3+ subset reduced the frequency of EBVSTs (figure 4A).
Supplemental material

Effects of CD45RA+ subsets on RAD-EBVSTs. EBVSTs were generated from the PBMC subsets indicated and their antigen specificity, measured in enzyme-linked immunospot assays as # cells that produce interferon-γ per 105 cells in response to EBV pepmixes on day 16. (A) CD45RA+ CD3+ (CD3+) and CD45RA+ CD3− (CD3−) fractions from three donors were added back to RAD-PBMCs before EBVST generation and their antigen specificities are compared with that of W-EBVSTs, RAD-EBVSTs, and RA+ EBVSTs. (B and C) CD45RA+ PBMCs from six EBV seropositive healthy donor buffy coats were sorted into TEMRAs (CD45RA+, CCR7−), TSCMs (CD45RA+, CCR7+, CD95+) and TN (CD45RA+, CCR7+, CD95−) using fluorescence-activated cell sorting and added back to RAD-PBMCs before EBV antigen stimulation. CD45RA+ PBMCs were generated by recombining the sorted TSCM (TS), TNAIVE (TN) and TEMRA (tE) subsets. This reconstituted CD45RA+ subset was added to RAD-PBMCs to generate W-PBMCs. (B) Fold expansion of EBVSTs generated from W-, RAD-, CD45RA+ subsets of RAD-PBMCs in the presence of the sorted CD45RA subsets (n=6). (C) Antigen specificity of EBVSTs for donors 1, 3, 4, and 6. (Donors 2 and 5 were not included in the analysis as the difference in antigen specificity between W-EBVSTs and RAD-EBVSTs was insignificant). Data shown are plotted as mean±SEM. EBV, Epstein-Barr virus; EBVSTs, EBV-specific T cells; PBMCs, peripheral blood mononuclear cells; SFC, spot forming cell; TEMRA, effector memory; TN, naïve T cell; TSCM, stem cell memory.
We then isolated CD45RA+ T-cell subsets (TEMRAs, TSCMs, and TN) based on their commonly associated phenotypic cell-surface molecules, CCR7 and CD95.27 30–32 38 First, we examined the expansion and specificity of EBVSTs derived from the isolated CD45RA+ subsets alone and combined with each other. T2 antigen stimulated TSCM showed the greatest expansion (~200-fold, not significant, online supplemental figures 6B,C). EBVSTs derived from TSCMs and TEMRAs, alone or combined demonstrated higher antigen specificity than those derived from TN, but lower than W-EBVSTs or RAD-EBVSTs (online supplemental figure 6C), but when combined with TN, their specificity was reduced, suggesting that TN subsets could inhibit the outgrowth and/or function of antigen-stimulated memory EBVSTs, and that the majority of EBV specificity observed in CD45RA+ EBVSTs is derived from TEMRAs and TSCMs.
To examine the effects of the isolated CD3+ CD45RA+ subsets on the proliferation and antigen specificity of RAD-EBVSTs, we added an equal number of each to RAD-PBMCs prior to EBV antigen stimulation. The addition of any CD3+ CD45RA+ subset/s inhibited the expansion of RAD-EBVSTs over the 16 days of culture (figure 4B), but only TN cells alone or in combination with TSCM or TEMRA decreased the antigen-specificity of RAD-EBVSTs from three of the four donors. (figure 4C). These experiments show that only TN PBMCs inhibit the proliferation and antigen specificity of EBVSTs.
RAD-EBVSTs maintain antigen specificity and proliferation
To investigate our concern that removal of the CD45RA+ CCR7+ CD95+ TSCM population from RAD-PBMC might remove a crucial long-lived EBVST population leaving more differentiated EBVSTs with reduced longevity,30 32 39 we investigated the effects of CD45RA depletion on the long-term antigen specificity proliferation and effector memory phenotype of RAD-EBVSTs. We observed no significant differences in the frequency of central memory-like cells (CD45RO+, CD62L+, CCR7+, and CD45RO+ CD62L+) populations on day 16 of culture (figure 5A), although a significantly higher proportion of RAD-EBVSTs were CD3+ CD4+ T cells, 50%±23% versus 32%±23% for W-PBMCs (p=0.002, figure 5B). The expression of LAG-3, TIM-3, KLRG-1, molecules associated with inhibitory transcriptional programming and T-cell exhaustion40 were reduced in RAD-EBVSTs compared with W-EBVSTs (p=0.005; online supplemental figure 7B,C).
