Article Text

Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
NK cells engineered to secrete IL-15 were found to be efficacious in clinical trials of patients with non-acute lymphoblastic leukemia (ALL) hematopoietic malignancies. However, their therapeutic potential in patients with B-ALL and the mechanisms underlying their efficacy in ALL were unknown.
WHAT THIS STUDY ADDS
We find that intrinsic blockade in type I interferon (IFN-I) production suppresses IL-15 expression and IL-15-induced NK surveillance in high-grade B-ALLs that overexpress the c-MYC oncoprotein. Importantly, MYC overexpression makes B-ALL cells sensitive to eradication by IL-15-producing NK cells.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our study rationalizes the extensive preclinical and clinical development of IL-15-secreting NK cell-based therapies for poor prognosis B-ALL subtypes that overexpress the difficult-to-drug MYC oncoprotein.
Introduction
B-cell acute lymphoblastic leukemia (B-ALL) is an aggressive form of childhood and adult lymphoid malignancy.1 With recent advances in targeted and cellular immunotherapies,2 3 90% of childhood B-ALL is curable. However, only 30%–40% of adults with B-ALL are cured.1 Specific pediatric and adult B-ALL subgroups including those with KMT2A or MYC/BCL2 rearrangements and hypodiploidy are challenging to treat.4 First-line chemotherapy, although effective in killing B-ALL cells, is toxic and worsens immunosuppression in many patients.5 Therefore, targeted strategies that concurrently kill leukemia cells while reversing immunosuppression in B-ALL patients must be developed. To this end, we must delineate the mechanisms dampening the anti-leukemia host immune responses in B-ALL.
Among immunosuppression pathways, we focus on the inactivation of IFN-I signaling and responses. The IFN-I pathway is one of the first lines of anticancer immune defenses, as exemplified in solid tumor models where genetic ablation of IFN-I receptor drives immunoediting and tumorigenesis.6 However, the biology and importance of IFN-I pathway in hematopoietic cancers including B-ALL is less understood.
Expression of IFN-I-stimulated genes (ISGs) was found to be differentially regulated across specific B-ALL subgroups.7 We and others found that targeted inactivation of driver oncogenes including MYC and treatment with epigenetic therapies induce autocrine production of IFN-Is from malignant B cells in vitro.8 9 However, these studies did not determine if expression of IFN-I signaling and ISGs are predictive of clinical outcome in B-ALL patients, the extent to which autocrine (B-cell) and paracrine (non-B-cell) IFN-I production, signaling, and/or responses are suppressed in B-ALL compared with their healthy counterparts, and whether such suppression could be reversed with IFN-I treatment.
Surprisingly, IFN-Is have not been used as single agent therapies for acute leukemia. IFN-Is were combined with bone marrow (BM) transplantation to prolong remission in patients with B-ALL.10 Recently, IFN-Is were found to enhance relapse-free survival (RFS) in acute myeloid leukemia (AML) patients who underwent allogeneic BM transplantation.11 We hypothesized that the poor efficacy of IFN-Is as a single agent therapy for acute leukemia results from reduction in IFN-I response from anti-leukemia immune subsets such as natural killer (NK) cells.12 We further postulated that the absence of IFN-I responding anti-leukemia immune cells in B-ALL is initiated by an intrinsic block in IFN-I production and/or response during primary leukemogenesis.
Here, we show that high expression of IFN-I response genes predicts favorable clinical prognosis in B-ALL patients. Using diagnosis samples from B-ALL patients and an aggressive MYC-driven B-ALL-prone transgenic mouse model with germline ablation of IFN-I signaling, we determine the mechanism(s) of suppression of IFN-I signaling during primary leukemogenesis and the consequences of this suppression on anti-leukemia immune surveillance. We observe that B-ALLs exhibit suppressed production of autocrine (B cell-derived) and paracrine (plasmacytoid dendritic cell (pDCs)-derived) IFN-Is. This intrinsic suppression in IFN-I production is sufficient for overt leukemogenesis. Among anti-leukemic immune subsets, NK cells are the most sensitive to suppression of IFN-I production in B-ALL due to the reduced production of the IFN-I-stimulated cytokine, interleukin (IL)-15, that is, produced by conventional DCs (cDCs) and is indispensable for NK-cell homeostasis.13 14 Suppression of IL-15 was more severe in B-ALL patients who expressed high levels of MYC.15
Translating our observations, we modified our novel CRISPRa-based NK-cell platforms16 to engineer soluble IL-15-producing NK cells that kill B-lymphoblasts and can potentially persist in B-ALL microenvironments devoid of IL-15 and IFN-Is. We find IL-15-producing allogeneic NK cells to be an effective approach to reverse the detrimental effects of natural suppression of IFN-I production in certain high-risk B-ALLs.
Methods
Patient samples
Deidentifiedhi diagnosis B-ALL patient BM mononuclear cells (BMMCs) and peripheral blood mononuclear cells (PBMCs) were obtained from the City of Hope Hematopoietic Tissue Biorepository and University of Pennsylvania Stem Cell and Xenograft Core after informed consent per Institutional Review Board policies. Age-matched healthy BMMCs were purchased from AllCells (Alameda, California, USA) and Stem Cell Technologies (Vancouver, Canada). Age-matched healthy PBMCs were isolated from buffy coats procured from the City of Hope Michael Amini Transfusion Medicine Center. Patient samples analyzed include a new cohort in online supplemental table S1 and a cohort reported in our recent publication.12
Supplemental material
Cell lines and cell culture
Mycoplasma-negative human cell lines (NK-92, K562, SEM, KOPN8, P493-6, VAL, and MHH-CALL4) were obtained from American Type Culture Collection or Deutsche Sammlung von Mikroorganismen und Zellkulturen and cultured in Roswell Park Memorial Institute (RPMI)-1640 with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin (complete RPMI, Invitrogen/Life technologies). NK-92 cells were cultured in the presence of recombinant human IL-2 (100 U/mL, R & D Systems). In P493-6 cells, MYC expression is tetracycline-regulated17 and MYC was inactivated by treating cells with 0.2 µg/mL doxycycline for 24 hours.
Mouse strains
To generate Eμ-Myc (hemizygous)/IFNAR1−/− mice, Eμ-Myc males were crossed with IFNAR1−/− females, and F1 progeny Eμ-Myc/IFNAR1+/− males were backcrossed with IFNAR1−/− females. Transnetyx conducted genotyping. Eμ-Myc and Eμ-Myc/IFNAR1−/− mice were monitored for visible symptoms of B-ALL including hunched posture, shortness of breath, visible lumps, and ruffled fur. Spleen and BM cells were harvested from euthanized B-ALL mice and cryopreserved in FBS containing 10% dimethyl sulfoxide and stored in liquid nitrogen.
