Article Text

Abstract
In the past decade, treatments targeting the immune system have revolutionized the cancer treatment field. Therapies such as immune checkpoint inhibitors have been approved as first-line treatment in a variety of solid tumors such as melanoma and non-small cell lung cancer while other therapies, for instance, chimeric antigen receptor (CAR) lymphocyte transfer therapies, are still in development. Although promising results are obtained in a small subset of patients, overall clinical efficacy of most immunotherapeutics is limited due to intertumoral heterogeneity and therapy resistance. Therefore, prediction of patient-specific responses would be of great value for efficient use of costly immunotherapeutic drugs as well as better outcomes. Because many immunotherapeutics operate by enhancing the interaction and/or recognition of malignant target cells by T cells, in vitro cultures using the combination of these cells derived from the same patient hold great promise to predict drug efficacy in a personalized fashion. The use of two-dimensional cancer cell lines for such cultures is unreliable due to altered phenotypical behavior of cells when compared with the in vivo situation. Three-dimensional tumor-derived organoids, better mimic in vivo tissue and are deemed a more realistic approach to study the complex tumor–immune interactions. In this review, we present an overview of the development of patient-specific tumor organoid-immune co-culture models to study the tumor-specific immune interactions and their possible therapeutic infringement. We also discuss applications of these models which advance personalized therapy efficacy and understanding the tumor microenvironment such as: (1) Screening for efficacy of immune checkpoint inhibition and CAR therapy screening in a personalized manner. (2) Generation of tumor reactive lymphocytes for adoptive cell transfer therapies. (3) Studying tumor–immune interactions to detect cell-specific roles in tumor progression and remission. Overall, these onco-immune co-cultures might hold a promising future toward developing patient-specific therapeutic approaches as well as increase our understanding of tumor–immune interactions.
- Tumor Microenvironment
- Translational Medical Research
- Therapies, Investigational
- Lymphocytes, Tumor-Infiltrating
- Immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- Tumor Microenvironment
- Translational Medical Research
- Therapies, Investigational
- Lymphocytes, Tumor-Infiltrating
- Immunotherapy
Introduction
Understanding the role of the immune system in oncogenesis and tumor progression has led to the development of multiple strategies that empower components of the immune system to attack neoplastic malignancies resulting in reduced disease load.1 As one of the most important inventions of the last decade, immune checkpoint inhibition (ICI) therapy has revolutionized the field of cancer treatment.2 Substantial data from multiple clinical trials confirm the excellent clinical efficacy of anti PD-1 and anti CTLA-4 drugs such as nivolumab and Ipilimumab in a subset of patients.3–7 Importantly, these therapies show high clinical efficacy in tumor types that harbor high mutational burden due to genetic instability.8 Such characteristics often lead to significant influx of immune cells at the tumor site due to neoantigen release.9 So far, several immunotherapeutic targets have shown promising results in treating tumors presented with such immunogenic signatures.
Other upcoming approaches targeting the immune system, such as therapeutic vaccination and cell transfer therapies, hold optimistic potential to induce long-lasting potent antitumor responses.10 Therapeutic vaccination based on various vaccine platforms, for example, synthetic long peptide (SLP) vaccines or mRNA-based vaccines, has demonstrated effective antitumor responses and tumor eradication in melanoma patients.11 Furthermore, significant immunogenicity has been demonstrated in end stage cervical cancer patients using SLP-based vaccines.12 More recently, mRNA cancer vaccines in combination with immune-checkpoint therapy are currently tested for efficiency and safety in multiple clinical trials.13 14
Despite the encouraging clinical potential of therapies targeting tumor immunology, approaches such as ICI especially for solid tumors are still limited. In fact, most strategies fail to induce long-lasting efficient cytotoxic responses in different types of cancers and clinical efficacy varies between patients.15 A brief overview of common immunotherapeutic approaches in different cancer types can be found in table 1. Immunotherapy resistance can be induced by various mechanisms among others neovascularization, metabolic alterations, insufficient antigen presentation and irreversible T cell exhaustion.15 16 Heterogeneity and treatment resistance of tumors also further disturb the early development of new immunotherapies that are associated with substantial costs.17 Moreover, intratumoral and intertumoral heterogeneity and varying immune response profiles between patients complicate the development of these therapies. Consequently, drugs that may have significant effects in only a few patients may be prematurely discarded. Therefore, predicting immunotherapy responses a priori is an important strategy to ensure cost-effective drug use. Considering the described issues and complications, taking into account patient-to-patient tumor-heterogeneity by developing personalized therapeutics, might benefit these patients greatly in terms of the timely interventions as well as cost-effectivity.18
Objective responses of varying immunotherapeutic strategies in multiple solid tumor types
For some cancers, strategies to predict responses to immune therapy have been consolidated in guidelines and implemented in routine clinical care, in particular for lung cancer.19 Generally speaking, however, drug response predictions are complex and to predictive biomarkers are often found in retrospect using genomic and molecular data of responding versus non-responding patients.20 At an earlier stage of drug development, to determine therapeutic efficiency during drug screening traditionally, two-dimensional (2D) cell lines are used.21 22 Recently, deep learning models trained on cell line were used as an alternative approach to screen chemotherapeutic drug responses and to generally predict treatment efficacy.23 While these cultures are cheap and easy to maintain, they do not include the hostile tumor microenvironment (TME) and its complexity.24 25 This lack can partly be circumvented by using in vivo animal models which constitute a more realistic TME.26 However, the patient-to-patient heterogeneity is not reflected in these genetically uniform models, complicating clinical translation of obtained efficacy results.
