Article Text

Abstract
The interplay between the immune system and tumor progression is well recognized. However, current human breast cancer immunophenotyping studies are mostly focused on primary tumors with metastatic breast cancer lesions remaining largely understudied. To address this gap, we examined exome-capture RNA sequencing data from 50 primary breast tumors (PBTs) and their patient-matched metastatic tumors (METs) in brain, ovary, bone and gastrointestinal tract. We used gene expression signatures as surrogates for tumor infiltrating lymphocytes (TILs) and compared TIL patterns in PBTs and METs. Enrichment analysis and deconvolution methods both revealed that METs had a significantly lower abundance of total immune cells, including CD8+ T cells, regulatory T cells and dendritic cells. An exception was M2-like macrophages, which were significantly higher in METs across the organ sites examined. Multiplex immunohistochemistry results were consistent with data from the in-silico analysis and showed increased macrophages in METs. We confirmed the finding of a significant reduction in immune cells in brain METs (BRMs) by pathologic assessment of TILs in a set of 49 patient-matched pairs of PBT/BRMs. These findings indicate that METs have an overall lower infiltration of immune cells relative to their matched PBTs, possibly due to immune escape. RNAseq analysis suggests that the relative levels of M2-like macrophages are increased in METs, and their potential role in promoting breast cancer metastasis warrants further study.
- Metastatic breast cancer
- Breast cancer
- Macrophages
- M2 macrophages
Li Zhu, Jessica L. Narloch and Sayali Onkar are Co-first authors.
- BOM
- Metastatic breast tumor in bone
- BRM
- Metastatic breast tumor in brain
- DE
- Differential expression
- ecRNA-seq
- Exome-capture RNA sequencing
- FFPE
- Formalin fixed paraffin embedded
- GI
- Gastrointestinal tract
- GIM
- Metastatic breast tumor in gastrointestinal tract
- GSVA
- Gene set variation analysis
- GTEx
- Genotype-Tissue Expression
- H&E
- Hematoxylin & Eosin
- HR
- Hormone receptor
- MET
- Metastatic breast tumor
- MFS
- Metastasis-free-survival
- mIF
- Multiplexed immunofluorescence
- OVM
- Metastatic breast tumor in ovary
- PBT
- Primary breast tumor
- SPM
- Survival-post-metastasis
- ssGSEA
- Single-sample gene set enrichment analysis
- sTIL
- Stromal tumor infiltrating lymphocyte
- TIL
- Tumor infiltrating lymphocytes
- TME
- Tumor microenvironment
- TNBC
- Triple negative breast cancer
Open AccessThis article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Statistics from Altmetric.com
Li Zhu, Jessica L. Narloch and Sayali Onkar are Co-first authors.
Introduction
Breast cancer is a highly heterogenous disease affecting 1 in 8 women in the US, and the most commonly diagnosed cancer in women worldwide. Despite recent improvements in overall survival rates, it is still the second leading cause of mortality due to cancer in women [1]. In the last two decades, significant progress has been made in the detection and treatment of primary breast tumors as a result of enhanced understanding of disease biology and the tumor microenvironment (TME). The breast TME represents a complex interaction between tumor cells, endothelial cells, fibroblasts, and a variety of pro- and anti-tumor immune cells capable of tipping tumor biology toward tumor growth and progression or immune rejection. During tumor growth, cancer cells can be detected and eliminated by the immune system, but some cancer cells may exploit several mechanisms to evade destruction by the immune system, enabling them to escape immune surveillance and progress through the metastatic cascade. For breast cancer, the most common sites of distant organ metastases include bones, lungs, liver and brain with ovaries and gastrointestinal tract (GI) being affected less frequently [2].
The interplay between the immune system and tumor development is now well recognized in a variety of tumor types, including the triple negative (TNBC) and HER2+ subtypes of breast cancer [3, 4]. However, existing immunophenotyping studies focus mainly on primary tumors, with the role of immune cells in metastatic progression remaining largely understudied. While numerous studies have now documented cellular and genomic evolution of breast cancers during metastasis [5, 6], very little is known about the co-evolution of immune cells and the TME. This study focused on addressing this gap in our understanding by performing immunophenotyping on two datasets: a) Pan-MET, transcriptomic profiles of 50 pairs of patient-matched primary (PBTs) and metastatic breast tumors (METs) in brain (BRM), ovary (OVM), bone (BOM) and gastrointestinal tract (GIM); and b) BRM-sTIL, a multi-institutional cohort of 49 patient-matched pairs of PBTs and BRMs with stromal tumor infiltrating lymphocytes (sTILs) percentages quantified by pathologic evaluation of hematoxylin & eosin (H&E) staining. Using gene expression signatures as surrogates for TILs, we discovered quantitative differences in immune cell profiles between PBTs and METs in the first dataset (Pan-MET). Those differences were confirmed using multiplexed immunofluoresence (mIF) in three pairs of PBT/OVMs and PBT/BRMs each. Consistent results were observed by comparing the sTILs percentages in additional PBT/BRM pairs in a second dataset (BRM-sTIL). Higher immune cell recruitment to the TME also showed weak association with better survivals in both datasets. Our study demonstrates the potential of using bioinformatics tools to investigate the evolution of the immune TME in breast cancer metastasis, and identifies M2-like macrophages as a potential therapeutic target for metastatic breast cancer.
