Article Text

Abstract
Background Cancer vaccines require adjuvants to induce effective immune responses; however, there is no consensus on optimal adjuvants. We hypothesized that toll-like receptor (TLR)3 agonist polyICLC or TLR4 agonist lipopolysaccharide (LPS), combined with CD4 T cell activation, would support strong and durable CD8+ T cell responses, whereas addition of an incomplete Freund’s adjuvant (IFA) would reduce magnitude and persistence of immune responses.
Patients and methods Participants with resected stage IIB-IV melanoma received a vaccine comprised of 12 melanoma peptides restricted by Class I MHC (12MP), plus a tetanus helper peptide (Tet). Participants were randomly assigned 2:1 to cohort 1 (LPS dose-escalation) or cohort 2 (polyICLC). Each cohort included 3 subgroups (a-c), receiving 12MP + Tet + TLR agonist without IFA (0), or with IFA in vaccine one (V1), or all six vaccines (V6). Toxicities were recorded (CTCAE v4). T cell responses were measured with IFNγ ELIspot assay ex vivo or after one in vitro stimulation (IVS).
Results Fifty-three eligible patients were enrolled, of which fifty-one were treated. Treatment-related dose-limiting toxicities (DLTs) were observed in 0/33 patients in cohort 1 and in 2/18 patients in cohort 2 (11%). CD8 T cell responses to 12MP were detected ex vivo in cohort 1 (42%) and in cohort 2 (56%) and in 18, 50, and 72% for subgroups V0, V1, and V6, respectively. T cell responses to melanoma peptides were more durable and of highest magnitude for IFA V6.
Conclusions LPS and polyICLC are safe and effective vaccine adjuvants when combined with IFA. Contrary to the central hypothesis, IFA enhanced T cell responses to peptide vaccines when added to TLR agonists. Future studies will aim to understand mechanisms underlying the favorable effects with IFA.
Trial registration The clinical trial Mel58 was performed with IRB (#15781) and FDA approval and is registered with Clinicaltrials.gov on April 25, 2012 (NCT01585350). Patients provided written informed consent to participate. Enrollment started on June 24, 2012.
- Melanoma
- Peptide vaccine
- Incomplete freund’s adjuvant
- Toll-like receptor
- Lipopolysaccharide
- polyICLC
- Clinical trial
- CD8 T cells
- Immune response
- ELIspot
- 12MP
- 12 melanoma peptides
- APC
- Antigen-presenting cell
- CV
- Coefficient of variation
- DLT
- Dose-limiting toxicity
- EU
- Endotoxin Unit
- IFA
- Incomplete Freund’s Adjuvant
- IVS
- In vitro stimulation
- LPS
- Lipopolysaccharide
- LR
- Likelihood of chi-square test statistic
- MTDC
- Maximum tolerated dose combination
- PS
- Performance status
- SIN
- Sentinel immunized node
- Tet
- Tetanus helper peptide
- TLR
- Toll-like receptor
- V0
- No IFA
- V1
- IFA with first vaccine
- V6
- IFA with all six vaccines
Open AccessThis article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Statistics from Altmetric.com
- Melanoma
- Peptide vaccine
- Incomplete freund’s adjuvant
- Toll-like receptor
- Lipopolysaccharide
- polyICLC
- Clinical trial
- CD8 T cells
- Immune response
- ELIspot
Introduction
Resistance to checkpoint blockade immunotherapy is commonly attributed to a lack of pre-existing T cell responses to cancer antigens. Thus, there is compelling need for methods to induce antitumor immunity. Cancer vaccines targeting either mutated neo-antigens or shared tumor antigens may accomplish this; however, a critical limitation of cancer vaccine technology is lack of consensus on optimal vaccine adjuvants, which are required to induce functional immune responses. Clinical trials to test adjuvants are more feasible with shared antigen vaccines than with mutated neo-antigens because neo-antigen vaccine composition varies for each patient, whereas the composition of a shared antigen vaccine is consistent across the study population.
The most common adjuvant for peptide vaccines in melanoma has been an incomplete Freund’s adjuvant (IFA). Peptide vaccines incorporating IFA have induced circulating T cell responses [1–3], but some are weak and transient [4]. Recent studies in mice have shown negative effects of IFA as a vaccine adjuvant [5, 6] and have suggested instead that an optimal adjuvant for short peptide vaccines is a TLR agonist plus an agonistic CD40 antibody, which induced strong and durable T cell responses and tumor control [5]. A goal of the present trial was to evaluate a similar approach in patients with melanoma. A multipeptide vaccine (12MP) has previously been found to be both safe and immunogenic [7, 8]. When this trial was initiated, agonistic CD40 antibodies were not available for clinical use. Instead, we used an alternative approach to support licensing of antigen presenting cells (APC) through CD40. Activated CD4 T cells upregulate CD40L; so, we included a peptide from tetanus toxoid known to activate CD4 T cells at the vaccine site and draining node [9–11]. We have shown that a modified form of the p2 peptide of tetanus toxoid residues 830–844 (AQYIKANSKFIGITEL, Tet) induces strong CD4 T cell responses in patients [8, 12]; so, inclusion of this peptide may offer an alternative to CD40 antibodies. Thus, the present study was designed to evaluate the safety and immunogenicity of vaccinating with a mixture of 12 short melanoma peptides (12MP) plus a tetanus helper peptide, combined with TLR agonists. To assess whether IFA interferes with vaccine activity, the study also included treatment arms with IFA.
The central hypotheses were that the TLR agonists may be safe and effective vaccine adjuvants and that decreasing use of IFA may further enhance the magnitude and persistence of the immune responses. Specific goals were: a) to determine the safety of intradermal and subcutaneous injection of the TLR4 agonist lipopolysaccharide (LPS) as a vaccine adjuvant with a multipeptide vaccine, b) to obtain preliminary data on whether administration of a multipeptide vaccine plus each of 2 TLR agonists is immunogenic with or without IFA, c) to obtain preliminary data on whether addition of either of two TLR agonists improves the persistence of circulating CD8 T cell responses to vaccination with a multipeptide vaccine, and d) to determine the local and systemic toxicities of administration of a multipeptide vaccine with each of 2 TLR agonists, and with or without IFA.