Cytometry
For flow cytometry surface staining, cells were thawed in complete RPMI and stained with fluorochrome-tagged surface antibodies and Ghost Dye UV 450 for 30 min on ice, followed by acquisition on a BD FACSymphony cytometer. For IFNα2b detection, cells were rested in complete RPMI for 3 hours at 37°C and treated with 3 µM class C CpG oligodeoxynucleotides (CpG ODN) for 2 hours at 37°C. After adding eBioscience Protein Transport Inhibitor Cocktail (1X) for 3 hours, cells were incubated overnight in 100 µL/mL of 20 mM EDTA at 4°C. The next day, cells were washed, surface stained, fixed and permeabilized using BD fix/perm buffer kit and incubated with IFNα2b antibody for 1 hour at room temperature (RT). To stain pSTAT1, cells were incubated at 37°C for 30 min, stimulated with IFNβ for 15 min, and fixed with 1% paraformaldehyde (Biolegend). Cells were permeabilized with 0.5X Perm Buffer IV (BD Biosciences), washed, and stained with anti-pSTAT1 antibody for 1 hour at RT. Data were acquired on BD FACSymphony cytometer and analyzed using FlowJo V.10.7.1.
Mass cytometry was carried out as before.12 18 Thawed samples, split into ‘unstimulated’ and ‘stimulated’ conditions, were rested overnight. ‘Stimulated’ cells were incubated with Phorbol 12-myristate 13-acetate (PMA)/Ionomycin for 4 hours. During incubation, brefeldin A and monensin (MilliporeSigma) were added in both conditions. After incubation, 2 mM EDTA was added for 15 min. Washed cells were stained for surface proteins and intracellular cytokines, followed by DNA staining with Cell-ID Intercalator-Ir (Standard BioTools). Data were normalized using MATLAB (https://github.com/nolanlab/bead-normalization/releases) and analyzed using Cytobank (Beckman Coulter,19). Online supplemental tables S2 and S3 contain cytometry antibodies.
Adoptive transfer of NK cells into B-ALL-bearing mice
Syngeneic NK cells were isolated from healthy UBC-GFP mice using Miltenyi Biotec mouse NK-cell isolation kit per manufacturer’s instructions. On onset of visible signs of B-ALL, 7×105 NK cells or PBS was intravenously injected into Eμ-Myc mice and leukemia-free survival was measured.
NK-cell proliferation and cytotoxicity
CRISPRa-engineered NK-92 cells were generated as described previously.16 To measure NK-cell proliferation, control-single guide RNA and IL-15-single guide RNA (sgRNA) transduced NK-92 cells were cultured in triplicates without (100,000 cells/well) or with 100 IU/mL IL-2 (50,000 cells/well) in complete RPMI-1640 media in a flat bottom 96-well-plate for 96 hours. After excluding dead cells using trypan blue, live cells were counted using a hemocytometer.
To measure NK cytotoxicity, targets were labeled with 2.5 µM carboxyfluorescein succinimidyl ester–violet dye and cocultured with either CRISPRa control-sgRNA or IL-15-sgRNA transduced NK-92 cells at an effector-to-target ratio of 10:1 in complete RPMI. After 5 hours, cells were stained with 7-aminoactinomycin D (7-AAD). Cytotoxicity was measured on BD FACSymphony flow cytometer and analyzed using FlowJo V.10.7.1. Specific cytotoxicity = ((7-AAD+target cell frequency in coculture with effector cells−7-AAD+ target cell frequency alone)/(100–7-AAD+ target cell frequency alone))×100.
ELISA
1×106 control-sgRNA and IL-15-sgRNA transduced NK-92 cells were stimulated with PMA (5 ng/mL) and ionomycin (0.5 µg/mL) for 24 hours at 37°C. IL-15 level was measured in culture supernatant using high-sensitivity human IL-15 ELISA kit (#41702) from PBL Assay Science as per manufacturer’s instructions.
Ex vivo treatment of mouse splenocytes with IFN-Is
2×106/mL splenocytes were treated for 8 hours with 10,000 IU/mL IFNβ (R&D Systems) in complete RPMI, washed, followed by downstream analysis.
Administration of IFNβ to B-ALL-prone mice
Tail vein peripheral blood of Eμ-MYC B-ALL-prone mice aged ~7–20 weeks was drawn to measure frequencies of immune cells before administration of IFNβ. Mice were then treated intraperitoneally with 50,000 IU/100 µL of IFNβ or 100 µL PBS for 9 days. On day 9, mice were euthanized and circulating immune cells were collected by cardiac puncture for flow cytometry.
Lentivirus production and transduction
Firefly luciferase lentivirus was produced from Lenti-X 293T human embryonic kidney cells (Takara Bio) by transfecting with plasmids containing luciferase (pLenti CMV Puro LUC, w168-1; Addgene), packaging (psPAX2; Addgene), and envelope (pMD.2G; Addgene) genes using lipofectamine 2000 (Invitrogen) in serum free media for 3 hours. Cells were supplemented with addition of high glucose complete Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) media containing 100 IU/mL penicillin, 100 µg/mL streptomycin, 10% FBS, 25 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.2, 1 mM sodium pyruvate and 0.1 mM nonessential amino acids. Next day, media was removed, and cells were incubated in complete DMEM containing 20 mM sodium butyrate (Alfa Aesar) for 5 hours. After 5 hours of incubation, media was changed back to complete DMEM without sodium butyrate. Next day, viral culture supernatant was collected, passed through a 0.45 µm filter, and concentrated with Lenti-X Concentrator (Takara Bio) per manufacturer’s instructions. Concentrated lentivirus was resuspended in 1 mL complete RPMI media. A 500 µL of concentrated virus was mixed with 500 µL of complete RPMI media containing 2×106 P493-6 cells and 8 µg/mL polybrene (Millipore Sigma). After 48 hours, cells were washed, cultivated, and selected in puromycin (1 µg/mL)-containing complete RPMI.
B-ALL cell line-derived xenograft mouse model to test the efficacy of CRISPRa-IL-15-producing human NK cells
1×106 P493-6 cells were luciferase-labeled and injected intravenously into 4–6 weeks NOD-SCIDIL-2Rγ−/− (NSG) mice. Engraftment of P493-6 cells was confirmed by bioluminescence imaging (BLI) using Lago-X (spectral instruments imaging). D-luciferin monosodium salt dissolved in PBS was injected intraperitoneally at 2.5 mg/mouse 12 min before BLI. Mice were kept under general anesthesia (3%–5% isoflurane) during BLI. After verifying B-ALL engraftment on day 9, mice were intravenously injected with either CRISPRa control or IL-15-producing NK-92 cells (7×106 NK cells per mouse). BLI was performed twice a week to measure disease progression.
Immunoblotting
Cells were lysed in RIPA buffer (Sigma) supplemented with 1% protease inhibitor ‘cocktail’ (Pierce). Proteins were then separated by electrophoresis through 4%–20% TGX gradient gels (BioRad) and were transferred to polyvinylidene fluoride membranes (Immobilion; Millipore). For the detection of human proteins by immunoblot analysis, we used primary antibodies listed in online supplemental table S5 and western enhanced chemiluminescence (BioRad). Blots were developed in ChemiDoc MP imaging system (BioRad).
Quantitative real-time PCR (qPCR)
RNA was extracted using NucleoSpin RNA Plus kit *(MACHERY-NAGEL) per manufacturer’s instructions. cDNA was synthesized using SuperScript IV Reverse Transcriptase (Invitrogen). qPCR was conducted using Power SYBR Green PCR master mix (ThermoFisher Scientific) and QuantStudio V.7 flex real-time PCR system (Applied Biosystems). Online supplemental table S4 lists qPCR primers.