Supplemental material
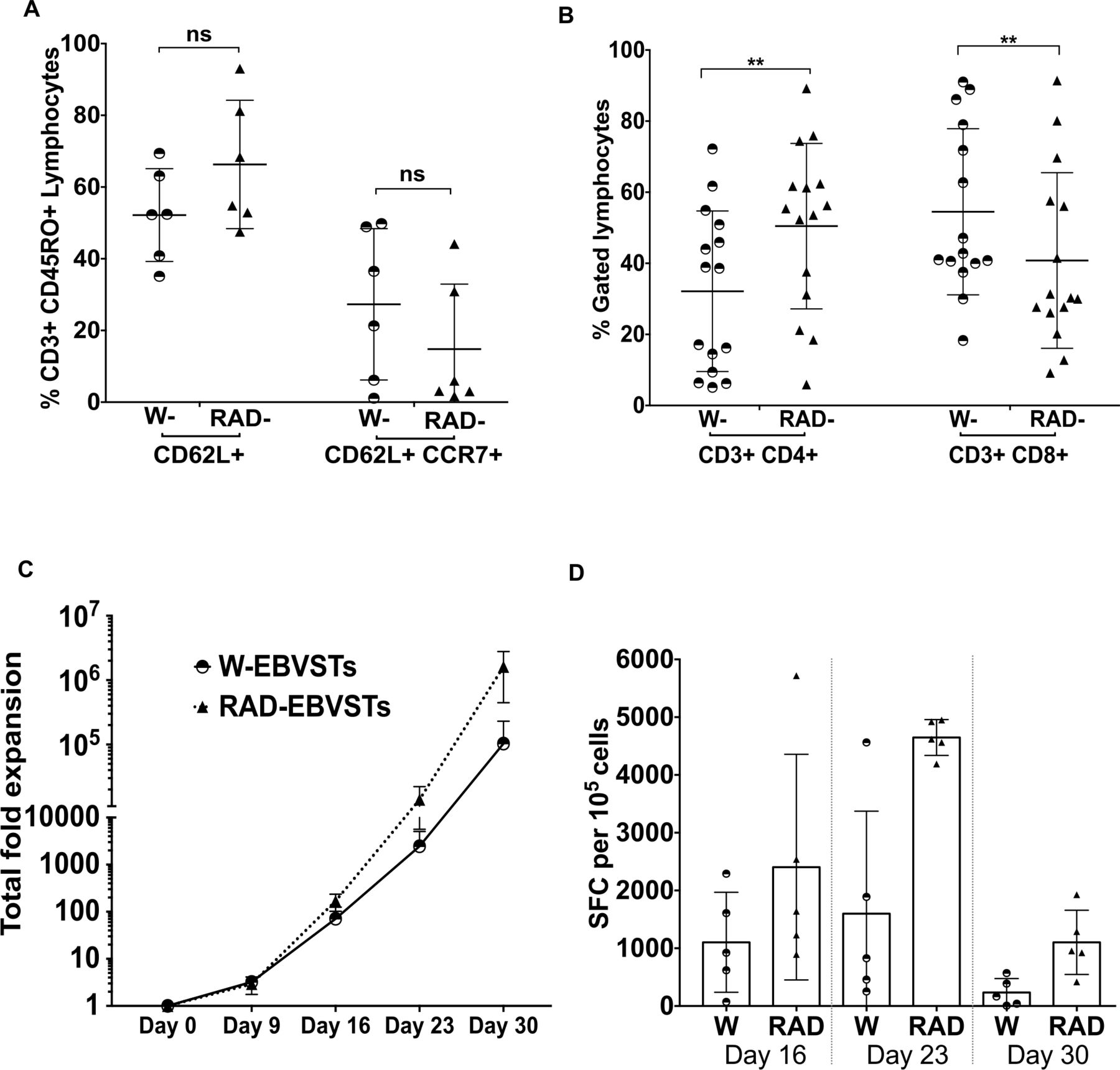
Differentiation, phenotype, and longevity of RAD-EBVSTs. EBVSTs were phenotyped on day 16 of culture for (A) TCM differentiation markers CD45RO+ CD62L+ and CD45RO+CCR7+, CD62L+, n=6 and (B) CD4 and CD8 ratios (n=15). To compare their proliferation (C) and antigen specificity (D) over a longer time period, we restimulated the cell on days 16, and 23. Antigen specificity of EBVSTs was evaluated at the end of each antigen stimulation by enzyme-linked immunospot assay. (Statistical comparisons were determined by paired two-tailed Student’s t-test. p<0.05, (0.12 (ns, non-significant), 0.033(*), 0.002(**), <0.001(***). Data shown are plotted as mean±SEM. EBVSTs, Epstein-Barr virus-specific T cells; SFC, spot forming cell; TCM, Central memory T-cells.