Materials and methods
Patient eligibility
Patients at least 18 years of age, expressing HLA-A1, −A2, −A3, −A11 or -A31 were eligible if they had biopsy-proven Stage IIB-IV melanoma rendered clinically free of disease by surgery, other therapy or spontaneous remission. Patients with Stage III-IV melanoma with definite or equivocal findings of persistent metastatic disease could be eligible if they did not meet RECIST criteria for measurable disease. Also required were ECOG performance status (PS) 0–1, and adequate organ function.
Vaccine components and treatment regimen
All participants were vaccinated with MELITAC 12.1 (100mcg of each of 12 Class I MHC restricted melanoma peptides (12MP) [7] and 200mcg of a tetanus helper peptide [12] (Additional file 1: Table S1)). Vaccines were administered with either of two TLR agonists and with or without IFA (Fig. 1a). The IFA used was Montanide ISA-51VG adjuvant (Seppic, Inc., Puteaux, France). PolyICLC (lot PJ2515-1-10, 2.0 mg/ml dry weight) was provided by the Cancer Research Institute/Ludwig Institute for Cancer Research (New York), who purchased it from Oncovir (Washington, DC). LPS was provided by Dr. Anthony Suffredini (Drug Master File Number BB-MF7294) at the National Cancer Institute (NCI) and was vialed and tested by Cambrex BioScience (Walkersville, MD) under oversight of the Biopharmaceutical Development Program, SAIC-Frederick, Inc., NCI-Frederick, Frederick, MD. Each vial contained lyophilized solid representing 10,000 endotoxin units (EU) of E. coli O:113 Reference Endotoxin Lot CC-RE-LOT 3 (1mcg endotoxin). Upon reconstitution in 5 mL water, it contained 2000 EU/mL in 1% Lactose, 0.1% PEG-6000. Regimens were administered half-subcutaneously and half-intradermally in one skin location that is rotated to different extremity sites on days 1, 8, 15, 36, 57 and 78.
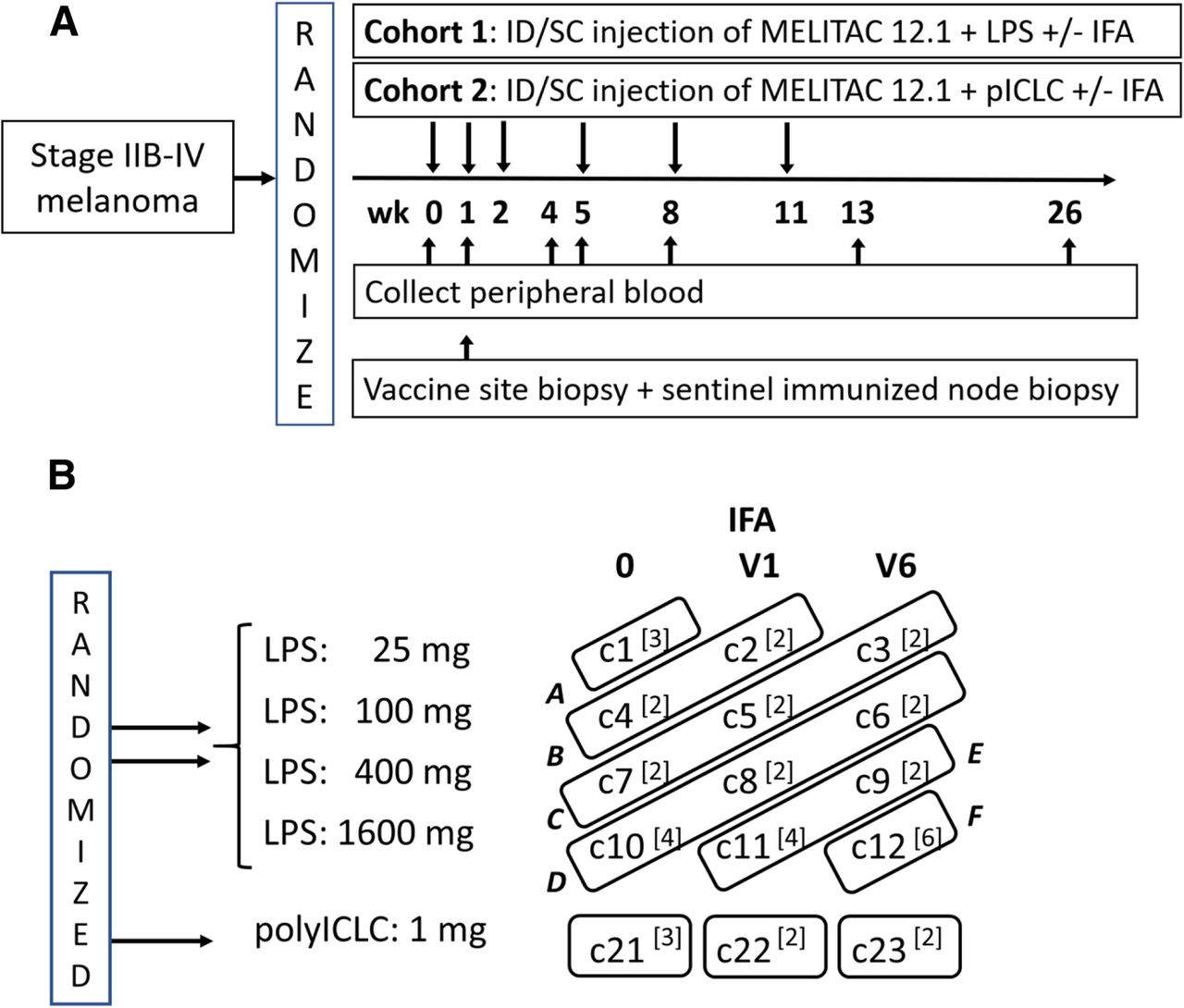
Clinical trial design. The schema for the clinical trial is shown in a. The zones for dose escalation of LPS in cohort 1 (A-F) are shown in b. The study combinations are numbered c1 - c12 for cohort 1 and c21-c23 for cohort 2 as shown
Study design
This was an early phase trial designed to determine the maximum tolerated dose combination (MTDC) of LPS and IFA from among twelve possible combinations in cohort 1 and the MTDC of polyICLC and IFA from among three possible combinations in cohort 2, and to obtain preliminary data on immune response for all the combinations under study. Eligible patients were randomly assigned 2:1 to cohort 1 or cohort 2 (Fig. 1). The 12 combinations in cohort 1 included 4 dose levels of endotoxin (25, 100, 400, 1600EU) administered in three vaccine regimens (12MP + Tet + LPS) and i) without IFA (V0), ii) plus IFA in the first vaccine only (V1), or iii) plus IFA in all six vaccines (V6); (Fig. 1b, Additional file 1: Table S2). The 3 combinations in cohort 2 included 1 dose level of polyICLC (1 mg) administered similarly for each of the three adjuvant regimens and, V0 or V1 or V6; (Fig. 1b, Additional file 1: Table S2). Toxicities were recorded (CTCAE v4). Blood was collected weeks 0, 1, 4, 5, 8, 13, and 26 (Fig. 1a). One week after the first vaccine, a vaccine site-draining lymph node was harvested under local anesthesia, using techniques reported [13], and 4 mm punch biopsies of that vaccine site were obtained (Fig. 1a).