Materials and methods
Details of methods are available in Additional file 1.
Data
Pan-MET dataset
Exome-capture RNA sequencing (ecRNA-seq) of patient-matched PBTs and METs were collected from brain, bone, ovary and GI, as previously reported in [7–9]. Clinical and pathological information of all samples are available in Additional file 2: Table S1. Formalin fixed paraffin embedded (FFPE) tissue sections of three pairs of PBT/BRMs and PBT/OVMs each were retrieved from the Pitt Biospecimen Core for multiplex staining.
BRM-sTIL dataset
Sample tissues of 49 pairs of patient-matched PBTs and BRMs were collected from four participating academic institutions (Duke University Medical Center, University of North Carolina Medical Center, University of Pittsburgh, Massachusetts General Hospital) for H&E staining. Clinical and pathological information is available in Additional file 2: Table S2. 15 pairs of PBT/BRMs overlap between the Pan-MET and BRM-sTIL (Additional file 2: Table S3).
Immune level quantification
We inferred the immune abundance from RNAseq data using single-sample gene set enrichment analysis (ssGSEA, i.e. immune score in ESTIMATE) [10], gene set variation analysis (GSVA) [11] and deconvolution methods --- CIBERSORT [12] and TIMER [13]. In addition to the samples in Pan-MET dataset, we also evaluated immune level in normal tissue samples achived from Genotype-Tissue Expression (GTEx) Project. H&E stained sections in BRM-sTIL dataset were manually counted for percent sTILs using standard criteria developed by the international TILs working group [14]. Each slide was independently reviewed by two study personnel (JLN and CL) to minimize inter-observer variability. When the sTILs differed by 10% or more, the study pathologist (AH) made the final determination.
Results
METs have lower total immune abundance than patient-matched PBTs
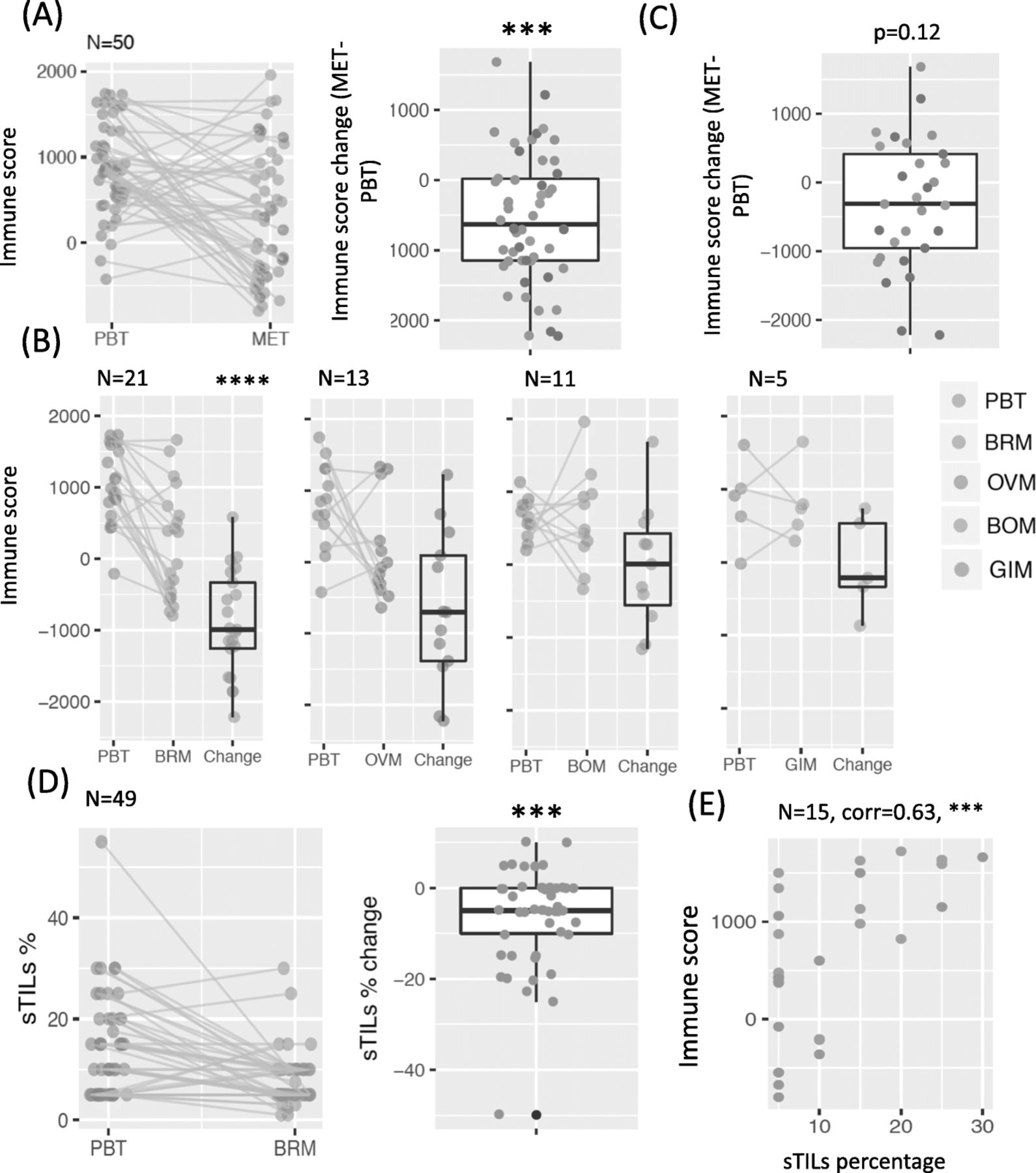
We estimated total immune abundance using RNAseq from 50 pairs of patient-matched PBTs and METs. For multiple METs that were matched to the same PBT, we first took the average. In general, METs showed a significantly lower total immune score compared to patient-matched PBTs (Fig. 1a; p < 0.001). The decrease in immune score was observed in METs collected from various sites, but was especially apparent in BRMs (p < 0.0001, Fig. 1b). Removing BRMs and combing all other METs, we noted a non-significant trend to decreased immune score in METs (p = 0.12, Fig. 1c). However, it should be noted that the small number of samples makes conclusions in the non-brain METs challenging. Validating the finding of decreased immune cells in brain METs, pathologic assessment of sTILs in an additional cohort of 49 patient-matched PBTs and METs revealed that BRMs also showed a significant decrease in the percentage of sTILs compared to patient-matched PBTs (p < 0.001, Fig. 1d). When grouping PBT/MET pairs by hormone receptor (HR) status and HER2 status, both datasets revealed a trend of decreased immune abundance in all subtypes, with TNBC subtype having the most significant decrease (p < 0.01, Additional file 2: Figure S1). Similar results were observed when we treated those METs matched to the same PBT as METs in different pairs (Additional file 2: Figure S2). While the total immune score only estimates the overall immune abundance in the bulk sample from RNAseq, and the sTILs percentage was carefully counted as the immune cell percentage in the stroma, the two measurements of immune abundance were significantly correlated (p < 0.001) for the 15 pairs of PBT/BRMs within both data sets (Fig. 1e). A lesser degree of agreement was only observed in tumors with extreme low sTILs (5%), possibly due to unstable estimates by both methods when the immune component is limited.

