Article Text

Abstract
Background Previous data have reported that the growth of established tumors may be facilitated by postsepsis disorder through changes in the microenvironment and immune dysfunction. However, the influence of postsepsis disorder in initial carcinogenesis remains elusive.
Methods In the present work, the effect of postsepsis on inflammation-induced early carcinogenesis was evaluated in an experimental model of colitis-associated colorectal cancer (CAC). We also analyzed the frequency and role of intestinal T regulatory cells (Treg) in CAC carcinogenesis.
Results The colitis grade and the tumor development rate were evaluated postmortem or in vivo through serial colonoscopies. Sepsis-surviving mice (SSM) presented with a lower colonic DNA damage, polyp incidence, reduced tumor load, and milder colitis than their sham-operated counterparts. Ablating Treg led to restoration of the ability to develop colitis and tumor polyps in the SSM, in a similar fashion to that in the sham-operated mice. On the other hand, the growth of subcutaneously inoculated MC38luc colorectal cancer cells or previously established chemical CAC tumors was increased in SSM.
Conclusion Our results provide evidence that postsepsis disorder has a dual effect in cancer development, inhibiting inflammation-induced early carcinogenesis in a Treg-dependent manner, while increasing the growth of previously established tumors.
- immunology
- oncology
- tumor microenvironment
- cytokines
- T-lymphocytes
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Colorectal cancer (CRC) is a major health issue worldwide and inflammation has been directly linked to CRC development and tumor progression.1 2 Among many other factors, proinflammatory mediators and increased oxidative stress were postulated as initiator mechanisms of carcinogenesis in CRC.3 This is supported by the increased risk of CRC in patients with inflammatory bowel disease and by the reduced incidence of polyps and CRC after the long-term use of cyclooxygenase inhibitors.4
Beyond local inflammation, additional systemic immune modifications, such as immunosuppression, are expected to modify cancer development.5 Sepsis is a life-threatening organ dysfunction caused by a deregulated host response to an infectious agent.6 Patients who survive to severe sepsis commonly develop a profound anti-inflammatory response, which predisposes to secondary infections,7 higher mortality, and possibly cancer.8 ,9 10 In the experimental setting, accumulating evidence have demonstrated that regulatory T cells (Treg) mediate the immune paralysis in the late-phase sepsis.11
Tregs are crucial for intestinal homeostasis through the regulation of local immune response.12 The adoptive transfer of Tregs inhibits the development of experimental colitis-associated colorectal cancer (CAC), and CRC associated with APC mutation.13 14 Accordingly, the systemic ablation of Tregs in experimentally established CAC attenuates tumor growth through the expansion of CD8+ T cells.15 Altogether, these findings shed light on a potential dual role of Tregs in CRC carcinogenesis.
The question that comes up is whether postsepsis disorder may interfere with initial inflammation-induced CRC carcinogenesis and whether Treg expansion during postsepsis may interfere in this scenario. The present study shows that postsepsis disorder decreases inflammation-induced colorectal tumors in a Treg-dependent manner while promoting the growth of previously established colorectal tumors.
Material and methods
Mice
Six-week to 8-week-old male C57BL/6 mice and Foxp3DTR knock-in mice (depletion of regulatory T cell (DEREG); Jackson Laboratory, USA) mice were bred and housed in the laboratory animal facility of the Ribeirao Preto Medical School (Sao Paulo, Brazil) and were kept in appropriate cages in temperature-controlled rooms with 12 hours dark–light cycles. They received sterilized food and acidified water ad libitum.
Sepsis and postsepsis model
Polymicrobial sepsis was induced by cecal and ligation puncture (CLP), as described previously.16 To save approximately 50% of mice after severe sepsis, the animals were treated with ertapenem (20 mg/kg, i.p., Merck Research Laboratory, Whitehouse Station, NJ) 6 hours after surgery, and every 12 hours for 3 days. The controls were sham operated, and they also received treatment with ertapenem (figure 1A).