In cohort 1, dose escalation was conducted using a two-stage method for dose-finding for combinations of agents [14]. The first stage was designed such that participants were treated in groups of size 2 until a participant experienced a dose-limiting toxicity (DLT), after which a model-based allocation (stage 2) began. The escalation plan for the first stage was based on grouping dose combinations into “zones,” which are shown in Fig. 1b and detailed in Additional file 2: Supplemental Text. With this dose escalation design, participants were accrued and assigned to other open combinations within a zone, but escalation did not occur outside the zone until a minimum 3-week follow-up period was observed for the first 2 participants accrued to a combination. The second stage modeling strategy using the continual reassessment model (CRM) [15] was planned but not realized since no participants in cohort 1 experienced a DLT. Additional design details are provided in Additional file 1: Supplemental Text. For cohort 2, with only 3 possible combinations of interest, the goal was to accrue 3 patients per combination in increasing magnitude conditional on 1 or fewer DLTs being observed and then to randomly accrue up to 3 additional patients per combination (Fig. 1b).
A DLT was defined as any unexpected adverse event that was possibly, probably, or definitely related to treatment and (1) ≥ Grade 1 selected ocular adverse events, (2) ≥ Grade 2 allergic reactions, (3) ≥ Grade 3 non-hematologic/non-metabolic toxicities, and (4) ≥ Grade 3 hematologic/metabolic toxicities. Grade 2 nausea and Grade 3 fatigue lasting ≤3 days after vaccination were expected toxicities, and injection site ulceration was expected in a subset of patients but vaccine site ulceration of 2 cm diameter or greater was considered a DLT.
Expansion
To assess the impact of including IFA (or not) on the immunologic parameters, the goal was to accrue up to 6 patients at the highest levels of LPS considered safe for each level of IFA. The choice of 6 patients per final combination was chosen to provide improved estimates of variability.
ELIspot assays
T cell responses were measured with IFNγ ELIspot either directly ex vivo, after cryopreservation (direct) or after in vitro sensitization (IVS). Responses to the 12 class I MHC-restricted melanoma peptides are mediated by CD8 T cells specifically [16–26], and responses to the tetanus peptide are mediated by CD4 T cells [12, 27]. Therefore, total PBMC were used for ELIspot assays, and responses per CD8 and CD4 counts were calculated based on their proportion of total PBMC as determined by flow cytometry as previously reported [8, 28, 29]. Methods for the IVS ELIspot assay have been reported [28]. For direct ELIspot assays, 200,000 PBMC were plated per well, and pulsed with synthetic peptide (10mcg/ml), in quadruplicate. Controls included irrelevant peptides, a mixture of viral peptides (CEF peptide pool), PMA-ionomycin and PHA. Evaluation of T-cell responses was based on the following definitions:
Nvax = number T-cells responding to vaccine peptide; Nneg = number T-cells responding to maximum negative control; Rvax = Nvax/Nneg. For evaluations of PBMC, a patient was considered to have a T-cell response to vaccination (binary yes/no), by direct ELIspot assay only if all the following criteria were met: (1) Nvax exceeded Nneg by at least 20/100,000 CD4 or CD8 cells (0.02%), where CD8 and CD4 counts were based on flow cytometry of PBMC. (2) Rvax ≥ 2, (3) (Nvax–1SD) ≥ (Nneg + 1SD), and (4) Rvax after vaccination ≥2 x Rvax pre-vaccine, as described in our prior analyses [8, 28]. The same criteria applied for IVS ELIspot assays except that the threshold for criterion (1) was higher at 30/100,000 CD8 cells. Fold-increases less than one were set to one to indicate no response and to prevent overinflating adjusted fold-increases. Continuous measures of immune response denoted as fold-increase must satisfy conditions (1)–(3) and were defined as the amount of Rvax.
Interassay coefficients of variation (CVs) were calculated for the response of 2 normal donors to the CEF peptide pool: for the high responder, mean number of spots per 100,000 cells was 250, and CV was 30%, and for the low responder, mean was 40 and CV was 44%.