Patient-derived organoids as a novel model to study immune oncology
In the past decade, patient derived three-dimensional (3D) organoid cultures have been developed and consist of complex multicellular structures grown from epithelial tumor cells isolated from tumor biopsies typically grown in an extracellular matrix such as Matrigel.27 The development of tumor organoids has advanced the field of in vitro molecular cell research as it presents a more realistic model of the tumor tissue than the known 2D grown tumor cell lines used in tumor modeling.22 25 Because organoids are likely to reflect the genetic characteristics of the parental cancer they are considered to be a promising model in precision medicine.28 Accordingly, organoids have been established for most solid types of cancer and a plethora of studies addressing their usefulness for guiding patient treatment are currently being performed (table 2).
Organoid establishment rate and established therapeutic screening
Apart from their potential usefulness for guiding oncological treatment of the individuals from which the material was obtained, organoids allow for complex cell–cell interactions and may thus better capture oncological disease when compared with more conventional experimental systems. Organoids also share many of the characteristics with the parent tumor such as, similar histological features and expression of stem cell, epithelial and mesenchymal markers as well as resemblance of the tumor transcriptome.29 Initiating conventional 2D cell cultures from clinical material is difficult and by inference cells capable of such initiating are not fully representative of the initial cancer. In addition, competition between cells in the culture flask will provoke phenotypic changes. Furthermore, 2D cultures suffer from a paucity of cell–cell interactions but because of their flat layout excellent nutrient availability, thereby limiting nutrient—and oxygen competition and metabolic alterations which are considered a crucial hallmark for cancer formation and progression.25 30 Considering the factors mentioned above, patient-derived organoids might also be a viable tumor model to predict patient-specific responses to immunotherapy. We must emphasize that little research on this specific topic has been performed, indicating the importance of further research. Next to the high relevance with the patient-specific cancer scenario, working with these organoids would be more cost-effective and ethical sound compared with in vivo animal models.
Encouraging is that recent studies have demonstrated the value of chemotherapeutic drug screening on organoids in several types of solid cancers including bladder cancer, glioblastoma and cholangiocarcinoma (table 2). While regular chemotherapeutic drugs were mainly designed to target rapid-dividing cancer cells, immunotherapeutic drugs target the complex interaction between tumor cells and immune cells via varying mechanisms and thus cannot be screened on organoids alone.31 Since organoids still lack an autologous immune component, which would be necessary to determine the patient-specific immunotherapeutic responses. The development of xenograft mouse models with a humanized immune system has shown its importance in immunotherapeutic screening. However, most systems remain allogenic due to difficulties in obtaining hematopoietic CD34+ stem cells from the same donor which is the common used sourced in such systems.32 Therefore, new strategies to co-culture organoids and autologous immune components are currently being developed. Indeed, these strategies might prove beneficial for high-throughput prediction of patient-specific immunotherapeutic responses.33 Moreover, co-cultures may further provide critical insights in the cellular interactions in the TME, possibly revealing new therapeutic targets and aid biomarker and neoantigen discovery for vaccine development.34
Organoid-immune co-culture establishment to study tumor–immune interactions
Co-cultures can be created in various ways depending, design of the experimental approach also being dependent on the scientific question to be answered. All patient derived co-cultures start with the digestion of primary tumor material obtained via surgical resection or varying biopsy procedures, for example, fine-needle aspiration biopsy.35 36 A recent study demonstrated that the success rate of organoid initiation differs between tumor types with an overall success rate of 36.8% for 13 different tumor types.37 Until, high success rates of around 90% are found in melanoma and glioblastoma.38 39 Briefly, fresh tumor tissue is digested and cultured in a basement membrane extract hydrogel dome (eg, Matrigel, Cultrex BME) that resembles the collagen rich basement membrane extracellular environment found in human tissues.40 This complex structure allows for cellular growth in a 3D way ensuring cell–cell interactions that mimics in vivo tissue including all downstream effects of these interactions, for example, cell signaling, metabolic alterations, and cell proliferation.41 While medium components differ between organoid subtypes, certain growth factors, such as Noggin, R-spondin-1, and Wnt3a, are universally used to ensure optimal organoid proliferation.42 Typically, organoids are grown in a basal membrane hydrogel, medium components are able to penetrate the hydrogel and can then be taken up by organoids in a setting that mimics nutrient uptake in vivo where cells bordering vascularization have the closest proximity to nutrients. Epithelial cells harboring the hypoxic core of the organoid have limited nutrient access and are prone to early cell death.43 A patient with colorectal cancer (CRC)-based study showed that patient-derived organoids can be expanded in a relatively short period of time with doubling rates between 3.5 and 5.25 days.44 This rapid proliferation might indicate a potential role in time efficient high-throughput drug screening. In addition, this opens a window of opportunity for personalized organoid immune co-cultures.