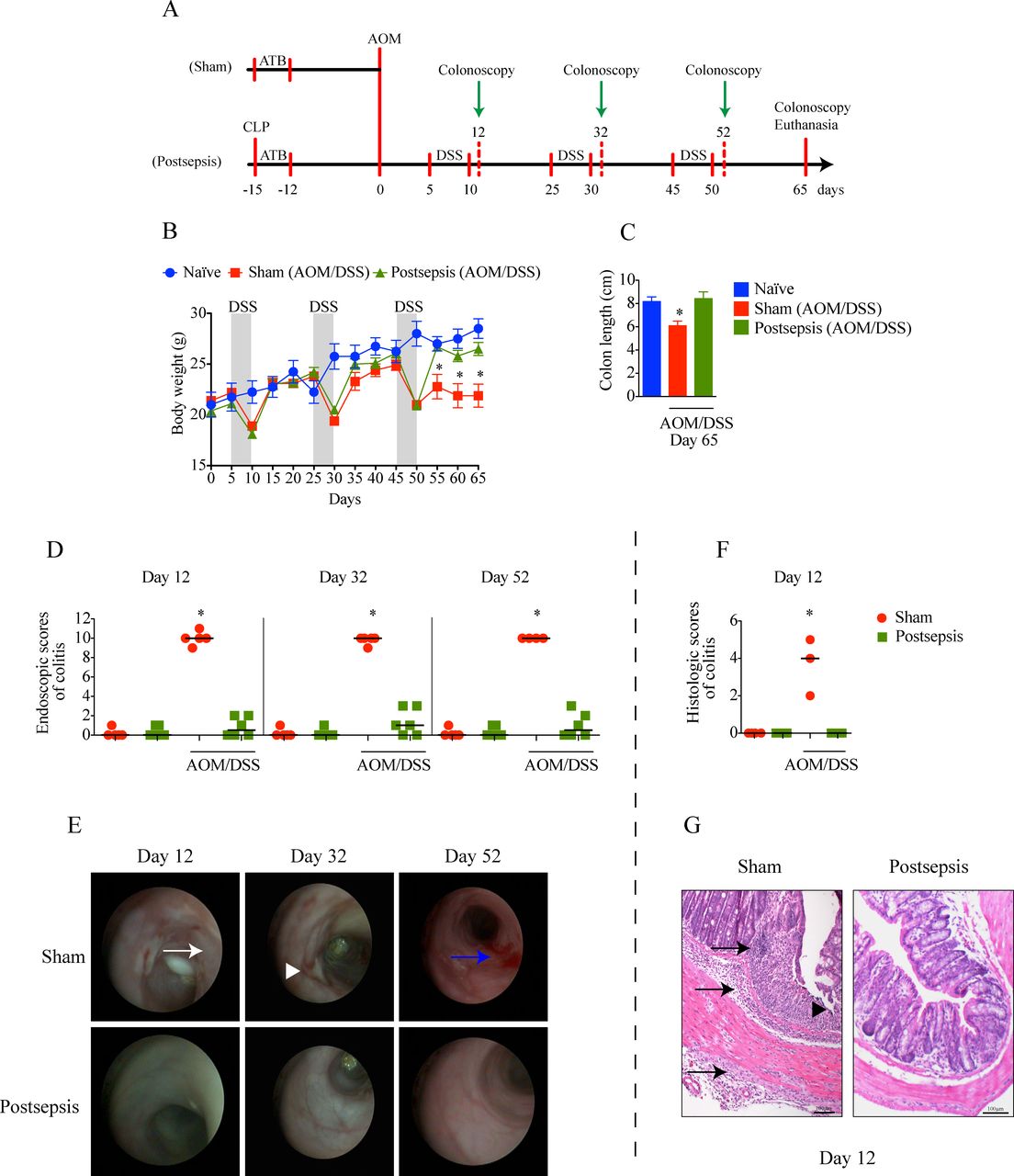
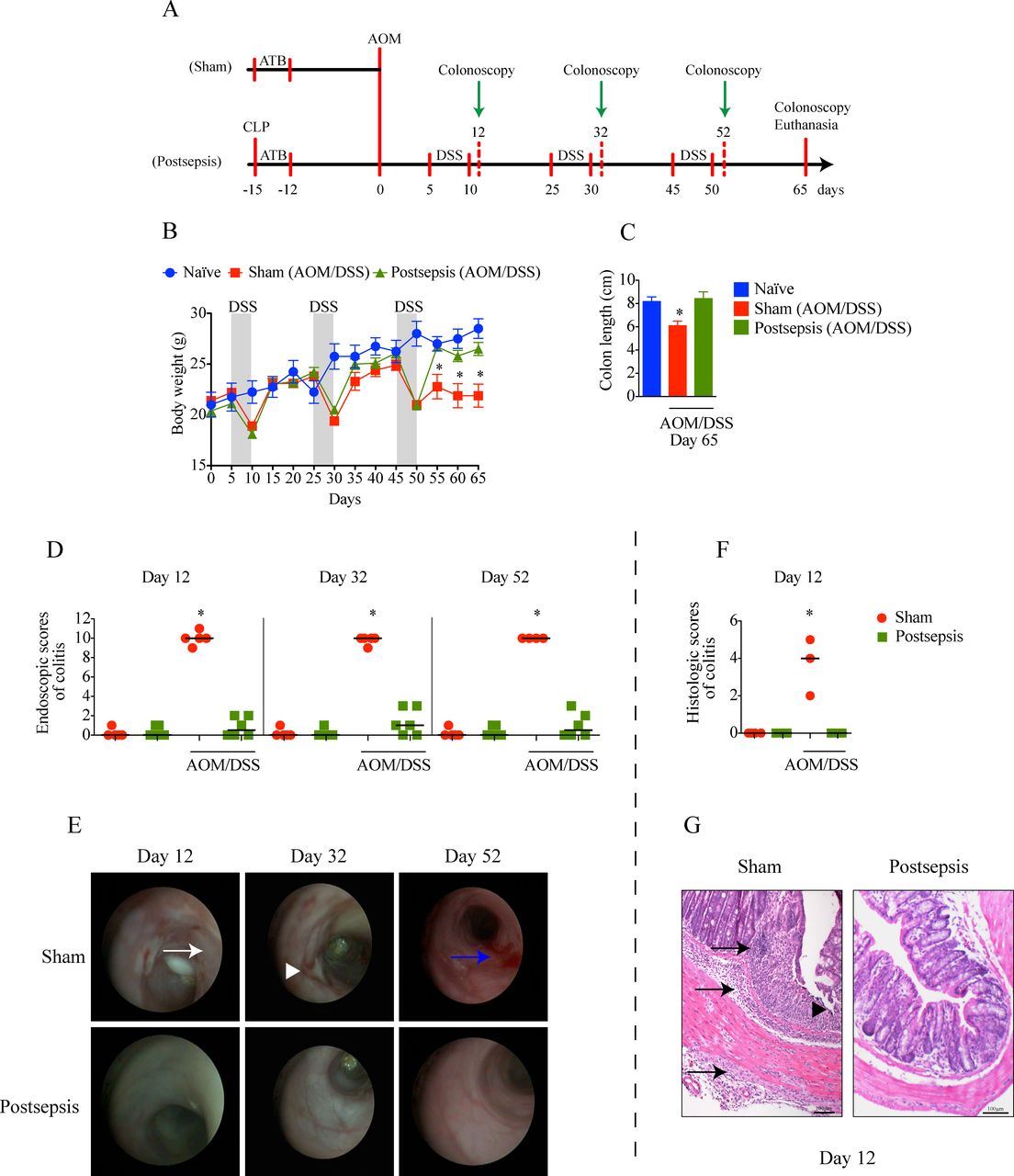
Postsepsis state prevents the development of azoxymethane (AOM)/dextran sodium sulfate (DSS)-induced colitis. (A) Schematic schedule of the administration of AOM and DSS in sham and postsepsis mice. (B) Temporal weight change of naive mice (blue, n=4), AOM/DSS-treated sham mice (red, n=10) and AOM/DSS-treated postsepsis mice (green, n=10). (C) Comparison of colon length at day 65 in naïve mice (n=4), AOM/DSS-treated sham (n=10) and postsepsis mice (n=10). (D) Endoscopic scores of colitis at days 12, 32, and 52 post-AOM administration. (E) Representative endoscopic images from the distal colon of sham (red circles) and postsepsis mice (green squares) at days 12, 32 and 52. (F) Histological scores of colitis at day 12 post-AOM administration. (G) Photomicrographs of representative (H&E staining) colons of sham-operated and postsepsis mice at day 12. White arrow: thick wall; white arrowhead: ulcer; blue arrow: bleeding; black arrow: transmural leukocyte infiltration; black arrowhead: erosion. The experiments were repeated three times. Data demonstrate a representative experiment. Data, mean±SEM. * (B) p=0.0006, (C) p=0.0076, (D) p<0.0001, (F) p<0.0001. CLP, cecal and ligation puncture; ATB, antibiotic.
AOM/DSS protocol
CAC was induced as described before with minor alterations.17 Azoxymethane (AOM)/dextran sodium sulfate (DSS) is a protocol composed of two hits. At first, AOM transforms some cells into a malignant phenotype, and DSS promotes colitis and CAC. Fifteen days after CLP, the mice were injected with the carcinogen AOM (10 mg/kg, i.p.; Sigma Chemical Co. St. Louis, Missouri, USA). After 5 days, the mice received drinking water supplemented with 2% DSS (Sigma Chemical Co.; MW, 36–50 kDa) for 5 days. Each cycle of DSS was repeated three times every 15 days. The mice were euthanized at day 65 (figure 1A). The colon length was registered as an indirect measure of colitis. Samples of colon tissue were harvested for histological analysis, cytokine measurement via ELISA, and Treg quantification via flow cytometry.
Colonoscopy
The animals were anesthetized with isoflurane (1 mL/mL, Cristalia, Brazil) and were submitted to a warm saline enema prior to colonoscopy. A high-resolution mouse video endoscope (TELE PACK VET X, Karl Storz) was used for monitoring colitis and local tumorigenesis, following a described protocol.18 The material consists of a camera, a light source, a monitor, a HOPKINS Forward-Oblique Telescope 30° (diameter 1.9 mm, length 10 cm) and a protection sheath. Colonoscopies were performed by a blinded examiner.
Colitis grading and tumor development quantification
Colitis was scored by four parameters (perianal findings, wall transparency, intestinal bleeding, and focal lesions), and the sum of scores ranged from 0 to 12 (0 being the absence of colitis and 12 being the most severe colitis).18
Tumor development was assessed by measuring the number and size of neoplastic lesions. Tumor load was determined by size, as described: score 1, just detectable; score 2, tumor covering up to one-eighth of the colonic circumference; score 3, tumor covering up to one-fourth of the colonic circumference; score 4, tumor covering up to half of the colonic circumference; score 5, tumor covering more than half of the colonic circumference (figure 2A).19
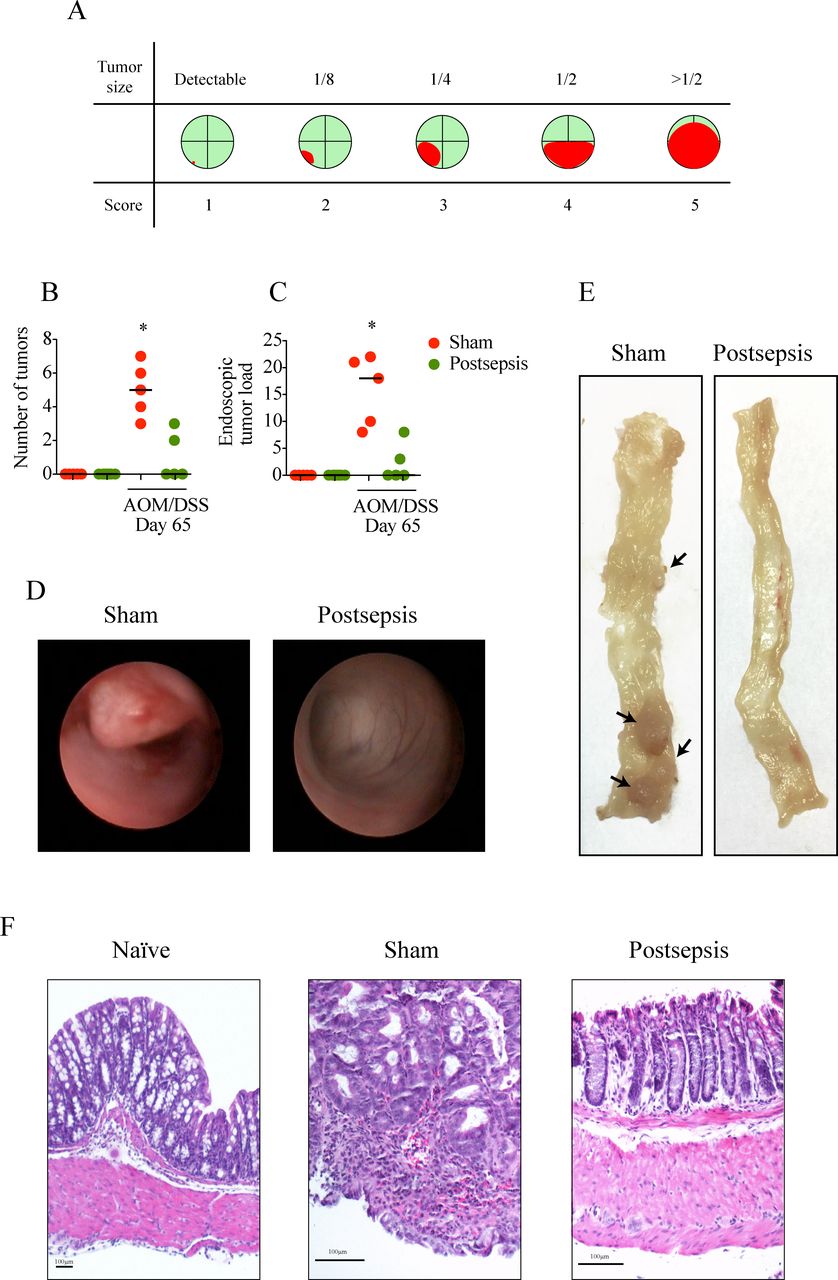
Colitis-associated cancer is impaired in sepsis-surviving mice. (A) Schematic schedule of the tumor-load scoring system. The red color indicates how polyps/tumors occupy the intestinal lumen as seen in colonoscopy. (B) Endoscopic number of colonic tumors and (C) tumor load per mice in sham (red circles) and postsepsis mice (green circles) following azoxymethane (AOM)/dextran sodium sulfate (DSS)-induced colitis (n=5 per group). (D) Representative endoscopic images from the distal colon of sham (left) and postsepsis mice (right) at day 65. (E) Representative images of colons of sham (left) and postsepsis mice (right) evidencing tumor development only in the sham group. (F) Photomicrographs of representative (H&E staining) tumor samples from sham and postsepsis mice. The experiments were repeated three times. Data demonstrate a representative experiment. Data, mean±SEM. *, p<0.0001.
