Article Text

Abstract
Background We assessed the efficacy and safety of avelumab, an anti-programmed death ligand 1 (PD-L1) antibody, in patients with previously treated metastatic adrenocortical carcinoma (mACC).
Methods In this phase 1b expansion cohort, patients with mACC and prior platinum-based therapy received avelumab at 10 mg/kg intravenously every 2 weeks. Continuation of mitotane was permitted; however, mitotane levels during the study were not recorded. Tumor response was assessed by Response Evaluation Criteria In Solid Tumors v1.1.
Results Fifty patients received avelumab and were followed for a median of 16.5 months. Prior treatment included ≥2 lines in 74.0%; mitotane was continued in 50.0%. The objective response rate (ORR) was 6.0% (95% CI, 1.3% to 16.5%; partial response in 3 patients). Twenty-one patients (42.0%) had stable disease as best response (disease control rate, 48.0%). Median progression-free survival was 2.6 months (95% CI, 1.4 to 4.0), median overall survival (OS) was 10.6 months (95% CI, 7.4 to 15.0), and the 1-year OS rate was 43.4% (95% CI, 27.9% to 57.9%). In evaluable patients with PD-L1+ (n = 12) or PD-L1− (n = 30) tumors (≥5% tumor cell cutoff), ORR was 16.7% vs 3.3% (P = .192). Treatment-related adverse events (TRAEs) occurred in 82.0%; the most common were nausea (20.0%), fatigue (18.0%), hypothyroidism (14.0%), and pyrexia (14.0%). Grade 3 TRAEs occurred in 16.0%; no grade 4 to 5 TRAEs occurred. Twelve patients (24.0%) had an immune-related TRAE of any grade, which were grade 3 in 2 patients (4.0%): adrenal insufficiency (n = 1), and pneumonitis (n = 1).
Conclusions Avelumab showed clinical activity and a manageable safety profile in patients with platinum-treated mACC.
Trial registration Clinicaltrials.gov NCT01772004; registered January 21, 2013.
- Avelumab
- Adrenocortical carcinoma
- PD-L1
- Phase 1
- JAVELIN solid tumor
- Neuroendocrine tumors
Christophe Le Tourneau and Christopher Hoimes contributed equally to this work.
- ACC
- Adrenocortical carcinoma
- AE
- Adverse event
- ALT
- Alanine aminotransferase
- AST
- Asparate aminotransferase
- CCNE1
- Cyclin E1
- CI
- Confidence interval
- CNS
- Central nervous system
- CR
- Complete response
- ECOG PS
- Eastern Cooperative Oncology Group performance status
- GGT
- γ-glutamyltransferase
- HR
- Hazard ratios
- mACC
- Metastatic adrenocortical carcinoma
- mTOR
- Mammalian Target of Rapamycin
- NE
- Not evaluable
- NF1
- Neurofibromin 1
- ORR
- Objective response rate
- OS
- Overall survival
- PD-1
- Programmed death 1
- PD-L1
- Programmed death ligand 1
- PFS
- Progression-free survival
- PRKAR1A
- Protein kinase cAMP-dependent type 1 regulatory subunit alpha
- RECIST
- Response Evaluation Criteria In Solid Tumors
- RPL22
- Ribosomal protein L22
- SAE
- Serious adverse event
- TERF2
- Telomeric repeat binding factor 2
- TERT
- Telomerase reverse transcriptase
- TKI
- Tyrosine-kinase inhibitor
- TRAE
- Treatment-related adverse event
- VEGF
- Vascular endothelial growth factor
- ZNRF3
- Zinc and ring finger 3
Open AccessThis article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Statistics from Altmetric.com
Christophe Le Tourneau and Christopher Hoimes contributed equally to this work.
Background
Adrenocortical carcinoma (ACC) is a rare endocrine tumor with an incidence of approximately 0.7 cases/1 million population per year [1]. Genomic studies indicate that mutations in defined driver genes, including zinc and ring finger 3 (ZNRF3), telomerase reverse transcriptase (TERT), protein kinase cAMP-dependent type I regulatory subunit alpha (PRKAR1A), ribosomal protein L22 (RPL22), telomeric repeat binding factor 2 (TERF2), cyclin E1 (CCNE1), and neurofibromin 1 (NF1), may have a major role in the etiology of ACC [2], and that ACC has a moderate overall mutation burden compared with other tumor types [3]. Patients with ACC have a poor prognosis, with an estimated 5-year survival rate for metastatic ACC (mACC) of < 20% [4–6]. Therapeutic options for mACC are limited and include surgery; platinum agents, mitotane, streptozocin, and other systemic therapies; and locoregional radiotherapy [7]. Although cytotoxic treatments are widely administered, they are associated with limited efficacy and high toxicity, and targeted agents have not shown clinically meaningful activity in this disease [8]. Thus, there is an urgent need for new treatment options.