Statistics
Statistical tests are two tailed. Exact p values are provided if significant (p<0.05) or trending toward significance (0.05<p<0.1). Mann-Whitney U test was used for pairwise comparisons between mouse cohorts. Kaplan-Meier survival p values were calculated using log-rank test. Sample size was calculated using ‘cpower’ function in R package. Each biological qPCR sample was run in three technical replicates. Cell-line experiments were repeated three times for reproducibility.
Results
Intrinsic suppression of IFN-I production and response in B-ALL predicts poor clinical outcome
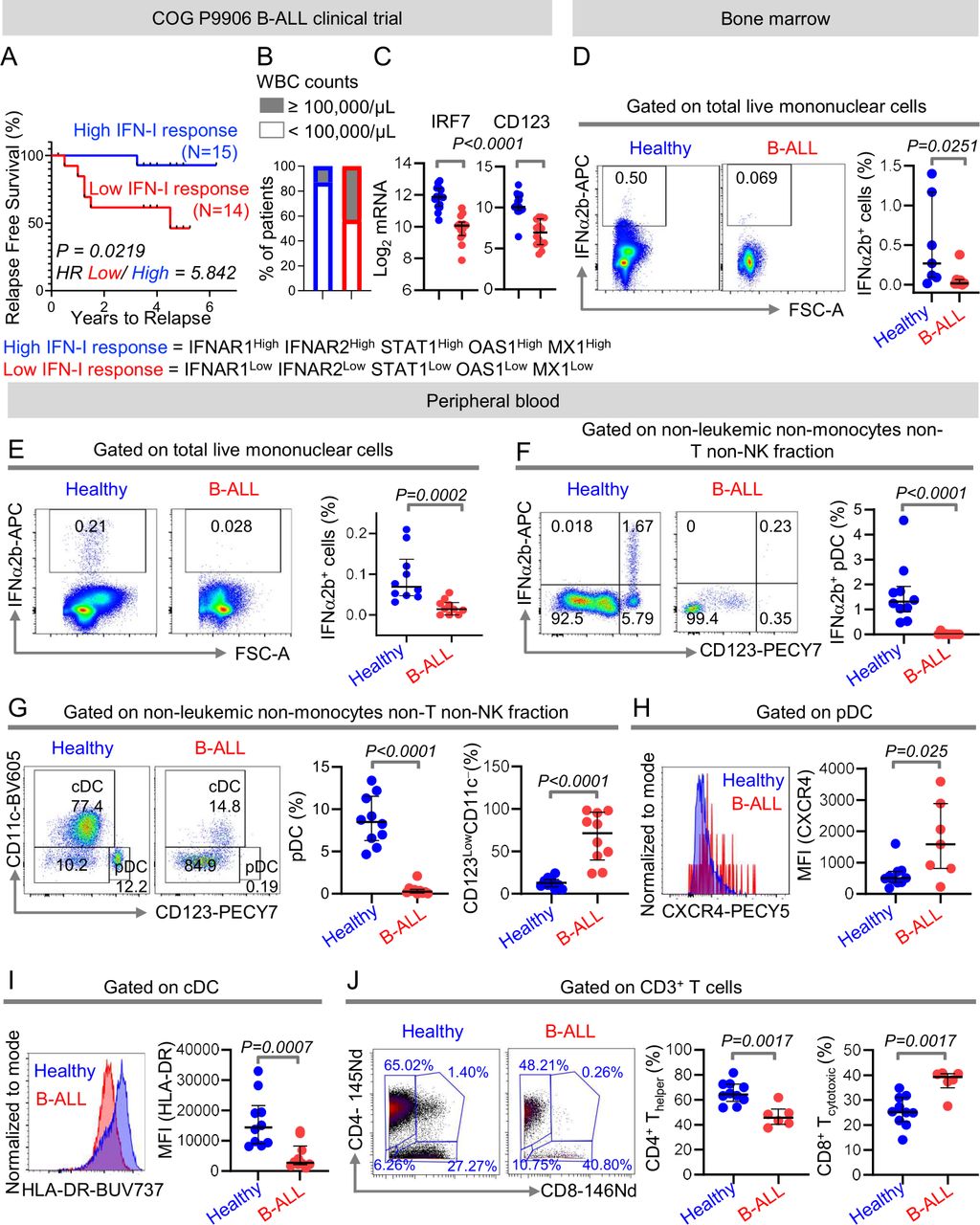
We hypothesized that expression of IFN-I signaling and response genes, being a measure of anti-leukemia immune surveillance, can predict clinical outcome in patients with B-ALL. To determine the importance of IFN-I signaling in B-ALL, we compared RFS probabilities of 207 children with high-risk B-ALL from the Children’s Oncology Group P9906 clinical trial20 21 divided into two groups based on their median expression of IFN-I signaling and response genes IFNAR1, IFNAR2, STAT1, MX1, and OAS1 at diagnosis. Patients with higher than median expression of all IFN-I signaling/response genes (n=15) had significantly longer RFS probabilities as compared with their IFN-I responselow counterparts (n=14) (figure 1A). Patients with high IFN-I response were ~3 times less likely (13%) to have white blood cell (WBC) counts of ≥ 100,000/μL that often correlate with poor clinical prognosis, compared with their low IFN-I response counterparts (43%) (figure 1B). Compared with better prognosis IFN-I responsehigh counterparts, poor prognosis IFN-I responselow B-ALL patients had reduced expressions of interferon regulatory factor 7 (IRF7), the gene indispensable for IFN-I production22 and CD123, a marker highly expressed by IFN-I producing immune cells23 24 (figure 1C). Consistent with the above, patients with high concomitant expression of IFN-I response genes, IRF7, and CD123 (n=13) fared better and were more likely to have WBC counts of < 100,000/μL at diagnosis compared with patients with low expression of IFN-I signaling genes, IRF-7, and CD123 (n=9) (online supplemental figure S1).

Intrinsic suppression of IFN-I production and response in B-ALL predicts poor clinical outcome. (A)Comparison of relapse-free survival probabilities of COG P9906 B-ALL patients separated into two groups that is, IFNAR1HighIFNAR2HighSTAT1HighOAS1HighMX1High (‘High IFN-I Response’, n=15) and IFNAR1LowIFNAR2LowSTAT1LowOAS1LowMX1Low (‘Low IFN-I Response’ n=14) based on the median transcript expression of these genes. (B)Stacked bar charts comparing the proportions of COG P9906 B-ALL patients with WBC counts ≥100,000/μL or WBC counts <100,000/μL within the ‘High IFN-I Response’ and ‘Low IFN-I Response’ cohorts. (C)Comparison of IRF7 and CD123 transcript levels within the ‘High IFN-I Response’ and ‘Low IFN-I Response’ cohorts. (D–F)Comparison of IFNα2b+ cells in class C CpG ODN-stimulated total BMMCs (D),total PBMCs (E),and within the HLA-DR+ non-B, non-monocytes, non-T, and non-NK immune cell fraction of PBMCs (F)between B-ALL patients (n=7 BMMC, n=10 PMBC) and healthy donors (n=7 BMMC, n=10 PMBC) by flow cytometry. (G)Comparison of peripheral blood pDC frequencies within the HLA-DR+ non-B, non-monocytes, non-T, and non-NK immune cell fractions between B-ALL patients (n=10) and healthy donors (n=10) by flow cytometry. (H)Median fluorescence intensity (MFI) of CXCR4 expression on peripheral blood pDCs of B-ALL patients (n=7) and healthy donors (n=10) by flow cytometry. (I)MFI of HLA-DR expression on peripheral blood cDC of B-ALL patients (n=10) and healthy donors (n=10). (J)Analysis of frequencies of CD4+ and CD8+ T cells within non-leukemic pan T-cell fraction of B-ALL patients (n=6) and healthy donors (n=10) using mass cytometry. For all flow cytometry experiments, one representative histogram or dot plot from each group is shown. Survival was calculated by Kaplan-Meier method and p value calculated by log-rank test. All other comparisons between any two groups were conducted using Mann-Whitney U test. Exact p values are provided whenever significant. B-ALL, B-cell acute lymphoblastic leukemia; BMMC, bone marrow mononuclear cell; cDC, conventional dendritic cell; PBMC, peripheral blood mononuclear cell; pDC, plasmacytoid DC; WBC, white blood cell.