Lower immune abundance in metastatic breast tumors (METs) compared to primary breast tumors (PBTs) (a) Total immune score in PBT/MET pairs in Pan-MET dataset, together with the paired changes (MET-PBT). b Paired changes of total immune score removing BRMs in (a). c Total immune score grouped by MET sites. d Stromal tumor infiltrating lymphocytes (sTILs) percentages of 49 pairs of PBT/BRMs in BRM-sTIL dataset. e Spearman’s correlation between sTILs percentages and total immune score for 15 pairs of PBT/BRMs overlapped by Pan-MET and BRM-sTIL. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05 from two-sided Wilcoxon signed rank test in (a-d) and correlation test in (e)
In addition, we also observed that METs had significantly lower expression of immune checkpoint molecules that downregulate immune response — including CD274 (PD-L1), PDCD1 (PD-1), CTLA4, but not VSIR (Additional file 2: Figure S3) — possibly due to fewer total immune cells. We also tested for differentially expressed (DE) genes between matched PBT/BRMs (ER+ and ER- separately), PBT/OVMs (ER+ only) and PBT/BOMs (ER + only) to eliminate possible confounding effect from ER status. Pathway enrichment analysis of DE genes (adjusted p < 0.05) from matched PBT/BRM, both ER+ and ER-, identified immune related pathways, such as KEGG_primary_immunodeficiency, as one of the top significantly enriched pathways (Additional file 3: Table S4, Additional file 4: Table S5). Several immune related pathways were also significantly enriched in PBT/OVM and PBT/BOM comparisons, but they were not among the top 50 significant list (Additional file 5: Table S6, Additional file 6: Table S7).
Taken together, both transcriptomic data and pathological assessment showed that METs have lower immune abundance than patient-matched PBTs.
METs have higher percentage of M2-like macrophages relative to the total immune abundance
We inferred the abundance of each immune cell population by two types of methods — enrichment analysis and deconvolution method. To validate those approaches, we first compared the GSVA scores of four common immune cell populations defined by both Davoli et al. [15] and Tamborero et al. [16]. The correlations ranged from 0.4 to 0.85 (Additional file 2: Figure S4), indicating overall high consistency. For further validation, we applied four methods; namely GSVA using the immune signatures from Davoli and Tomborero, and two methods of deconvolution (CIBERSORT and TIMER) to a publicly available single cell RNA-seq dataset [17], in which immune cell percentages were available using cell markers. Based on the correlations, the estimated levels of B cell, T cell, and macrophages by immune signatures from Davoli and Tamborero, and deconvolution method TIMER, were in general most highly correlated with actual abundance of corresponding cell types, although some signatures were not quite specific, such as CD4+ mature T cell and CD8+ effector T cell in Davoli signatures. CIBERSORT estimates showed lower correlations as expected, because the actual percentages were calculated based on three cell types, while CIBERSORT considered 22 cell types (Additional file 2: Figure S5).