When initiating co-cultures of organoids with immune cells, different sources of immune cells can be used. Tumor-infiltrating lymphocytes (TILs) are most representative of the in vivo tumor situation due to their origin from the TME that is to be mimicked and hence their exposure to tumor-associated antigens (TAAs), proteins and soluble factors secreted by either tumor or tumor-associated cells.45 TIL can be isolated from the remaining cancer tissue that was used to generate organoids, isolation starts with mechanical disruption usually followed by enzymatic digestion. Separation of CD45+ cells from tumor debris is often performed using Ficoll separation or CD45+ cell selection using magnetic beads.46 Alternatively, autologous peripheral blood mononuclear cells (PBMCs) may be used for co-cultures that are more easily obtainable from blood samples using Ficoll separation. A further source of autologous immune cells is generating these from patient-derived induced pluripotent stem cells, but this has not yet been attempted in the context of organoids.47
Organoid-immune co-cultures can be constituted in various ways depending on the scope of research. Autologous co-cultures will be most similar to the in vivo situation. However, setting up autologous co-cultures often finds it limitations in tissue availability especially if TILs are the desired immune source. Rather, an allogeneic system can be generated in which the immune cells are derived from a different donor than the organoids.48 One must be aware that such allogeneic systems will induce a potent immune response by HLA mismatched immune activation overshadowing specific responses against TAAs.49
Regardless if organoids and immune cells are autologous or allogeneic, most extensively studied are so-called direct co-cultures that include organoids grown in close proximity to the tumor immune infiltrate (figure 1). If adequately setup such a system may allow for checkpoint-ligand binding, recognition of epitopes by T cell receptors and nutrient competition.50 Direct co-cultures can be constructed in varying ways such as submerged dome culture to which immune components are added. Previous work demonstrated that the dense structure of hydrogel domes, however, hardly allows for immune cell infiltration.48 For this reason, most co-cultures are rather suspension cultures in which immune cells are grown in the same medium as the organoids but without the full hydrogel dome.33 This specific requirement remains a complicating factor for efficient culture conditions. Typically, organoids and immune cells are first cultured in their own distinct medium before starting the co-culture. For immune cells typical media used are: RPMI 1640, DMEM or MEM. Organoids are cultured in medium that contains specific growth and stem cell factors necessary for the used organoid subtype. Both media contain specific substances that allow for efficient growth of each particular cell type. Organoids in general are unable to expand in the media typically used for immune cells, due to the lack of stem cell signaling factors. Immune cells can proliferate in organoid expansion medium, however, some culture components consistently used in organoid cultures may be less tolerated. For example, we recently showed nicotinamide that is beneficial to organoid expansion diminished immune cell proliferation.48 Organoids from different tissue origin types have varying needs regarding type of growth factors. So, no universal expansion medium for neither immune cells nor organoids is available yet. Therefore, for each co-culture/disease setting, precise experiments to discover an optimal culture medium in which immune cells are not harmed and organoids are still able to proliferate should be performed prior to attempting a co-culture.