The above findings suggested that B-ALL may be associated with an intrinsic defect in IFN-I production and consequently, in IFN-I mediated responses. Therefore, we compared the proportion of IFN-I (IFNα2b)-producing cells in ODN-stimulated tissue-matched and age- matched samples from healthy donors and B-ALL patients (online supplemental table S1) using high-dimensional flow cytometry (online supplemental figure S2A,B). We observed significant reductions in total IFN-I+ cells in BMMCs and PBMCs of B-ALL patients (figure 1D,E). Hence, the ALL microenvironment has suppressed IFN-I production.
We determined which IFN-I-producing immune subset(s) are suppressed in B-ALL. First, we measured IFN-I-producing cells in B-ALL PBMC samples, all of which had CD19+ leukemia, allowing distinction of leukemic and anti-leukemic immune fractions. Frequencies of IFNα2b+ and total pDCs, the ‘professional’ and highest IFN-I producer, were reduced within the non-leukemic immune fraction in stimulated B-ALL compared with healthy PBMC (figure 1F,G). Reduction in pDCs was compensated by an increase in the CD123lowCD11c− fraction (figure 1G). Residual pDCs in B-ALL patients expressed increased CXCR4 (figure 1H), a marker that correlates inversely with pDC’s IFN-I production potential.25 Next, we measured frequencies of total pDCs and IFNα2b+ within non-leukemic fraction only in CD19+ BM B-ALLs and found it to be significantly reduced compared with healthy donors (online supplemental figure S3A,B). Cytometry was not sensitive to detect IFN-I in stimulated B cells. We infer that the B-ALL microenvironment harbors an intrinsic defect in IFN-I production.
Our recent findings of impaired NK-cell maturation and cytotoxicity in B-ALL patients,12 provide a biological foundation for our hypothesis that IFN-I-driven immune responses are suppressed in B-ALL. Because IFN-Is drive immune homeostasis, we predicted that reduced IFN-I production in B-ALL may suppress immune surveillance beyond NK cells. Therefore, we analyzed phenotypes of non-NK immune subsets as readouts of dampened IFN-I production in B-ALL by flow and mass cytometry (online supplemental figure S4). We found reduced expression of major histocompatibility class II (MHCII), a downstream target of IFN-I response,26 in antigen-presenting DCs (figure 1I) and B cells (online supplemental figure S5) of B-ALL patients. Significantly reduced frequencies of CD4+ T helper cells within the CD3+ T-cell fraction (figure 1J) lead us to speculate that suppressed IFN-I production in B-ALL may impair antigen presentation (MHCII) and subsequent T helper-cell multiplication and anti-leukemia immune responses.27 28 Overall, IFN-I production is impaired in B-ALL patients and such impairment is associated with suppression of anti-leukemia immune surveillance and poor prognosis.
Intrinsic suppression of IFN-I production is sufficient to drive overt B-cell leukemogenesis
In solid tumor mouse models, germline deletion of the IFN-I receptor, IFNAR1, accelerates tumorigenesis.6 29 Whether IFNAR1 deletion accelerates B-cell leukemogenesis and the extent to which intrinsically suppressed IFN-I production in B-ALL (figure 1) impacts this process is unknown. To address this, we generated B-ALL-prone mice lacking both copies of the IFNAR1 (IFNAR1−/−/Eμ-MYC). Eμ-MYC was chosen because: (1) it models high-risk B-ALL driven by the ‘difficult-to-drug’ MYC oncogene,30 31 and (2) we showed MYC transcriptionally represses IFN-I production in malignant B cells in vitro,9 suggesting intrinsic suppression of IFN-I production in Eμ-MYC mice.
Contrary to observations in solid tumors,6 germline deletion of IFNAR1 did not accelerate B-ALL development (figure 2A). We predicted that unaltered latency to overt leukemogenesis after IFNAR1 deletion in Eμ-MYC mice may be caused by intrinsic suppression of IFN-I production. To test this, after validating the germline ablation of IFNAR1 in spleen and BM WBCs by quantitative real time PCR (qPCR) (figure 2B, online supplemental figure S6 and S7A), we compared splenic pDC frequencies and counts in healthy, Eμ-MYC, and IFNAR1−/−/Eμ-MYC mice. As predicted, pDCs were reduced to the same extent in B-ALL-bearing mice with or without the IFNAR1 compared with their healthy counterparts (figure 2C).

Intrinsic suppression of IFN-I production is sufficient to drive overt B-cell leukemogenesis. (A)Comparison of leukemia‐free survival between IFNAR1+/+ B-ALL-bearing (n=33) and IFNAR1−/− B-ALL-bearing (n=15) Eμ-Myc mice. (B)Quantitation of IFNαR1 and IFNαR2 transcript expression by qPCR in MACS sorted splenic B- and non-B cell fraction of wildtype (normal, B cell fraction, n=12; non-B cell fraction, n=11), IFNAR1+/+ Eμ-Myc B-ALL-bearing (B cell fraction, n=11; non-B cell fraction, n=10) and IFNAR1−/− Eμ-Myc B-ALL-bearing (B cell fraction, n=6; non-B cell fraction, n=5) mice. (C)Comparison of splenic pDC numbers and representative flow cytometry plots of normal (n=14), IFNAR1+/+ Eμ-Myc B-ALL-bearing (n=12) and IFNAR1−/− Eμ-Myc B-ALL-bearing (n=10) mice. (D–F)Quantitation of transcripts of IFNβ1, IFNα1 and IFNα2 in splenic B cells (D),and STAT1 and MX1 in splenic B- and non-B cell fractions (E, F)by qPCR. (G)Fold change in induction of STAT1 and MX1 transcripts after IFNβ stimulation in splenic WBCs of normal (n=9) and IFNAR1+/+ Eμ-Myc B-ALL-bearing (n=10) mice. (H)Histogram overlays and scatter plot showing the MFI and fold increase in MFI after IFNβ stimulation in splenic WBC from normal (n=5) and IFNAR1+/+ Eμ-Myc B-ALL-bearing mice (n=5). Ubiquitin (UB) was used as housekeeping gene in qPCR. For all flow cytometry experiments, one representative dot plot from each group is shown. Survival was calculated by Kaplan-Meier method and p value calculated by log-rank test. All other comparisons between any two groups were conducted using Mann-Whitney U test. Exact p values are provided whenever significant (<0.05) or trending to significance (0.05<p<0.1). B-ALL, B-cell acute lymphoblastic leukemia; MFI, median fluorescence intensity; pDC, plasmacytoid dendritic cell; WBCs, white blood cells.