Comparing patient-matched PBTs and METs, the GSVA score and abundance estimate from deconvolution methods for most immune cell populations were significantly lower in METs (Fig. 2a-c). Adjusting for total immune abundance, most immune cell populations were still lower, but M2-like macrophages were significantly higher in METs (Fig. 2d). Since CIBERSORT provides an empirical p value testing the null hypothesis that a particular sample does not contain any of the 22 cell types, we removed 16 pairs with at least one sample with p > 0.05, M2-like macrophages were still higher in METs, but there was only a trend to significance (Additional file 2: Figure S6). Significant increment was also observed in the ratio of the relative percentages of M2 and M1, indicating dominant level of M2 over M1 (Fig. 2e). When separating PBT/MET pairs to different MET sites or HR/HER2 subtypes, the results were generally consistent (Additional file 2: Figure S7-S8). Due to the lack of adjacent normal tissues, it is impossible to fully eliminate the effect contributed by the different cellular composition of the normal tissues. However, when comparing the percentage of M2-like macrophages in normal tissues with RNAseq data downloaded from GTEx, we observed that M2 macrophages was lower in normal brain and small intestine and similar in ovary (normal bone tissue is not available in GTEx) compared to normal breast, suggesting that the increased M2 macrophage in METs was not due to the presence of normal tissues (Additional file 2: Figure S9).