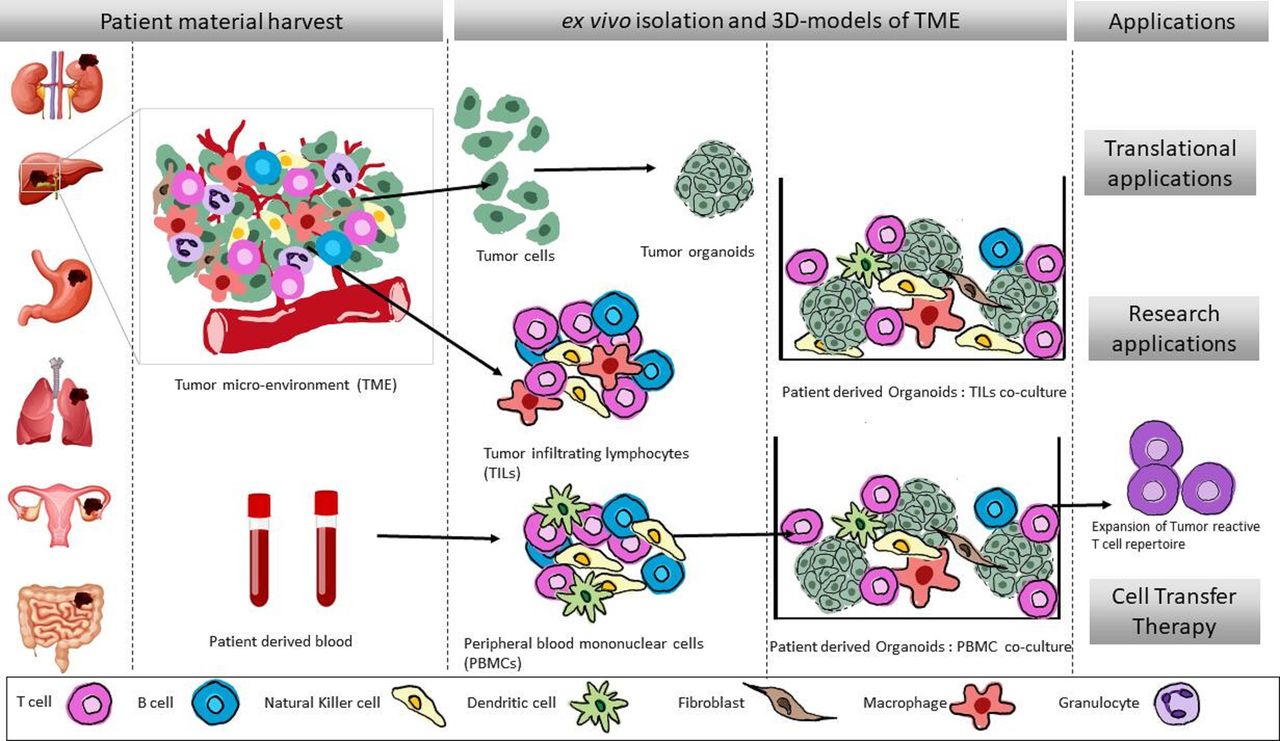
Generation of patient-specific ex vivo three-dimensional models of TME representation. Schematic overview of the generation of patient-specific autologous oncoimmune co-culture models. Tumor tissues are harvested postsurgical resections to isolate TILs as well as generation of organoid. Additionally, autologous PBMCs can be isolated from the blood of the patient. These tumor organoids and immune cells are used further to reconstitute the TME ex vivo in the co-culture setup. These oncoimmune co-cultures can further be used various clinical and translational research applications.
As said immune cells cannot penetrate the hydrogel dome in which organoids thrive. Suspension cultures can be supplemented with a small percentage of hydrogel (10%). Even in these low percentages organoids are, at least in Matrigel, able to sustain their 3D structure without alterations in shape and size. However, less organoids are able to grow out leading to absolute lower organoid numbers in these settings.40 48 51 Although, most ECMs are derived from a foreign non-human source, mainly mouse sarcoma cell lines, allogenic immune reactions targeting ECM are typically not seen which may possibly be caused by high conservation in these proteins shared between multiple species.52 Still, one study suggests to enzymatically break down the ECM before co-culturing using dispase II in order to prevent any a specific T cell activation.53 While there are seemingly no immune responses targeting ECM, little research has been performed on the role of ECM components regarding immune cell survival, proliferation, etc. While ECM used in organoid culturing mainly contains the major basement membrane proteins: laminin, collagen, entactin and perlecan, tumor-derived factors such as TGF-B and matrix metalloproteinases are also present.54 These factors are known to modulate the immune system and reduce the antitumor immune response indicating that co-cultures might be affected by the components found in Matrigel and BME.55 56 In a