Histological analysis
All colonic lesions (inflammation, polyps and tumors) were fixed in phosphate-buffered formalin and unblocked in paraffin. Tissue sections (4.0 µm) were prepared from the paraffin-embedded tissue blocks, stained with H&E, and evaluated in a blinded fashion by an experienced pathologist, MDB.
Colitis was scored from 0 to 6 according to cell infiltration and tissue damage.20 Focally increased numbers of inflammatory cells in the lamina propria were scored as 1, inflammatory cells extending into the submucosa as 2 and transmural infiltration as 3. For tissue damage, mild lymphoepithelial lesions were scored as 1, erosions as 2, and extensive mucosal damage as 3.
All polyps and suspected lesions were microscopically evaluated and were classified as follows: normal colon, polyp without dysplasia, adenoma with low-grade dysplasia, adenoma with high-grade dysplasia, and invasive adenocarcinoma.
Western blotting assay
Following euthanasia 3 days after AOM injection, colon samples were harvested in radioimmunoprecipitation assay buffer (Thermo Scientific, USA) supplemented with a protease (Roche) and phosphatase (Calbiochem) inhibitors. The protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Scientific, USA). Equivalent amounts of protein (30 µg) in each extract were separated by electrophoresis on polyacrylamide gel followed by transfer to blotting membrane (GE Healthcare Life Sciences, GE). Next, membranes were incubated at 4°C overnight antibodies against γH2A.X (ab81299; 1:5000; ABCAM, USA) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (ab181602; 1:10,000; ABCAM, USA). Next, the membrane was incubated with a secondary antibody conjugated with peroxidase. Chemiluminescence reactions on membranes were imaged using the ChemiDocTM XRS+System (BioRad, Life Technologies, Carlsbad, California, USA).
Real-time PCR qRT-PCR was conducted for specific genes related with DNA repair e cell proliferation (ATM, ATR, MGMT (O-6-methylguanine-DNA methyltransferase), ALKB1, P53, KI67 and βCAT (β-catenin)). Total RNA was obtained using RNeasy Mini Kit (Qiagen, DE) and total RNA was reverse transcribed using high-capacity cDNA RT Kit (Applied Biosystem) according to manufacturer’s instructions. cDNA was used as template for qPCR of genes of interest using Taqman Universal Master Mix II with UNG (Applied Biosystems). TaqMan probes were acquired from Thermo Fisher Scientific (USA): Atm (Mm01177457), Atr (Mm01223626), MGMT (Mm00485014), ALKB1 (Mm012968822), Trp53 (Mm01731290), Ki67 (Mm01278617), β-Catenin (Ctnnb1; Mm00483039_m1) and Gapdh (Mm99999915_g1). Data were collected and analyzed using an Eppendorf Mastercycler RealPlex2 system (Eppendorf, DE). The fold change was calculated using the 2−∆∆Ct method (Gapdh as a housekeeping gene).
Cytokine quantification
On days 0, 12, 52 and 65, the mice were euthanized, and the colons were collected and homogenized in a lysis buffer containing protease inhibitors. Interferon (IFN)-γ, interleukin (IL)-1β, IL-6, keratinocyte chemoattractant (KC), tumor necrosis factor (TNF), IL-33 and tumor growth factor (TGF)-β in intestinal mucosa were quantified using the Milliplex Map Kit (Cat. MCYTOMAG-70K, Merck, USA), according to the manufacturer’s recommendations. The assay was run on a Luminex MagPix instrument (Luminex Corporation, USA).
Isolation of colonic intraepithelial lymphocytes (IELs)
Colonic IELs were isolated as described previously,21 with minor alterations. Briefly, the colons were flushed with phosphate-buffered saline (PBS) to remove feces, opened longitudinally, and cut transversally into 1.0 cm fragments. The tissue fragments were washed with PBS, were placed in RPMI-1640 containing EDTA 0.5 M and DTT 1 M for 30 min at 37°C with rotation and were vortexed thoroughly. To obtain IELs, the cell suspension was passed through a 40 µm cell strainer and was washed twice with cold culture medium.
Flow cytometry analysis
The IELs were stained with fluorochrome-labeled antimouse CD4 (RM4-5, BD Biosciences, USA). Intracellular detection of Foxp3 was performed using the Foxp3 Staining Kit from BD Bioscience with anti-Foxp3 (MF-23, BD Biosciences, USA), according to the manufacturer’s recommendations. The cells were quantified via flow cytometry on a FACSCanto (BD Biosciences, USA) and were blindly analyzed using the FlowJo software V.9 (FlowJo, LLC, USA).
In vivo depletion of Tregs
Aiming to deplete Tregs, we designed two different protocols using sham or postsepsis animals. In the first set of experiments, the mice received cyclophosphamide (CYP; 100 mg/kg, i.p., Baxter, USA) 1 day before the AOM injection. The second experiment consisted of two injections of diphtheria toxin (DTX; 0.5 mg/animal/day, i.p., Sigma, USA) starting 2 days before AOM in DEREG animals. Treg depletion was confirmed via flow cytometric analysis of the splenic tissue. Colitis and tumor formation were monitored via colonoscopy, as aforementioned.
Growth of previously established tumors in postsepsis mice
To address the potential dual role of the postsepsis state in carcinogenesis, CLP was performed in animals with tumors that were already established. In the first set of experiments, the mice were submitted to the AOM/DSS protocol and were later randomized to the sham operation or CLP group on day 65. All animals received ertapenem, as described above. Tumor development and growth were measured using serial colonoscopy. In another set of experiments, the mice were submitted to CLP or sham operation and were subcutaneously inoculated with 105 MC38luc cells derived from C57BL6 murine colon adenocarcinoma 15 days after surgery.22 Tumor growth was quantified via digital caliper or bioluminescence quantification (IVIS Lumina; Caliper Lifesciences, USA) following D-luciferin administration (150 mg/kg, i.p., Caliper Lifesciences, USA).
Statistical analysis
All results were expressed as the mean±SEM. The differences were assessed using Student’s t-test or analysis of variance, followed by Bonferroni’s correction when appropriate. Kaplan-Meier plots were used to estimate survival, using the log-rank test to calculate differences between groups. Statistical analysis was performed using the Prism V.5.0 software (GraphPad). Statistical significance was set at a level of p<0.05.