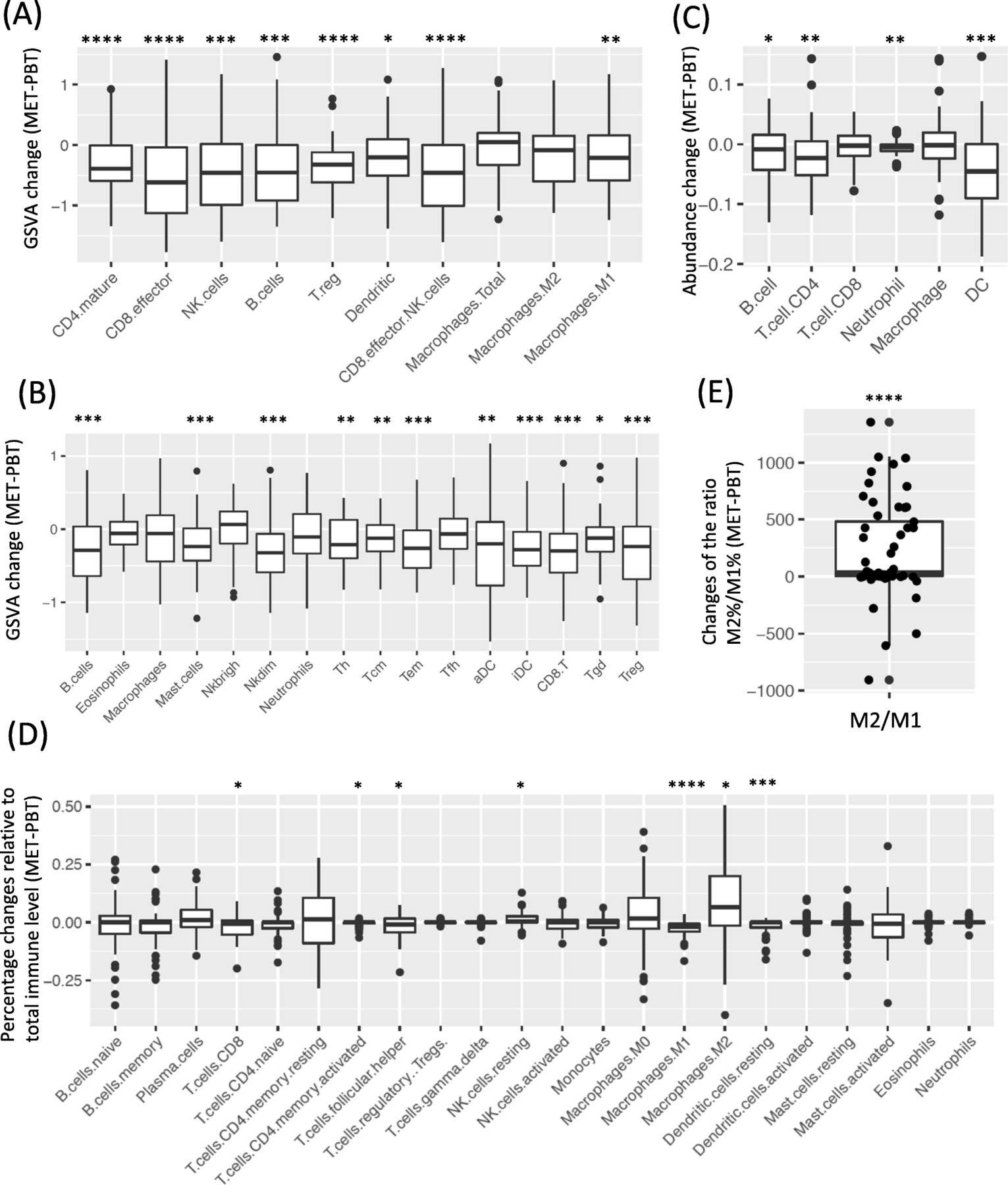
Paired comparison of the abundance of immune cell population in PBT/MET pairs in Pan-MET. a-b GSVA score changes (MET-PBT) of (a) Davoli signature and (b) Tamborero signature. c Abundance changes estimated by deconvolution method TIMER. d Changes of percentages relative to total immune level estimated by deconvolution method CIBERSORT. e Changes of the ratio of relative percentages of M2 and M1. ****FDR < 0.0001, ***FDR < 0.001, **FDR < 0.01, *FDR < 0.05 by Benjamini-Hochberg correction. Two-sided Wilcoxon signed rank test
Multiplexed immunofluoresence confirms the in-silico results
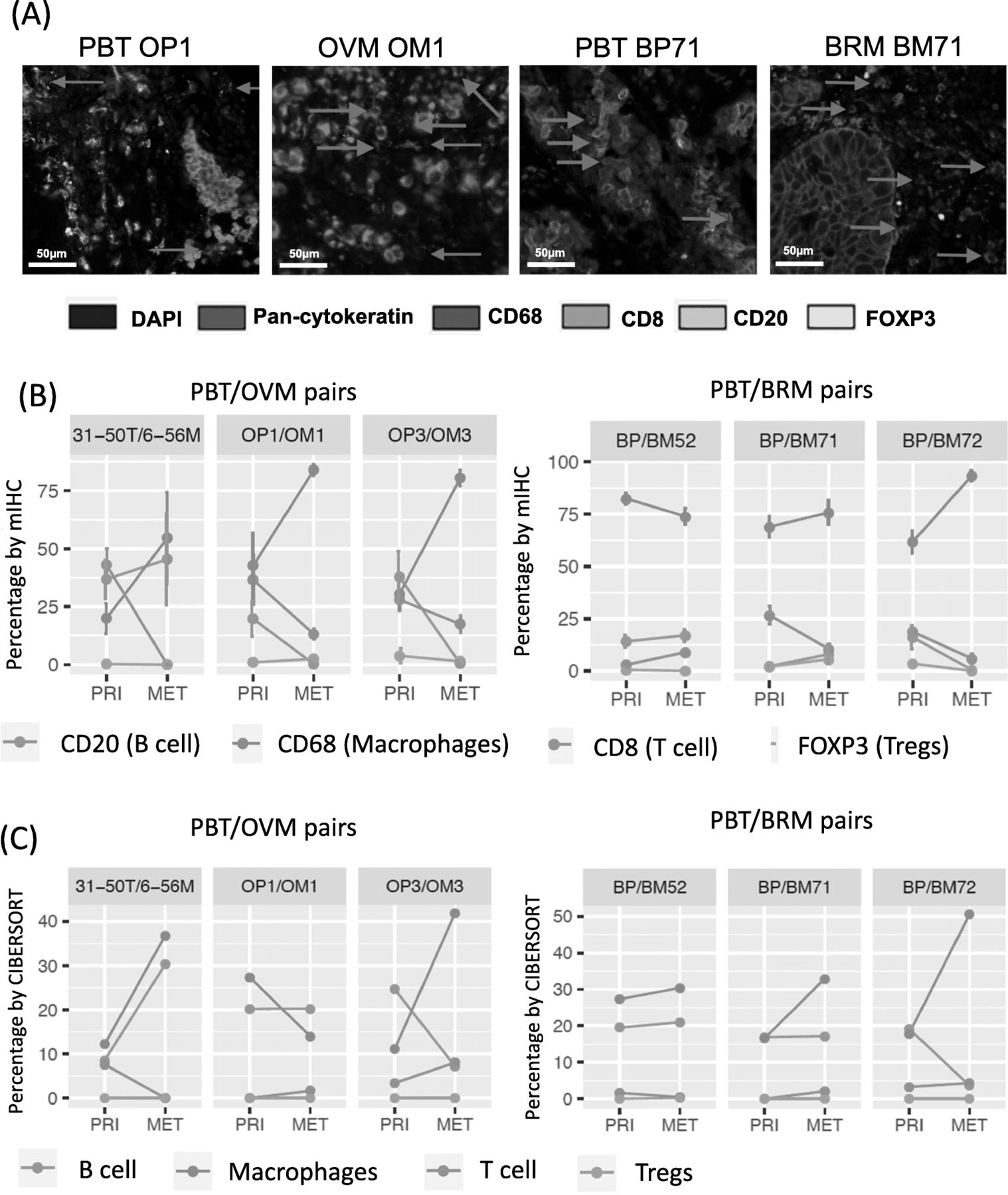
To further validate in silico results, we selected three pairs of PBT/BRMs and three pairs of PBT/OVMs, which were shown to have higher M2-like macrophages relative to the total immune abundance, for multispectral immunofluorescence (Fig. 3a). Three pairs of PBT/OVMs and two pairs of PBT/BRMs showed increased macrophages in METs, and the majority of METs had lower B cells and T cells (Fig. 3b), consistent with percentage estimated from CIBERSORT (Fig. 3c and Additional file 2: Figure S10).