direct co-culture cell–cell interaction is optimal, and therefore, organoid and immune interaction can be studied extensively focusing on both molecular mechanism and overall cell survival and immune cell expansion. Furthermore, after the co-culture, cells can be phenotyped and culture medium analyzed for the presence of both pro-inflammatory cytokines released by immune components such as IFNy and TNFa which are associated with antitumor immunity.57 Contrary, anti-inflammatory cytokines, such as IL-10 and TGF-B, released by both organoids and disrupted immune cells might point toward an immune suppressive microenvironment induced by organoids.56 Immune factors that affect tumor progression or remission that are of a soluble nature (eg, cytokines) could also be assessed using an indirect culturing approach in which immune cells are separated from tumor cells by means of a physical barrier preventing direct cell–cell interaction. Such barriers can be established using trans well insert with a pore size that ensures diffusion of cytokines but does not permit cell migration. Alternatively, supernatant derived from activated immune cells can be used to supplement growth medium to assess the effect of soluble immune components on tumors or vice versa.48
Screening for patient-specific antibody-based immune therapies using organoid: immune cell co-cultures
Swift developments in immune-organoid co-culturing might potentially change the way we perceive translational immunotherapeutic research with more emphasis on and respect for patient specificity. A form of therapy that might particularly benefit a personalized approach is immune checkpoint blockade (ICB) therapy, which relies on the interaction between immune checkpoints and their corresponding ligands on immune cells and tumor cells. As a result of interpatient and interdisease variability in the clinical efficacy of ICB therapy, new methods to predict patient-specific responses are urgently needed and organoids are of high interest for this purpose (figure 2). Furthermore, combinational