Article Text

Abstract
Background Tumor necrosis factor superfamily member 14 (TNFRSF14)/herpes virus entry mediator (HVEM) is the ligand for B and T lymphocyte attenuator (BTLA) and CD160-negative immune co-signaling molecules as well as viral proteins. Its expression is dysregulated with an overexpression in tumors and a connection with tumors of adverse prognosis.
Methods We developed C57BL/6 mouse models co-expressing human (hu)BTLA and huHVEM as well as antagonistic monoclonal antibodies (mAbs) that completely prevent the interactions of HVEM with its ligands.
Results Here, we show that the anti-HVEM18-10 mAb increases primary human αβ-T cells activity alone (CIS-activity) or in the presence of HVEM-expressing lung or colorectal cancer cells in vitro (TRANS-activity). Anti-HVEM18-10 synergizes with antiprogrammed death-ligand 1 (anti-PD-L1) mAb to activate T cells in the presence of PD-L1-positive tumors, but is sufficient to trigger T cell activation in the presence of PD-L1-negative cells. In order to better understand HVEM18-10 effects in vivo and especially disentangle its CIS and TRANS effects, we developed a knockin (KI) mouse model expressing human BTLA (huBTLA+/+) and a KI mouse model expressing both huBTLA+/+/huHVEM+/+ (double KI (DKI)). In vivo preclinical experiments performed in both mouse models showed that HVEM18-10 treatment was efficient to decrease human HVEM+ tumor growth. In the DKI model, anti-HVEM18-10 treatment induces a decrease of exhausted CD8+ T cells and regulatory T cells and an increase of effector memory CD4+ T cells within the tumor. Interestingly, mice which completely rejected tumors (±20%) did not develop tumors on rechallenge in both settings, therefore showing a marked T cell-memory phenotype effect.
Conclusions Altogether, our preclinical models validate anti-HVEM18-10 as a promising therapeutic antibody to use in clinics as a monotherapy or in combination with existing immunotherapies (antiprogrammed cell death protein 1/anti-PD-L1/anti-cytotoxic T-lymphocyte antigen-4 (CTLA-4)).
- costimulatory and inhibitory T-cell receptors
- immunotherapy
- lymphocyte activation
- T-lymphocytes
- lymphocytes, tumor-infiltrating
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- costimulatory and inhibitory T-cell receptors
- immunotherapy
- lymphocyte activation
- T-lymphocytes
- lymphocytes, tumor-infiltrating
WHAT IS ALREADY KNOWN ON THIS TOPIC
Immunotherapies (IT) and immune checkpoint inhibitors (ICI) opened a new era in cancer treatment leading to major clinical benefits in only subgroups of patients pushing the research toward the development of another ICI.
TNFRSF14/HVEM is the ligand for B and T lymphocyte attenuator (BTLA) and CD160-negative immune co-signaling molecules as well as viral proteins and is overexpressed in tumors concurring to tumors adverse prognosis.
WHAT THIS STUDY ADDS
To study the therapeutic effect of antagonist HVEM monoclonal antibodies in an immunocompetent and dynamic environment, we developed C57BL/6 mouse models co-expressing human (hu)BTLA and huHVEM.
Anti-HVEM allowed the in vitro co-activation of T cells and its injection in mice induced the development of a marked T cell-memory phenotype with the development of tumor antigen-specific T cells contributing in HVEM+ tumor reduction or rejection.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Altogether, these results highlight the interest of anti-HVEM IT alone or in combination with another IT to further enhance antitumor immunity in clinic.
Introduction
Immunotherapies (IT) and immune checkpoint inhibitors (ICI) opened a new era in cancer treatment. ICI unleash antitumoral immune responses, leading to major clinical benefits in subgroups of patients. However, rate and duration of IT successes are variable among cancer types and patients. Indeed, in lung cancer antiprogrammed cell death protein 1 (anti-PD-1)/programmed death-ligand 1 (PD-L1) antagonistic antibodies offer a durable remission in 30% of patients.1 2 However, in colorectal cancer, the efficacy of anti-PD-1/PD-L1 was restricted to a small subset of patients presenting a high microsatellite instability status.3 4 Consequently, new IT targets and approaches are needed to achieve efficacy in a larger proportion of patients.
HVEM, or TNFRSF14, is a TNF-receptor family member largely expressed by healthy immune and non-immune cells and participates to immune homeostasis.5 6 HVEM is also upregulated in numerous solid and hematological malignancies such as melanoma,7 digestive cancers,8 9 or breast cancer.10 HVEM network of interactors is complex and induces either cell activation or inhibition.11 Indeed, HVEM binding to B and T lymphocyte attenuator (BTLA) and CD160 (BY55) triggers co-inhibitory signals whereas its binding to LIGHT (TNFSF14) and lymphotoxin-α are co-stimulatory . Similar to PD-1 and CTLA-4, BTLA is an important co-inhibitory receptor expressed by B and T cells.12 Therefore, targeting HVEM is a promising but complex IT strategy.13
In our study, we focused on lung and colorectal cancer that are two of the most worldwide common cancers in both men and women. In colorectal cancer, HVEM upregulation in malignant lesions is linked to tumor status and pathological stage, with an independent prognostic value.8 In lung cancer, HVEM expression seems to be a tumor driven mechanism, independent from the PD-L1 network that may contribute to immune escape.14
Our team previously described a monoclonal anti-HVEM monoclonal antibody (mAb) (anti-HVEM18-10) that preferentially inhibits HVEM interaction with BTLA and enhances γδ-T cells responses against lymphoma.15 Here, we further evaluate the CIS-activity and TRANS-activity of anti-HVEM18-10. CIS-activity represents the antibody binding on effector HVEM+CD4+ and HVEM+CD8+ T cells directly in the absence of huHVEM+ target cell lines. TRANS-activity is measured by checking immune cell activation in the presence of a HVEM+ cell line in co-culture. We also show the interest of anti-HVEM18-10 combination with other ICIs such as anti-PD-L1 mAb, although HVEM18-10 alone is sufficient to trigger T cell activation in the absence of PD-L1 expression. In order to better understand HVEM18-10 mAb effect in vivo, we developed an innovative syngeneic immunocompetent mouse model expressing human BTLA (huBTLA+/+) or both human BTLA and human HVEM (huBTLA+/+huHVEM+/+, (double KI (DKI)). Our experiments performed in both mouse models showed that anti-HVEM18-10 treatment was efficient to decrease tumor growth and strengthened local immune response. Moreover, the rechallenge of tumor-free mice (20% of treated mice) demonstrates that the immune response is durable with a marked memory T cells phenotype.
Altogether, our preclinical data validate anti-HVEM18-10 as a promising antibody to use in clinics alone or in combination with existing therapies (anti-PD-1/anti-PD-L1/anti-CTLA-4).
Methods
Antibodies
Anti-HVEM18-10 and anti-PD-L1 3.1 are homemade antibodies that have been described previously.15,16 Briefly, mAbs are murine IgG1 antihuman HVEM or PD-L1 mAb, produced as ascites and purified by protein A binding and elution with the Affi-gel Protein A MAPS II Kit (Bio-Rad, Marnes-La-Coquette, France). Mouse IgG1 isotype control was purchased from Miltenyi Biotec (Bergisch Gladbach, Germany).
Transcriptomic expression of HVEM and PD-L1
To study the transcriptomic expression of HVEM and PD-L1 data were extracted from The Cancer Genome Atlas (TCGA) database containing 526 lung adenocarcinoma samples and 388 colorectal carcinoma samples. Tumor cell lines gene expression values were extracted from the Cancer Cell Line Encyclopedia. Data were normalized and expressed as log2(TPM+1) of raw value.
Co-culture and proliferation assays
After thawing and overnight resting in RPMI supplemented with 10% fetal calf serum, peripheral blood mononuclear cells (PBMCs) were stained with Cell Trace Violet (Thermo isher) according to manufacturer’s instructions. Co-culture assays were there performed during 72 hours and described in online supplemental material.
Supplemental material
Knockin mouse model
Generation and genotyping of B6-Tnfrsf14tm1Ciphe and BtlaTm1Ciphe are described in online supplemental material. Mice were bred and maintained under specific pathogen-free conditions at the CIPHE animal facility.
Experimental tumor growth experiments
Single-cell suspensions of MC-38 or MC-38huHVEM colorectal cancer cells were injected (0.5×106 for neo challenge, 2×106 for rechallenge) subcutaneously in the right flank of huBTLA+/+huBTLA+/+ mice. Mice bearing tumor between 50 and 100 mm3 were then randomized at day 9 and mice were injected with an isotype control (2 mg/kg or 10 mg/kg) or anti-HVEM18-10 (2 mg/kg or 10 mg/kg) or CTLA-4 antibody (2 mg/kg) every 3–4 days with for a total of six injections.