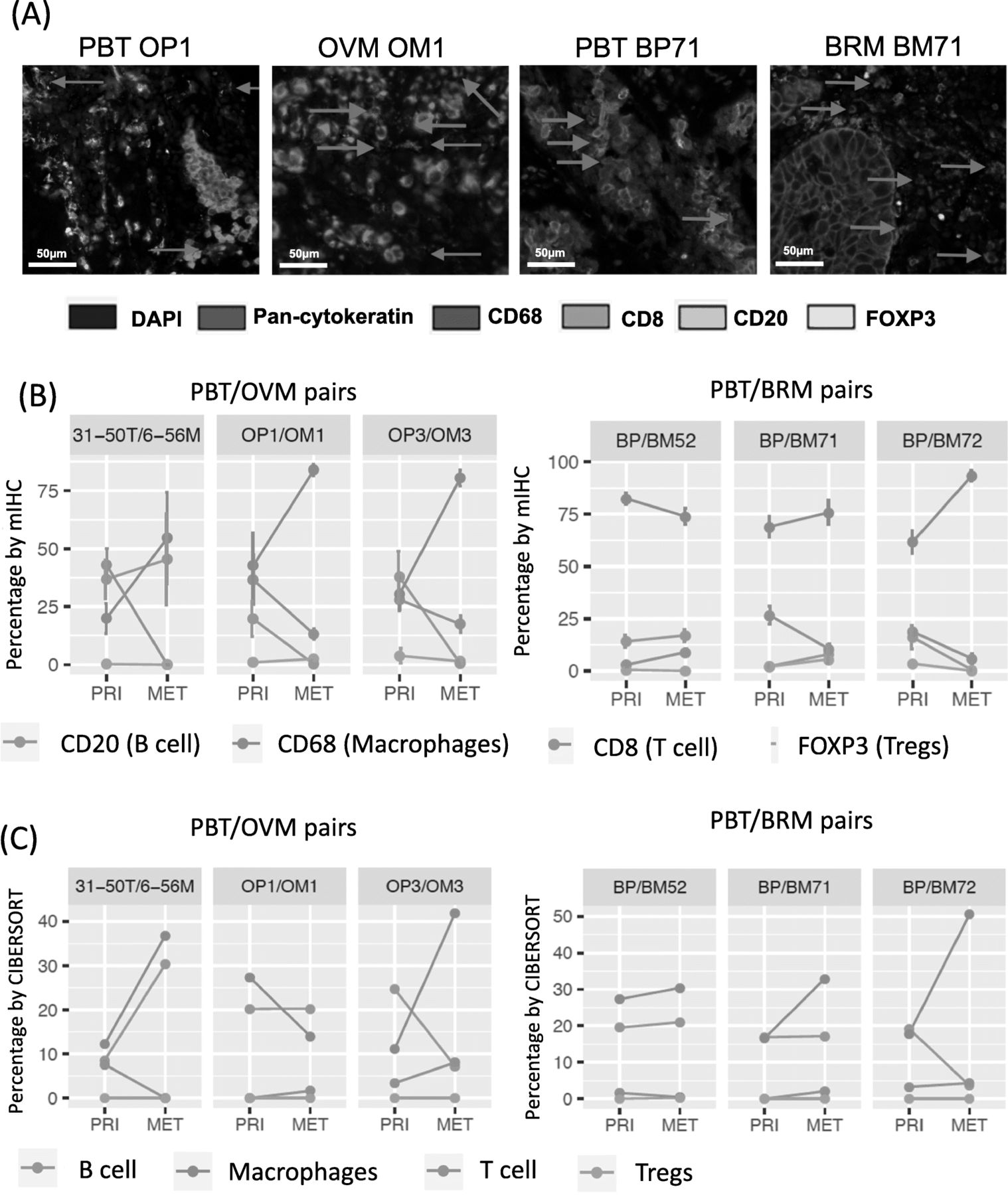
Multispectral immunohistochemical (mIHC) staining of selective pairs in Pan-MET. a mIHC staining images of one pair of PBT/OVMs and PBT/BRMs. b Percentage (by cell) of each immune cell population denoted by markers using mIHC staining. c Relative percentages of corresponding immune cell populations estimated by CIBEROSRT
Hormone receptor (HR) positive tumors are associated with lower total immune abundance
To examine the contribution of each clinical variable, we tested the association between immune level (at PBT, MET and their changes) and all clinical variables available (Additional file 7: Table S8, Additional file 8: Table S9). Both the RNAseq and the sTIL dataset revealed that HR+ PBTs have significantly lower immune scores than HR- PBTs (Fig. 4a). Further, HR+ METs tended to have a smaller decrease in immune abundance compared to PBTs, although this was only significant in the BRM-sTIL dataset. However, stratifying tumors by HR and HER2 status revealed that METs in all categories had lower immune level than paired PBTs (Additional file 2: Figure S1), indicating that decreased immune is not entirely due to HR status. On the other hand, therapies were also strongly associated with the immune level, but they were highly related to tumor subtypes − 94% of ER+ cases received endocrine therapy; 64% HER2+ cases and 6% HER2- patients received HER2 treatment; 87% of all cases received chemotherapy. Due to the heterogeneity of the treatments, and the association with subtype, it is not possible to correct for this confounding variable.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Association of immune abundance with clinical variables and survivals. a Association between immune score and sTILs with clinical variables. b Association between survivals and immune score of PBT/BRM pairs in (b) Pan-MET dataset and (c) BRM-sTIL dataset. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05 from Wilcoxon signed rank and Kruskal-Wallis test in (a) and log-rank test in (b)-(c)
Higher immune abundance is weakly associated with longer time to development of BRMs and longer survival post BRMs
We hypothesized that immune level of PBT may be associated with metastasis-free-survival (MFS), while immune level of MET and its change from PBT to MET are potentially associated with survival-post-metastasis (SPM). Combining all PBT/MET pairs into one cohort, immune score was not significantly associated with MFS or SPM (Additional file 2: Figure S11), likely due to the confounding effect of different MET sites on outcome. Considering PBT/BRM pairs had the largest sample size, we tested the potential association between immune score and survival specifically in PBT/BRMs. In the pan-MET dataset, there was a trend in association between higher immune levels in PBTs and longer time to development of BRMs (i.e. MFS) (Fig. 4b). However, such a trend was not observed between SPM with immune levels in BRM or immune level change between PBT and BRM (Fig. 4b). In the BRM-sTIL dataset, higher sTILs percentage in PBT was not associated with MFS. Instead, there was a trend toward an association between a higher sTILs percentage in MET and longer SPM (Fig. 4c). We did not observe significant associations between the relative level of M2-like macrophage and survivals (Additional file 2: Figure S12).