We compared expression of IFN-I and ISG transcripts in BM and splenic WBCs of healthy, Eμ-MYC, and IFNAR1−/−/Eμ-MYC mice. Sufficient splenic cells were available to measure IFN-Is and ISGs in the leukemic (B-cell) and non-leukemic (non-B-cell) fractions separately (online supplemental figure S6). For BM, bulk WBCs were used. IFN-I transcript expression was reduced to the same extent in splenic leukemic fraction and total BM WBCs, in IFNAR1+/+ and IFNAR1−/− B-ALL-bearing mice (figure 2D, online supplemental figure S7B). IFN-I expression in the non-leukemic splenic fraction containing pDCs was unaltered, likely because pDCs form a very small proportion of this fraction and other cells in this fraction do not produce IFN-Is (online supplemental figure S8A). Expression of IFN-I signaling (STAT1), and response genes (MX1, OAS1) was suppressed independently of the IFNAR1 genotype in splenic leukemic and non-leukemic fractions and BM of B-ALL-bearing mice (figure 2E,F, online supplemental figure S7C and S8B,C).
To determine whether exogenous IFN-Is can rescue ISG suppression in B-ALL, we compared the expressions of STAT1, MX1, and activation/phosphorylation of STAT1 after ex vivo stimulation of primary splenic samples from healthy and B-ALL-bearing Eμ-MYC mice. IFN-I stimulation increased expression and activation of IFN-I pathway genes to the same extent (or more) in B-ALL spleens compared with healthy spleens (figure 2G,H). Hence, the natural defect in IFN-I pathway in B-ALL lies in the first step, that is, in IFN-I production and this defect precludes any downstream suppression of IFN-I responses, such as ablation of IFNAR1.
Reduced IFN-I production in B-ALL is sufficient to suppress IL-15 and systemically impair NK surveillance
We compared indicators of IFN-I-mediated anti-leukemia immune surveillance in spleen and BM of healthy, Eμ-MYC and IFNAR1−/− Eμ-MYC mice by flow cytometry (online supplemental figure S9). Consistent with the identical suppression of the IFN-I pathway and latency to overt leukemia in Eμ-MYC and IFNAR1−/− Eμ-MYC mice (figure 2), no significant enhancement in suppression of immune subsets was seen after genetic ablation of the IFNAR1 in Eμ-MYC mice. For example, B-ALL-bearing mice with and without IFNAR1 exhibited similar reduction in numbers of splenic NK1.1+NKp46+ NK cells, CD3+ pan T cells, CD4+ Th cells, CD8+ Tc cells as compared with normal mice (figure 3A, online supplemental figure S10A). While splenic cDC numbers were unaltered between healthy and B-ALL bearing mice, the frequency of cDCs expressing MHCII was reduced in B-ALL-bearing mice with or without IFNAR1 (online supplemental figure S10A,B). In BM, only NK cells were highly sensitive to the suppressed IFN-I production and were unaffected by IFNAR1 deletion. In contrast, T-cell numbers were reduced in BM of B-ALL-bearing mice only on IFNAR1 ablation (figure 3B, online supplemental figure S11).

Reduced IFN-I production in B-ALL is sufficient to suppress IL-15 and systemically impair NK surveillance. (A, B)Comparison of NK cell counts and representative plots of NK frequencies within the non-B, non-T and Gr1 negative cell fractions of the spleen (A)and bone marrow (B)of normal (spleen, n=14; bone marrow, n=12), IFNAR1+/+ Eμ-Myc B-ALL-bearing (spleen, n=12; bone marrow, n=8) and IFNAR1−/− Eμ-Myc B-ALL-bearing (spleen, n=10; bone marrow, n=8) mice. (C, D)Comparison of frequencies of NK subsets within the four-stage NK-cell effector maturation pathway in spleen (C)and bone marrow (D)of normal (spleen, n=14; bone marrow, n=12), IFNAR1+/+ Eμ-Myc B-ALL-bearing (spleen, n=12; bone marrow, n=8) and IFNAR1−/− Eμ-Myc B-ALL-bearing (spleen, n=10; bone marrow, n=8) mice. (E)Comparison of leukemia‐free survival between Eμ-Myc B-ALL-bearing mice treated with vehicle control or with syngeneic NK cells (n=6, each group). (F–H)Quantitation of IL-15 transcript expression by qPCR in MACS sorted splenic non-B cell fraction (F)and total bone marrow (G)of wildtype (normal, spleen, n=11; bone marrow, n=7), IFNAR1+/+ Eμ-Myc B-ALL-bearing (spleen, n=10; bone marrow, n=7) and IFNAR1−/− Eμ-Myc B-ALL-bearing (spleen, n=5; bone marrow, n=6) mice. (H)Fold change in induction of IL-15 transcript in IFNβ-stimulated splenic WBCs of normal (n=9) and IFNAR1+/+ Eμ-Myc B-ALL-bearing (n=10) mice. Ubiquitin (UB) was used as housekeeping gene in qPCR. For all flow cytometry experiments, one representative dot plot from each group is shown. All other pairwise comparisons between any two groups were conducted using Mann-Whitney U test. Exact p values are provided whenever significant (<0.05) or trending to significance (0.05<p<0.1). B-ALL, B-cell acute lymphoblastic leukemia; NK, natural killer; WBCs, white blood cells.
Because NK cells are consistently reduced independently of the IFNAR1 status in two different leukemia microenvironments, we focused on IFN-I-driven NK responses. IFN-Is positively impact NK cell effector maturation both by directly binding to the IFNAR1/2 on NK cells32 33 and indirectly through induction of IL-15 expression in cDCs.13 Frequencies of the cytotoxic and more mature CD11b+CD27+ effector NK subset were significantly reduced and those of less cytotoxic, CD11b−CD27−and CD11b−CD27+ immature NK fractions were significantly increased in spleen and BM of B-ALL mice independent of IFNAR1 genotype (figure 3C,D). These observations suggested that NK cell-based therapies would be attractive to overcome the intrinsic suppression of IFN-I production in B-ALL. As predicted, administration of syngeneic healthy NK cells prolonged leukemia-free survival of B-ALL-bearing Eμ-MYC mice compared with their counterparts who received no NK cells (figure 3E, online supplemental figure S12).