Cancer cells can exploit the programmed death 1 (PD-1)/programmed death ligand 1 (PD-L1) immune checkpoint pathway to promote an immunosuppressive environment and allow tumor growth. Specifically, PD-L1 expressed on tumor cells binds to PD-1 expressed on activated T cells, resulting in downregulation of antitumor immune responses [9, 10]. Monoclonal antibodies targeting the PD-1/PD-L1 axis have shown clinical activity in multiple tumor types [11]. PD-L1 is expressed in ACC tissues throughout various stages of disease, providing a rationale for assessing anti–PD-1/PD-L1 antibodies in mACC [12].
Avelumab is a human IgG1 monoclonal antibody with a wild-type Fc region that was designed to specifically bind and block PD-L1 [13]. Unlike other anti–PD-L1/PD-1 antibodies in clinical practice, avelumab is unique in that preclinical models show that it can induce antibody-dependent cellular cytotoxicity of tumor cells [14, 15]. Thus, avelumab may engage both the adaptive and innate immune systems. Avelumab treatment has been associated with durable responses in several tumor types [16–18], and avelumab has been approved in various countries for the treatment of patients with metastatic Merkel cell carcinoma and in the United States and Canada for treatment of locally advanced or metastatic urothelial carcinoma that progressed during or after platinum-containing chemotherapy [19]. Here we report results from a phase 1b cohort of patients with previously treated mACC who received avelumab.
Methods
Study design and treatment
JAVELIN Solid Tumor (ClinicalTrials.gov ID NCT01772004) in an international, multicenter, phase 1, open-label trial designed to investigate the clinical activity, safety, and pharmacokinetics of avelumab in patients with metastatic solid tumors, with expansion in selected tumor types. Findings from the dose-escalation portion of this study (phase 1a) were reported previously [13]. In this phase 1b dose-expansion cohort, eligible patients were aged ≥18 years and had an Eastern Cooperative Oncology Group performance status (ECOG PS) of ≤1; histologically or cytologically confirmed mACC with ≥1 measurable lesion; ≥1 line of prior systemic therapy for metastatic disease (including ≥1 platinum-based therapy); and relapsed, refractory, or progressive disease following last line of treatment. Patients receiving mitotane were permitted to continue treatment during the trial; however, details relating to ongoing mitotane treatment was not systematically collected and mitotane levels during the study were not recorded. Patients were also required to have adequate hematologic, hepatic, and renal function and availability of a fresh or archival tumor specimen. Exclusion criteria included prior treatment with a T cell-targeting immune checkpoint inhibitor, other cancer diagnosis within 5 years, rapidly progressive disease, CNS metastases, known autoimmune disease, or ongoing steroid treatment (except for patients with adrenal insufficiency, who could continue treatment at a physiological replacement dose).
The trial was conducted in accordance with the ethics principles of the Declaration of Helsinki and the International Council for Harmonisation Guidelines on Good Clinical Practice. The protocol was approved by the institutional review board or independent ethics committee of each center. All patients provided written informed consent before enrollment.
Procedures
Avelumab (EMD Serono, Rockland, MA) 10 mg/kg was administered intravenously over 60 mins every 2 weeks until progression, unacceptable toxicity, or other protocol-based criteria for withdrawal occurred. Dose modifications were not permitted. Treatment was discontinued following: 1) any grade 4 adverse event (AE) except for single laboratory values out of the normal range that were unrelated to study treatment, without clinical correlate, and resolved within 7 days with medical management; 2) any grade 3 AE except for: (i) transient (≤6 h) influenza-like symptoms or pyrexia controlled with medical management; (ii) fatigue, local infusion-related reaction, headache, nausea, or emesis that resolved to grade ≤ 1 within 24 h; (iii) single laboratory values out of the normal range that were unrelated to study treatment and without clinical correlate (excluding grade ≥ 3 increase in liver enzyme concentrations) that resolved to grade ≤ 1 within 7 days; (iv) tumor flare phenomena (local pain, irritation, or localized rash at a tumor site); (v) grade ≥ 3 amylase or lipase abnormality not associated with symptoms or clinical manifestations of pancreatitis; or 3) increase in Eastern Cooperative Oncology Group performance status to ≥3 not resolved to ≤2 by Day 14 of the following cycle. Grade 2 AEs were managed by dose delays; events that did not resolve to grade ≤ 1 by the end of the next treatment cycle or recurred led to permanent discontinuation of avelumab (except for hormone insufficiencies that could be managed by replacement therapy). A premedication regimen of diphenhydramine and acetaminophen was administered 30 to 60 min before all avelumab infusions.
Safety was assessed at each biweekly trial visit and included assessment of AEs, physical examination, and clinical laboratory tests. AEs and laboratory abnormalities were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.0. A serious AE (SAE) was defined as any untoward event that was life-threatening, required hospitalization, resulted in disability, was a congenital anomaly, or resulted in death. Immune-related AEs were identified using a prespecified list of Medical Dictionary for Regulatory Activities terms.
Clinical activity was assessed by investigators using Response Evaluation Criteria In Solid Tumors (RECIST) v1.1 [20] and modified immune-related response criteria [21]. Radiographic tumor assessments were completed at baseline and then every 6 weeks. For patients who had a partial response (PR) or complete response (CR), a confirmatory computed tomography or magnetic resonance imaging scan was completed ≥28 days later, preferably at the scheduled 6-week interval. PD-L1 expression was assessed using a proprietary immunohistochemistry assay (Dako PD-L1 IHC 73–10; Dako, Carpinteria, CA) [18].