Statistical analysis of immunologic analyses
Primary immunologic analyses were based upon eligible patients, and maximal immune response was based upon responses in the blood through week 26. For hypothesis-testing, patients who discontinued protocol therapy prior to collection of all blood samples for allergic reactions or adverse events, disease progression, or noncompliance were considered immune response failures if no response was observed in evaluable samples. Immune response was a binary indicator of whether or not the criteria listed above were met, and immune response rates were calculated as the proportion of participants with an immune response. Point estimates and 90% confidence intervals were calculated for all summary parameters. Permutation tests [30] were used to assess differences in number of T-cells responding to vaccine peptide adjusting for negative control (i.e., Nvax-Nneg) over the first 12 weeks across groups defined by combinations of LPS dose, inclusion of IFA and inclusion of polyICLC. P-values were based upon 2000 randomly generated permutations and a p-value cutoff of 10% was used to indicate statistically significant results. Negative binomial regression was used to assess count data and contrasts were used to test specific hypotheses with p-values computed from the likelihood ratio chi-square test statistic (LR).
Results
Clinical characteristics
Total enrollment was 53 participants; however, 2 participants did not receive study treatment. Thus, demographic, safety, and immunologic summary data are reported for 51 patients who were enrolled and treated. These included 33 males (65%) and 18 females (35%). Most patients had ECOG PS of 0 (90%) and stage III disease at registration (78%). Additional details are provided in Additional file 1: Table S3.
Toxicities and adverse events
Treatment related adverse events (AE) were limited to grades 1–3, with only one grade 3 (Additional file 1: Table S4). Two participants experienced DLTs, both in cohort 2 (polyICLC). One treated on the V1 sub-arm had grade 3 skin ulceration and was taken off study after 3 vaccines. One on the V6 sub-arm experienced several grade 2 toxicities, none of which individually met predefined criteria for a DLT, but which in aggregate were felt to be dose-limiting. This patient was taken off treatment after 4 vaccines. Overall, no study combinations were estimated to be too toxic for patient accrual.
CD8 T cell response to 12MP
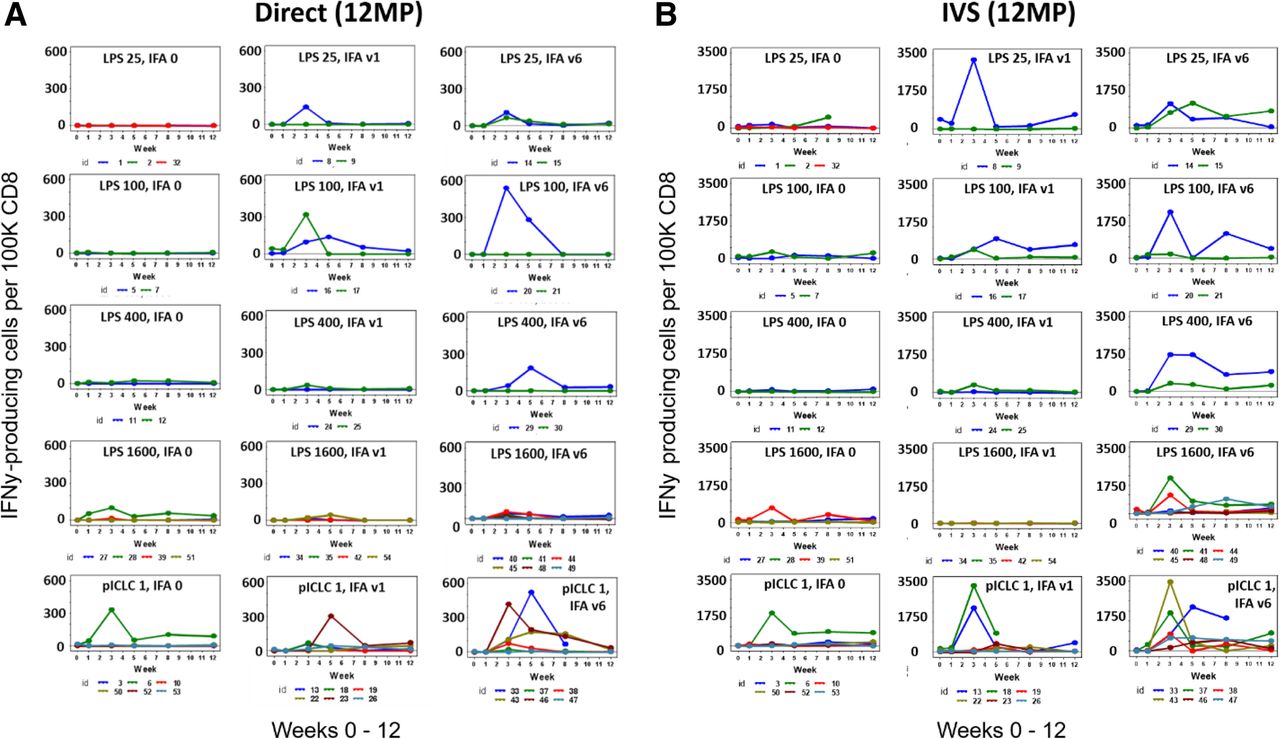
T cell responses to 12 peptide epitopes were evaluated both against the pool of 12 peptides (12MP pool), as well as each peptide individually, using IFNγ ELIspot assays. As described in the methods section, pre-existing immune responses were not considered responses to vaccination: in the uncommon cases with pre-existing immune responses, response to vaccination required at least a 2-fold increase over pre-existing response. The primary comparisons among study groups were made for weeks 0–12, as these data were consistently available (Fig. 2a). Responses to 12MP were detected ex vivo for 47% of patients overall, with the magnitude exceeding 600 spots/105 CD8 T cells for some patients (Fig. 2a). Responses per study cohort and sub-arm are summarized in Additional file 1: Table S5 and per patient in Additional file 1: Table S6. Ex vivo T cell responses to 12MP were detected in 14 of 33 patients (42%) in cohort 1 (LPS) and in 10 of 18 patients (56%) for cohort 2 (polyICLC). Overall, for study arms with no IFA; IFA V1, and IFA V6, CD8 T cell responses to 12MP were detected ex vivo in 18, 50, and 72% of patients, respectively. Similarly, the sum of CD8 T cell responses to each of the 12 peptides was assessed after IVS, and these plots are shown in Fig. 2b.