Tumor preparation for mass cytometry and immunoprofiling
Tumors were resected from huBTLA+/+ and DKI mice which received anti-HVEM18-10, anti-CTLA-4, or isotype treatment. Tumors were collected and digested using the Tumor Dissociation Kit (Miltenyi Biotech). Single cells suspension was then prepared and stained then acquired on a Helios mass cytometer (Cy-TOF, Fluidigm) and analyzed using the OMIQ software platform (OMIQ). Protocol is detailed in online supplemental method.
Unsupervised CyTOF data clustering and phenotypic analysis
CyTOF data files were exported (Helios program, Fluidigm), debarcoded and live cells were gated in FlowJo (Treestar, BD). Live cell.fcs files were exported and analyzed using OMIQ online platform (OMIQ).17 Detailed protocol for data clustering and phenotypic analysis is described in online supplemental method. All used panels are shown in tables 1–4.
Flow cytometry panel for HVEM expression on T cells subsets
Flow cytometry panel for proliferation assay
Panel for tumor infiltrating lymphocytes exploration with mass cytometry (Ext238)
Flow cytometry panel for lymph node phenotyping
Results
HVEM is highly expressed on T cells and the blockade of HVEM in CIS enhances T cell activation
We first studied the effect of anti-HVEM18-10 blockade directly on T cells (CIS-blockade). PBMCs were isolated from healthy donors and stimulated for 72 hours with anti-CD3 in combination with anti-HVEM18-10 or an IgG1 control mAbs. We found that HVEM was highly expressed on activated CD4+ and CD8+ T cells (figure 1A,B). HVEM expression is increased on stimulation in CD4+ T cells (figure 1A), this effect was marked on CD4+ effector memory T cells (figure 1C). HVEM expression did not differ on CD8+ T cell, on stimulation or memory subtypes (figure 1B,D). HVEM is highly expressed on less differentiated T cells subsets (naïves and central memory (CM)) and tends to be less expressed on more differentiated subsets (effector memory (EM), EMRA[effector memory cells re-expressing CD45RA). Thus, HVEM expression was significantly upregulated on CD4+ EM, but not other T cell subsets following activation. T cell proliferation and activation marked by the expression of CD25 significantly increased on anti-HVEM co-stimulation compared with control (figure 1E–H). Taken together, our results show that CIS-HVEM blockade enhances T cell activation.

HVEM is highly expressed on T cells and the CIS-HVEM blockade enhance T cells activation. (A–H) Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors, and cultured for 72 hours with anti-CD3 (OKT3) stimulation alone (orange bars), with OKT3 stimulation and anti-HVEM18-10 treatment (blue bars/lines), or without any stimulation (black bars). HVEM expression was assessed in resting (black bars) and activated (orange bars) CD4+ (A), CD4+ T cells subsets (naïve, central memory (CM), effector memory (EM), and EMRA) (B), CD8+ (C), CD8+ T cells subsets (D) by flow cytometry on healthy donors (n=3). (E–H) PBMCs were incubated with OKT3 and anti-HVEM18-10 antibody (blue bars/lines) or a control monoclonal antibody (mAb) IgG1 (orange bars/lines). Proliferation profile of T cells was assessed by flow cytometry detecting Cell Trace Violet (CTV) (E, G for CD4+ and CD8+ T cells, respectively) or CD25 staining (F, H for CD4+ and CD8+ T cells, respectively). (E–H) One representative plot is shown for each T cell subset. Bar plots are the mean±SEM of four different healthy donor samples. (A–D n=3, E–H n=4). *P<0.05; **p<0.01; ***p<0.001 (Student’s t-test). ns, not significant.
HVEM expression is higher than PD-L1 in human lung and colorectal cancers
Next, we decided to study the expression status of HVEM on tumor from different cancers. Transcriptomic data from the TCGA containing 525 adenocarcinoma samples and 300 colorectal carcinoma samples were extracted, and the expression of HVEM and PD-L1 was examined (figure 2A,B). We found that HVEM was highly expressed in both cancers. While PD-L1 expression was high in lung cancer and low in colorectal cancers. Interestingly, HVEM expression is greater than that of PD-L1 expression in both cancers (figure 2A,B). Next, we studied HVEM and PD-L1 transcriptomic expression (from the cancer cell line encyclopedia) and cell surface expression by cytometry in lung and colorectal cancer cell lines (figure 2C,D). HVEM and PD-L1 are heterogeneously expressed in lung cancer cell lines, we selected two lung cancer cell lines that expressed both genes (NCIH2291) or in large majority HVEM compared with PD-L1 (NCIH2405) for subsequent experiments (figure 2C). The vast majority of colorectal cancer cell line highly expressed HVEM whereas PD-L1 was expressed in lower amount (figure 2D). Reflecting these observations, we selected HVEM+PD-L1− HT29 cell line for subsequent experiments (figure 2D). These results were confirmed in NCIH2291 (HVEM+ PD-L1+), NCIH2405 (HVEM+ PD-L1−), and HT29 (HVEM+PD-L1−) by flow cytometry assessment of PD-L1 and HVEM surface expression (figure 2E,F).
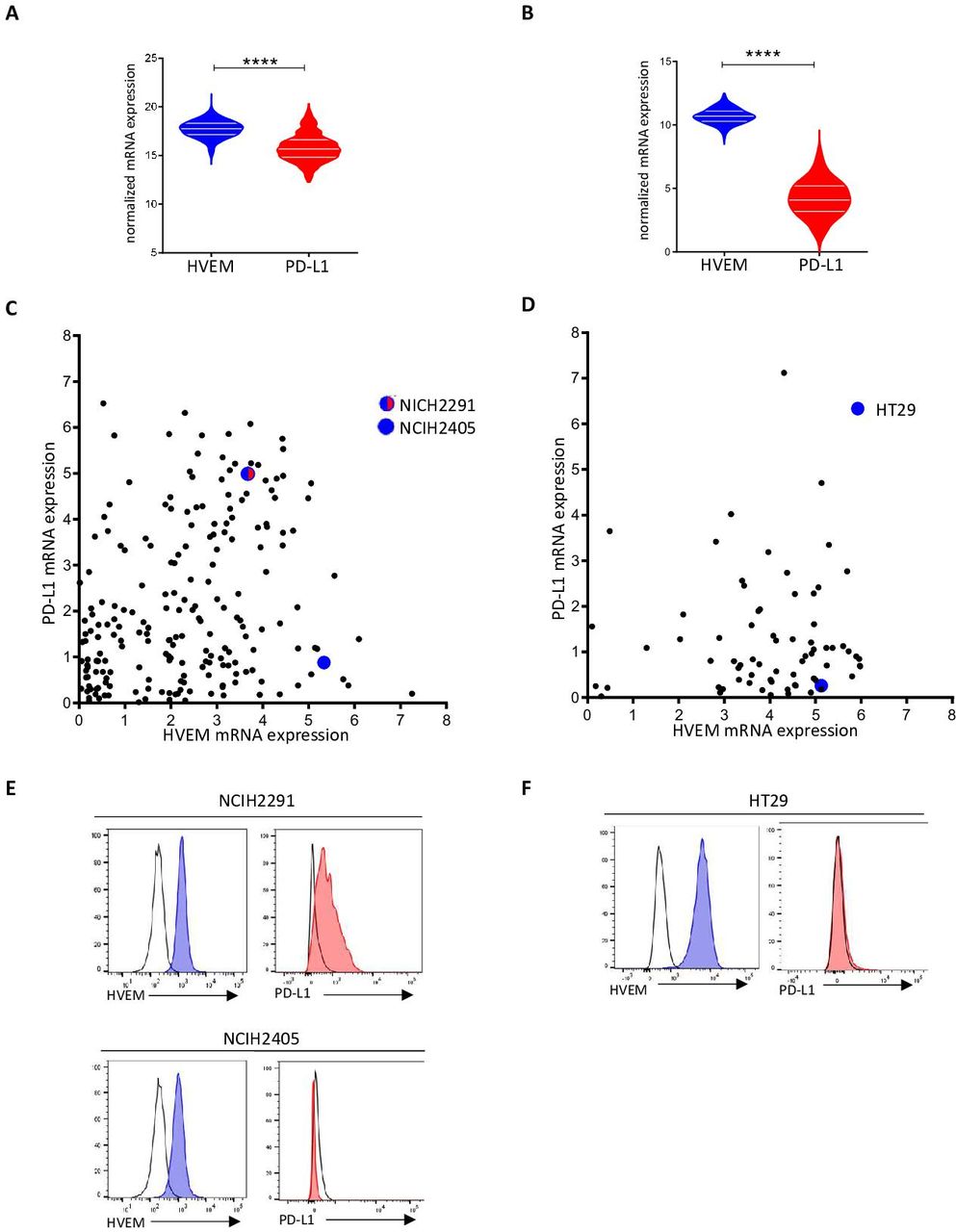
HVEM is more expressed than programmed death-ligand 1 (PD-L1) in lung and colorectal cancers. HVEM and PD-L1 transcriptomic expression was analyzed in 526 lung adenocarcinoma samples (A) and 388 colorectal carcinoma samples (B). Normalized expression data were extracted from The Cancer Genome Atlas (TCGA) database (****p<0.0001). HVEM and PD-L1 transcriptomic expression was analyzed in lung (C) and colorectal (D) cancer cell lines. (C, D) Data are extracted from the Cancer Cell Line Encyclopedia (CCLE) and expressed in log2(TPM+1). HVEM and PD-L1 phenotypic expression on selected lung (E) (NCIH2291 and NCIH2405, respectively) and colorectal (F) cancer cell line (HT29). HVEM staining in dark gray and control isotype in light gray. mRNA, messenger RNA.