In a transgenic mouse model of IFN-Ilow, MYC-inducible primary T-ALL, we showed that administration of IFN-Is restores NK surveillance and improves survival and NK cells mediate IFN-I’s anti-leukemic effects.9 To determine the extent to which reduced IFN-I production drives NK suppression in B-ALL, we administered IFNβ to B-ALL-prone Eμ-MYC mice aged 7–20 weeks for 9 days. In each mouse, we measured the fold change in percentages of NK cells, NK-cell effectors, and B-cells (ALL) before and after administration of PBS (control) or IFNβ. Administration of IFNβ to B-ALL prone mice reduces the leukemia progression and increases the frequencies of total NK cells and cytotoxic CD27+CD11b+ NK-cell effectors in circulation (online supplemental figure S13). Changes we find induced in NK subsets after IFNβ administration phenocopy those shown to be induced by IL-15 treatment.34
IL-15 is an IFN-I-induced cytokine indispensable for NK-cell survival, proliferation, effector maturation, and immune surveillance.13 35 IL-15 expression in splenic non-leukemic fraction and total BM of both IFNAR1+/+ and IFNAR1−/− B-ALL-bearing mice was equally reduced (figure 3F,G), strengthening our observations that IFNAR1 loss does not worsen NK suppression in B-ALL. Next, we determined the extent to which ex vivo addition of IFN-Is to splenic leukocytes from B-ALL mice can stimulate IL-15 transcription in B-ALL. IFN-I treatment induced IL-15, although to a lesser extent in the B-ALL microenvironment compared with their healthy counterparts (figure 3H). We speculate that the inability of ex vivo IFN-I stimulation to induce IL-15 production to the same extent in B-ALL and normal splenic microenvironments (figure 3H) could result from alterations in IFN-I-responsive, IL-15-producing cDCs in B-ALL.
Overall, natural suppression of IFN-I production during B-leukemogenesis is sufficient to systemically suppress IL-15 expression and impair NK surveillance. NK cell-based therapies16 36 37 thus represent a good therapeutic strategy to overcome the intrinsic suppression of IFN-I and IL-15-induced NK surveillance in B-ALL.
Expression of IL-15 inversely correlates with MYC expression in human B-cell malignancies
We showed suppressed NK-cell numbers and effector maturation in patients with B-ALL.12 Together with the intrinsic suppression of IFN-I production in B-ALL patients (figure 1), our findings12 suggested that the IFN-I-induced, NK surveillance-activating cytokine, IL-15,13 35 may be reduced in patients with B-ALL. Unfortunately, antibodies to compare human IL-15 protein expression in healthy individuals and B-ALL patients are unavailable.38 As a surrogate for IL-15 expression, we compared frequencies of potential IL-15-producing cDCs within the non-leukemic fraction of B-ALL patients and healthy donors. cDCs were significantly reduced in PBMC of B-ALL patients (figure 4A). Reduced pDC-derived IFN-Is and cDC-derived, IFN-I-induced cytokine, IL-15, suggest IL-15-producing NK cells may be therapeutically efficacious in B-ALL.36 39

Expression of IL-15 inversely correlates with MYC expression in human B-cell malignancies. (A)Comparison of peripheral blood cDC frequencies within the HLA-DR+ non-B, non-T, and non-NK immune cell fraction of B-ALL patients (n=10) and healthy donors (n=10) by flow cytometry. (B)Linear regression analyzing the correlation between MYC and IL-15 transcript expression and plots of MYC and IL-15 mRNA expression in B-ALL patients with three major subtypes of leukemia from three independent data sets: EGAS00001003266 (ETV6::RUNX1 (n=187); ETV6::RUNX1-like (n=42); Ph/Ph-like (n=482), MYC-BCL2-driven /KMT2A-rearranged /Hypodiploid (n=237); 672 children and 240 adults, 36 unknown), the COG P9906 GSE11877 (ETV6::RUNX1 (n=3); others including Ph-like (n=155); KMT2A-rearranged (n=21); all pediatric) and in MILE GSE13159 (MYC driven+MLL (n=83); ETV6::RUNX1 (n=94); Ph+ (n=122)) data sets. (C)Comparison of transcript levels of MYC and IL-15 in classical MYC-driven Burkitt’s lymphoma/leukemia and non-MYC-driven B-cell lymphoma types from GSE132929: (Burkitt’s lymphoma (n=59) and non-Burkitt’s lymphoma (n=231). Non-MYC-driven (non-Burkitt’s) lymphomas include diffuse large B-cell lymphoma (n=95), follicular lymphoma (n=65), unspecified high-grade B-cell lymphoma (n=4), mantle cell lymphoma (n=43), double hit lymphoma (n=1), and marginal zone lymphoma (n=23)). B-ALL, B-cell acute lymphoblastic leukemia; cDC, conventional dendritic cell; NK, natural killer.
We asked whether IL-15 is differentially suppressed across the B-ALL subtypes with widely divergent outcomes. To this end, we compared the expression of IL-15 transcripts in major subgroups of childhood and adult B-ALL from three independent datasets.4 20 21 40–42 IL-15 expression was significantly reduced in patients with MYC or BCL2 translocations, KMT2A-rearrangements, and hypodiploidy in comparison to Ph+, Ph-like, and ETV6::RUNX1 subtypes (figure 4B). Expression of IL-15 correlated negatively with MYC expression in B-ALL subtypes (figure 4B). The converse relationship between IL-15 and MYC expression was validated in B-cell lymphomas: classical MYC-driven burkitt’s lymphoma (BL) expressed lower IL-15 than other subtypes (figure 4C). These observations are concordant with our previous findings that MYC directly represses IFN-I signaling.9 We infer that strong suppression of IL-15 in MYC-overexpressing human B-ALLs might render them particularly sensitive to the administration of NK cell-based therapies, as shown in mice in figure 3E.
CRISPRa-engineered IL-15-secreting NK cells eradicate MYC-overexpressing B-ALLs in vitro
IL-15-producing NK therapies were found to be effective in preclinical studies and clinical trials of AML, chronic lymphocytic leukemia (CLL), and BL.36 39 43 We investigated whether IL-15-producing NK cells are also efficacious in eradicating human B-ALL, specifically B-ALL subtypes characterized by high MYC expression. To engineer IL-15-producing NK cells for proof-of-concept studies, we used CRISPRa to transcriptionally activate IL-15 in the NK-cell line, NK-92.16 CRISPRa-mediated transcriptional activation of IL-15 in human NK cells has advantages over lentiviral overexpression.36 43 First, it is easier to introduce a ~20 nt sgRNA to activate IL-15 transcription by CRISPRa as opposed to transducing NK cells with an overexpression vector encoding IL-15. Second, IL-15 transcript levels increase by ~3-fold and secreted IL-15 is 2 pg/mL in CRISPRa IL-15 NK cells (figure 5A), which is much lower than that seen in NK cells engineered to overexpress IL-15.36 43 Hence, cellular toxicity caused by excessive production of IL-1543 can be avoided in CRISPRa.