T cell responses over time (weeks 0–12). CD8 T cell responses to 12MP are shown for each patient from direct ELIspot assays (a), and from IVS ELIspot assays (b). Direct assay data represent response to pooled 12MP; IVS ELIspot data represent sum of responses to each of the 12 individual peptides. Response magnitude is shown as the number of IFNγ-secreting cells, less negative controls, per 100,000 CD8 cells. Values are shown as zero if they did not meet criteria for positivity
Patterns of immune response over time were compared across study groups by modeling the data in PBMC across all time points through week 12. This method is a statistically robust assessment of differences in response patterns between groups over time (Table 1). In cohort 2 (polyICLC), direct ELIspot responses to 12MP were higher if IFA was given for all vaccines compared to no IFA (V6 vs V0, p = 0.036). This was evident also for cohort 1 (p = 0.065) and for analysis across both cohorts (p = 0.036). The CD8 response to 12MP also was higher with polyICLC than with the highest dose of LPS, among patients receiving IFA with all 6 vaccines (p = 0.031). Similarly, the ex vivo and IVS CD8 responses to the sum of individual peptides in 12MP were higher for polyICLC than for LPS1600 and for V6 than V0 in multiple comparisons (Table 1). Thus, for both direct and IVS ELIspot assessments, polyICLC was a more effective adjuvant than LPS, and inclusion of IFA in all vaccines significantly enhanced CD8 T cell response rates to defined melanoma antigens.
MEL58 ELIspot data comparisons across time (weeks 0–12)
We also evaluated immune responses in the sentinel immunized nodes (SINs), but the SINs were harvested early (week 1), and responses were not detected ex vivo. However, among 34 patients evaluated for immune response in the SIN after in vitro stimulation, 11 (32%) had an immune response. These included 18% (4/18) after vaccines with LPS, and 58% (7/12) after vaccines with pICLC. Immune responses in the SIN were observed in 27% (3/11) without IFA, and in 35% (8/23) with IFA (V1 or V6). These SIN responses are shown for all patients in Additional file 1: Table S6.
A pre-existing T cell response to 12MP was detected ex vivo in only 1 patient (#53), who did not develop a vaccine-induced T cell response. In IVS ELIspot assays, 3 (6%) had small pre-existing responses (patients 1, 14, 28), of whom 2 developed vaccine induced responses to 12MP ex vivo, and 2 had responses to 12MP after in vitro stimulation. As specified in the methods, a vaccine-induced T cell response was reported only if there was additional response of at least 2x any pre-existing response.
For the two patients in cohort 2 who came off early for DLTs, immune response data are shown in Additional file 1: Figure S1, where T cell responses to multiple peptides were evident in both.
CD4+ T cell responses to tetanus peptide
T cell responses to the tetanus helper peptide were assessed in direct ELIspot assays. Overall, the permutation tests found no significant differences in response patterns to tetanus peptide among cohorts or study arms. Individual plots of these data for all patients are shown in Additional file 1: Figure S2. T cell responses to the tetanus peptide for any time point were observed in 58% (90% CI:[42, 72]) of patients on cohort 1 and 72% (90% CI:[50, 88]) on cohort 2, and in 24% (90% CI:[8, 46]), 75% (90% CI:[52, 91]), and 89% (90% CI:[69, 98]) of patients in subgroups V0, V1, and V6, respectively (Additional file 1: Table S5).
Magnitude and breadth of CD8 T cell responses
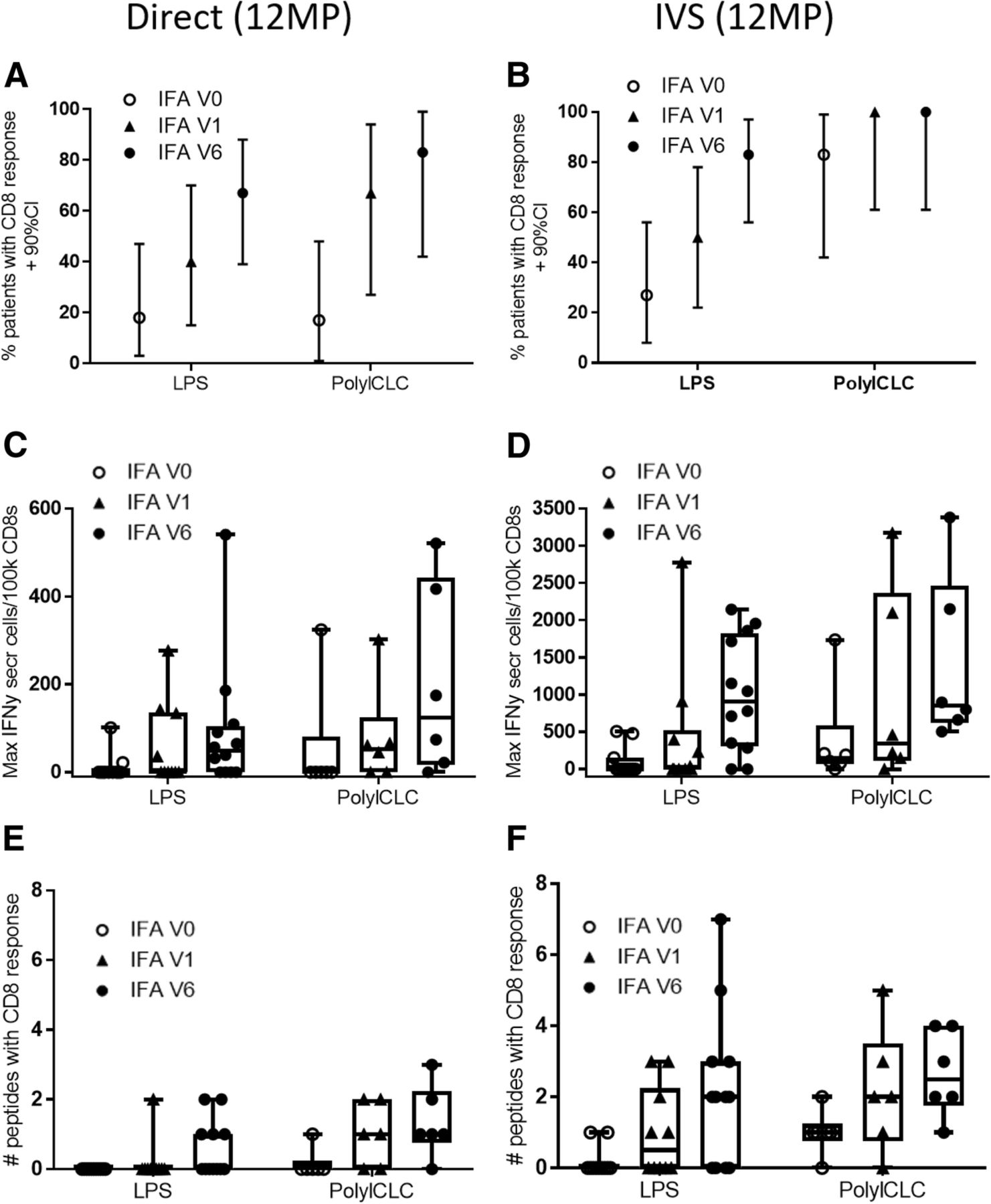
In addition to modeling the immune response across the study population, the fractions of patients with CD8 T cell responses were assessed directly. Immune response rates to 12MP increased with increasing IFA use, both in direct and IVS ELIspot assays (Fig. 3a, b). Similar findings were evident with direct ELIspot based on the sum of responses to the individual peptides (Additional file 1: Table S5), with higher responses for cohort 2 than cohort 1. When IFA was included, the maximum number of IFNγ-secreting cells was higher in direct (Fig. 3c) and in IVS ELIspot assays (Fig. 3d). Similarly, the fold-increase in the T cell responses to 12MP was also higher with inclusion of IFA (data not shown). Immune responses were detected to a broader range of peptides when IFA was included in ex vivo assays (Fig. 3e) or in IVS assays (Fig. 3f).