Outcomes
Primary endpoints for the whole JAVELIN Solid Tumor trial are the occurrence of dose-limiting toxicities during the first 3 weeks of treatment in the phase 1a dose-escalation part (reported previously) [13] and confirmed best overall response adjudicated by an independent review committee in specified expansion cohorts. Prespecified endpoints assessed in this cohort included best overall response per investigator assessment (defined as best response per RECIST v1.1 obtained among all tumor assessments after the start of treatment with avelumab until documented disease progression), immune-related best overall response, duration of response (defined as the time from first documented PR or CR until progressive disease or death, whichever occurred first), progression-free survival (PFS), overall survival (OS), evaluation of PD-L1 expression, and safety (including incidence and severity of AEs). Change in the sum of target lesion diameters from baseline over time was evaluated in patients with baseline tumor assessments and ≥ 1 postbaseline assessment.
Statistical analysis
Enrollment of 50 patients was planned for this cohort; this sample size was selected to provide point estimates and 95% Clopper-Pearson CIs for an objective response rate (ORR; proportion of patients with a PR or CR) of 10% (95% CI, 3.3% to 21.8%) in the case of 5 responders, and 20% (95% CI, 10.0% to 33.7%) in the case of 10 responders. Safety and clinical activity were analyzed in all patients who received ≥1 dose of avelumab. Time-to-event endpoints were estimated with the Kaplan-Meier method, and CIs for the median were calculated using the Brookmeyer-Crowley method. P values for association between categorical variables were determined using the Fisher exact test.
Results