Anti-HVEM18-10 enhances T cells responses against lung cancer cell line NCIH2291
Knowing the expression of HVEM among tumor cell lines allowed us to further test anti-HVEM effect. Co-cultures of PBMCs and the lung cancer cell line NCIH2291 (HVEM+ PD-L1+) were performed during 72 hours in the presence of anti-CD3 in combination with an IgG1 control or anti-HVEM18-10. The addition of anti-HVEM18-10 to the co-culture drastically increased proliferation ratio and CD25 expression of CD4+ and CD8+ T cells compared with control (figure 3A–D). Moreover, the addition of anti-HVEM18-10 to the co-culture also drastically increased the amount of procytotoxic cytokines tumor necrosis factor (TNF)-α and interferon (IFN)-γ as measured by ELISA (figure 3E,F). These results confirm an effect of the blockade of HVEM on CIS-T cell activation and TRANS -activation as well.

Anti-HVEM18-10 enhances T cells responses against NCIH2291. Adherent lung cancer cell line NCIH2291 was seeded in wells 24 hours before the experiment. Then we cultured peripheral blood mononuclear cells from healthy donors for 72 hours with OKT3 stimulation and treated or not with anti-HVEM18-10 antibody (blue bars/lines). (A–D) Proliferation profile of T cells by Cell Trace Violet (CTV) staining (A for CD4+ and B for CD8+ T cells) and CD25 expression (C for CD4+ and D for CD8+ T cells). (E, F) Tumor necrosis factor (TNF)-α (E) and interferon (IFN)-γ (F) secretion was measured by ELISA in culture supernatant. Bar plots are the mean±SEM of different healthy donor samples (A–D n=13; E–F n=10). *P<0.05; ***p<0.001; ****p<0.0001 (Student’s t-test).
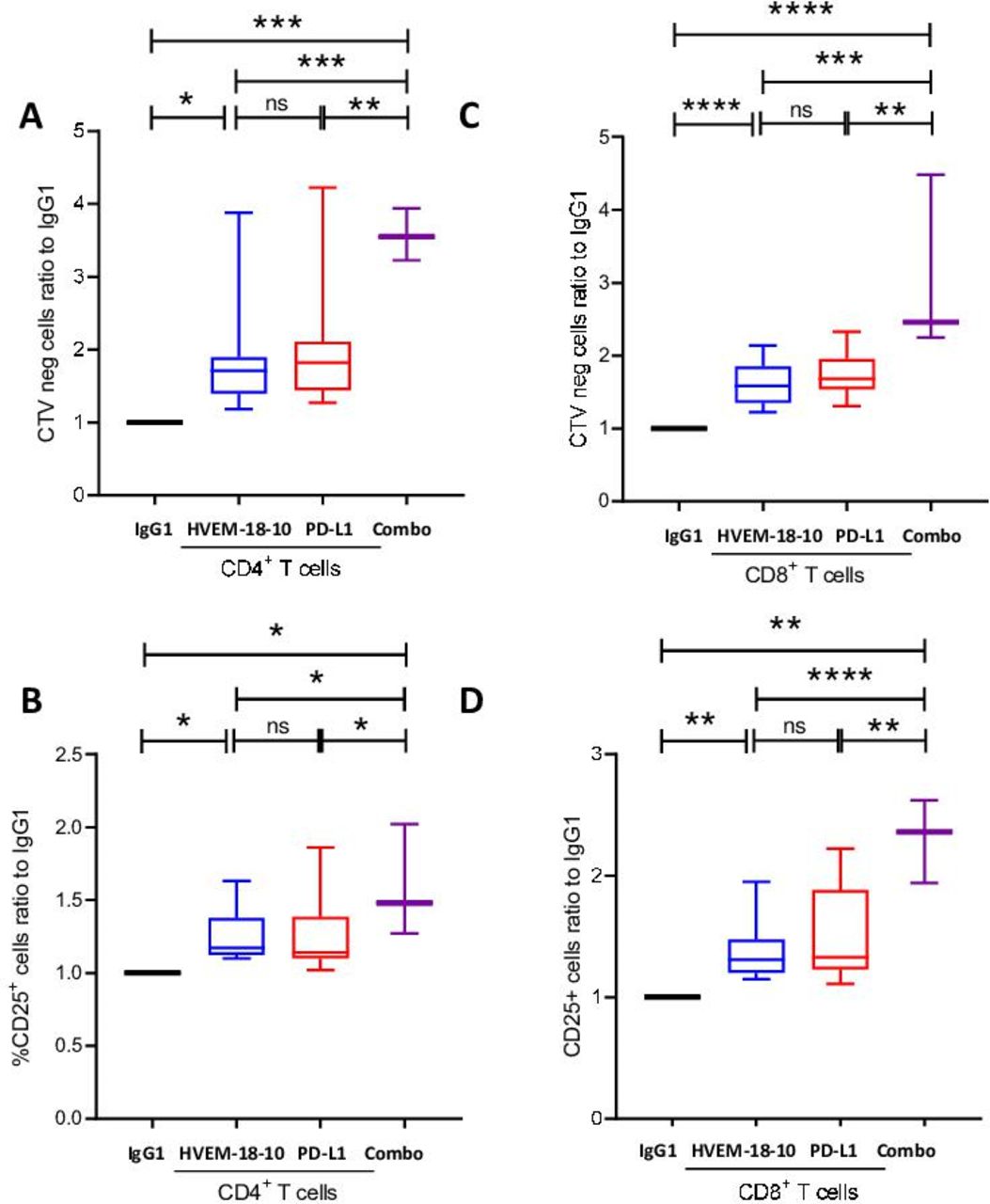
Anti-HVEM18-10 triggers T cell activation, proliferation, and synergizes with anti-PD-L1
Anti-PD-L1 antibody has been a real game changer in IT to treat patients with lung cancer.18 Therefore, we thought to test whether the combination of anti-HVEM18-10 to anti-PD-L1 could improve T cell responses in co-culture experiments with HVEM and PD-L1-positive lung tumor cell line. PBMCs were primed with suboptimal anti-CD3 dose, then activated with anti-HVEM18-10, anti-PD-L1, both antibodies (combo), or IgG1 control during 72 hours (figure 4A–D). The addition of anti-HVEM18-10 (blue bar) was sufficient to improve T cells proliferation and CD25 expression compared with control. Likewise, the anti-PD-L1 (red bar) also increased T cell activation and proliferation ratio compared with control. Interestingly, the combination of anti-HVEM18-10 and anti-PD-L1 (purple bar) in culture showed even greater effect on T cell proliferation and CD25 expression compared with the separate conditions. These results highlight the potential of anti-HVEM18-10 and anti-PD-L1 combination to strengthen T cell activation.

Anti-HVEM18-10 synergizes with anti-programmed death-ligand 1 (PD-L1) to enhance T cells responses against lung cancer cell line NCIH2291. Adherent lung cancer cell line NCIH2291 were cultured with peripheral blood mononuclear cells from healthy donors for 72 hours with CD3 stimulation and treated with anti-HVEM18-10 antibody (blue bars/lines) or not (black bars/line) (A–D) proliferation profile of T cells by Cell Trace Violet (CTV) staining (A for CD4+ and C for CD8+ T cells) and CD25 expression (B for CD4+ and D for CD8+ T cells). Bar plots are the mean±SEM of different healthy donors samples (n=13 for IgG1, anti-HVEM, and anti-PD-L1 conditions, n=3 for combo condition). *P<0.05; **p<0.01; ***p<0.001; ****p<0.0001 (Student’s t-test). ns, not significant.