Frequency and Magnitude of T cell responses to 12MP by ELIspot assay ex vivo (a, c, e), and after IVS (b, d, f). The proportion and 90% confidence interval (CI) of patients with a response to 12MP pool are shown in panels A and B, for each cohort and subgroup. The magnitude of these responses (maximum number of spots per 1 × 105 CD8 T cells) is shown in (c) and (d), where each symbol represents the maximum response for a patient. If the values did not meet criteria for a response, they are shown as zero. Boxplots represent 25th to 75th percentiles, with tails showing the full range, except outliers. The number of peptides to which a response was detected is shown for each patient with a response ex vivo (e) and after IVS (f)
Persistence and durability of the CD8 immune responses
Durability of CD8 T cell responses was assessed by number of time points with positive responses to 12MP after start of vaccine treatment (weeks 1 or later) and by the percent of participants evaluated who had T cell responses at week 26 (wk26). Median numbers of time points with ex vivo responses to 12MP, for V0, V1, V6, respectively, were 0, 0, and 1.5 for LPS and 0, 1.5, and 2.5 for polyICLC. For IVS assays, those values were 0, 0.5, and 4, for LPS, and 1.5, 2, and 4 for polyICLC (Fig. 4a, b), representing significant increases overall from V0 to V6 (LR p = 0.022 and p < 0.001 for ex vivo and IVS, respectively) but not for V0 to V1 (LR p = 0.4 and p = 0.3 for ex vivo and IVS, respectively).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Persistence and durability of the CD8 T cell responses to 12MP. Persistence of the T cell responses to 12MP are shown in a (ex vivo) and b (IVS) as the number of PBMC dates in which a response was detected (after week 0). The maximum possible is 6 (after baseline). Durability of the T cell response for 3 months after the last vaccine is shown as the proportion of patients with 90% confidence interval (CI) with response detected at d183 (of those evaluated ex vivo (c) and after IVS (d). Also for group 23 (pICLC, V6), the measured immune response magnitudes are shown through week 26 ex vivo (e) and IVS (f)
Persistence of T cell responses in PBMC at wk26 was evaluable by ex vivo ELIspot (n = 30) and in IVS ELIspot (n = 40) assays. At this late time point, CD8 T cell responses to 12MP were detected ex vivo in 13%, and after IVS in 48%. Ex vivo responses at wk26 were detected only in cohort 2 patients who had IFA included (V1 and V6) (Fig. 4c). After IVS, responses were detected wk26 in 14, 42, and 86% of patients in V0, V1, and V6 subgroups, respectively (n = 14, 12, 14, respectively) (Fig. 4d). The increase for V6 versus V1 versus V0 overall was significant for IVS assay results only (LR p < 0.001) Thus, persistent responses were significantly enhanced with inclusion of IFA in all 6 vaccines, compared to use of TLR agonists alone, and were similar with either TLR agonist, though they may be slightly more common with polyICLC than with LPS.
Immune response rates summarized by HLA type
Patients expressing HLA-A1, A2, A3, or other A3 supertype alleles (A11, A31), were represented in each cohort, and CD8+ T cell responses were identified among patients expressing each HLA subtype (Additional file 1: Tables S6 and S7). There were differences in immunogenicity among the individual peptides, as previously observed [7, 8, 28]. By IVS ELIspot, the highest response rates were to the HLA-A2 peptide IMD (gp100209–217 (2M)) (68%), HLA-A1 peptide DAE (tyrosinase240-251S) (59%), HLA-A3 peptide SLF (MAGE-A196–104) (43%), and the HLA-A2 peptide GLY (MAGE-A10254–262) (52%) (Additional file 1: Table S8). For 9/12 peptides, the immune response rates were higher in Cohort 2 than in Cohort 1, and for 2 of them the immune response rates were 0 in both; only one peptide (YMD) had an immune response rate marginally higher in Cohort 1 (29% vs 25%). No apparent differences in durability of immune response were observed among different HLA alleles (Additional file 1: Table S9).
Clinical outcome
Overall survival and disease-free survival were high for the entire study population. The study was not powered to investigate changes in overall and disease-free survival among study groups, but they appear similar thus far. (Additional file 1: Figure S3).