To assess longer-term persistence, we cultured W-EBVST and RAD-EBVST beyond 30 days using weekly antigen restimulation. Although both populations continued to expand, RAD-EBVSTs showed ~one log higher expansion (figure 5C). The frequency of antigen-specific T cells increased until the third stimulation, then decreased after the fourth (S4), However, RAD-EBVSTs sustained their advantage over W-PBMCs with 1103±557 IFN-γ SFCs compared with 234±240 SFC per 105 cells for W-EBVSTs after S4 (figure 5D). In summary, CD45RA depletion improves in vitro persistence and antigen specificity, without adverse phenotypic effects.
CD45RA depletion enhances the in vivo antitumor activity of EBVSTs
To compare the antitumor activity of W-EBVSTs and RAD-EBVSTs in our murine model, we implanted LCLs subcutaneously into NSG mice. Following tumor engraftment, we adoptively transferred autologous W-EBVSTs or RAD-EBVSTs and measured antitumor efficacy and T-cell persistence. Although there was no significant difference in T-cell bioluminescence between mice receiving W-EBVSTs or RAD-EBVSTs (figure 6A), tumors responded more rapidly in mice receiving RAD-EBVSTs and reached a volume of only 16.17±18.31 mm3 by day 11, compared with 136.35±41.85 mm3 (p<0.001) in those receiving W-EBVSTs (figure 6B). Both groups of mice ultimately cleared the tumor but on day 11 in response to RAD-EBVSTs compared with day 35 for W-EBVSTs; (figure 6B). This difference was reproduced in experiments in which the tumor cells expressed FF-luc. Figure 6C,D show tumor bioluminescence over time, with tumor volume reaching only 514±91 mm3 on day 12 after RAD-EBVST infusion compared with 1376±200 mm3 after W-EBVSTs; p<0.001) (figure 6E). Tumor bioluminescence in the left upper-limb region, consistent with the metastatic spread, was seen only in control mice and those receiving W-EBVSTs (figure 6C).

EBVSTs generated from RAD-PBMCs have more rapid in vivo antitumor activity than W-EBVSTs. EBV-transformed human B lymphoblastoid cell-line (LCL) tumors (2.5×106–3×106) were suspended in 200 µL Matrigel and injected subcutaneously into NSG mice. Eight days later, when tumors were palpable, autologous W-EBVSTs or RAD-EBVSTs were infused by tail vein injection. The antitumor efficacy of EBVSTs from two healthy donors was measured in two separate experiments, in which either the EBVSTs or the tumor cells were rendered bioluminescent by expression of FF-luc from a retrovirus vector. In the first experiment, mice received 5×106 FF-luc expressing EBVSTs, (A) Bioluminescence imaging of FF-luc expressing EBVSTs (n=6/group) at the time points indicated after EBVST injection. (B) Tumor volume as measured using calipers. (n=6/group). In the second experiment, tumors were labeled with FF-luc and mice received 1×106 EBVSTs (n=5 per group). (C) Bioluminescence imaging of FF-luc expressing EBV-LCL tumor. Mice that did not receive EBVSTs (controls) were euthanized on day 19 due to high tumor volume. (D) Graphical analysis of total tumor radiance flux in each group. (E) Graphical representation of total tumor volume as measured using calipers. Multiple t-test analysis p<0.05, (0.12 (ns, non-significant), 0.033(*), 0.002(**), <0.001(***). Data shown are plotted as mean±SD. Statistical significance was determined using the Holm-Sidak method, with alpha=0.05. Each row was analyzed individually, without assuming a consistent SD. EBVSTs, Epstein-Barr virus-specific T cells; FF-luc, Firefly luciferase.