Interestingly, we next tested whether anti-HVEM18-10 could enhance T cell response against PD-L1− lung and colorectal cancer cell line (online supplemental figure 1). Anti-HVEM18-10 was sufficient to improve CD4+ and CD8+ T cell activation with a marked increase of CD25 expression and proliferation in co-culture with either lung or colorectal cell line, validating the use of anti-HVEM18-10 with PD-L1− tumors for future IT treatments.
huBTLA+/+ and DKI mice show wild-type-like hematopoietic cell proportions
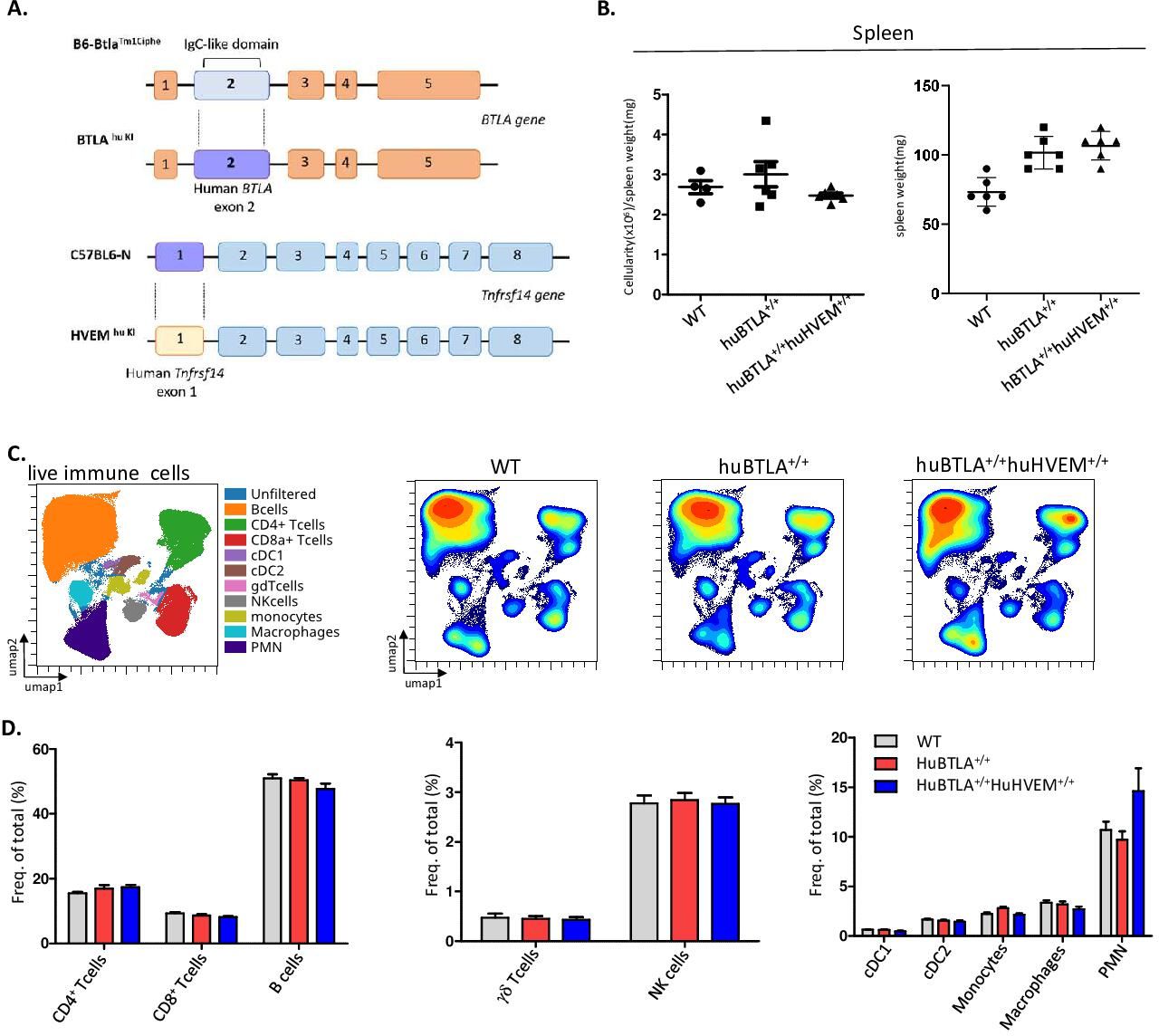
To test whether the effect of anti-HVEM18-10 in vivo occurs via CIS-blockade or TRANS-blockade using preclinical tumor models, we developed KI mice model expressing either huBTLA+/+ or both huBTLA+/+/huHVEM+/+ or DKI (figure 5A). To produce huBTLA+/+ mice, exon 2 from wild-type mouse was substituted by human exon 2. Likewise, HVEM+/+ mice were developed by replacing exon 1 with human HVEM gene (tnfrsf14-001) exon 1. These mice were then crossed to obtain DKI mice. The presence of the insertion was confirmed by PCR and cytometry. The expression of human/mouse HVEM on T cells, DC (dendritic cells), Ly6C+ cells and neutrophil and BTLA on T cells and B cells was examined by cytometry (online supplemental figure 2A,B). Both HuBTLA+/+ and DKI mice showed the same number of cells in the spleen (figure 5B) and same proportion of T cells by cytometry. Other lymphoid and myeloid cells are also found in the same proportions in both models (figure 5C). Indeed, the frequencies of B cells, γδ-Tcells and natural killer cells were similar among mice models. Similarly, myeloid cell frequencies (PMN[Polymorphonuclear], cDC1, cDC2, macrophages, monocytes) remained unchanged among these models (figure 5D). Gating strategies for different cells subset are shown in online supplemental figure 2C. These observations allowed further in vivo tumor response studies with these new mouse models.
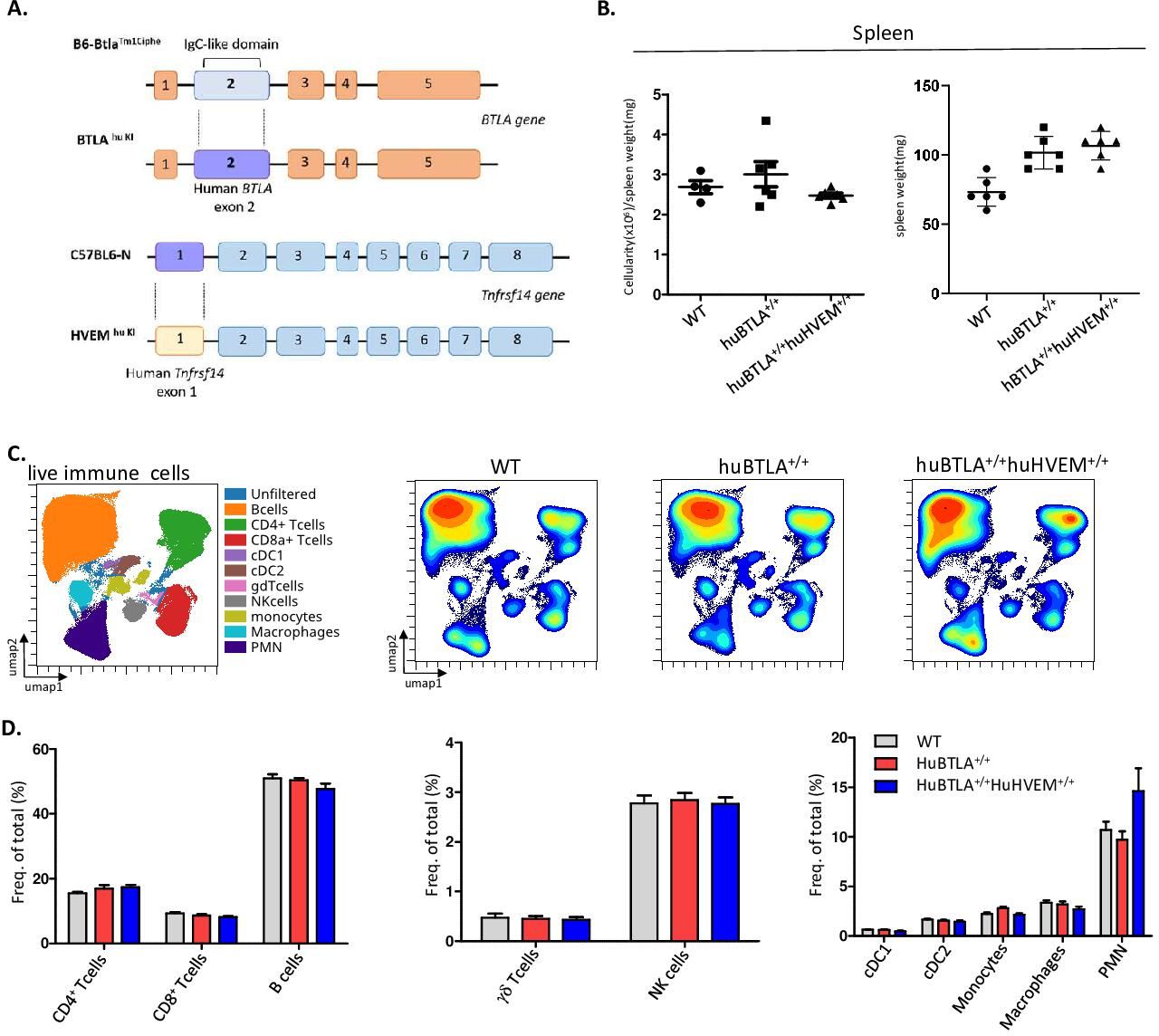
Human (hu) B and T lymphocyte attenuator (BTLA) and double knockin (DKI) mice show the same hematopoietics cell homeostasis. (A) Representative scheme of huBTLA+/+huBTLA+/+ and huHVEM+/+ genetic modification. For huBTLA+/+huBTLA+/+, murine exon 2 was replaced by huBTLA exon 2 and for huHVEM+/+murine exon 1 was replaced by huHVEM gene exon 1. (B) Numbers of total cell per spleen weight for wild-type (WT), huBTLA+/+huBTLA+/+, and huHVEM+/+ is represented (left) spleen weight (mg) is represented on the right for six control (Ctrl), huBTLA+/+, and six huHVEM+/+huBTLA+/+ mice. (C) uniform manifold approximation and projection (UMAP) showing the distribution of immune cell subset within WT, huBTLA, and DKI mice. (D) Immune cell subset frequency was quantified in the three mice groups. Density UMAP representing major and minor cell subsets (bottom). NK, naturak killer.