Between September 9, 2014, and the data cutoff date of December 31, 2016, 50 patients were enrolled in 6 countries. Median age was 50 years (range, 21 to 71 years) (Table 1). Patients were heavily pretreated; the median number of prior lines of systemic therapy was 2 (range, 1 to 6), and 37 patients (74.0%) had received ≥2 prior lines. Date of disease progression before enrollment was documented in 49 patients, and median time since last disease progression was 0.92 months (range, 0.33 to 8.61 months). Twenty-five patients (50%) continued to receive concurrent mitotane. Median duration of avelumab treatment was 3.4 months (range, 0.5 to 24.8 months), and median follow-up was 16.5 months (range, 11.7 to 27.6 months). Patients received a median 7 doses of avelumab (range, 1 to 52). At data cutoff, 5 patients (10.0%) remained on treatment. Reasons for treatment discontinuation were disease progression (n = 32 [64.0%]), AE (n = 5 [10.0%]), death (n = 3 [6.0%]), withdrawal of consent (n = 2 [4.0%]), protocol noncompliance (n = 1 [2.0%]), and other (n = 2 [4.0%]).
Patient Demographics and Baseline Characteristics
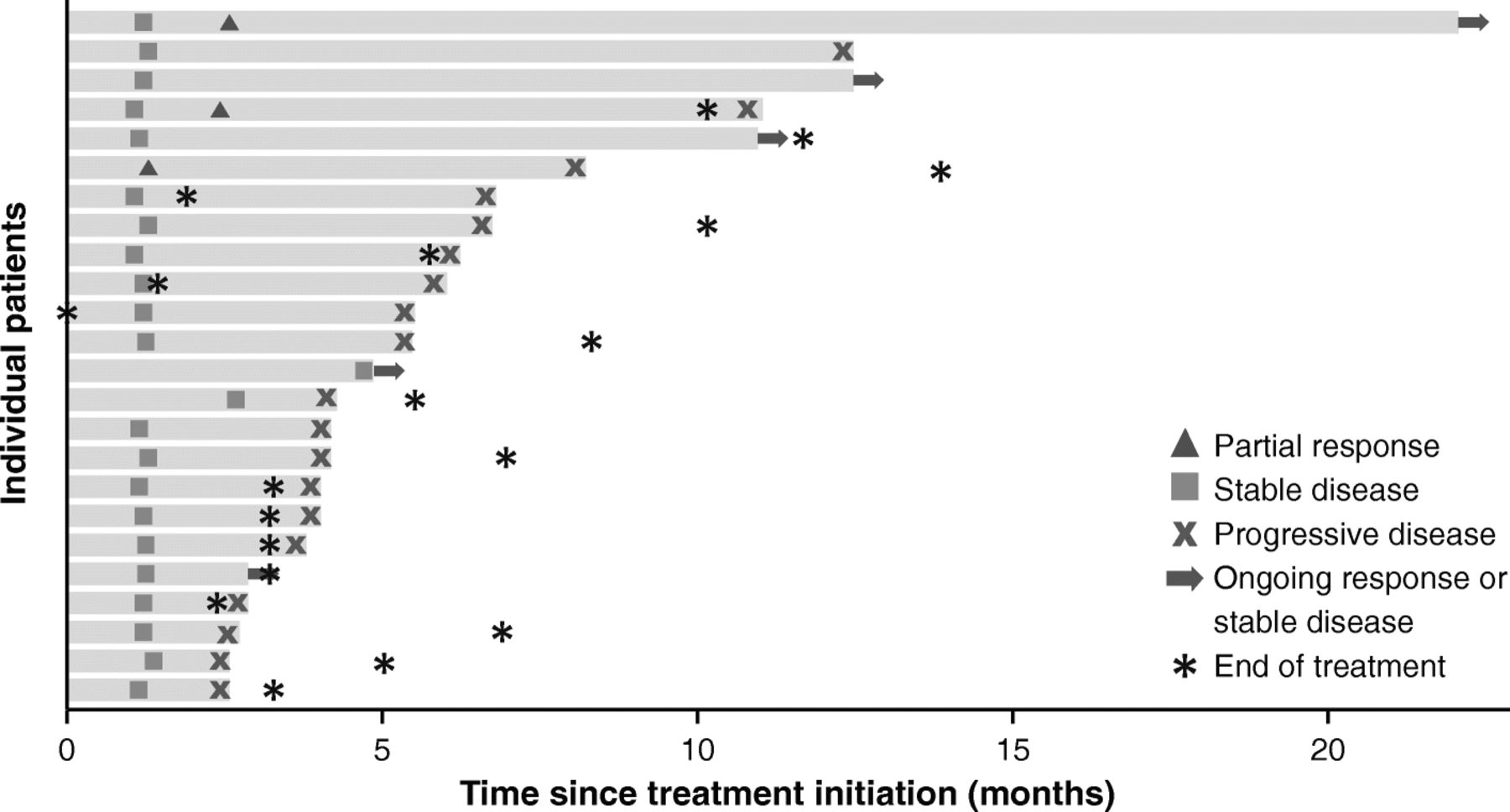
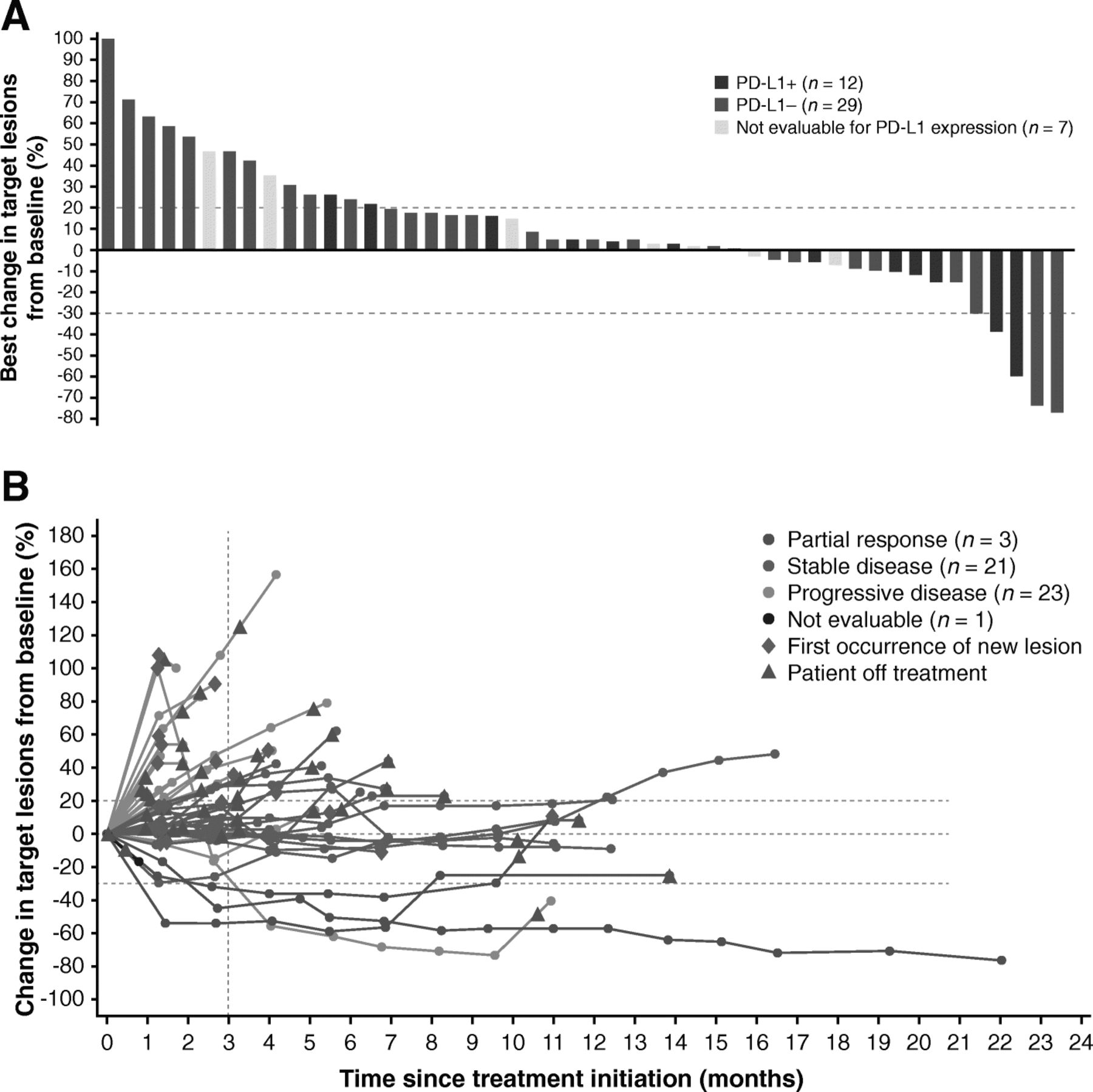
Of 50 patients enrolled, 3 had confirmed PRs, and the ORR was 6.0% (95% CI, 1.3% to 16.5%) (Table 2). Of the 3 responding patients, 2 were receiving concomitant mitotane (both had prior progression while receiving mitotane), and 2 had PD-L1+ tumors (based on 1% or 5% expression cutoffs). In patients who had received 1 (n = 13), 2 (n = 18), or ≥ 3 (n = 19) prior lines of systemic therapy since diagnosis, ORR was 15.4%, 5.6%, and 0%, respectively. Response was ongoing in 1 patient, who had a duration of response of 19.4 months at data cutoff (Fig. 1); this was a man aged 63 years with a PD-L1− tumor who had received prior treatment with cisplatin, etoposide, and mitotane and had target lesions in the liver and lymph node (Additional file 1: Figure S1). The patient received avelumab with concurrent mitotane, had a PR documented at the second assessment (week 13), and remained on treatment without progression throughout 22.0 months of follow-up. Twenty-one patients had a best response of stable disease, resulting in a disease control rate of 48.0%, which was the same in patients with