CRISPRa-engineered IL-15 secreting NK cells eradicate MYC-overexpressing B-ALLs in vitro. (A)Fold change in IL-15 transcript levels by qPCR in dCas9-VP64-GFP+ NK-92 cells transduced with control sgRNA-RFP (Control NK) or IL-15 sgRNA-RFP (IL-15 NK) and levels of secreted IL-15 by ELISA in culture supernatant of control NK and IL-15 NK cells stimulated with PMA and Ionomycin for 24 hours. (B)Cell counts of control and IL-15 NK cells cultured in the presence and absence of rhIL-2 (100 U/mL) for 96 hours. (C, D)Specific cytotoxicity of control and IL-15 NK cells by flow cytometry using (C)K562, (D)SEM (KMT2A-rearranged), KOPN8 (KMT2A-rearranged), MHH-CALL4 (hypodiploid, CRLF2-rearranged), P493-6, and VAL as target cell lines. Effector: target=10:1. (E)Specific cytotoxicity of control and IL-15 NK cells by flow cytometry against MYC-overexpressing (MYCON) and MYC-inactivated (MYCOFF) P493-6 cells. MYC was inactivated by treatment of P493-6 cells with 0.2 µg/mL of doxycycline for 24 hours. p=0.0108, control (MYCON) vs IL-15 NK (MYCON); p=0.0037, control (MYCOFF) vs IL-15 NK (MYCOFF). All experiments were conducted in three technical and three biological replicates. One representative of three biological replicates of each experiment is shown. Comparisons between any two groups were conducted using Student’s t-test (A–E).Exact p values are provided whenever significant (<0.05) or trending to significance (0.05<p<0.1). B-ALL, B-cell acute lymphoblastic leukemia; NK, natural killer; ns, not significant.
CRISPRa-IL-15-NK cells are comparable in their properties and functionality to NK cells overexpressing IL-15.43 Transcriptional activation of IL-15 improved the proliferation of IL-2-dependent NK-92 cells in the presence and absence of IL-2 (figure 5B). CRISPRa-IL-15-NK cells killed NK-sensitive erythroleukemia target cell line K-562 more effectively compared with control sgRNA-modified counterparts (figure 5C).
Because MYC-overexpressing B-ALL express the lowest IL-15, we measured the efficacy of our CRISPRa-IL-15-NK cells in killing MYChigh B-ALL cells in vitro. CRISPRa-IL-15-NK cells lysed 4/5 MYChigh B-ALL samples at significantly higher efficiency as compared with CRISPRa-control sgRNA-NK cells. One MYC/BCL2-rearranged B-ALL line was already highly sensitive to NK cell-mediated clearance, and the addition of IL-15 did not improve lysis (figure 5D). Control sgRNA and CRISPRa-IL-15-NK cells did not kill allogeneic PBMCs isolated from healthy donors, indicating that our engineered NK cells specifically kill ALL targets, and not normal immune cells (online supplemental figure S14).
Next, we determined the therapeutic potential of IL-15-producing NK cells in MYClow B-ALLs, known to have more intact IFN-I signaling than their MYC-overexpressing counterparts9 (online supplemental figure S15). To this end, we compared the cytotoxicities of CRISPRa control and IL-15-NK cells against MYC-overexpressing (MYCON) and MYC-inactivated (MYCOFF) malignant B cells.17 MYCOFF (IFN-Ihigh) B-lymphoblasts were less sensitive to eradication by both control and IL-15 NK cells compared with their MYCON (IFN-Ilow) counterparts. However, the enforced expression of IL-15 in NK cells significantly enhanced its potential to kill both MYCON (IFN-Ilow) and MYCOFF (IFN-Ihigh) B-lymphoblasts (figure 5E). Thus, MYC overexpression is at least one factor that sensitizes B-lymphoblasts to NK cell-based therapies. CRISPRa-engineered IL-15 NK cells thus represent a potentially safe and particularly efficacious approach to treat poor prognosis MYC-overexpressing, IFN-Ilow B-ALLs.
CRISPRa-engineered IL-15-secreting NK cells slow down progression of MYC-overexpressing B-ALL in vivo
We strengthened the therapeutic potential of IL-15-producing NK cells in poor prognosis MYC-overexpressing B-ALL by comparing the in vivo efficacy of CRISPRa-engineered IL-15 human NK cells and control NK cells in a cell-line derived xenograft model of human MYC-driven B-lymphoblasts. We transplanted luciferase-labeled MYC-overexpressing malignant human B cells into immune deficient NSG mice. After confirming B-lymphoblast engraftment by BLI, we administered CRISPRa-control or CRISPRa-IL-15 NK cells and compared disease progression between the two groups by BLI (figure 6A). CRISPRa IL-15-secreting NK cells significantly slowed down progression and spread of engrafted human B-lymphoblasts compared with control NK cells in vivo (figure 6B,C). Thus, IL-15-producing NK cells warrant extensive preclinical and clinical studies for the treatment of high-risk B-ALL subgroups that overexpress MYC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CRISPRa-engineered IL-15-secreting NK cells slow down progression of MYC-overexpressing B-ALL in vivo. (A)Experimental design for comparing the in vivo therapeutic efficacies of CRISPRa-engineered IL-15 producing and control NK cells in a luciferase-labeled human cell line-derived xenograft (CDX) of MYC-driven B-cell malignancy (P493-6). Schematic was created using BioRender.com. (B)Bioluminescence imaging of leukemia progression in NOD-SCID IL2Rγ−/− (NSG) CDX recipients before (day 9 post-CDX transplantation) and after treatment with control and IL-15 NK cells (days 12 and 15 post-CDX transplantation) (n=6 mice each in control NK and IL-15 NK arms). (C)Quantification of fold change in full body bioluminescence (photons/sec/cm2/sr) on days 12 and 15 post B-lymphoblast transplantation in control-NK (n=6) and IL-15-NK- treated (n=6) CDX recipients. P values were calculated by Mann-Whitney U test. Exact significant p values (<0.05) are provided. B-ALL, B-cell acute lymphoblastic leukemia; NK, natural killer.
Discussion
Via these studies, we provide new evidence for the natural suppression of IFN-I production during primary murine and human B-cell leukemogenesis. In addition to reduced IFN-I transcription by malignant B cells seen here and in our earlier studies,9 we find pDC-derived paracrine IFN-I production is reduced in patients and mice with B-ALL. This blockade in the first step of the IFN-I pathway is sufficient to drive leukemogenesis and overrides any additional downstream disruption of the IFN-I pathway such as, ablation of IFNAR1 shown here. The clinical relevance of IFN-I suppression in human B-ALL is underscored by the favorable prognosis of B-ALL patients who express high levels of ISGs.
Our comparisons in IFNAR1+/+ and IFNAR1−/− B-ALL-bearing mice suggest that suppression of IFN-I production is likely caused by B-lymphoblasts. We previously showed that MYC suppresses IFN-I production by malignant cells and blocks NK surveillance in an MYC-inducible T-ALL mouse model.9 Similarly, MYC-induced signaling in B-lymphoblasts may perturb cytokines required for pDC differentiation in B-ALL. Another explanation for reduction of pDCs in B-ALL microenvironments could be the MYC-driven accumulation of B-lymphoid precursors thereby suppressing the lymphoid lineage pDCs.44 However, reduction in myeloid pDCs cannot be ruled out especially given the plasticity between the lymphoid and myeloid pDCs.44 How oncogenic pathways perturb pDC homeostasis remains to be explored.
Among non-leukemic host immune cells, NK cells are maximally impacted by the suppressed IFN-I production in B-ALL likely because of the reduced synthesis of IL-15 by IFN-I-responding cDCs. While ex vivo treatment of primary B-ALL samples with IFN-Is fully rescues proximal IFN-I signaling, its partial induction of IL-15 was possibly due to absence or functional suppression of cDCs. Further studies are needed to delineate the mechanisms underlying the suppression of cDCs in B-ALL. As with suppression of NK surveillance shown by us,12 we speculate that oncogenic pathways in leukemia cells may drive suppression of cDCs in ALL.