Blocking trans-BTLA-HVEM binding in vivo is sufficient to decrease solid tumor growth
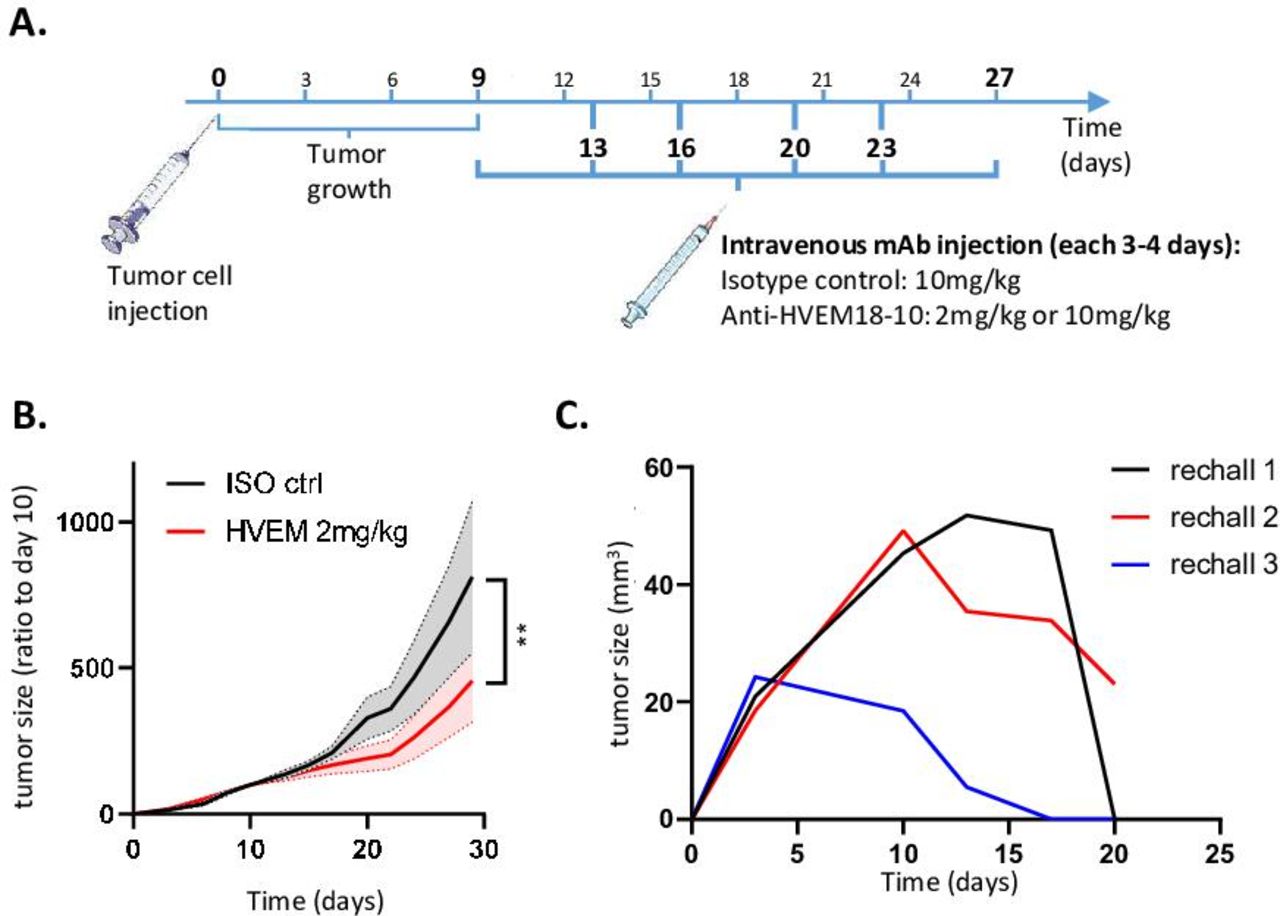
We then decided to challenge huBTLA+/+ mice with a colorectal cancer cell line MC-38 that is not expressing constitutively HVEM; 0.5×106 MC-38 tumor cells were injected subcutaneously in the right flank of huBTLA+/+ mice. Mice bearing tumor between 50 and 100 mm3 were then randomized at day 9 and mice were injected with anti-HVEM18-10 or isotype control every 3–4 days with for a total of six injections (online supplemental figure 3B). As envisioned, tumor growth of parental MC-38 (HVEM-) injected with an isotype control or with anti-HVEM18-10 at 10 mg/kg did not show any significant decrease in tumor growth. We then transduced huHVEM in MC-38 cells and selected a clone that that was able to bind effectively anti-HVEM18-10 (namely: MC-38huHVEM; online supplemental figure 3A). MC-38huHVEM tumor cells were injected subcutaneously as previously described. Here, we observed a decrease in tumor growth at any dose of anti-HVEM18-10 used 2 and 10 mg/kg (figure 6B; online supplemental figure 3C). Moreover, 2 mice out of 10 in the 2 mg/kg dose and 1 out of 10 in the 10 mg/kg dose fully rejected the tumors. These three tumor free mice were rechallenged with fourfold more concentrated MC-38huHVEM tumors cell inoculate (ie, 2×106 cells) in contralateral flank. All animals were monitored during 21 days postchallenge to ensure immunological memory persistence. In two of these three mice, the tumor was not measurable at day 21. However, after the euthanasia of these three mice at day 21, tumor mass was absent in their left flank revealing the presence of a scar tissue instead (figure 6C). Altogether, these data confirmed the in vivo efficiency of anti-HVEM18-10 to block trans-BTLA-HVEM binding.

Blocking trans-B and T lymphocyte attenuator (BTLA)-HVEM binding in vivo is sufficient to decrease solid tumor growth. (A) Scheme representing in vivo tumor experiments settings. (B) Measure of tumor growth showed as ratio to size of tumor at randomization day. Colorectal cancer cells MC-38huHVEM were injected (0.5×106) at day 0 in human (hu)BTLA+/+ mice and isotype (ISO) control (ctrl) (black) or anti-HVEM18-10 antibody at 2 mg/kg (red) concentration were injected. (C) Tumor-free mice from B and C measure of tumor growth after MC-38huHVEM tumor cells rechallenge (2×106). (B–C) n≥7; (D) n=3. **p<0.01 (two-way analysis of variance). mAb, monoclonal antibody.