Results
Postsepsis disorder prevents AOM/DSS-induced colitis
In a previous report, our group showed that postsepsis disorder increases the tumor growth of subcutaneously inoculated B16 melanoma cells.10 To investigate the role of the postsepsis condition on the early phases of the inflammation-induced carcinogenic process, we compared the effects of AOM/DSS in sepsis-surviving or sham-operated mice (figure 1A). As depicted in figure 1B, all animals developed weight loss after each cycle of DSS. However, the development of colitis was observed only in the sham-operated group, as quantified by the reductions in colon length (figure 1C) and bowel inflammation and determined both by colonoscopy (figure 1D,E) and microscopic analysis (figure 1F,G). Sham-operated animals submitted to AOM/DSS cycles presented significant thickening, bleeding, erosions, and ulcers by colonoscopy analysis (figure 1E) and edema, transmural leukocyte infiltration and erosion according to microscopic analysis (figure 1G). Notably, none of the above-mentioned parameters were detected in the sepsis-surviving group. Collectively, these results indicate that the postsepsis state impairs the development of AOM/DSS-induced colitis.
CAC development is impaired in sepsis-surviving mice
Next, tumor development in sham-operated mice was compared with tumor development following sepsis induction. Consistent with our previous data, a significant impairment in the development of CRC was found in sepsis-surviving animals at day 65 after AOM administration (figure 2). The tumor burden (mean number of tumors per colon, figure 2B) and tumor load (figure 2C) were significantly higher in the sham-operated mice than in the sepsis-surviving mice. Notably, 5/5 mice (100%) that were administered AOM/DSS developed tumorous lesions, as identified via colonoscopy (figure 2D) and via gross examination following necropsy (figure 2E). On the other hand, only 40% of postsepsis mice exhibited tumorous lesions (Fisher’s exact test—p<0.05).
In terms of histopathological features, 25% of sham-operated animals developed invasive adenocarcinoma, one quarter exhibited adenomas with low-grade dysplasia, and 50% presented adenomas with high-grade dysplasia (figure 2F, online supplementary table S1). Conversely, only 25% of sepsis-surviving mice exhibited adenomas with low-grade dysplasia. Importantly, these animals did not develop adenomas with high-grade dysplasia or invasive adenocarcinomas (figure 2F, online supplementary table S1). In summary, these observations demonstrate that the postsepsis condition might modify the colonic environment, and it has a profound impact on the colitis-associated initiation of CRC.
Supplemental material
Impairment of CRC development in postsepsis mice is associated with reduced DNA damage and colonic inflammation
Given our previous results and because DNA damage and proinflammatory mediators have been described as crucial for cancer initiation,23 24 we hypothesized that the immunosuppression observed postsepsis may interfere with colon carcinogenesis by local inhibition of AOM-induced DNA damage and the inflammatory response.
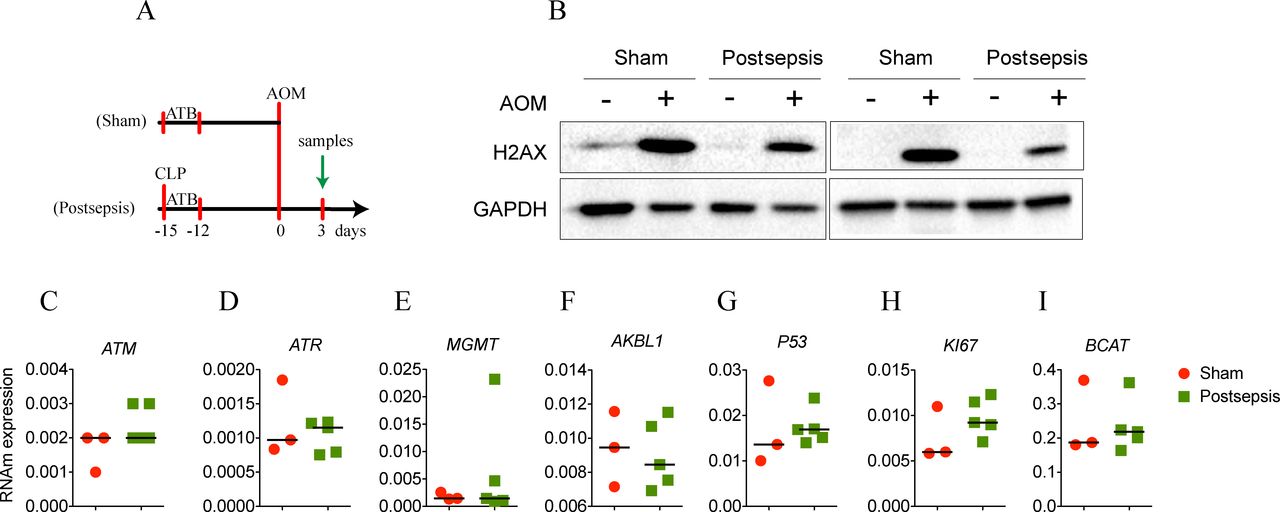
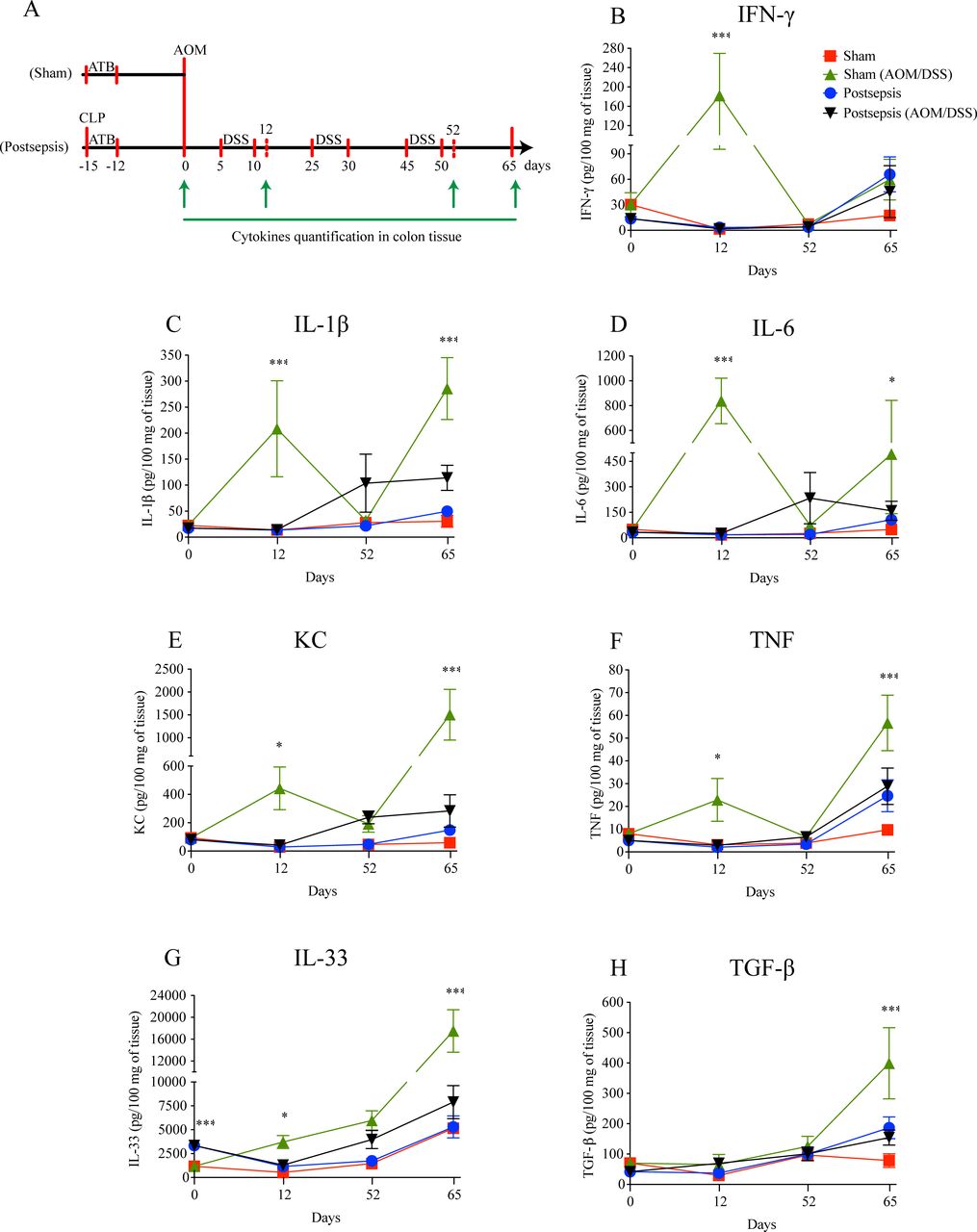
To address this hypothesis, at first, we quantified the amount of histone phosphorylated (γ-H2AX) after the single injection of AOM (figure 3A). As depicted in figure 3B, postsepsis state reduced AOM-induced DNA damage in colon. Since this result did not appear to be related to non-inflammatory gene alterations (figure 3C, D, E, F, G, H, I), we performed a screening of cytokines and chemokines throughout different phases of colitis development (figure 4). By the end of the first DSS cycle (day 12), we found higher levels of proinflammatory cytokines in colonic samples isolated from the sham-operated group compared with those obtained from the sepsis-surviving animals (IFN-γ, IL-1β, IL-6, KC, TNF, and IL-33). These mediators were still elevated at day 65 following AOM treatment (figure 4B, C, D, E, F, G). Of note, the anti-inflammatory cytokine TGF-β was increased in the colon tissues of sham mice only at day 65 of the AOM/DSS protocol (figure 4H). Together, these findings support the idea that the anti-inflammatory postsepsis environment negatively regulates CAC development.