Our results partly explain why IFN-Is are effective when administered to acute leukemia patients who undergo allogeneic BM transplantation10 11 45: intrinsically suppressed IFN-I production in B-ALL may reduce IFN-I-dependent anti-leukemia immune subsets, including NK cells and cDCs, thereby rendering single agent IFN-I therapy ineffective. We maintain that IL-15-producing NK cells, represent a novel, effective, and affordable approach to achieve the combined therapeutic benefits of IFN-I administration and BM transplantation for several reasons: (1) graft versus leukemia effects in allogeneic transplants are known to be largely mediated by NK cells,46 (2) allogeneic NK cell-based therapies do not induce graft-vs-host disease, cytokine release syndrome, or neurotoxicity,47 and (3) and we find suppressed IL-15 production and NK surveillance to be the most striking consequences of IFN-I loss in B-ALL.
Our studies suggest that CRISPRa-engineered IL-15-producing NK cells represent an efficacious and potentially safe approach to reverse the detrimental effects of suppressed IFN-I, IL-15 production and NK surveillance in high-risk B-ALL (graphical abstract). While IL-15-producing NK cells were shown to be effective in preclinical studies of AML, CLL, and BL,36 39 43 we are the first to delineate the biology underlying its efficacy in B-ALL and demonstrate its ability to eradicate B-ALL subtypes with highest MYC and lowest IL-15 expression including MYC/BCL2-rearranged, KMT2A-rearranged, and hypodiploid. We find that MYC overexpression sensitizes B-ALLs to NK cell-mediated killing. Hence, IL-15-producing NK cells may be a viable alternative to drugging MYC in these high-grade B-ALL subtypes.48
The lack of reliable cytometry antibodies for mouse and human intracellular IL-1538 and mouse IFN-Is precluded us from measuring these proteins in specific immune subsets in the B-ALL microenvironment. This problem was mitigated for CRISPRa-IL-15-NK cells by measuring secreted IL-15 and conducting phenotypic (proliferation) and functional (cytotoxicity against NK-sensitive erythroleukemia) validation of these cells.
Because of the relative ease in constitutively expressing dCas9-VP64,16 NK-92 lymphoma cells49 were used instead of primary NK cells to engineer CRISPRa IL-15-NK cells. CRISPRa-engineered and other primary IL-15-producing NK cell-based therapies therefore await further preclinical and clinical development. In this study, we did not measure the efficacy of IL-15-secreting human NK cells in prolonging survival of mice bearing B-ALL xenografts. Such studies would require repeat injections of the IL-15 NK cells or their further manipulation to increase persistence in human ALL xenograft-bearing mice. Nevertheless, our current mechanistic findings will accelerate the development of primary IL-15-producing NK therapies37 50 for sustaining remission of hard-to-treat human B-ALL.
Supplemental material
Data availability statement
Data are available in a public, open access repository. Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information. GSE numbers are indicated for all analyses involving previously published microarray and RNA sequencing data sets in figure legends. The following information has been provided for the deidentified B-ALL patient samples used in our study: Patient I.D., age, gender, tissue type, cytogenetics, translocation or mutation status, disease status, the originating institution, and frequencies of IFNalpha2b+ cells wherever available. For mass and flow cytometry-based immune cell profiling, we have provided details of markers, labels, and gating strategy in Supplemental Information. We have also provided detailed protocols for all experiments and analyses in the Methods Section of the manuscript. Renewable materials can be obtained by directly contacting the corresponding author at sswaminathan@coh.org.
Ethics statements
Patient consent for publication
Ethics approval
Mice were purchased from Jackson Laboratories and maintained in the City of Hope’s laboratory animal resource facility under approval of Institutional Animal Care and Use Committee (IACUC 19032). This study is classified as non-human subjects research under the City of Hope IRB19373.
Acknowledgments
We thank the Hematopoietic Tissue Biorepository (HTB) of City of Hope, the Bass Center for Childhood Cancer and Blood Diseases at Stanford University, and the Stem Cell and Xenograft Core (SCXC) at the University of Pennsylvania for providing us primary B-ALL patient specimens. We thank the staff at the Human Immune Monitoring Center at Stanford University and at the Analytical Cytometry Core of the City of Hope. We thank Dr. Mark Davis (Stanford University) for access to PBMCs from an influenza vaccine study and Dr. Eleanora Heisterkamp (City of Hope) for providing us K562 cells for conducting in vitro cytotoxicity experiments. Schematics were created using BioRender.com.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @anilsharma085, @sswamin17
Contributors AK conducted most of the experiments, analyzed data, and interpreted the results. ATK conducted experiments and analyzed clinical data from publicly available B-ALL datasets. CD conducted CyTOF-based immune profiling of patient samples, analyzed data, and interpreted the results. SA assisted AK with NK cell therapy adoptive transfer experiments in primary mouse models. ASO, SJL and AC assisted AK with CRISPR-based NK-cell modifications. TM and MH collected, processed and curated samples from consented B-ALL patients used for flow cytometry and CyTOF. KMS, NJL, MC, SKT, LG and GM provided access to fully deidentified coded samples with associated clinical annotation collected on IRB-approved research protocols. JY and MAC provided guidance on NK-cell biology. KMS, NJL, MC, SKT, GM, LG, CH, SA, STR, SI and ZG provided scientific input on ALL biology, immune microenvironment in ALL, and/or IFN-I signaling. CW-C provided guidance on CRISPR. All authors edited the manuscript. SS, SJF and HTM provided scientific guidance, administrative, technical, and material support. SS conceived the study, developed the experimental methodology, interpreted the results, wrote the manuscript, and supervised the study. SS is responsible for the overall content as guarantor.
Funding This work was supported by the following grants: Special Fellow (LLS 3366-17) and Translational Research Program (LLS 6624-21) Awards from The Leukemia and Lymphoma Society (SS), American Society of Hematology Scholar Award (SS), PhRMA Foundation Research Starter Grant in Drug Discovery (SS), Childhood Cancer Research Grant from the Andrew McDonough B+ Foundation (SS), City of Hope Chancellor’s Research Grant (SS), P50 CA107399-12 National Cancer Institute-City of Hope Lymphoma SPORE Career Enhancement Program Pilot Award from the National Institutes of Health (PIs: SJF and Larry Kwak, Pilot Awardee: SS), a Research Start-Up Budget from the Beckman Research Institute of the City of Hope (SS), and 1U24CA224309 from the National Institutes of Health (HTM). The Hematopoietic Tissue Biorepository and Analytical Cytometry Core at the City of Hope are Shared Resource Cores supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572. SKT is a Scholar of the Leukemia and Lymphoma Society and holds the Joshua Kahan Endowed Chair in Pediatric Leukemia Research at the Children’s Hospital of Philadelphia. SI is supported by the Israel Cancer Research Fund, City of Hope and Professorship award. CH is supported by NCI 5K22CA251649-02, Rally Foundation for Childhood Cancer, V Foundation for Cancer Research, and the Leukemia Research Foundation.
Disclaimer The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests The work described in this study is covered by pending US and PCT patent applications assigned to City of Hope and/or Stanford University with inventors SS, AK, ATK, CW-C, SJL, CD and HTM. MAC and JY are cofounders of CytoImmune Therapeutics.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.