Anti-HVEM treatment decreases tumor growth, exhausted CD8+ T cell, KLRG1- regulatory T cell infiltrate and increases EM conventional CD4+ T cell infiltrate
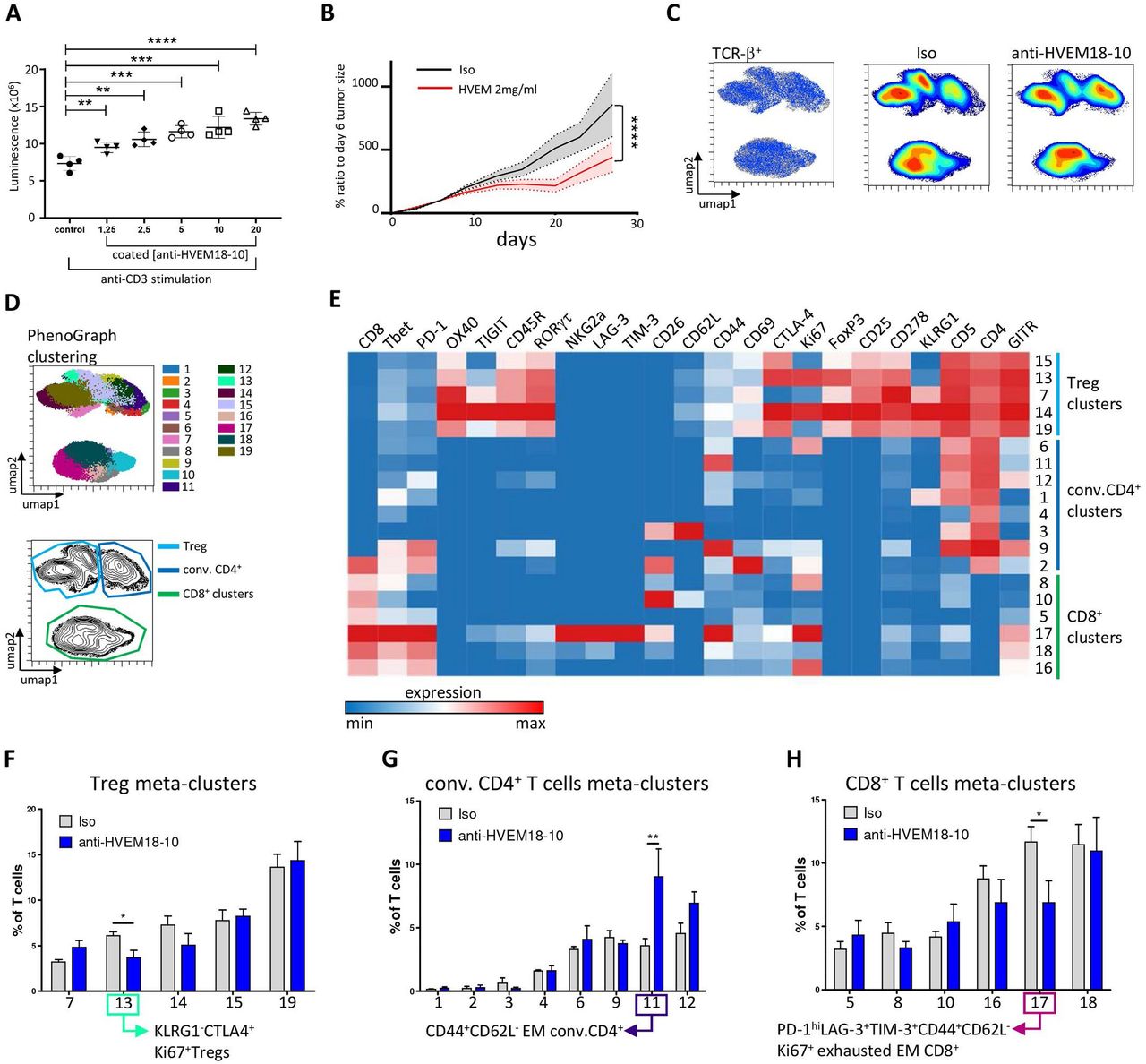
Then, we investigated the effect of anti-HVEM on DKI lymphocytes (figure 7A). The proliferation of sorted spleen T cells was assessed after 72 hours of stimulations with coated anti-CD3 and anti-HVEM18-10 or isotype control at increasing concentration. The addition of anti-HVEM18-10 increased DKI T cells proliferation in a dose-dependent manner similarly to human T cells (figure 1), validating in vitro this new mouse model (figure 7A). The effect of anti-HVEM18-10 in vivo was then studied using DKI mice. DKI mice were challenged with 0.5×106 MC-38huHVEM tumor cells which were injected subcutaneously as described previously. We observed a decrease in tumor growth when anti-HVEM18-10 was injected at 2 mg/kg (figure 7B). Moreover, 3 mice out of 12 in the anti-HVEM18-10 group completely rejected the tumors and were later investigated in rechallenge experiments. To understand the immune mechanisms underlying tumor reduction after anti-HVEM18-10 treatment, tumor infiltrating lymphocytes (TILs) phenotypes were investigated in anti-HVEM18-10-treated mice and isotype controls by mass cytometry. Tumor infiltrating T cells was mapped using UMAPs in anti-HVEM18-10 and isotype controls treated mice. Density analysis of UMAPs showed phenotypic variations between conditions (figure 7C), that were investigated by unsupervised analysis using T cell subset markers (online supplemental figure 4A–C). Next, a clustering of tumor infiltrating T cells was performed using the well-described PhenoGraph algorithm and the 19 identified clusters were projected on the UMAP highlighting the heterogeneity of intratrumoral T cells. Five clusters belonging to regulatory T (Treg) cells (CD4+CD25+FoxP3+), eight clusters belonging to conventional CD4+ T cells (conv. CD4+ T cells/non-Tregs) and six clusters belonging to CD8+ T cells were identified (figure 7D,E). Interestingly, while T cell clusters were very heterogeneous, only three clusters were significantly altered in anti-HVEM18-10 compared with isotype control treated mice (figure 7E–H and online supplemental figure 4B). These clusters were manually gated for validation in online supplemental figure 4D,E. First, cluster 13, a subset of KLRG1-CTLA-4+Ki67+ Tregs, was statistically decreased in anti-HVEM18-10 condition (figure 7F and online supplemental figure 4E). Cluster 11, composed of EM, CD44+CD62L- CD4+ conv. T cells, was significantly increased under anti-HVEM18-10 treatment (figure 7G and online supplemental figure 4E). In this cluster, EM CD4+ T cells did not exhibit marks of immunosuppression or exhaustion such as high levels of PD-1, TIM-3, or T cell immunoreceptor with Ig and ITIM domains (TIGIT). Therefore, these EM CD4+ T cells may favor antitumoral immune response. Third, cluster 17, an EM (CD44+CD62L-) CD8+ T cell subsets showed higher expression of PD-1 and expressed T-cell immunoglobulin and mucin containing protein-3 (TIM-3), Lymphocyte-activation gene 3 (LAG-3), and CTLA-4. Therefore, we labeled these EM CD8+ T cells as exhausted. Interestingly, these exhausted CD8+ T cells decreased under anti-HVEM18-10 treatment (figure 7H and online supplemental figure 4E). Taken together, our results demonstrate a loss in immunosuppressive T cell subsets (KLRG1- Tregs, and exhausted EM CD8+ T cells) and an increase in EM CD4+ T cells. These phenotypic changes contribute to the reduction of tumor growth and correspond to phenotypes observed in previous studies showing tumor reduction following immunotherapeutic treatments in mouse models.19 20

Anti-HVEM treatment decreases tumor growth, exhausted CD8+ T cell, KLRG1- regulatory T (Treg) cells infiltrate, and increases effector memory (EM) conventional (conv.) CD4+ T cell infiltrate. (A) T cells from double knockin (DKI) mice or wild-type (WT) were cultured with CD3 alone or with anti-HVEM18-10 at increasing concentration. T cells proliferation assessed by luminescence using Cell titerglo. (B) Measure of tumor growth showed as ratio to size of tumor at randomization day. Colorectal cancer cells MC-38huHVEM were injected (0.5×106) at day 0, and isotype control (Iso ctrl) (black) or anti-HVEM18-10 antibody at 2 mg/kg (blue) concentration were injected. (C) UMAP analysis of anti-HVEM18-10 (blue) or Iso ctrl (Iso, gray). Density analysis revealed phenotypic modification between anti-HVEM treatment and Isotype treatment. (D) PhenoGraph clustering was performed on tumor infiltrating