Discussion
There is no consensus on best adjuvants to support strong and durable T cell responses to cancer antigens. Our prior work has demonstrated that vaccines using peptides emulsified in IFA can induce CD8 T cell responses in 70–80% of patients based on ex vivo IVS ELIspot assays, and can also induce CD4 T cell responses in most patients, while also supporting induction of peptide-specific antibody responses [31]. The immune responses can exceed 5% of circulating CD8 T cells after vaccination with peptides in IFA alone [1, 28]. However, some T cell responses with IFA are transient and not all patients develop strong responses [4]. Thus, there is interest in enhancing T cell responses to vaccines.
Concerns about use of IFA have been raised by murine studies, which showed that peptide vaccination in IFA induced inflammation at vaccine sites that selectively recruited and depleted peptide-specific T cells, thereby negatively impacting tumor control [5, 6]. Multiple investigators have induced strong and durable CD8 T cell responses to short peptides in mice using adjuvants combining a TLR agonist and an agonistic CD40 Ab [5, 32, 33]; however, this approach has not yet been evaluated in humans. The Mel58 clinical trial was designed to test whether vaccination with minimal epitope melanoma peptides in a TLR agonist, combined with helper T cell activation, would be more effective at inducing durable T cell responses than use of the same adjuvant preparation combined with IFA. However, in contrast to our underlying hypothesis, we found that circulating CD8 T cell responses to minimal epitopes were greater in magnitude and durability when IFA was included, especially when IFA was included in all 6 vaccines.
The trial tested agonists for both TLR3 and TLR4. TLR4 agonists have also been studied as vaccine adjuvants, but the classic TLR4 agonist, LPS, has long been considered too toxic for human use. However, the present formulation of GMP grade endotoxin has a strong safety profile [34–38]. Human experience with it, administered systemically, either by intravenous injection or by inhalation, is that it causes systemic inflammatory responses that are transient and very well-tolerated up to 2500 EU per dose [34, 35, 39]. LPS is known to activate innate immunity, however; to our knowledge, it has not been previously been used as a vaccine adjuvant. In the present study, we escalated from 25 EU to 1600 EU, with and without IFA, and there were no DLTs. Thus, these data support the safety of bacterial LPS as a vaccine adjuvant. Interestingly, one patient had skin hypopigmentation (patient 27, LPS 1600, V0) though no T cell response to 12MP or tetanus peptide ex vivo, but positive to tyrosinase (DAEK) with in vitro stimulated ELIspot assay in PBMC and SIN (Additional file 1: Table S4). The goal of the rapid dose-escalation was to define safety at the maximal tolerated dose, up to 1600 EU. Since that dose was found safe, most patients in Cohort 1 were enrolled at 1600 EU dose, limiting the ability to determine which of the 4 LPS doses is most immunogenic. Within this constraint, no significant difference in immunogenicity was observed among the LPS doses.
The TLR3 agonist polyICLC has been studied in preclinical models and in clinical trials [40–42], with favorable safety and immunogenicity profiles, and, when combined with IFA, it has been shown to enhance CD4, CD8, and antibody responses to long NY-ESO-1 peptides compared to IFA alone [3]. Sabbatini et al. did observe marked injection site reactions in 2 patients treated with NYESO1 long peptides plus IFA and 1.4 mg polyICLC, and discontinued treatment early for 4 of 11 patients. Considering this, we employed a lower dose (1 mg) in our study [3]. We observed injection site reactions that met stopping criteria for 1 of 6 patients in arm 22 (polyICLC, V1) and that contributed to the overall DLT for one of 6 patients in arm 23 (polyICLC, V6). However, these were not serious adverse events, which resolved after stopping treatment. These DLTs did not meet stopping criteria for any sub-arm of cohort 2. Thus, the regimen is considered safe; however, prominent local injection site reactions can be expected. Overall, the data support polyICLC as an effective vaccine adjuvant when combined with IFA, for inducing CD8 T cell responses to minimal peptides, with an acceptable safety profile. This regimen appears marginally better than LPS plus IFA, which was very well-tolerated, but also supported immune responses. Other TLR agonists have been shown to enhance T cell responses to peptides in vaccines, in particular TLR9 agonist CpG-B (7909, PF-3512676) [43, 44]. Thus, a range of TLR agonists have value in combination with IFA as vaccine adjuvants.
For an adjuvant to have maximum benefit, it has to generate an antigen depot, to activate APC, and to provide co-stimulation through CD4 T cell help [45]. The present study provided an antigen depot with the water-in-oil emulsion with IFA, TLR agonists to activate APC, and a tetanus peptide that is very effective at inducing CD4 helper T cell responses. Activation of CD4 helper T cells will induce CD40L expression, which in turn can license APC and enhance their antigen presentation. We have not formally tested the impact of CD40L expression by tetanus-reactive CD4 T cells but have found in this trial that T cell responses to the tetanus helper peptide was greater with inclusion of IFA (V1 or V6) than without it (V0). Thus, the impact of IFA may include both a direct effect on the CD8 T cell response and an indirect effect, through activation of CD4 T cells, and subsequent APC activation.