Postsepsis disorder reduces the azoxymethane (AOM)-induced DNA damage. (A) Schematic schedule of DNA-damage evaluation in the model of colitis-associated colorectal cancer. (B) Representative images of western blotting assay of histone phosphorylated (γ-H2AX). The gene expression of (C) ATM, (D) ATR, (E) MGMT, (F) AKBL1, (G) P53, (H) KI67, and (I) BCAT was investigated with no differences between sham and postsepsis mice. The experiments were repeated twice. Data demonstrate a representative experiment. Data, mean±SEM. CLP, cecal and ligation puncture; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
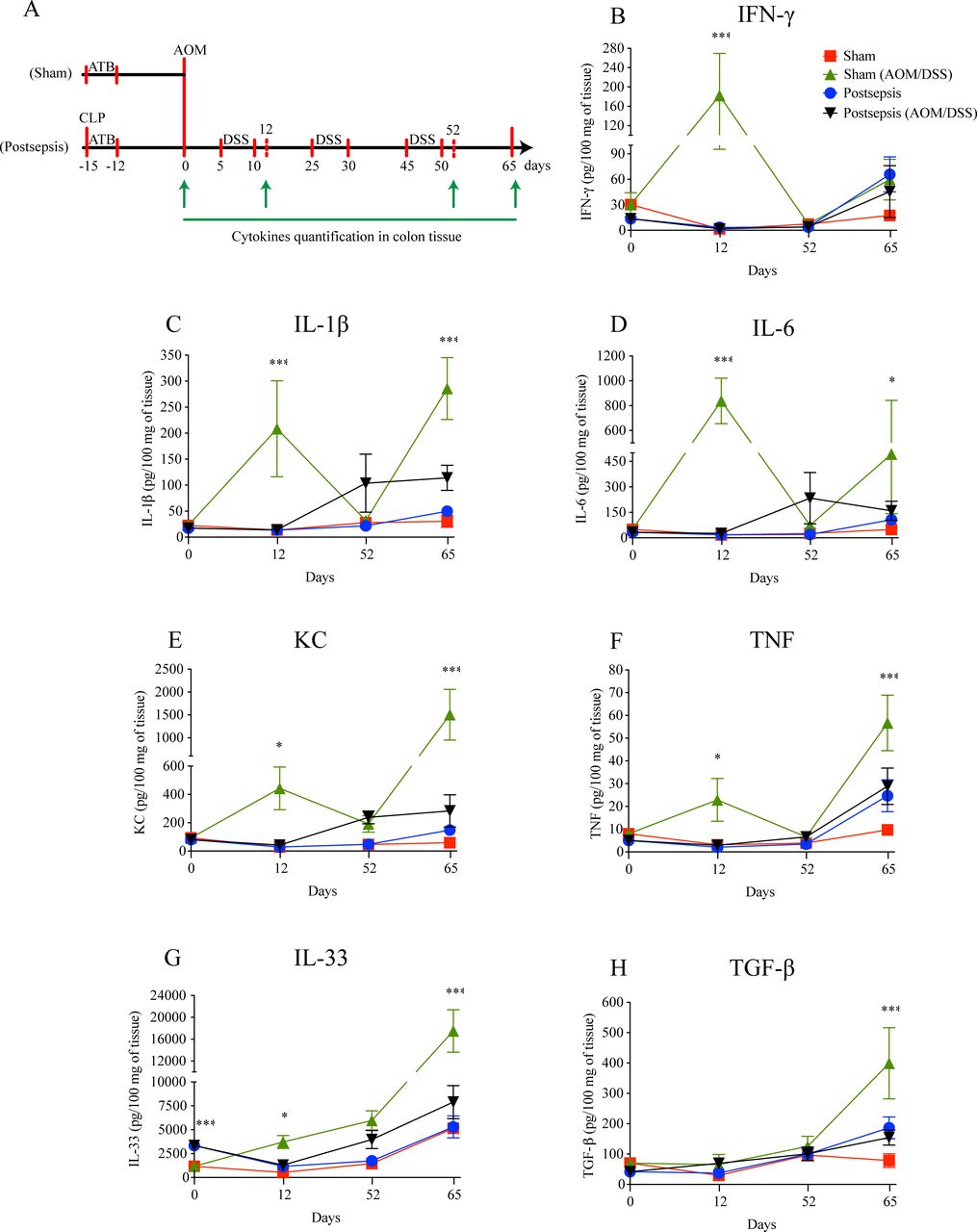
Postsepsis disorder impairs the increase of inflammatory cytokines after colitis induction. (A) Schematic schedule of cytokine quantification in the bowel wall following the model of colitis-associated colorectal cancer. (B) Interferon (IFN)-γ, (C) interleukin (IL)-1β, (D) IL-6, (E) keratinocyte chemoattractant (KC), and (F) tumor necrosis factor (TNF) concentrations peak at day 12 in the sham mice but not in the postsepsis mice. The concentrations of regulatory cytokines (G) IL-33 and (H) tumor growth factor (TGF)-β are increased at day 65 only in the sham mice but not in the postsepsis mice (n=5 per group). The experiments were repeated twice. Data demonstrate a representative experiment. Data, mean±SEM. ***, p<0.0001. *, (D) p=0.0410, (E) p=0.0385, (F) p=0.0097, (G) p=0.0230. AOM, azoxymethane; CLP, cecal and ligation puncture; DSS, dextran sodium sulfate.