or without concomitant mitotane. Using immune-related response criteria, ORR was 6.0% (95% CI, 1.3% to 16.5%), and the disease control rate was 58.0% (Additional file 1: Table S1). Of 48 patients evaluable for change in tumor size, 16 (33.3%) had a reduction of any level vs baseline, including a reduction of ≥30% in 4 patients (8.0%) (Fig. 2a). One patient had an initial assessment showing progressive disease (~ 100% increase vs baseline and new lesion at week 7) but later assessments showed a decrease vs baseline (nadir − 74%; Fig. 2b).
Confirmed Best Overall Response Based on RECIST v1.1

Time to and duration of response or stable disease (n = 24). Bars represent individual patients with partial response or stable disease. At time of data cutoff, partial response was ongoing in 1 patient

a Best change and b change over time from baseline in sum of target lesion diameters in evaluable patients (n = 48). In a bars for individual patients are color-coded according to tumor PD-L1 status based on a 5% PD-L1 expression cutoff in tumor cells. In b lines for individual patients are color-coded based on confirmed best overall response
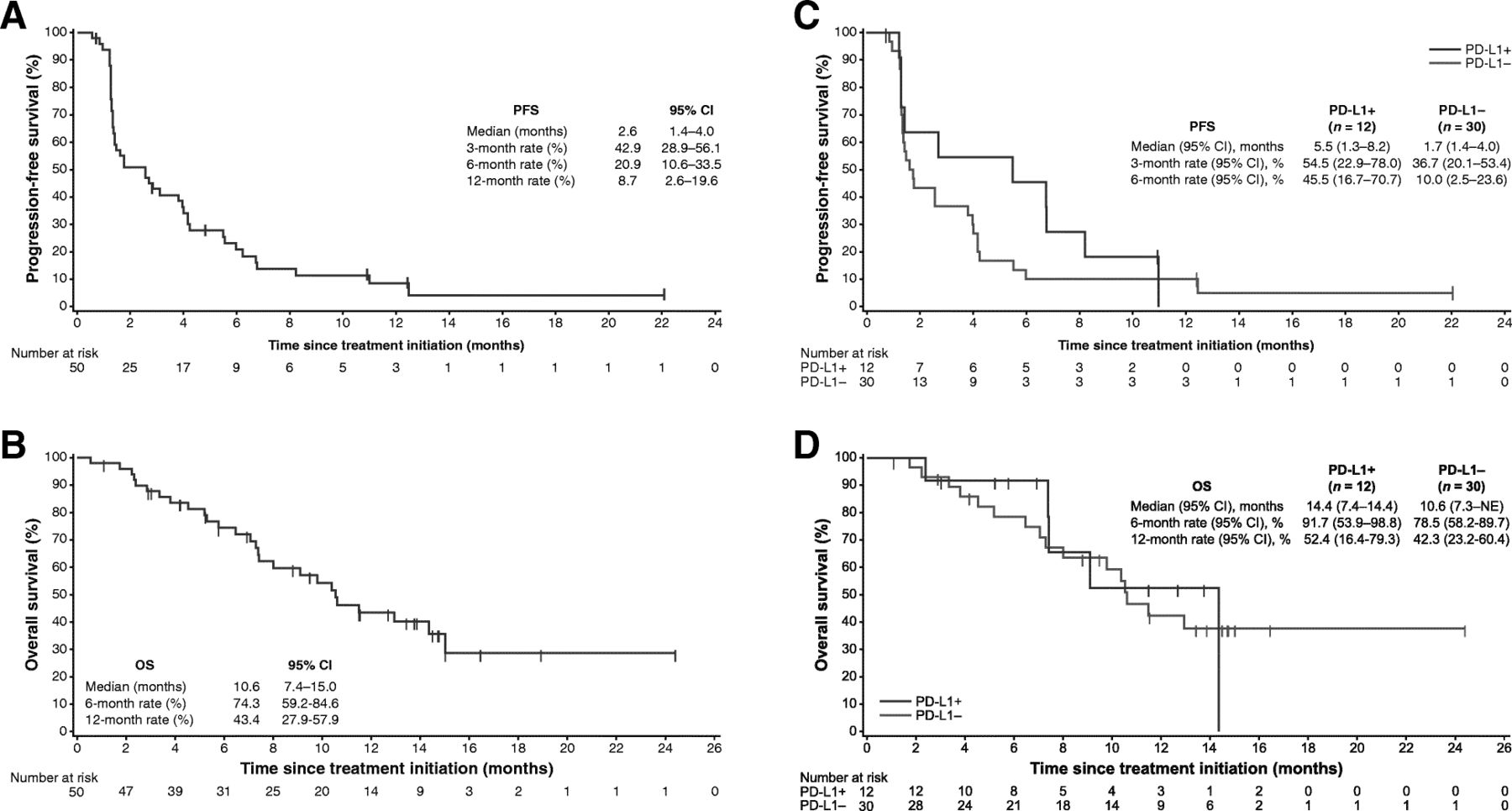
Median PFS was 2.6 months (95% CI, 1.4 to 4.0), and PFS rates at 6 months and 1 year were 20.9% (95% CI, 10.6% to 33.5%) and 8.7% (95% CI, 2.6% to 19.6%), respectively (Fig. 3a). Using immune-related criteria, median PFS was 3.8 months (95% CI, 2.4 to 5.5). Median OS was 10.6 months (95% CI, 7.4 to 15.0), and the 1-year OS rate was 43.4% (95% CI, 27.9% to 57.9%) (Fig. 3b).

{kind=link}
{kind=link}
{kind=link}
a PFS and b OS in the overall population (N = 50), and c PFS and d OS based on PD-L1 expression on tumor cells (5% cutoff) in evaluable patients (n = 42). NE, not evaluable