lymphocytes (TILs) from anti-HVEM18-10 and Iso ctrl-treated mice and represented as a UMAP. Nineteen meta-clusters were identified among T cells (top). Treg (light blue), conv. CD4+ T cell (dark blue), and CD8+ T cell (green) cluster gating on a UMAP (bottom). (E) Heatmap representation of marker expression among the 19 identified PhenoGraph meta-clusters. Cluster were associated with Tregs (light blue), conv. CD4+ T cells (dark blue), and CD8+ T cells (green). Bar charts represent cluster variation between anti-HVEM18-10 treatment compared with Iso ctrl in Tregs (F), conv. CD4+ T cells (G), and CD8+ T cells (H). *P<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Anti-HVEM-therapy builds a tumor-specific memory T cell response associated with tumor antigen responsive T cells
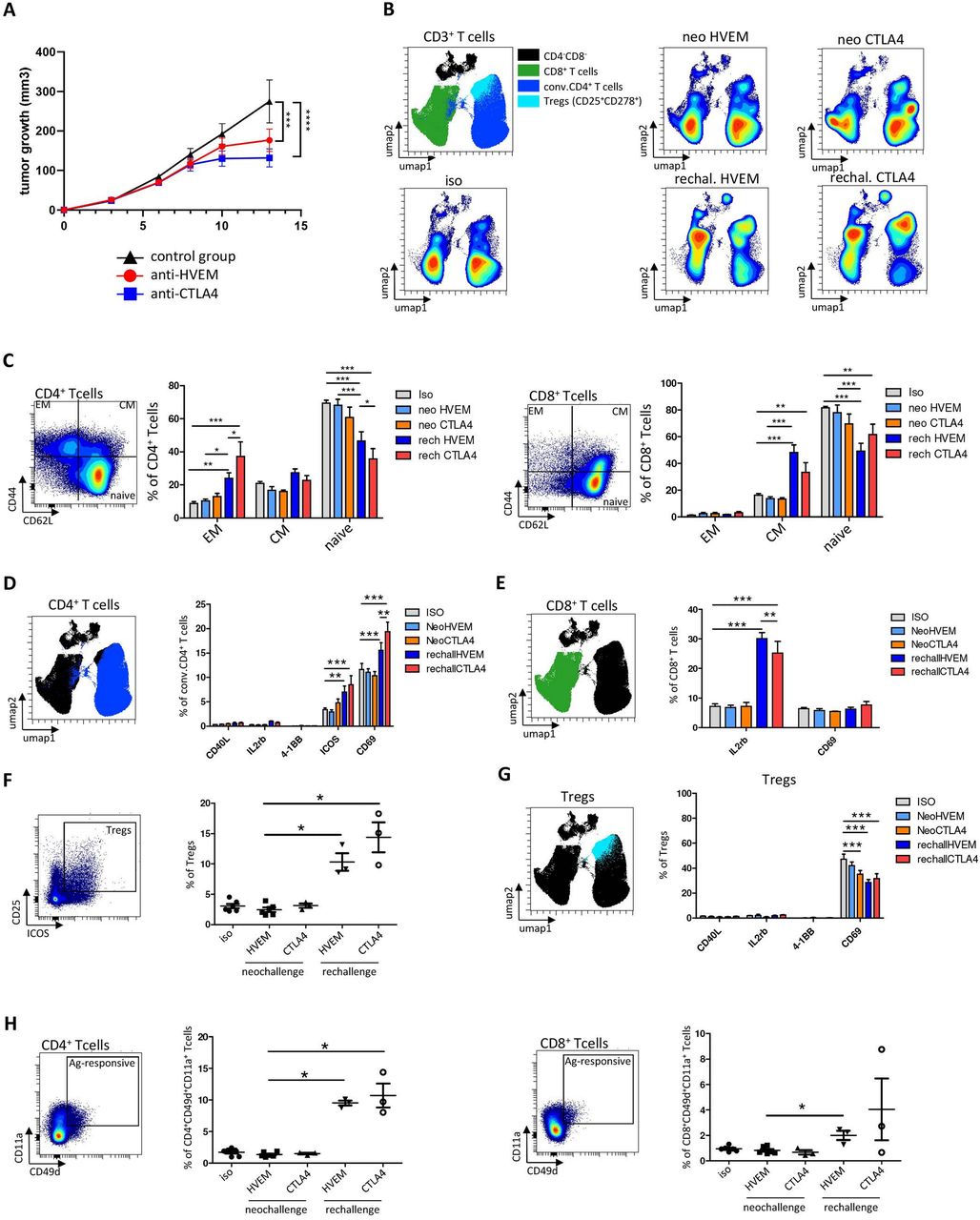
We next decided to compare in vivo the effect of anti-HVEM response to a well-known checkpoint inhibitor anti-CTLA-4 on tumor growth and T cell activation. DKI mice were challenged with 2×106 MC-38huHVEM tumor cells as previously described, then injected with an isotype control, anti-HVEM18-10 (2 mg/kg), or anti-CTLA-4 (2 mg/kg) at days 7 and 10. Tumor infiltrating and draining lymph node (LN) T cell phenotype was analyzed at day 14. As expected, the injection of anti-HVEM18-10 or anti-CTLA-4 decreased tumor growth and led to total tumor rejection in three mice under anti-HVEM and anti-CTLA-4 treatment (figure 8A and online supplemental figures 5A–C). Hence, tumor-free mice respectively from anti-HVEM and anti-CTLA-4 treatment were rechallenged with 6×106 cells MC-38huHVEM tumors cells in contralateral flank. Interestingly, tumors did not grow in these mice 14 days after rechallenge. Therefore, we hypothesized that primary tumor rejection built an anti-tumor T cell memory response that led to rechallenge rejection. Hence, we compared T cell response in rechallenged mice following anti-HVEM18-10 and anti-CTLA-4 treatments. Draining LN were resected and dissociated then T cell phenotypes were assessed by flow cytometry. T cell mapping was drastically modified after rechallenge compared with neo-challenge as shown by density UMAPs (figure 8B). Then, memory T cell subsets were gated (using CD44 and CD62L expression) and neo-challenged and rechallenged mice treated with anti-HVEM or anti-CTLA-4 were compared. We found that rechallenged treated mice either for anti-HVEM18-10 or anti-CTLA-4 were enriched in EM CD4+ T cells (CD44+CD62-L-) compared with their neo-challenged counterpart and isotype control (figure 8C, left). This increase was significantly superior in rechallenged CTLA-4-treated compared with rechallenged HVEM-treated mice (figure 8C, left). Noteworthy, naïve CD4+ T cells decreased after rechallenge. Concerning CD8+ T cells, we found that rechallenged mice with anti-HVEM18-10 or anti-CTLA-4 showed more CM CD8+ T cells (CD44+CD62-L+) compared with their neo-challenged counterpart and isotype control (figure 8C, right and online supplemental figure 5D). This increase tended to be superior in rechallenged HVEM-treated compared with rechallenged CTLA-4-treated mice. Again, naïve CD8+ T cells decreased as well on treatments. Taken together, these results show an enrichment in memory CD8+ and CD4+ T cells in rechallenged conditions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Anti-HVEM18-10 therapy builds a memory T cell response associated with CD4+ and CD8+ tumor antigen responsive T cells. (A) Tumor growth profile overtime following, isotype control (black), anti-CTLA-4 (blue), or anti-HVEM18-10 (red) treatment. (B) Lymph nodes (LN) were dissociated and cell stained for flow cytometry. UMAPs represent the phenotypic distribution of T cells within LN in isotype control mice (n=6), anti-HVEM18-10 (n=6), or anti-CTLA-4 (n=3) treated neo-challenged mice or anti-HVEM18-10 (n=3) or anti-CTLA-4 treated rechallenged mice (n=3). (C) Study of effector memory (EM), central memory (CM), and naïve conventional (conv.) CD4+ and CD8+ T cells among neo-challenged and rechallenged conditions (D) Study of immune checkpoint expression in conv. CD4+ T cells among neo-challenged and rechallenged conditions. (E) Study of immune checkpoint expression in CD8+ T cells among neo-challenged and rechallenged conditions. (F) Regulatory T (Treg) cells frequency among neo-challenged and rechallenged conditions. (G) Study of immune checkpoint expression in Tregs among neo-challenged and rechallenged conditions. (H) Study of CD49d+CD11a+ tumor-specific CD4+ and CD8+ T cells in neo-challenged and rechallenged conditions. *P<0.05; **p<0.01; ***p<0.001.