therapies, for example, chemotherapy and ICB, might be tested using organoids for their clinical efficacy. Sato et al showed a successful attempt at long-term expansion of human organoids.42 Subsequently, Voabil et al developed a patient derived tumor fragment platform to assess the early immunological response to immunotherapy and demonstrated the patient-specific response to correlate with the native TIL composition of the tumor.58 Together, this further supports the need for such tumor-immune co-culture models to screen multiple therapeutics and combinations of therapeutics in a patient-specific setting. While some early attempts of constructing patient-specific organoids and peripheral immune components show promising results in screening patient specific, therapy efficacy, the native TILs, especially ICI responding exhausted T cells are missed in such models.59 A recent study showed TIL expansion and T cell reactivity against the SIY tumor antigen after addition of anti PD-1 antibody in human and mouse organoid cultures,60 demonstrating the potential use of such novel approaches toward personalized therapy.
{kind=link}
{kind=link}
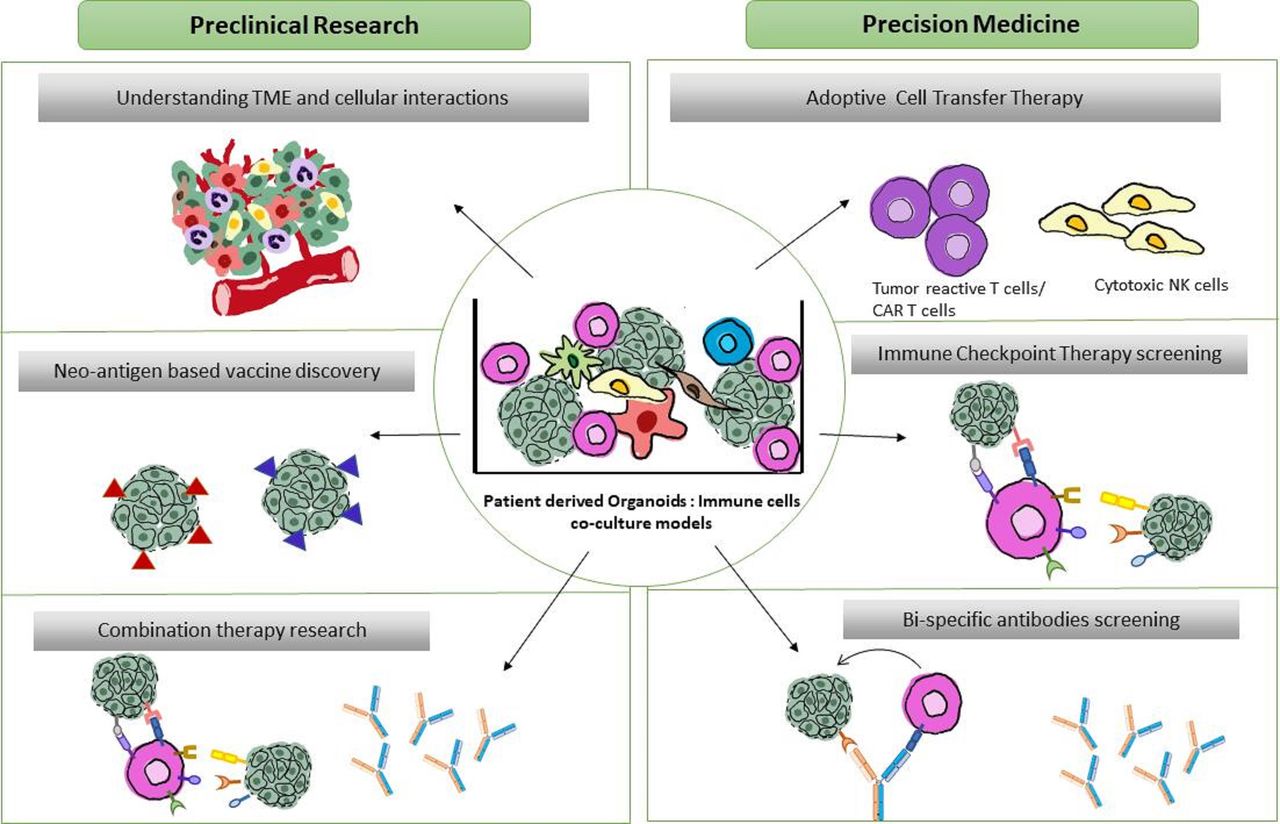
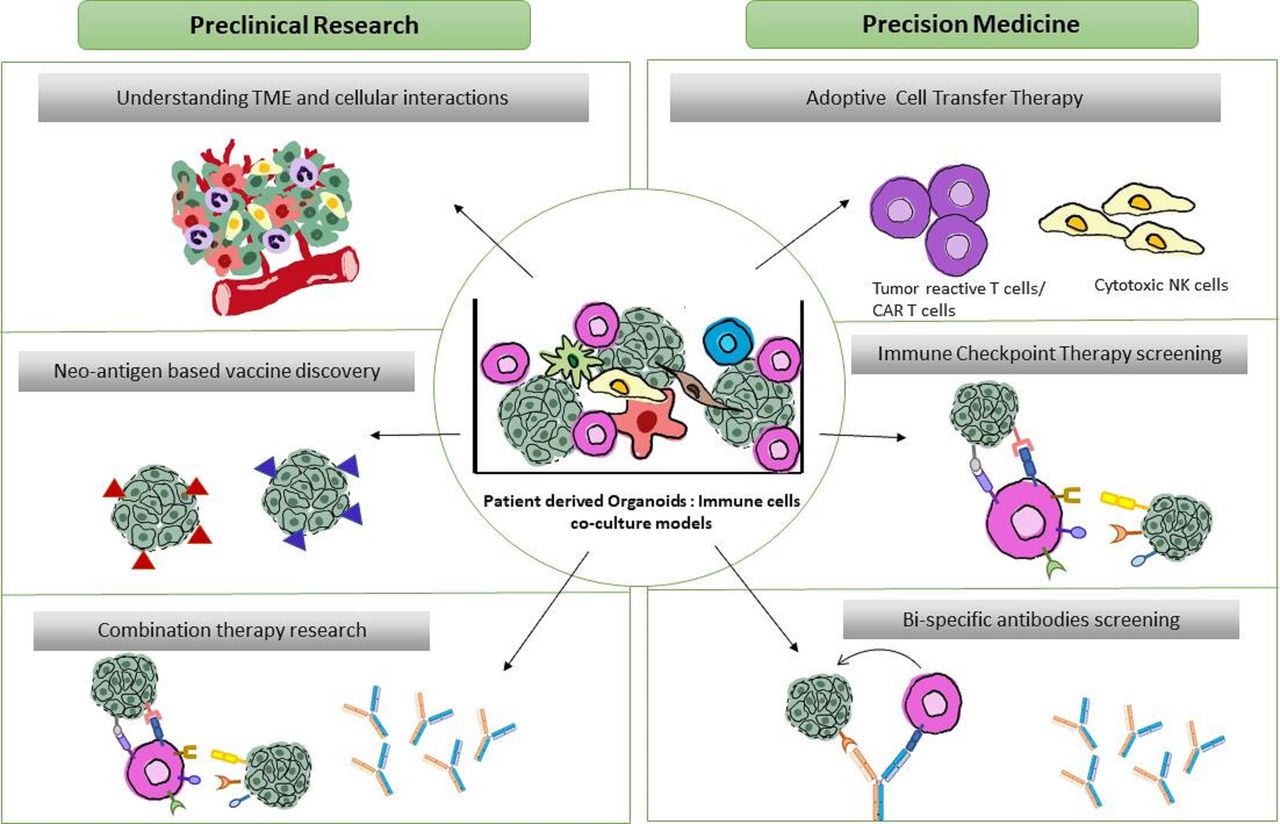
Translational and fundamental applications of patient-derived organoid-immune co-cultures. Schematic overview of proposed co-culture applications. Preclinical applications (left) focus on (1) understanding the TME dynamics and oncoimmune landscape (2) Neoantigen discovery in a patient-specific manner for potential cancer vaccine developments (3) screening of combinational therapies of existing ICI-therapies and bispecific antibodies. Precision medicine applications (right) include (1) Personalized expansion of tumor reactive lymphocytes for adoptive cell transfer therapies (2) Patient-specific high throughput screening of immune checkpoint therapies and (3) screening and developing targeted bispecific antibodies directed to tumor and immune cells in the close proximity within the tumor. TME, tumor microenvironment.