PD-L1 expression was evaluable in tumor samples from 42 patients (84%). In patients with PD-L1+ (n = 12) or PD-L1– (n = 30) tumors based on a 5% PD-L1 expression cutoff, ORR (95% CI) was 16.7% (2.1% to 48.4%) vs 3.3% (0.1% to 17.2%) (P = .192), median PFS was 5.5 (1.3 to 8.2) vs 1.7 (1.4 to 4.0) months (HR, 0.66 [95% CI, 0.32 to 1.39]), and median OS was 14.4 (7.4 to 14.4) vs 10.6 (7.3 to not evaluable) months (HR, 0.91 [95% CI, 0.33 to 2.49]) (Fig. 3c and Fig. 3d). Findings were similar based on analyses of PD-L1 expression using a 1% cutoff (Additional file 1 : Table S2 and Figure S2). Analyses of higher tumor cell PD-L1 cutoffs and PD-L1 expression on tumor-infiltrating immune cells were not informative because few patients had PD-L1+ tumors (0 to 5 patients).
All 50 patients (100.0%) had an AE of any grade; 41 patients (82.0%) had a treatment-related AE (TRAE) (Table 3). The most common TRAEs of any grade were nausea (n = 10; 20.0%), fatigue (n = 9; 18.0%), hypothyroidism (n = 7; 14.0%), and pyrexia (n = 7; 14.0%). Five patients (10%) had an infusion-related reaction; all were grade 1 or 2. Eight patients (16.0%) had a grade 3 TRAE and no patient had a grade 4 or 5 TRAE. Of patients with or without concomitant mitotane treatment during avelumab treatment (n = 25 in each group), 6 (24.0%) vs 2 (8.0%) patients had a grade 3 TRAE, respectively. Increase in alanine aminotransferase level (n = 2) was the only grade 3 TRAE that occurred in more than 1 patient; both patients received concomitant mitotane. Treatment-related grade 3 elevations in liver enzyme levels occurred only in patients who received concomitant mitotane. Twelve patients (24.0%) had an immune-related TRAE of any grade; those occurring in ≥1 patient were hypothyroidism (n = 7), adrenal insufficiency (n = 3), and pneumonitis (n = 2). Two patients (4.0%) had a grade 3 immune-related TRAE: adrenal insufficiency (n = 1) and pneumonitis (n = 1). Thirty-two patients (64.0%) had an SAE; in 4 patients (8.0%), the SAE was treatment related: pneumonitis (n = 1), adrenal insufficiency and transaminitis (n = 1), thyroiditis (n = 1), and pyrexia (n = 1). Six patients (12.0%) had AEs that led to death, but none were treatment related: disease progression (n = 4), failure to thrive (n = 1), and respiratory failure (n = 1).