Discussion
It is now well appreciated that immune cells are a critical component of the TME. Studies of the breast TME have largely focused on tumor mutational and transcriptional landscapes in primary breast cancers, and with more recent attention to metastatic tumors. Our study is novel in two main regards: (1) we examined two cohorts of matched PBTs and METs, one of which includes METs in different sites, allowing us to discern site-specific immune changes from primary to metastatic disease and (2) we evaluated immune abundance by both gene expression analysis and H&E staining, and observed overall high consistency. Our data demonstrate the potential of using bioinformatics tools to investigate the immune contexture of both primary and matched metastatic tumors when tumor lesions may not be available for staining.
Our paired patient-matched comparison revealed a decrease in immune cells from primary to metastatic breast cancer, which is consistent with limited existing studies [18–20]. In-silico analysis of the Pan-MET dataset, validated by mIF staining, highlights the potential enrichment of M2-like macrophages as the tumor cells metastasize to various sites, especially brain and ovary. This is consistent with the growing body of literature that has shown macrophages to be one of the key players in establishment of distant METs [21–23]. Our survival analysis suggests enhanced MFS and SPM in patients with higher immune cell recruitment to primary and metastatic tumors, although the significance of these findings were not consistent between the Pan-MET and BRM-sTIL, possibly due to small sample size and/or sample heterogeneity.
This work has multiple important strengths. First, it utilizes established genomic data sets for elucidating the immunobiology of matched PBTs and METs. Second, it is one of the larger studies of a cohort of patient-matched PBTs and METs. Third, it effectively integrates state-of-the-art genomic analyses with multiplexed immunohistochemistry conducted in a subset of tumors to confirm results. Our study also has several limitations. First, due to the scarcity of patient-matched pairs of primary and metastatic breast cancer, our sample set remains somewhat small relative to studies of primary breast tumors alone. Second, RNAseq analysis was performed on bulk tumor samples, and thus gene expression cannot be attributed to specific cells. Although we attempted to reduce such bias by normalizing the immune score against the non-tumor cell percentage (with consistent conclusions), single cell RNA-sequencing may be needed to completely resolve uncertainties related to cellular heterogeneity. Third, in our mIF studies, the percentage of all immune cells within the tumor was often below 10%. Given these limited numbers of immune cells, our results should be interpreted with caution. Despite these limitations, our study clearly highlights an opportunity to utilize existing data to shed light on the co-evolution and involvement of immune cells in the progression of a primary tumor and its metastatic cascade within an individual patient. It also nominates M2-like macrophages as a potential target for therapeutic immune manipulation of the metastatic cascade.