Depletion of CD45RA positive lymphocytes restores T2 antigen responsiveness in patient with lymphoma T cells
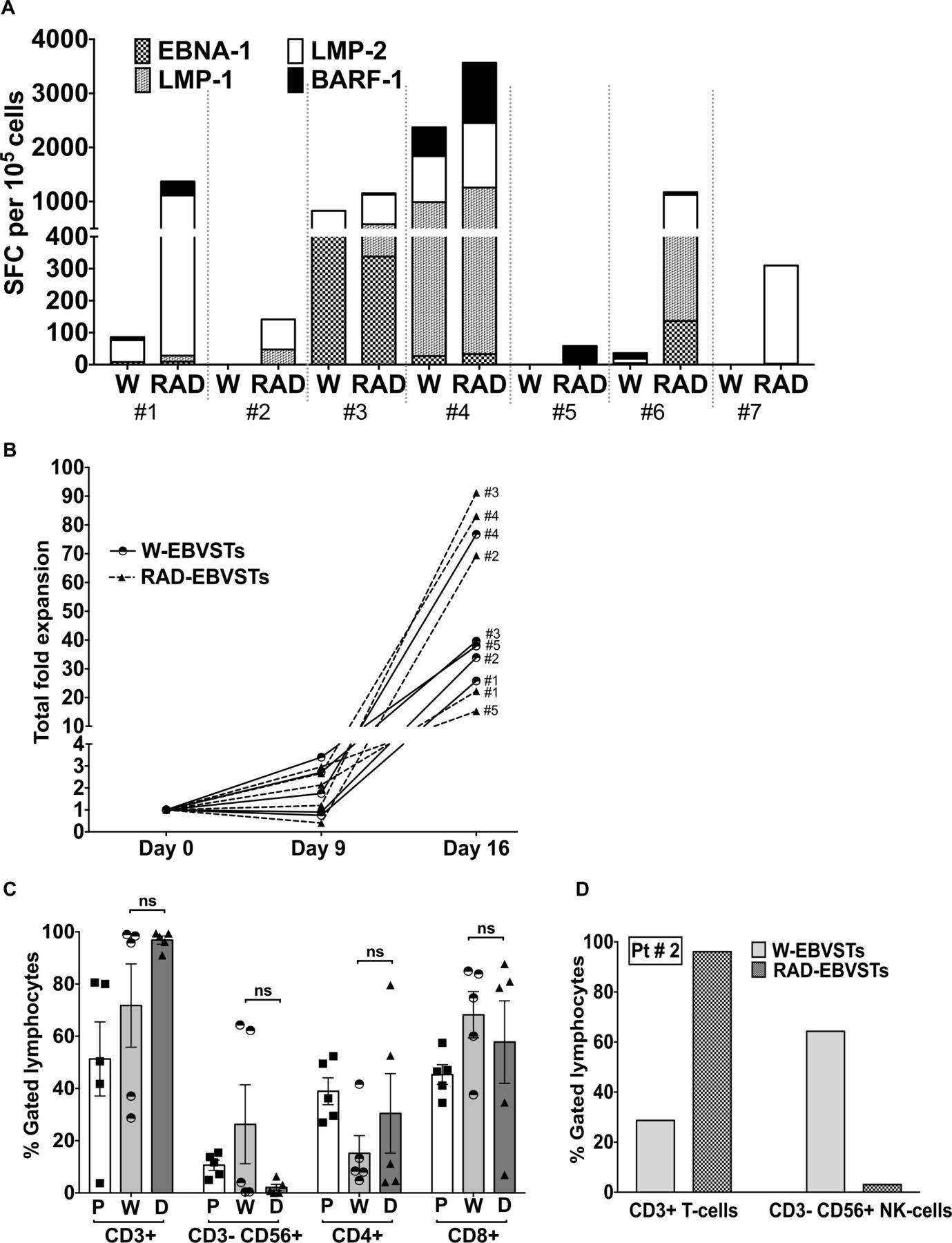
Finally, we determined if CD45RA depletion of PBMCs would facilitate the generation of more potent antigen-specific T cells from patients with EBV+ lymphoma. RAD-EBVSTs from 7/7 patients showed a substantially increased frequency of EBV T2 antigen-specific T cells (figure 7A), even in three patients whose W-PBMCs failed to respond: patient #2: 142 SFCs versus 0 SFCs; patient #5: 58 versus 0 SFCs; and patient #7: 310 versus 0 SFCs (RAD-EBVSTs vs W-EBVSTs) (figure 7A). The fold expansion of EBVSTs from three of five patients was also increased by CD45RA depletion (figure 7B). As observed in healthy donors (online supplemental figure 2C), CD45RA depletion reduced the frequency of NK cells in EBVST for all patients (figure 7C). Though not statistically significant overall, this decrease was pronounced in patients with a high frequency of NK cells as shown for patient 2 (Pt#2, 3.11% NK cells in RAD-EBVSTs vs 64.32% in W-EBVSTs; figure 7D). Thus, CD45RA-depletion overcame major challenges to the manufacturing of EBVSTs from patients with lymphoma and restored antigen responsiveness in T cells that failed to respond within the W-PBMC fraction.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Depletion of CD45RA positive lymphocytes restores EBV antigen responsiveness in lymphoma patient PBMCs. EBVSTs were generated from W-PBMCs or RAD-PBMCs from the cryopreserved PBMCs of seven patients with EBV+ lymphoma who had been procured for our clinical trial (NCT 01555892), including three whose EBVSTs had failed the manufacturing release criteria of >20 IFN-γ SFCs per 105 cells, and two with low antigen specificity. (A) Frequency of IFN-γ SFCs per 105 EBVSTs in response to EBNA-1, LMP-1, LMP-2, and