TRAEs by Concomitant Mitotane Treatment (any grade in ≥10% of patients in either group or grade 3 in any patient)
Discussion
In this single-arm phase 1b study, which to our knowledge is the largest prospective trial of a checkpoint inhibitor in mACC, avelumab showed antitumor activity with an acceptable safety profile in a platinum-treated population. Three patients (6.0%) had an objective response, including patients with PD-L1+ and PD-L1− tumors, and all of whom had received only 1 (n = 2) or 2 (n = 1) prior lines of treatment. This suggests that the activity of avelumab might be greatest in patients with limited pretreatment, although the small patient numbers in this study prevent any definitive conclusion. Reasons for improved response in patients with less pretreatment may include a smaller tumor burden, decreased proportion of treatment-resistant cells within the tumor, and reduced immunosuppression associated with multiple prior lines of chemotherapy. Although the ORR and median PFS were modest in this heavily pretreated population, the disease control rate was 48.0%, median OS was 10.6 months, and the 1-year OS rate was 43.0%. No association was seen between concomitant mitotane treatment and clinical activity of avelumab, although the absence of detailed patient data relating to ongoing mitotane treatment, including drug levels, is a limitation of the study.
Apart from the current study, other data reported in ACC with anti–PD-L1/PD-1 agents are preliminary findings from phase 2 studies of nivolumab and pembrolizumab in patients with previously treated advanced ACC. Of 7 patients who received nivolumab, 5 had a best response of disease progression and 2 were awaiting evaluation [22]. Of 11 patients who received pembrolizumab, 2 had a PR, 1 achieved stable disease, and the 6-month PFS rate was 27% [23]. In addition, in the phase 1a study of avelumab in patients with various advanced malignancies, a PR occurred in a patient with ACC [13].
The manageable safety profile of avelumab seen in patients with mACC was consistent with experience in other tumor types [16–18]. Patients receiving concomitant mitotane had a higher rate of grade 3 TRAEs than those not receiving mitotane (24.0% vs 8.0%), particularly liver enzyme elevations. This reflects the known toxicity profile of mitotane, which includes hepatic, gastrointestinal, neurological, and hematologic AEs [24]. However, our study showed that the tolerability of avelumab and mitotane in combination is acceptable.
Current treatment options for patients with mACC are highly limited. In the first-line setting, response rates with mitotane monotherapy are estimated to be approximately 10% to 30%, although data from prospective trials are lacking [7]. In a randomized phase 3 study of mitotane combined with either etoposide, doxorubicin, and cisplatin or streptozocin in patients with unresectable ACC without prior treatment (except mitotane), the ORR was 23.2% vs 9.2% (P < .001), the disease-control rate was 58.3% vs 31.4% (P < .001), median PFS was 5.0 vs 2.1 months (P < .001), median OS was 14.8 vs 12.0 months (P = .07), and serious AEs occurred in 58.1% vs 41.6% of patients [25]. In a phase 2 trial of gemcitabine plus metronomic fluoropyrimidine as second-/third-line treatment in patients with advanced ACC who were receiving ongoing mitotane treatment (n = 28), the ORR was 7.1%, disease control rate was 46.4%, median time to progression was 5.3 months, and median OS was 9.8 months; grade 3/4 AEs were leukopenia (21.4%), thrombocytopenia (3.5%), and mucositis (3.5%) [26]. Thus, the results from our study indicate that avelumab has comparable clinical activity and may be better tolerated than existing treatment options for this hard-to-treat cancer. A randomized phase 2 study in non-small-cell lung cancer has shown that combining an anti–PD-1 antibody with platinum-based chemotherapy increased the ORR and prolonged PFS vs chemotherapy alone [27]. This suggests that studies in ACC of avelumab in combination with chemotherapy or as maintenance therapy after first-line induction chemotherapy are warranted.
Targeted molecular therapies have been explored in patients with previously treated advanced ACC, but efficacy has been poor or nonexistent. In a phase 3 trial of linsitinib (insulin-like growth factor receptor 1 and insulin receptor inhibitor) vs placebo in 139 patients with previously treated ACC, of whom 55% had received prior cisplatin, ORR was 3.3% vs 0%, disease control rate was 32.2% vs 34.7%, median PFS was 44 vs 46 days (P = .30), and median OS was 323 vs 356 days (P = .77) [28]. Treatment with erlotinib (tyrosine kinase inhibitor [TKI] of epidermal growth factor receptor) plus gemcitabine in 10 patients resulted in tumor-size reduction in 1 patient, whereas 8 patients had progressive disease at first evaluation [29]. In other studies, no patient treated with combinations of bevacizumab (anti-VEGF antibody) plus capecitabine [30] or sorafenib (multi-TKI) plus paclitaxel [31] had an objective response or stable disease. In a study of axitinib (VEGF receptor inhibitor), none of 13 patients had an objective response, 4 patients showed decreased tumor growth, and median PFS was 5.5 months [32]. Among 35 evaluable patients who received sunitinib (multi-TKI), 14.2% had stable disease as best response and median PFS was 2.8 months [33]. In a study assessing cixutumumab (anti-insulin growth factor 1 receptor antibody) plus temsirolimus (mTOR inhibitor), 11 of 26 patients (42.3%) had a best response of stable disease [34]. Thus, the activity of immunotherapy may be more promising than seen to date in studies of targeted therapies.