Intraepithelial Tregs are increased in the colon of postsepsis mice
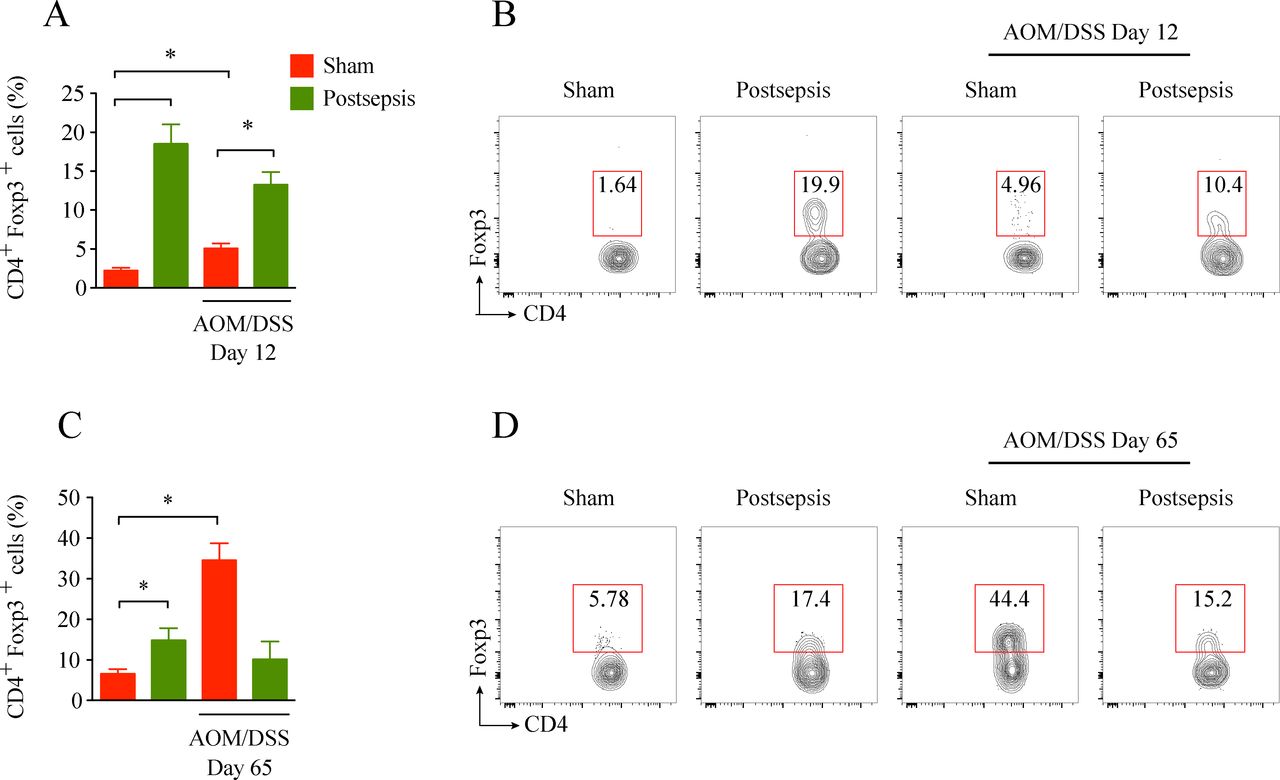
T regulatory cells are crucial players during the immunosuppression condition observed after sepsis resolution.11 To investigate their participation in the impairment of early colorectal carcinogenesis during postsepsis disorder, the frequency of colonic intraepithelial Tregs was measured in our experimental model. As depicted in figure 5 and in accordance with previous reports,9 11 we found a higher frequency of colonic intraepithelial T CD4+ regulatory cells in the sepsis-surviving mice than in the sham-operated mice. A higher percentage of Tregs cells was observed in the colon of postsepsis mice after the first cycle of DSS (day 12 after AOM administration, figure 5A, B). On the other hand, Tregs were found in higher frequencies among the sham-operated mice when the colon tumors were already established (day 65; figure 5C, D). Collectively, these results suggest that Treg expansion following sepsis resolution is related to the anti-inflammatory colonic environment and the impairment of the initiation of CRC.

Intraepithelial Tregs are increased at baseline in the bowel of postsepsis mice. (A) Intraepithelial lymphocyte (IEL) infiltration of Tregs in postsepsis mice at the baseline, and after dextran sodium sulfate (DSS) cycle in sham mice (n=5 per group). The graph is representative of two independent experiments. (B) Representative dot plots of flow cytometric analysis performed at day 12. (C) IEL infiltration of Tregs in postsepsis mice at day 65 and in sham mice that were administered azoxymethane (AOM)/DSS (n=5). (D) Representative dot plots of flow cytometric analysis performed at day 65. The experiments were repeated twice. Data demonstrate a representative experiment. Data, mean±SEM. (A) *, p=0.0002, **, p=0.0571, ***, p=0.0253. (B) *, p=0.2904 (not significant), **, p=0.0002, ***, p=0.0013.
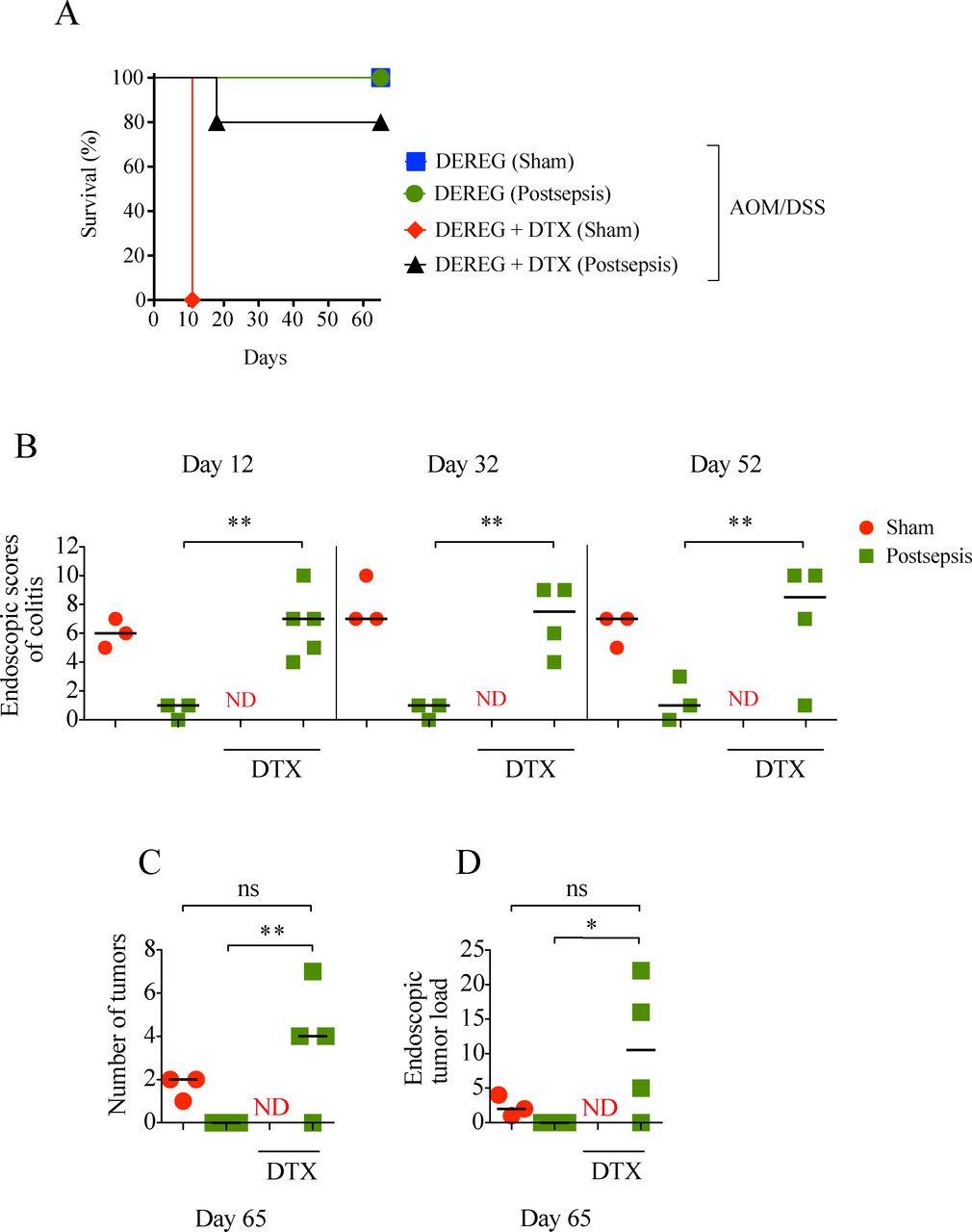
Treg depletion restores colonic inflammation and fosters cancer development in postsepsis mice
We next sought to investigate the participation of Tregs in colitis impairment and directly tested its impact on CAC initiation in postsepsis disorder. By taking advantage of DEREG harboring Treg subsets tagged with a human DTX receptor, we were able to efficiently ablate these cells on in vivo DTX administration.15
As depicted in figure 6A, in contrast to the sepsis-surviving group, the sham-operated animals depleted of Tregs did not survive after the first cycle of DSS (figure 6A), likely due to severe colitis, as seen on necropsy. The depletion of Tregs in the sepsis-surviving mice restored their competence to develop colitis and colonic tumors, in a similar fashion to sham-operated mice (figure 6B, C, D). Accordingly, the pharmacological depletion of Tregs through low-dose CYP just before the AOM/DSS protocol restored the development of colitis and colorectal tumors in the sepsis-surviving mice (see online supplementary figure S1). In summary, these results support the idea that colonic Treg expansion after sepsis resolution promotes a local anti-inflammatory response and negatively impacts colitis-associated colorectal carcinogenesis.