BARF-1 pepmixes on day 16. Each patient is divided by a dotted line and denoted by #(Number) (B) Total fold expansion of W-EBVSTs and RAD-EBVSTs from day 0 to 16. (C) T-cell and NK-cell phenotype of W-PBMCs on day 0 (P) before antigen stimulation, and of W-EBVSTs (W) and RAD-EBVSTs (D) on day 16 (n=5). Data shown are plotted as mean±SEM (D) CD3+T cells and CD3–CD56+NK cells in W-EBVSTs and RAD-EBVSTs from pt #2. EBV, Epstein-Barr virus; EBVSTs, EBV-specific T cells; IFN, interferon; NK, natural killer; PBMC, peripheral blood mononuclear cell; SFC, spot forming cell.
Discussion
VSTs provide a safe and effective therapy for virus-associated diseases in immunocompromised patients and can be effective against viral malignancies. Increasing the frequency and repertoire of antigen-specific T cells in VST products increases their preclinical potency and should enhance their clinical efficacy. Using EBVSTs as our model, we showed that removal of CD45RA-negative T cells from PBMCs prior to initiation, robustly and reproducibly improved the rate of EBVST expansion, the frequency of antigen-specific T cells, and the number of EBV antigens recognized, while decreasing the expression of exhaustion markers. TCR analysis revealed a selective expansion in RAD-EBVSTs, of clonotypes that expanded poorly or not at all in W-EBVSTs, suggesting that the CD45RA expressing fraction of PBMCs inhibits the antigen-driven outgrowth of memory clonotypes. This inhibitory activity mapped to the naïve fraction (TN) of PBMCs since other CD45RA+ PBMCs such as TSCM, TEMRA, and CD3-negative PBMCs had no inhibitory activity. RAD-EBVSTs produced more rapid control of autologous EBV+ lymphomas in our murine xenograft model and CD45RA depletion of PBMCs enabled the expansion of EBVSTs from patients with EBV+ lymphoma whose PBMCs were otherwise refractory to stimulation.