In the murine studies that have shown negative effects of IFA on CD8 T cell responses to short peptides, an alternative vaccination approach using a water-soluble adjuvant preparation, including TLR7 agonist imiquimod and CD40 antibody induced more durable immune responses and better tumor control [5]. Also, strong CD8 responses have been induced in mice by co-administration of peptides, CD40 Ab, and PolyIC [46]. Clinical grade human agonistic antibodies to CD40 were not available at the time of the present clinical trial. Thus, the vaccine regimen included the tetanus helper peptide to enhance CD4 help via CD40L expression. It will be valuable to reconcile the favorable findings from this trial in light of the unfavorable findings with use of IFA in murine models. There are several differences in the experimental setting for the murine studies and that of the present clinical trial. These include dose and volume differences in the vaccine, differences in T cell frequency, inclusion of helper peptide in the human trial, as well as potential differences between human and mouse. In the murine studies, 100 mcg of peptide was given in a 100 mcl emulsion with IFA [5, 6]. In the human trial, 100 mcg of each peptide was given in a 1 ml emulsion with IFA. Considering the impact of the large depot in the mouse, volume of that depot is likely relevant to the observed findings. A mouse typically weighs about 25 g; thus, 100 mcl represents 1/250th of the mass of the mouse. For a 70 kg human, the 1 ml emulsion used in the clinical trial represents 1/70,000th of the mass of the patient. This 280-fold v/v difference is dramatic: If the patients had been administered a 280 ml emulsion, a much more dramatic vaccine site effect might be anticipated. Also, the murine studies used adoptive transfer of 1 × 106 activated antigen-reactive T cells which represents about 50% of circulating CD8 T cells [47]. This exceeds the pre-treatment frequency of antigen-specific T cells in humans, probably by at least 2 logs, and also exceeds what is induced over time with vaccination. Thus, the administration of a high dose of antigen-reactive T cells into a massive IFA depot may explain in part the difference between the experimental findings in the mouse and what is observed in this clinical trial. Also, this trial included T cell help, in the form of a tetanus helper peptide, which was not included in the murine studies. We have found that vaccination with IFA plus TLR agonist and inclusion of CD4 help induced a high rate of T cell responses ex vivo, durable in most patients for at least 6 months.
In prior work, we observed transient responses by circulating T cells [4, 28] and that T cells accumulate at sites of vaccination with peptides in IFA [48, 49]. These observations, in light of murine data on IFA as an adjuvant [5], suggested a decline of responsive circulating T cells due to accumulation at vaccine sites.. Alternatively, the transient responses observed with direct ELIspots may be explained by reversion of effector T cells to memory, especially after the vaccine sequence is completed. As such, they may not be detected as effectors ex vivo but are functional after restimulation. In support of this, we observed stimulated responses out to wk. 26 in 85% of the evaluable patients with V6 IFA (Fig. 2b), compared to 50 and 14% respectively with V1 and V0. Therefore repeated doses of IFA may support durable memory responses rather than accumulation and depletion at vaccine sites.
New strategies for vaccination against mutated neoantigens have promise for enhancing immune repertoires; however, the clinical trials of neoantigen vaccines published to date have all used different vaccine adjuvant strategies, and most of the T cell responses induced were detectable only after in vitro stimulation [50–52]. Thus, enhanced strategies for vaccination remain a high priority for the field. The present study suggests that a TLR agonist alone may not be sufficient for induction of a strong T cell response to a peptide vaccine, and that inclusion of IFA with a helper peptide remains an effective strategy. Future trials should test whether addition of a CD40 antibody plus TLR agonist at the vaccine site can further enhance T cell responses in patients, with or without IFA.
Conclusions
This clinical trial was designed to test whether vaccination with 12 short melanoma peptides in combination with TLR agonists polyICLC or LPS with IFA was safe and immunogenic in melanoma patients. Only 2 DLTs were observed, in different sub-arms of cohort 2 (polyICLC): no treatment combination met stopping criteria. A driving hypothesis was that inclusion of IFA with TLR agonists would be less effective in generating a durable T cell response. However, in contrast to our hypothesis, peptide-specific CD8 T cell responses were more durable and of greater magnitude when IFA was included as an adjuvant, regardless of whether it was combined with polyICLC or LPS. Furthermore, our study suggests that, overall, polyICLC may induce marginally better CD8 T cell responses than LPS. Future studies will aim to understand mechanisms underlying the favorable effects with IFA.
Funding
NCI R01 CA057653 (CLS); 5K25CA181638 (NW); P30 CA044579 (Biorepository and Tissue Research Facility, Office of Clinical Research, and Biostatistics Shared Resource); gifts from Alice and Bill Goodwin and the Commonwealth Foundation for Cancer Research, and Clinical and Laboratory Integration Project grant from the Cancer Research Institute (CLS, MM).
Acknowledgments
We thank the NIH Clinical Center (Anthony Suffredini) and the Cancer Research Institute/ Ludwig Institute for Cancer Research for kindly providing the LPS and polyICLC, respectively.
Authors’ contributions
GRP, KAC, NAW, NV, MES and CLS designed the study. WWG, EMG, DHD and CLS were involved in treatment of patients. KTS, NVG and DHD executed experiments to generate ELIspot results. MM, GRP and CLS analyzed the results. Lastly, MM and CLS drafted the manuscript and/or substantially revised it. All authors read and approved the final manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Notes
Ethics approval and consent to participate
The clinical trial Mel58 was performed with IRB (#15781) and FDA approval and is registered with Clinicaltrials.gov on April 25, 2012 (NCT01585350). Patients provided written informed consent to participate.
Consent for publication
Not applicable.
Competing interests
C.L. Slingluff Jr. reports receiving commercial research grants from Merck, GlaxoSmithKline, 3 M, and Celldex; has ownership interest (including patents) in several of the peptides used in the 12MP vaccine with UVA Licensing and Ventures Group; and is a consultant/advisory board member for Immatics, Curevac, and Polynoma. No potential conflicts of interest were disclosed by the other authors.
- 12MP
- 12 melanoma peptides
- APC
- Antigen-presenting cell
- CV
- Coefficient of variation
- DLT
- Dose-limiting toxicity
- EU
- Endotoxin Unit
- IFA
- Incomplete Freund’s Adjuvant
- IVS
- In vitro stimulation
- LPS
- Lipopolysaccharide
- LR
- Likelihood of chi-square test statistic
- MTDC
- Maximum tolerated dose combination
- PS
- Performance status
- SIN
- Sentinel immunized node
- Tet
- Tetanus helper peptide
- TLR
- Toll-like receptor
- V0
- No IFA
- V1
- IFA with first vaccine
- V6
- IFA with all six vaccines
References
Footnotes
Electronic supplementary material The online version of this article (10.1186/s40425-019-0625-x) contains supplementary material, which is available to authorized users.