The need for new screening methods is mainly due to the lack of consistent and reliable biomarkers which complicates efficient treatment. Votanopoulos et al generated a co-culture consisting of organoids and autologous lymph nodes. The addition of PD-1 inhibitors pembrolizumab or nivolumab led to decreased cell viability in some of the melanoma organoids. Interestingly, this organoid response correlated in 85% of the cases with clinical response of the patient. Although not thoroughly tested, this system allows for screening of checkpoint inhibitors that exert their function outside the TME such as CTLA-4 inhibitors which target T cells in the draining lymph node.39 The use of organoid-immune co-cultures in anti PD-1 killing assays are also demonstrated in lung cancer organoid-immune co-cultures although in this study staphylococcal enterotoxin B was used as a super antigen by crosslinking major histocompatibility complex II (MHC-II) molecules with T cell receptors, limiting its translation to a more subtle antigen-specific setting.61 Next to screening of mono-ICI, combinations of ICI and conventional chemotherapeutics or targeted therapy (eg, VEGF inhibitor), could be assessed on co-cultures, optimizing each patients’ therapeutic approach individually. Furthermore, new innovative antibodies targeting one or multiple immune checkpoints or receptors could be analyzed for their therapeutic efficacy in a patient-specific setting. Examples of these drugs that could also greatly benefit from testing on co-cultures are targeted cytokine strategies and bispecific antibodies targeting receptors on both tumor and immune cells, most commonly T cells with one molecule. Efficiently closing the distance between effector T cells and tumor cells, such bispecific antibodies may promote rapid localized tumor clearance while minimizing systemic immune activation.62 A recent study demonstrated the application of such a bispecific antibody in a high-grade serous ovarian cancer organoid-immune co-culture. While anti PD-1 treatment is capable of shifting immune cell phenotype from an exhausted or naïve state to an activated effector state quantified by increased IFNγ production by CD4+T cells, CD8+T cells and NK cells. This effect is more pronounced when a bispecific antibody targeting PD-L1 and PD-1 is used. Moreover, these results correlate with in vivo findings, illustrating that organoid-immune co-cultures might, in the future replace or at least supplement in vivo models.63 While personalized screening might improve efficient drug use, understanding resistance mechanism and enhancing the effects of ICB might improve overall therapy efficiency. A CRC organoid-immune co-culture demonstrated that organoids resistant to anti-PD-1-associated immune killing had significantly higher levels of myeloid-derived suppressor cells (MDSC).64 These findings correlate with co-cultures in patients with gastric cancer that demonstrated the unresponsiveness of PD-L1 positive organoids to anti PD-1 therapy in the presence of MDSC. Depletion of MDSC in culture conditions led to enhanced immune-associated organoid killing following anti-PD-1 therapy.65 The use of these systems might, therefore, not only benefit individual patients but also generate broader knowledge on therapeutic resistance mechanisms.
Use of organoid-immune co-cultures for screening and generation of immune cells for adoptive cell transfer therapies
While antibody-based therapies might be the obvious candidate for personalized screening using co-cultures, the potential of this approach reaches further. Currently, cell transfer therapies such as chimeric antigen receptor (CAR) lymphocytes that recognize cell surface cancer antigens are being developed. Most promising results are observed in hematological malignancies, but clinical potential might also exist for solid tumors.66 67 One study used co-cultures to demonstrate CAR NK cell responses against TAAs in CRC despite low expression levels. They observed responses against healthy organoids from some patients as well, thereby possibly identifying patients that will endure severe side effects.68 Similar results were obtained in co-cultures with CAR T cells targeting mutant antigen EGFRvIII on glioblastoma organoids.38 CAR T cells were able to rapidly clear EGFRvIII+ organoids in a highly specific manner. Highly specific cytotoxicity is also demonstrated in CAR T cell screening on bladder organoids where T cells engineered to target MUC1 could specifically kill MUC1+69 organoids. These studies demonstrating the potential for CAR lymphocyte-organoids co-cultures in patient-specific therapy screening. In addition to therapy testing, the patient-specific aspect of organoids could potentially be exploited to expand tumor reactive T cells for cell transfer therapy applications in a completely personalized manner. This application is supported by genetic display and neo/cancer antigen expression data comparing parent tumors and ex vivo organoids demonstrating high similarity.70–72 As such, tumor reactive immune cells could potentially grow out from TILs or PBMC cultured in close proximity to organoids. Recently, Dijkstra et al demonstrated the expansion of tumor reactive T cells from paired PBMC, using microsatellite instable CRC and non-small cell lung cancer (NSCLC) organoids, quantified by organoid-specific killing.53 Their methods rely on tumors that harbor high mutational burden and that are therefore prone to neoantigen expression for which reactive T cells are circulating.73 Upregulation of CD107a and IFNy secretion in CD8+ T cells was observed in 50% of MHC-I high CRC T cell organoid co-culture. Co-cultures with NSCLC showed similar responses, also in patients in which tumor reactivity could not be observed before co-culturing. Killing assays that use these expanded tumor reactive PBMCs demonstrated tumor-specific responses in a CD8+ T cell-dependent manner that were absent in co-cultures with healthy organoid controls.53 A similar setup using pancreatic cancer organoids and autologous PBMCs was also able to drive T cell expansion. These results were not observed in healthy pancreatic organoids, implying T cell reactivity against TAAs. The total number of expanded T cells varied between patients, indicating that patient heterogeneity may influence immune recognition.74 These results indicate that co-cultures might potentially facilitate personalized cell transfer therapies by generation of tumor reactive T cells within a limited period using relatively easily obtainable material. While tumor reactive T cell expansion using organoid co-culture models has only been showed in a limited number of cancer types, it might potentially be used as a platform in multiple solid tumors paving the way for personalized cell transfer therapy.