Despite improvements in manufacturing,33 41 the generation of VSTs with high virus specificity is frequently difficult due to the competitive outgrowth of T cells and NK cells that lack virus specificity. This is particularly true for tumor virus-specific T cells in patients with cancer, likely because their tumors exert an inhibitory effect.25 42 In these patients, the cytokines and improved media formulations used to increase the success and speed of manufacture, also support the expansion of bystander T cells that did not receive cognate antigen stimulation.
While not expanding greatly in EBVSTs from healthy donors, NK cells comprised up to 90% of EBVSTs from the patients with lymphoma in our clinical trial. Although NK cells may have some activity against lymphoma, they poorly survive cryopreservation,43 hence are unlikely to contribute greatly to the antitumor effect. Since circulating NK cells express CD45RA,44 this problem was eliminated by CD45RA depletion. We initially hypothesized that outgrowth of TNs in response to cytokines IL-7 and IL-1545–47 was responsible for the high frequency of T cells lacking EBV-specificity in W-EBVSTs. However, the relatively low expansion of CD45RA+ EBVSTs compared with W-EBVSTs and RAD-EBVSTs in response to antigen stimulation, as well as the maintenance in W-EBVSTs of large clonotypes that dominated PBMCs, suggests that the bulk of the bystander cells are unstimulated memory T cells lacking EBV specificity.
RAD-PBMCs showed an increased CD4:CD8 ratio that was carried into the derived EBVSTs. Since W-EBVSTs were often dominated by CD8+T cells, RAD-EBVSTs had a more balanced phenotype. CD4+T cells play a critical role in sustained antitumor efficacy,1 16 48 and both CD4 and CD8 T cells are cytotoxic to EBV-infected target cells.49 Further, since major histocompatibility complex class-I downregulation is common in lymphoma, the recognition of antigen presented on HLA class II molecules by cytotoxic CD4+T cells is likely beneficial, resulting in IFN-γ production that in turn upregulates HLA class I molecules.50
CD45RA depletion removes not only TNs but also terminally differentiated TEMRAs, potentially suppressive Tregs,51 and long-lived TSCM while leaving a majority of antigen-presenting monocytes in RAD-PBMCs. Removal of TSCMs, the earliest form of T-cell memory with the greatest capacity for self-renewal and proliferation was a concern.30 32 39 However, it has been reported that TSCMs do not expand unless isolated from PBMCs, so they were unlikely to contribute to EBVSTs from W-PBMCs. This phenomenon was clearly illustrated in () online supplemental file 6, in which the high proliferation and antigen-specificity of EBVSTs derived from isolated TSCM subsets was dramatically reduced in the presence of TN or any other subset. Notably, TSCMs did not continue to proliferate within antigen-stimulated RA+ EBVSTs, again supporting our suggestion that TNs inhibit memory T-cell expansion. These observations diminished our concerns that depleting antigen-specific TSCMs could contribute to decreased persistence. Our concern that removal of TSCMs might drive an exhausted and differentiated phenotype39 40 45 was also unfounded, since expression of the early differentiation markers, CD62L and CCR7, were unchanged, while exhaustion markers including TIM-3, KLRG-1 and LAG-3 were decreased and RAD-EBVSTs proliferated to a greater extent over 30 days and better maintained their specificity. While isolated TSCMs might indeed be better starting material in translation to the clinic, we are constrained by the lack of clinical grade antibodies for selection.