Funding
We acknowledge funding support from Breast Cancer Research Foundation (AVL, SO, LAE), National Cancer Institute of the National Institutes of Health (P30CA047904), Fashion Footwear Association of New York (FFANY), the Nicole Meloche Memorial Breast Cancer Research Foundations, the Penguins Alumni Foundation, the Shear Family Foundation, TL-1 CTSA Pre-Doctoral Training Grant (5TL1TR001116–03), Duke Cancer Institute Core Grant (P30CA014236), and Magee-Womens Research Institute and Foundation. AVL and SO are recipients of Scientific Advisory Council awards from Susan G. Komen for the Cure, and are Hillman Foundation Fellows. LAE is a Hillman Scholar for Innovative Cancer Research.
Acknowledgments
This project used the Pitt Biospecimen Core, and the UPMC Hillman Cancer Center Tissue and Research Pathology shared resource which is supported in part by NIH/NCI award P30CA047904. We acknowledge the support of the UPMC Cancer Registry and in particular Louise Mazur for clinical data abstraction. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from: the GTEx Portal on 11/17/2017.
Supplementary information
Authors’ contributions
LZ, JLN and GB performed data analysis. SO and MJ performed mIF staining and analyzed staining result. CL, NN and UC validated sTIL counting. AH oversaw TIL counting. RK, KPG oversaw mIF staining. PKB, and CKA contributed tumor samples for sTIL counting. ACS, DAAV, LAE and PCL contributed to the interpretation of the results. GCT oversaw data analysis. SS, KLB, SO and AVL generated study concept and oversight. All authors read and approved the final manuscript.
Availability of data and materials
Data and code for all bioinformatic analyses are available on https://github.com/lizhu06/TILsComparison_PBTvsMET.
Notes
Ethics approval and consent to participate
Ethics approval and consent to participate were approved under the University of Pittsburgh IRB # PRO11100645, 14040193, 1500502, 1602030.
Consent for publication
Not applicable.
Competing interests
PKB receives Speaker’s Honoraria from Genentech-Roche and Merck; research funding from Pfizer and Merck; and is a consultant in Eli Lilly, Merck, and Angiochem. CA receives research funding and serves an uncompensated advisory role for Novartis, Merrimack, Lily, Seattle Genetics, Nektar, Tesaro. CA receives research funding from Merck, Tesaro, PUMA, and G1-Therapeutics. CA has a compensated consultant role with Merck, PUMA and Eisai, and uncompensated advisory role with Genentech. CA receives royalties from UpToDate, Jones and Bartlett. ACS receives Susan G. Komen Foundation Career Catalyst Research award (CCR15329745). DAAV owns stock in TTMS, Tizona, Oncorus, BMS; patents licensed in Tizona, BMS, Astellas; grants from BMS; Scientific Advisory Board in Tizona, Oncorus, F-Star, Pieris; and consultancy in Crescendo, Intellia, MPM, Onkaido, Servier. LE reports grants and non-financial support from Roche/Genentech, Corvu; grants and personal fees from AstraZeneca; grants from EMD Serono; personal fees from Syndax, Amgen, Medimmune, AbbVie, Gristone Oncology, Peregrine, Celgene, eTHeRNA; non-financial support from Bristol-Myers Squibb; grants, personal fees and non-financial support from Aduro Biotech, Bayer, Replimune, Novartis, Macrogenics, Vaccinex, Maxcyte; personal fees and other from Molecuvax; grants from Merck; and member, FDA Advisory Committee on Cell Tissue and Gene Therapies; member, Board of Directors for Society of Immunotherapy of Cancer. PCL owns stock in Amgen (AMGN) and his spouse KP-G has consulted for Bayer/Loxo. KLB is an employee of Eli Lilly. SO is a member of the Scientific Advisory Panel for NSABP.
- BOM
- Metastatic breast tumor in bone
- BRM
- Metastatic breast tumor in brain
- DE
- Differential expression
- ecRNA-seq
- Exome-capture RNA sequencing
- FFPE
- Formalin fixed paraffin embedded
- GI
- Gastrointestinal tract
- GIM
- Metastatic breast tumor in gastrointestinal tract
- GSVA
- Gene set variation analysis
- GTEx
- Genotype-Tissue Expression
- H&E
- Hematoxylin & Eosin
- HR
- Hormone receptor
- MET
- Metastatic breast tumor
- MFS
- Metastasis-free-survival
- mIF
- Multiplexed immunofluorescence
- OVM
- Metastatic breast tumor in ovary
- PBT
- Primary breast tumor
- SPM
- Survival-post-metastasis
- ssGSEA
- Single-sample gene set enrichment analysis
- sTIL
- Stromal tumor infiltrating lymphocyte
- TIL
- Tumor infiltrating lymphocytes
- TME
- Tumor microenvironment
- TNBC
- Triple negative breast cancer
References
Footnotes
Supplementary information Supplementary information accompanies this paper at 10.1186/s40425-019-0755-1.