Next, we investigated activation marker expression on CD4+ and CD8+ T cells from LN (figure 8D,E and online supplemental figure 5E). In CD4+ T cells no modification in the expression of CD40L, IL-2Rβ, or 4-1BB was observed. However, the expression of inducible costimulator (ICOS) in rechallenged mice after anti-HVEM18-10 or anti-CTLA-4 treatment increased compared with controls. Similarly, CD69+CD4+ T cells were more abundant in rechallenged mice compared with controls. Noteworthy, CD69 overexpression was greater in anti-CTLA4 rechallenged mice compared with anti-HVEM rechallenged mice (figure 8D). In CD8+ T cells, an overexpression of IL2-Rβ in rechallenged mice compared with controls was observed. Here, IL2-Rβ expression was greater in anti-HVEM18-10 rechallenged mice compared with anti-CTLA-4 rechallenged mice (figure 8E). This suggests a better response to IL-2 or IL-15 and consequently more CD8+ T cell activation. Noteworthy, we did not notice any difference in the expression of CD69 on CD8+ T cells (figure 8E). Next, the Treg population within LNs after rechallenge was investigated. Treg frequency increased after rechallenge in both anti-HVEM18-10 and anti-CTLA-4 mice compared with anti-HVEM18-10 neo-challenged group (figure 8F). Last, a decrease in the expression of CD69 in Treg was observed, suggesting less activation or recruitment of peripheral blood Treg (figure 8G). Taken together, these results show that rechallenged mice rejected secondary tumor inoculation thanks to a strong T cell memory response linked to an increased expression of IL-2Rβ and a recruitment of tumor responsive CD8+ and CD4+ T cells.
Recently, the CD11a+CD49d+ CD4+ and CD8+ T cells were reported as tumor antigen responsive T cells, which participate to antitumor response.21 Therefore, we gated CD11a+CD49d+ CD4+ and CD8+ T cells (figure 8D and online supplemental figure 5F) and showed that tumor antigen responsive CD4+ T cells increased on rechallenge with anti-HVEM18-10 and anti-CTLA-4 mice (figure 8H) compared with controls. Interestingly, tumor antigen responsive CD8+ T cells significantly increased after rechallenge with anti-HVEM18-10 only (figure 8H). Taken together, our results show that the antitumoral memory response that allowed tumor rejection relies on enrichment in CD4+ and CD8+ memory subpopulations, increase of IL-2Rβ/activation markers, and more importantly on tumor antigen-specific T cell subsets, which most likely allow secondary tumor rejection.
Discussion
HVEM is a molecular switch as its effect depends on the ligand involved that is largely expressed in lung and colorectal tumors. Here, we showed that anti-HVEM18-10 antagonist mAb potentiates in vitro T cell proliferation and activation by blocking preferentially the interaction with the inhibitory ligands of HVEM. Then, we developed an innovative mouse model expressing both human BTLA and human HVEM. Anti-HVEM18-10 injection in mice induced the development of a marked T cell-memory phenotype and tumor antigen-specific (CD49d+CD11a+) T cells contributing in HVEM+ tumor reduction or rejection, especially after rechallenge. Therefore, HVEM targeting is a great addition to the currently available arsenal of IT.
To assess the clinical relevance of HVEM targeting in lung and colorectal tumors, we screened HVEM expression public transcriptomic databases. We found that HVEM was largely expressed in these cancers and did not correlate with PD-L1 expression. Surprisingly, the literature reporting HVEM expression in lung and colorectal cancers remains sparse.8 14 22 A more detailed multiparametric study, like was shown for pancreatic cancer23 would participate to the improvement of tumor type stratification and define who could benefit from HVEM18-10 mAb treatment.
In our in vitro preclinical settings, we showed that the anti-HVEM18-10 mAb increased primary human αβ-T cells activity alone (CIS-activity) or in presence of HVEM-expressing lung or colorectal cancer cells in vitro (TRANS-activity). Thus, we observed an immune reactivation similarly to that triggered by anti-PD-1 or anti-CTLA-4 treatment, favoring the idea that HVEM18-10 mAb avoids HVEM interaction with its inhibitory ligands. Indeed, our model do not allow to appreciate fully the effect of the binding of HVEM18-10 mAb on huCD160 and huLIGHT interaction with HVEM. The same observation was made with anti-HVEM18-10, which allowed γδ-T cells activation, especially against HVEM+ lymphoma cells.15 In the light of these observations, the use of HVEM18-10 mAb to potentiate anti-tumor T cell responses in hematological cancer as well as in solid cancers is of great interest.
Interestingly, anti-HVEM18-10 synergized with anti-PD-L1 effects and maximized T cells responses when tumor cells expressed both targets. Therefore, anti-HVEM18-10 could be used in combination with other ICI therapies and reinforce the still growing arsenal of ICI combinations in ongoing trials anti-CTLA4/anti-PD-L124 anti-TIGIT/anti-PD-L1,25 and anti-BTN3A1/anti-PD-L1.26 Nevertheless, we showed that anti-HVEM18-10 alone is still sufficient to trigger efficient T cells responses in PD-L1− conditions. Thus, anti-HVEM18-10 may increase the target patient population independently of their PD-L1 status and PD-L1− cancers such as colorectal cancer may benefit from this new treatment.
Recently, anti-HVEM IT was investigated in a prostate cancer mouse model,27 where Aubert et al showed that anti-HVEM18-10 reduced the growth of a HVEM+ tumors in NOD.SCID.gc-null mice reconstituted with human T cells. In our study, we generated innovative immunocompetent mouse models expressing extracellular domains of huBTLA or both huBTLA and huHVEM. This, allowed preclinical experiments settings using human anti-HVEM18-10 in immunocompetent mice bypassing the use of human T cell reconstitution in nude mice. TIL phenotype showed a decrease in Treg infiltration, CD8+ T cells exhaustion, and an increase of EM CD4+ T cells, expanding results from Aubert et al in prostate tumors27 to colon cancer. In addition, anti-CTLA4 treatment showed similar phenotypic modifications, which was already reported in the literature.19 20 Our new mouse models allowed the study of therapeutic mAb directly in an immunocompetent and dynamic environment. To date, only one recent study described a KI mouse model expressing human PD-1 and CTLA-4 molecules.28 The number of these syngeneic mouse models will increase in the future being closer to the clinic.
In our settings, around 20% of the mice completely rejected the tumor enabling the study of memory response induced by the rechallenge. Within tumor draining LN, we found a specific T cell memory composition, marked by an increase in IL-2Rβ+ CM CD8+ T cells and EM CD4+ T cells in comparison to neo-challenged mice. More importantly, draining LN sheltered a population of CD49d+CD11a+ CD8+ and CD4+ lymphocytes, which were recently described as tumor antigen-specific T cells.29 In a previous collaborative study, we showed that the addition of anti-HVEM18-10 was sufficient to decrease prostate cancer tumor growth and this effect was reverted by CD8 depletion.27 Thus, the antitumoral effect triggered by anti-HVEM18-10 is mainly due to CD8+ T cells. Therefore, anti-HVEM18-10 treatment leads to a systemic, tumor antigen-specific, T cell memory response allowing the mice to control distal secondary tumor implantation, similar to CTLA4 or anti-PD-1 treatment in other studies.30–33
Conclusion
HVEM appears to be a very promising IT target for oncological and hematological malignancies. Anti-HVEM18-10 mAb treatment demonstrated that antitumor immune response was strengthened, delays tumor growth or eradicate tumors, and induces a memory immune response in a cutting-edge preclinical mouse model. Altogether, these results highlight the interest of anti-HVEM IT alone or in combination with another IT to further enhance antitumor immunity.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Animal experiments were performed in accordance with institutional committees and French and European guidelines for animal care with reference number APAFIS number 6IS1-20160n02339418 vS.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @MyeLau_Gorvel, @NUNESJacques1, @Geoffguittard
CD, LG and MM contributed equally.
Correction notice This article has been corrected since it was first published online. In the original article Daniel Olive was not listed as a co-corresponding author. This has now been corrected.
Contributors DO, FB, and HL conceived the study and designed the experiments with input from all authors. CD, LG, and MM performed most experiments. SP, CD, AZ, FA, and FF provided technical support. JAN, BM, LG, and GG contributed intellectual input and helped to interpret data. DO led the research program. CD, LG, GG, and DO prepared the manuscript with input from all authors. DO. is the guarantor of this study.
Funding This study was supported by the RHU PIONEER (ANR-17-RHU5-0007), France 2030. LG and CD are supported by the RHU PIONEER (ANR-17-RHU5-0007), France 2030. CIPHE is supported by the Investissement d’Avenir program PHENOMIN (ANR-10-INBS-07). GG was supported by The fondation Bristol-Myers Squibb and Canceropole PACA emergence funding A*MIDEX Institut Cancer et Immunologie (AMX-19-IET-001).
Competing interests DO is a cofounder and shareholder of Imcheck Therapeutics, Alderaan Biotechnology, Emergence Therapeutics, and Stealth IO. HL is a co-founder and scientific advisor of JC discovery. FB reports payment or honoraria for lectures, presentations, speaker bureaus, manuscript writing, or educational events from AstraZeneca, Bayer, Bristol-Myers Squibb, Boehringer Ingelheim, Eli Lilly Oncology, F Hoffmann-La Roche, Novartis, Merck, MSD, Pierre Fabre, Pfizer, and Takeda, outside the submitted work. The other authors do not declare any conflict of interest.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.