The enhanced outgrowth of EBV-specific memory T cells in the absence of CD45RA+ T cells has three possible, non-mutually exclusive explanations. First, EBV-specific memory T cells are selectively enriched in the RAD fraction of PBMC. Second, a more competitive outgrowth of non-EBV-specific bystander cells in W-EBVSTs, and third, a component of CD45RA+ PBMCs inhibits TCR-activated EBV-specific memory T-cell outgrowth. Neither memory T-cell enrichment nor competitive outgrowth of non-specific T cells in W-EBVSTs can fully explain the reduced expansion within W-PBMCs of the clonotypes that dominated RAD-EBVSTs. The third possibility is supported by the observation that EBVSTs from some patients with lymphoma could be expanded only if the RA+ fraction was removed prior to stimulation, and by the greater fold expansion of (likely EBV-specific) clonotypes in RAD-EBVSTs. The low in vitro expansion of CD45RA+ EBVSTs suggested that the CD45RA+ fraction was not the main source of bystander outgrowth, pointing rather to an inhibitory effect.
Clonal analysis of W-PBMC, RAD-PBMC, and RA+ PBMC fractions and their derived EBVSTs by TCR sequencing provided insights into the greater antigen-specificity of RAD-EBVSTs. The more selective expansion of clonotypes that expanded poorly or not at all in W-EBVSTs coupled with their greater antigen specificity also supported the inhibition theory. W-EBVSTs demonstrated less selective expansion with a majority of the top-ranking clonotypes in W-PBMCs constituting top-ranking clonotypes in their derived EBVSTs.
Observations made from TCR sequencing must be interpreted with caution due to sampling limitations, the inclusion of only the TCR Vβ chain sequencing, the purity of cell sorting, and because we do not know the antigen specificity of each clonotype. Although we refer to our EBV antigen-stimulated cells as EBVSTs, they are not 100% EBV-specific and non-EBV specific bystander cells are present in the cultures. In particular, we cannot make conclusions about the fate of TNs most of which circulate with a such low frequency that it is improbable that members of the same naïve clonotype would be present in the PBMC aliquots used for TCR analysis and those used for EBVST expansion.52 53 In our study, clonotypes shared between different PBMC subsets likely represent memory T cells, limited to TCM and TEM within RAD-PBMCs and TSCM and TEMRA within RA+PBMCs, although, recent studies have attributed a small proportion of abundant T-cell clones to the naïve repertoire.35 52 54
Interactions between T-cell subsets have been shown to affect T-cell differentiation, proliferation, and metabolism, and several groups have reported that memory T cells can influence TNs.45 55 56 Klebanoff et al showed that non-apoptotic Fas—FasL interaction between naïve and antigen-experienced memory cells induced precocious differentiation of activated TN cells resulting in impaired overall antitumor immunity.56 The inhibitory activity we observed resided in the CD3+ fraction of CD45RA+ PBMCs, and further subset separation implicated the TN subset. Since an equal number of cells from each sorted subset was added to RAD-PBMCs, we can rule out inhibitory effects resulting from the dilution of EBVSTs in the starting RAD-PBMCs. Although we did not identify the mechanism of inhibition, we did investigate the duration of sensitivity of RAD-PBMCs to CD45RA+ subsets, and found that RAD-PBMCs remained sensitive to the effects of CD45RA+ cells for at least 3 days after antigen stimulation in terms of antigen specificity, while the inhibitory effect on proliferation was lost after 3 days (preliminary data not shown). We speculate that inhibition of memory clonotypes by TNs may represent a physiological strategy to ensure the polyclonal response of TNs to new pathogens, by preventing the oligoclonal outgrowth of cross-reactive memory T cells lacking protective function. Further studies to evaluate this clonal inhibition of antigen-specific memory T cells are planned.