Supplemental material
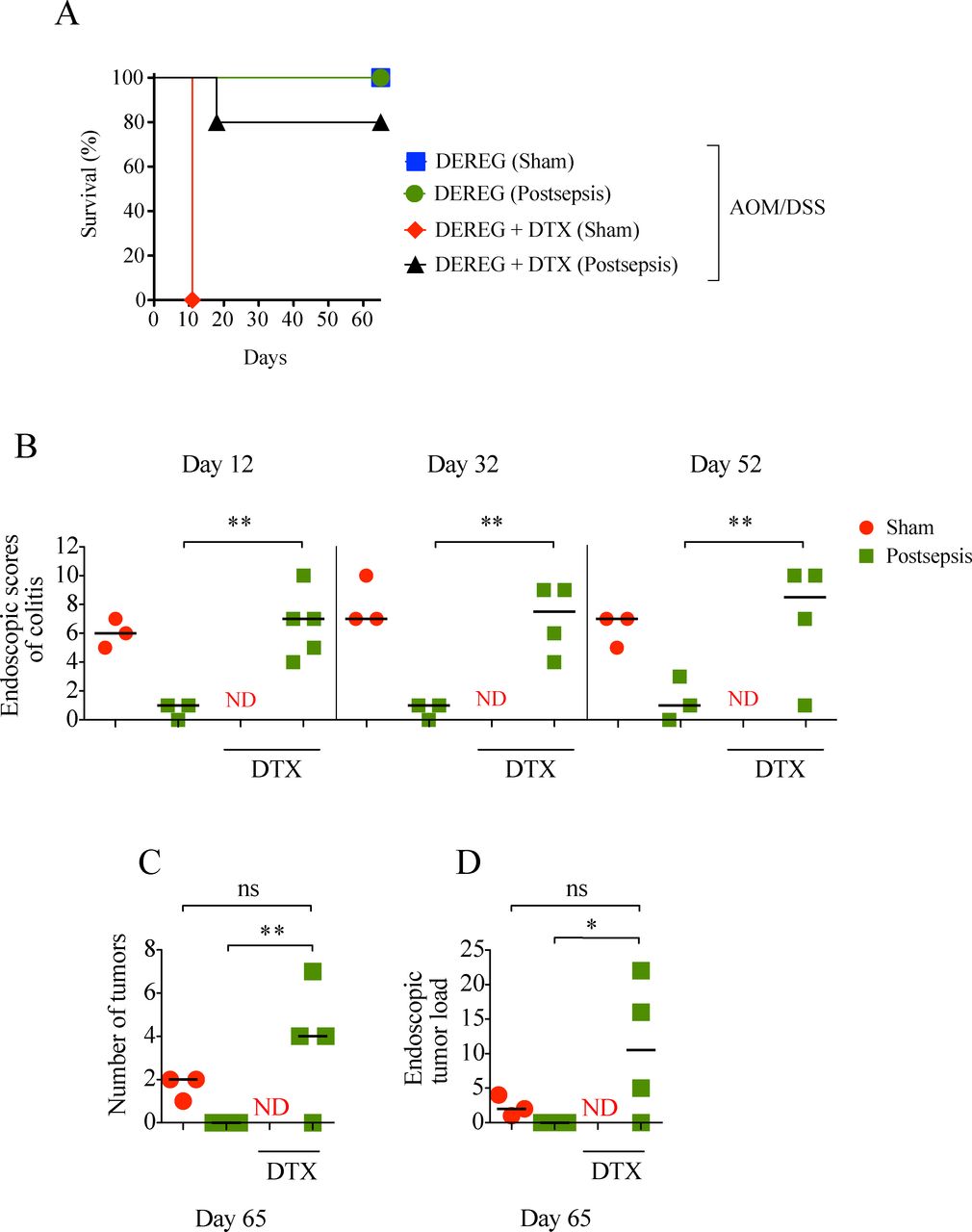
Treg depletion restores colon inflammation and facilitates cancer development in postsepsis mice. (A) Higher mortality in Treg-depleted sham mice after first dextran sodium sulfate (DSS) cycle (depletion of regulatory T cell (DEREG) sham, n=3; DEREG postsepsis, n=3; DEREG +diphtheria toxin (DTX) sham n=6; DEREG +DTX postsepsis, n=5). (B) Endoscopic scores of colitis at days 12, 32, and 52, evidencing colitis in Treg-depleted postsepsis mice. Endoscopic number of colonic tumors (C) and tumor load (D) showing that Treg-depletion postsepsis mice also developed tumors. The experiments were repeated twice. Data demonstrate a representative experiment. Data±SEM. (B) **, p=0.0015, p=0.0031, p=0.0406. (C) *, p=0.0075. (D) *, p=0.0450. ns, not significant; ND, not determined.
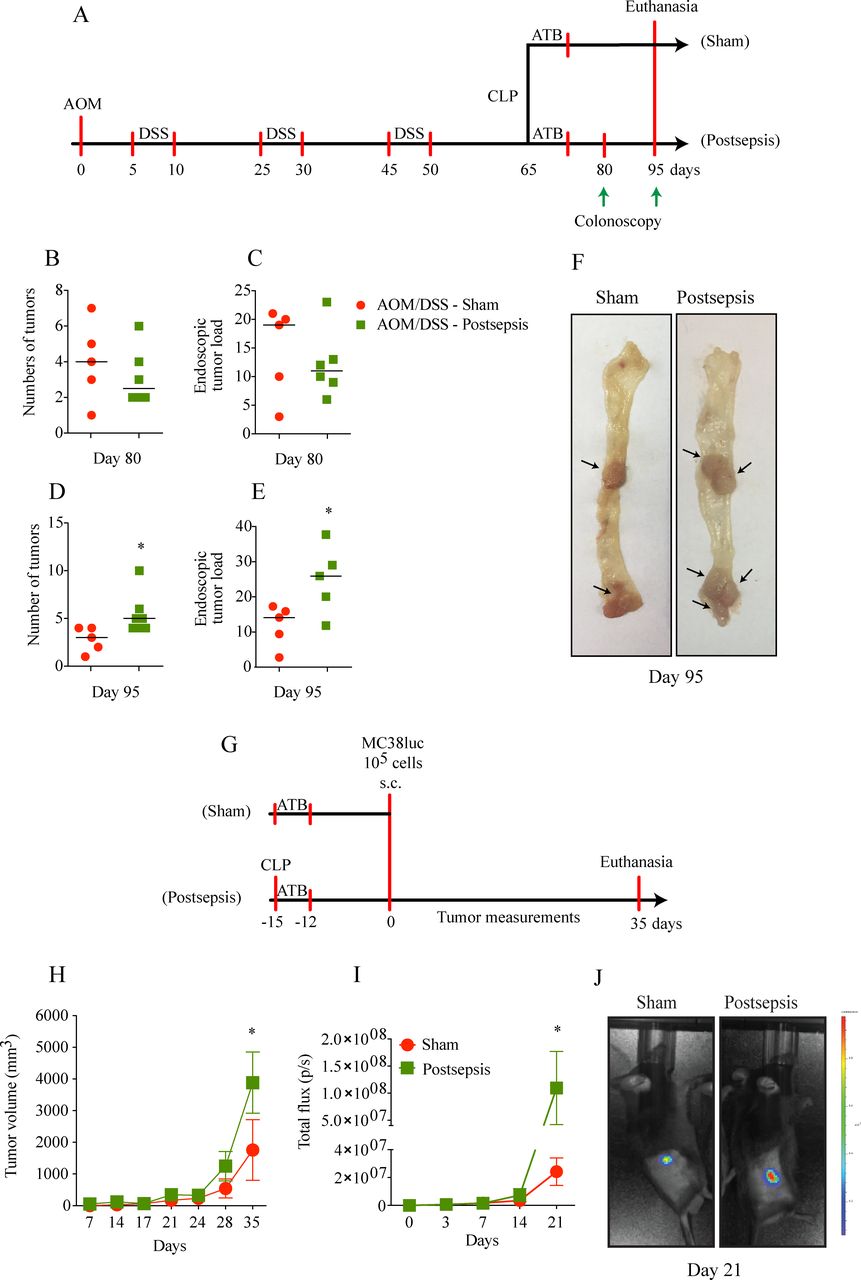
Postsepsis disorder is associated with increased growth of established tumors
Since the immunosuppression following sepsis resolution has been described as a relevant promoter of the growth of lung carcinoma and melanoma,9 10 the effect of postsepsis status was investigated in the context of previously established CAC. For this purpose, we first induced CAC development through the AOM/DSS protocol, which was followed by sepsis induction using the CLP model, as detailed in figure 7A. Notably, the mice were randomly distributed at day 65 of the protocol into 2 groups, the sham operated or postsepsis group, which had an equivalent tumor burden. After 15 days of CLP, both groups maintained a similar number of tumors and tumor load (figure 7B,C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tumor growth of established tumors is increased in the postsepsis state. (A) Schematic schedule of the azoxymethane (AOM)/dextran sodium sulfate (DSS) protocol, followed by cecal and ligation puncture (CLP). (B)–(E) Number of tumors and tumor load at days (B)–(C) 80 and (D)–(E) 95, respectively, showing increased tumor load in postsepsis group at day 95. (F) Representative images of colons of sham (left) and postsepsis mice (right) evidencing a higher tumor development in postsepsis group. Arrows point to the grossly observed tumors. (G) Schematic schedule of s.c. injection of MC38luc cells in sham and postsepsis mice. (H) and (I) Increased tumor growth in postsepsis group measured via (H) digital caliper and (I) bioluminescence (n=10 per group). (J) Representative animal from each group was analyzed at day 21 for the tumor bioluminescence quantification. The experiments were repeated twice. Data demonstrate a representative experiment. Data, means±SEM. *, (D) p=0.0363, (E) p=0.0336, (H) p=0.0089, (I) p=0.0307.