Current limitations and future prospective
Organoid-immune co-cultures have the potential to deepen our understanding of tumor immunology and might pave the way to more efficient personalized medicine. However, this approach is still in early development and technological challenges still limits preclinical applications. The most prominent limitation is the relatively low efficiency of organoid generation from tumor tissue. The average efficiency of organoid generation is 36.8%, which covers 13 different types of tumors, but can reach as low as 19% in prostate cancer.37 Low numbers of organoid establishment complicate their use in high-throughput drug screening. Efficiency can be increased by culturing in conditioned medium as used in, for example, CRC organoids. These tumors harbor mutually exclusive mutations in the wingless/Integrated (WNT)/B-catenin pathway leading to constitutive activation of this pathway. Therefore, WNT depleted medium can be used to stimulate organoid growth over healthy organoids. Unfortunately not every cancer has a shared mutational profile and is, therefore, eligible for conditioned medium usage.75 Further research is needed to increase organoid establishment efficiency. Furthermore, tissue availability and/or especially TIL yield, can limit co-culture setup. To circumvent this problem PBMC might be a more promising and easily obtainable source of immune cells. Still one must be aware that PBMCs do not mimic the phenotype and characteristics seen in TILs as they have not been exposed to local tumor-mediated immune modulation. While a co-culture setup better mimics tumor immune interaction with special emphasis on patient heterogeneity, it cannot fully capture the mechanics and interactions found in the tumor microenvironment. The in vivo situation is much more complex with paracrine signaling, autologous ECM and vascularization presence of cancer-associated fibroblasts of which recently co-cultures with liver cancer organoids have been established.76 77 Currently, novel technology such as tumor-on-chip is being developed, and it usage in (high throughput) drug screening explored, as nicely reviewed in several publications.78 79 Interesting is the inclusion of native (ECM, derived from tumor or from distant metastatic organs in these models.80 81 At this stage co-cultures cannot replace in vivo models entirely. However, the native ECM, as is captured in tissue slices or retrieved by decellularization technology, can be used as a scaffold for co-culture models to mimic the TME even more.82 The ECM is also known to play an important role in accessibility of drugs and is part of the complicated TME. Creating a hydrogel that is derived from this ECM even enables bioprinting which allows for including tumor cells and other cells that play a role in tumor progression and metastasis.83 84 Further research should be conducted to validate the use of these novel type tumor models in clinical settings and evaluate their use for screening purposes with patient response data.
Concluding remarks
Personalized medicine is becoming increasingly more important due to deepened knowledge on intertumoral and intratumoral heterogeneity. This complexity is acknowledged in the fact that immunotherapeutic strategies are often only efficient in a small subset of patients. Onco-immune co-cultures might be used to improve our understanding of tumor–immune interaction and more notably, as a tool to assess patient-specific responses prior to immune therapy. Further applications entail patient-specific transfer of expanded tumor reactive lymphocytes and neoantigen discovery for vaccine development.
Ethics statements
Patient consent for publication
Acknowledgments
The authors acknowledge Dr. Jaap Kwekkeboom for his contribution toward the development of these co-culture models.
References
Footnotes
Contributors JD conceptualized the review. LM and JD wrote the review. MMAV, SB, LJWvdL and MP provided valuable guidance and editing inputs.
Funding JD, MMAV, LJWvdL and SB received the TKI-LSH (Topconsortium Kennis en Innovatie-Life Sciences & Health) grant (TIL, EMC-LSHM17064).
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.