Our most clinically relevant finding was that removal of the CD45RA subset from lymphoma patient PBMCs enabled the expansion of EBVSTs that were otherwise unresponsive to stimulation. The enhanced proliferation of EBVSTs after CD45RA-depletion and the elimination of dendritic cells from the manufacturing process more than compensates for the ~15–20% cell loss of memory T cells during the depletion process. It had been proposed that in patient with lymphoma, T2-EBVSTs circulate with low frequency due to their elimination within the tumor.57 58 Our results suggest that they do circulate but with decreased ability to respond to stimulation. Removal of CD45RA+ T cells from PBMCs was beneficial for the specificity of T cells for a range of viruses including AdVs, BKV, and VZV, and can be achieved in a good manufacturing practice compliant manner. This may be of particular importance for the generation of T cells targeting other tumor viruses, like papillomaviruses, HTLV1, Kaposi’s sarcoma virus, and Merkel cell polyomavirus, and may also extend to memory T cells specific for non-viral tumor antigens in patients with cancer. The reduction by CD45RA depletion of TNs that are the major source of alloreactive T cells that cause graft versus host disease in the allogeneic setting, may also provide a safer platform for allogeneic T-cell therapies targeting tumors not only via their native TCRs, but also via CARs.59–61
In summary, we have established a clinically feasible platform to improve the potency of VSTs. Based on our findings, we have initiated three new clinical trials using CD45RA depleted PBMCs to manufacture VSTs for phase I/II trials; autologous EBVSTs for EBV-positive lymphoma (NCT01555892), allogeneic, ‘off-the-shelf’ CD30.CAR-EBVSTs in patients with relapsed or refractory CD30-positive lymphomas (NCT04288726) and multivirus-specific T cells for the treatment of virus infections after HSCT (NCT04013802).
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are included in the article or uploaded as supplementary information. Any other relevant data is available upon reasonable request to crooney@bcm.edu and sandhyas@bcm.edu. Any specific data analysis codes used for TCR sequencing data analysis can be available upon reasonable request to u235274@bcm.edu.
Ethics statements
Patient consent for publication
Ethics approval
All the data collected from human specimens are collected in accordance with informed consent using Baylor College of Medicine IRB-approved protocols (H7634, H7666, and H29617). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We would like to acknowledge members of the GMP and GLP group at the Center for Cell and Gene Therapy, Baylor College of Medicine, Houston, Texas, USA, for their help with organizing patient samples used for this study. We would also like to acknowledge Adaptive Biotechnologies for their help in TCR immunosequencing study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @me.sandhya
Correction notice This article has been corrected since it was first published. The spelling of Michael Schmueck-Henneresse in the author list has been corrected.
Contributors SS and CMR conceptualized the project, designed the research and experimental methodologies, and wrote the original draft of the paper. MS-H assisted with the project conceptualization. SS conducted the experimental investigation and designs. SS performed a majority of in-vitro and in-vivo studies. NUM assisted with the in-vitro studies and NUM, KSP, and TS assisted in in-vivo studies. BM and HZ helped with the clinical samples. SS conducted the data analysis and generated the associated figures. MW conducted the TCR immunosequencing data analysis and related figures. SS, CMR and HEH secured funding. SS and CMR wrote the initial draft, and SS, MS-H, TS, CMR, HEH, and MKB contributed to reviewing and editing the paper. SS and CMR performed the reviewers’ revision edits. CMR is the guarantor for this article.
Funding This work was supported by: CPRIT RP160283 - Baylor College of Medicine Comprehensive Cancer Training Program, NIH-NCI P50 CA126752, NIH-NLBI-HHSN268201600015I, American Society of Gene and Cell Therapy (ASGCT) Career Development Award 2019, Alex’s Lemonade Foundation Reach Award, A SCOR 7001-19 from the Leukemia and Lymphoma Society, and an SRA from Tessa Therapeutics, Singapore.
eCompeting interests SS and CMR are co-inventors of this approach of CD45RA depletion which has been patented and licensed to Tessa Therapeutics. MKB, and CMR have equity in Allovir, Marker Therapeutics and have served on the advisory boards of Tessa Therapeutics and Marker. MKB has equity in Tessa Therapeutics and has served on advisory boards for Walking Fish Therapeutics, CellGenix GmbH, Abintus, Allogene, Bellicum Pharmaceuticals, Bluebird Bio, Athenex, Memgen, Turnstone Biologics, Coya Therapeutics, TScan Therapeutics, Onkimmune, Poseida Therapeutics, AlloVir. CMR has received research support from Tessa Therapeutics. HEH has equity in AlloVir and Marker Therapeutics, has served on advisory boards for Tessa Therapeutics, Novartis, Gilead, GSK, Kiadis, and Fresh Wind Biotechnologies, and received research support from Tessa Therapeutics and Kuur Therapeutics.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.