In contrast to our previous findings in the cancer-initiation protocol, sepsis induction enhanced the number of polyps and the tumor load when colorectal tumors were previously formed (figure 7D, E, F). Assessment of tumor counting is limited by the fact that, during disease progression, two or more polyps/tumors may coalesce which can result in a reduction of the total number of polyps/tumors (figure 7D, E). Furthermore, in the model of subcutaneously implanted MC38luc cells, the postsepsis mice showed increased tumor growth compared with the sham-operated mice (figure 7H, I), which was confirmed by detecting the increased tumor burden following luciferin administration (figure 7J). Together, these findings indicate a potential dual role for postsepsis disorder in the cancer context, which on one hand promotes cancer development in previously established tumors and on the other hand impairs early inflammation-induced carcinogenesis.
Discussion
In the present work, we demonstrated that inflammation-induced carcinogenesis is hampered in postsepsis disorder in a Treg-dependent fashion, while the tumor growth of previously established CRC is increased. To our knowledge, this is the first evidence showing that inflammation-induced early carcinogenesis is impaired following sepsis resolution.
The postsepsis period is characterized by an anti-inflammatory condition,25 associated with high susceptibility to subsequent infections and/or other diseases, leading to consequent higher mortality rates.26 The long-term downregulation of the acquired immune response that occurs after sepsis episodes has been described in humans and experimental models.10 26 In the present study, a lack of an intestinal inflammatory response to DSS was observed after more than 60 days following sepsis induction, as shown by the reduced severity of colitis and reduced proinflammatory cytokine concentration in mice previously submitted to sepsis. Moreover, we have shown that sepsis-surviving subjects also presented a significant reduction in the inflammation-induced development of colorectal adenomas and adenocarcinomas.
Inflammatory bowel disease (ie, ulcerative rectocolitis and Crohn’s disease) has been implicated as a causative factor for CRC.27 The clinical relevance of inflammation in colon carcinogenesis is reinforced by the cancer-preventive effect of long-term intake of cyclooxygenase inhibitors, which suppress local eicosanoid production and other inflammatory parameters.28–30 Thus, the reduced DNA damage and production of proinflammatory cytokines during DSS-induced colitis and of other inflammatory events following sepsis may explain, at least in part, the reduction in AOM/DSS-induced CRC.
Many of the connecting mechanisms between CRC and inflammation are still under discussion. Resident cells surrounding the inflammatory site secrete inflammatory cytokines and other mediators, including eicosanoids and oxygen-derived and nitrogen-derived free radicals,31 which mediates several inflammatory events. Consequently, other leukocytes are recruited to the inflammatory site, where they release new waves of inflammatory mediators and free radicals, which could trigger carcinogenesis through inducing DNA damage in the epithelial cells and other cell types.31 Cytokines such as IFN-γ, IL-1β, IL-6, IL-8 and TNF were previously described to play a crucial role in the CRC carcinogenesis as well.32–37 In line with this view, we observed that the inhibition of AOM/DSS-induced CRC in sepsis-surviving mice was associated with a reduction of DNA-damage and inflammatory cytokines production.
It is noteworthy that we observed a significant expansion of intestinal Tregs when animals were previously submitted to sepsis. Accordingly, the previous literature has shown a systemic expansion of Tregs following acute sepsis resolution.11 IL-33, an alarmin found in epithelial cells and related to Treg polarization, is directly implicated in postsepsis-induced Treg expansion.38 In the present work, IL-33 was also found in increased concentrations in the postsepsis intestine before colitis induction
To evaluate if Tregs could be causative players in the reduction of early carcinogenesis observed in postsepsis mice, Tregs were depleted using DTX in DEREG mice or CYP in wild-type mice. The depletion of Tregs in control mice led to fulminant colitis and death, as previously described,15 while restoring the development of colitis, adenomatous intestinal polyps, and colorectal adenocarcinomas induced by the AOM/DSS protocol in the sepsis-surviving group.
These findings ultimately indicate that Tregs mediate the reductions in colitis and AOM/DSS-induced carcinogenesis in the postsepsis state. The mechanism through which Tregs reduce colitis is not the aim of the present study; however, the literature indicates that Tregs suppress the Th1 response by producing the anti-inflammatory cytokines TGF-β, IL-10 and adenosine, which induce T CD8+ exhaustion and stimulate suppressive cells, namely, myeloid-derived suppressor cells, and macrophage polarization towards an immunotolerant profile.39–42 In addition, Tregs promote tumor immune evasion and facilitate tumor growth and metastatic spread.43–45
The postsepsis state is related to a boost in Treg population and function,11 which mediates the increased growth of previously established tumors.9 Accordingly, depleting Tregs in mice with established tumors reduced the tumor growth and precluded the progression from adenomas to adenocarcinomas.15 This shed light on the fact that these cells may have a dual role during colorectal-cancer initiation and progression, that is, impairing early inflammation-inducing carcinogenesis at the same time as promoting the growth of well-established tumors.
Collectively, our results introduce the concept that the postsepsis state impairs the inflammation-induced early carcinogenesis of CRC through the expansion of intestinal Tregs (see online supplementary figure S2). We postulate that in the early phase of colorectal carcinogenesis, postsepsis disorder reduces local inflammatory-related cancer initiators and early promoters, leading to a global impairment of cancer initiation; however, during the late stages of cancer progression (ie, established tumors), postsepsis spurs tumor growth through reduced antitumor immunity and changes in the tumor microenvironment, as previously described.9 10 These results may translate into the clinical setting. Prospective clinical studies are highly expected to evaluate how inflammation-induced carcinogenesis occurs in patients who have recovered from severe sepsis and septic shock and how the modulation of Treg activity can be explored as a therapeutic or preventative strategy. In addition to contributing to a better understanding of the pathogenesis involved in colorectal carcinogenesis, clinical confirmation and the further evaluation of our findings might hereafter impact screening and the further development of chemoprevention strategies to help reduce the cancer burden in the general population.
Supplemental material
Acknowledgments
The authors are grateful to Ieda Regina dos Santos, Ana Kátia dos Santos, Marco Antônio de Carvalho, Giuliana Bertozzi Francisco, Diva Amábile Montanha de Sousa, Tadeu Franco Vieira and Sérgio Roberto Rosa for their technical support.
References
Footnotes
Contributors Conceived and designed the experiments: CAL, JMM, FQC and RAR. Performed the experiments: CAL, JMM, KAdL, CWW, LAN, MDF, CMSS, DFC, JYS, PRV and MDB. Analysed the data: CAL, JMM, KAdL, MDB, JCAF, VCCdL, FQC, VK and RAR. Contributed reagents/materials/analysis tools: VK, JCAF and FQC. Drafted the first version of the manuscript: CAL, JMM and FQC. All authors approved the final version of the paper.
Funding This work was supported by the São Paulo Research Foundation (FAPESP) under grant agreement no. 2013/08216-2 (Center for Research in Inflammatory Diseases).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The experimental protocols and procedures were previously approved by the local Ethics Committee (protocol number: 195/2013) and were in accordance with the Declaration of Helsinki and the Guide for Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.