Conclusions
In patients with mACC previously treated with chemotherapy, avelumab had moderate clinical activity either as monotherapy or with concurrent mitotane (50% of patients), particularly in those with limited pretreatment or PD-L1+ tumors, and with an acceptable safety profile. These findings provide a rationale for studies of avelumab-based combination treatment in patients with advanced ACC.
Acknowledgments
We thank the patients and their families, the investigators, co-investigators, and study teams at each of the participating centers and at Merck KGaA, Darmstadt, Germany, EMD Serono Research & Development Institute (Billerica, MA; a business of Merck KGaA, Darmstadt, Germany), and Quintiles (Durham, NC).
Authors’ contributions
Conception and design: CLT, AvH, VC, JLG. Provision of study materials or patients: CLT, CH, CZ, DJW, SB, RC, MW, JLG. Collection and assembly of data: All authors. Data analysis and interpretation: All authors. Manuscript writing: All authors. Final approval of manuscript: All authors.
Notes
Ethics approval and consent to participate
The trial was conducted in accordance with the ethics principles of the Declaration of Helsinki and the International Council for Harmonisation Guidelines on Good Clinical Practice. The protocol was approved by the institutional review board or independent ethics committee of each center. All patients provided written informed consent before enrollment.
Consent for publication
Not applicable.
Competing interests
C. Le Tourneau has served in consulting or advisory roles for Merck & Co., Bristol-Myers Squibb, Novartis, and AstraZeneca and has received honoraria from EMD Serono and AstraZeneca. C. Hoimes has served in consulting or advisory roles for Eisai and Seattle Genetics and has served on speakers bureaus for Bristol-Myers Squibb and Roche-Genentech. C. Zarwan has served in consulting or advisory roles for Perceptive Informatics and Revere Pharmaceutics. D.J. Wong has served in consulting or advisory roles for and has received honoraria from Bristol-Myers Squibb and has received research funding from Armo Biosciences, AstraZeneca/MedImmune, BioMed Valley Discoveries, Bristol-Myers Squibb, EMD Serono, KURA Oncology, Merck & Co., and Roche-Genentech. S. Bauer has served in consulting or advisory roles for Bayer, Blueprint Medicines, Deciphera, and Eli Lilly; has received honoraria from Bayer, Novartis, GSK, Pfizer, and PharmaMar; has received research funding from Blueprint Medicines, Incyte, and Novartis; and has received reimbursement for travel and accommodation expenses from PharmaMar. R. Claus has served in consulting or advisory roles for AbbVie, Gilead, Janssen Oncology, and Roche-Genentech and has received honoraria from Janssen Oncology and Roche. M. Wermke has served in consulting or advisory roles for AstraZeneca, Bristol-Myers Squibb, Novartis, and Roche-Genentech and has received research funding from Boehringer, Celgene, Novartis, and Pfizer. S. Hariharan reports employment with and stock ownership in Pfizer. A. von Heydebreck reports employment with and stock ownership in Merck KGaA. V. Kasturi reports employment with EMD Serono. V. Chand reports employment with EMD Serono and stock ownership in Bristol-Myers Squibb. J.L. Gulley has received research funding from Astellas Medivation, Bavarian Nordic, Celgene, EMD Serono, and Pfizer.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
- ACC
- Adrenocortical carcinoma
- AE
- Adverse event
- ALT
- Alanine aminotransferase
- AST
- Asparate aminotransferase
- CCNE1
- Cyclin E1
- CI
- Confidence interval
- CNS
- Central nervous system
- CR
- Complete response
- ECOG PS
- Eastern Cooperative Oncology Group performance status
- GGT
- γ-glutamyltransferase
- HR
- Hazard ratios
- mACC
- Metastatic adrenocortical carcinoma
- mTOR
- Mammalian Target of Rapamycin
- NE
- Not evaluable
- NF1
- Neurofibromin 1
- ORR
- Objective response rate
- OS
- Overall survival
- PD-1
- Programmed death 1
- PD-L1
- Programmed death ligand 1
- PFS
- Progression-free survival
- PRKAR1A
- Protein kinase cAMP-dependent type 1 regulatory subunit alpha
- RECIST
- Response Evaluation Criteria In Solid Tumors
- RPL22
- Ribosomal protein L22
- SAE
- Serious adverse event
- TERF2
- Telomeric repeat binding factor 2
- TERT
- Telomerase reverse transcriptase
- TKI
- Tyrosine-kinase inhibitor
- TRAE
- Treatment-related adverse event
- VEGF
- Vascular endothelial growth factor
- ZNRF3
- Zinc and ring finger 3
References
Footnotes
Funding This trial was sponsored by Merck KGaA, Darmstadt, Germany, and is part of an alliance between Merck KGaA, Darmstadt, Germany, and Pfizer, Inc., New York, NY, USA. Medical writing support was provided by ClinicalThinking, and funded by Merck KGaA and Pfizer.
Availability of data and materials The datasets used and/or analyzed during this study are available from the corresponding author on reasonable request. More information can be found at https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html.
Electronic supplementary material The online version of this article (10.1186/s40425-018-0424-9) contains supplementary material, which is available to authorized users.