Article Text

Abstract
Background Natural killer (NK) cells have potent antitumor activities. Nevertheless, adoptive transfer therapy of NK cells has gained very limited success in patients with solid tumors as most infused NK cells remain circulating in the peripheral blood instead of entering tumor sites. Chemokines and their receptors play important roles in NK cell distribution. Enhancing chemokine receptors on immune cells to match and be driven to tumor-specific chemokines may improve the therapeutic efficacy of NK cells.
Methods The CCR5-CCL5 axis is critical in NK cell homing to tumor sites. Thus, we analyzed CCR5 expression on NK cells from patients with cancer and healthy donors. We then upregulated CCR5 and CCL5 with lentiviruses and oncolytic viruses in NK and tumor cells, respectively. Animal experiments were also carried out to test the efficacy of the combination of oncolytic virus with NK cells.
Results In NK cells from patients with various solid tumors or healthy subjects, CCR5 was expressed at low levels before and after expansion in vitro. CCR5-engineered NK cells showed enhanced tumor infiltration and antitumor effects, but no complete regressions were noted in the in vivo tumor models. To further improve therapeutic efficacy, we constructed CCL5-expressing oncolytic vaccinia virus. In vitro data demonstrated that vaccinia virus can produce CCL5 in tumor cells while infectivity remained unaffected. Supernatants from tumor cells infected by CCL5-modified vaccinia virus enhanced the directional movement of CCR5-overexpressed NK cells but not green fluorescent protein (GFP)-expressing cells. More importantly, NK cells were resistant to the vaccinia virus and their functions were not affected after being in contact. In vivo assays demonstrated that CCL5-expressing vaccinia virus induced a greater accumulation of NK cells within tumor lesions compared with that of the prototype virus.
Conclusion Enhancement of matched chemokines and chemokine receptors is a promising method of increasing NK cell homing and therapeutic effects. Oncolytic vaccinia viruses that express specific chemokines can synergistically augment the efficacies of NK cell-based therapy.
- immunology
- oncology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Adoptive transfer of tumor-killing immune cells shows promise in treating malignant diseases.1 Among cytotoxic cells, natural killer (NK) cells inherently recognize and eradicate malignant cells, which have attracted attention for developing more efficient antitumor regimens.2 In treating hematological malignancies such as leukemia, NK cell infusion results in positive clinical responses3 4; however, NK cell-based therapies barely succeed in solid tumors.5 6 In patients with solid tumors, the intrinsic or infused NK cells mostly circulate in the peripheral blood instead of infiltrating into malignant lesions.7–9 The limited migration toward tumor tissues impairs the therapeutic effectiveness of NK cells. Therefore, overcoming the challenge of accessing malignant cells by killer cells is critical for developing clinically effective NK cell-based therapies.
Chemokines and their receptors are central players in determining the trafficking and distribution of various types of cells including NK cells.10 Chemokines bind to their cognate chemokine receptors and initiate the directional movement of cells to sites with higher concentrations of the chemoattractant.11 It is thus feasible to improve the tumor accumulation of NK cells by upregulating specific chemokines within tumor lesions and their cognate receptors on NK cells simultaneously. Reports from our group and others have shown that the chemotactic cytokine CCL5 is widely overexpressed in malignant lesions, directs infiltration of T cells and indicates better prognosis of patients with different types of tumors.12–15 As the most important receptor for CCL5, CCR5 is indispensable for the site-specific localization of immune cells. NK cells are also recruited by CCL5 via CCR5.16–18 In this context, we could manipulate CCL5 and its receptor CCR5 to alter the in vivo distribution of NK cells.
Lentiviruses are a powerful tool for gene modification in NK cells,19 using which we can construct CCR5-overexpressing NK cells. Additionally, oncolytic viruses (OVs) represent an attractive tool for local adaption of the tumor microenvironment as they can selectively replicate and produce exogenous proteins within tumor cells.20 In this report, we adopted the oncolytic vaccinia virus to increase CCL5 production within tumor tissues and tested the strategy of CCR5-transgenic NK cells in combination with chemokine-expressing OV for tumor treatment. Our study demonstrates that OV treatment and NK cell transfer produces a synergistic response that is effective in eradicating solid tumors.
Methods
Cell lines and reagents
The human colon cancer cell line HCT-116, human cervical cancer cell line HeLa, human breast cancer cell lines MCF-7 and MDA-MB-231, and human chronic myelogenous leukemia cell line K562 were obtained from the American Type Culture Collection (ATCC, Manassas, Virginia, USA). All cell lines were maintained in Dulbecco's Modified Eagle Medium (DMEM) or Roswell Park Memorial Institute-1640 (RPMI-1640) media (Thermo Fisher Scientific, Waltham, Massachusetts, USA) supplemented with 5% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific), 1% penicillin and streptomycin (Thermo Fisher Scientific) and antimycoplasma reagent (InvivoGen, San Diego, California, USA). Cells were passaged every 2–3 days to maintain appropriate densities.
NK cell isolation and expansion
The buffy coats of healthy volunteers were obtained from local blood centers. After mononuclear cells were isolated using Ficoll-Paque (Sigma-Aldrich, St. Louis, Missouri, USA), CD56+CD3- cells were purified using the NK Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Expanding procedures of NK cells were adapted from a previous study.21 Briefly, NK cells were suspended at 1×106 cells/mL in X-VIVO 10 media (Lonza, Basel, Switzerland) supplemented with 5% heat-inactivated FBS, 200 U/mL interleukin-2 (IL-2; R&D Systems, Minneapolis, Minnesota, USA), 10 ng/mL IL-15 (R&D Systems) and mitomycin-treated K562 cells at 1×106 cells/mL. During the 15 days of expansion, NK cells were passaged every 2–3 days with replenishment of cytokines and freshly prepared K562 cells.
Lentiviral vector construction and NK cell transduction
CCR5 was cloned from a cDNA library of human blood cells using PCR, after which the coding sequence was cloned into green fluorescent protein (GFP)-expressing pCDH lentivirus vector (System Biosciences, Mountain View, California, USA). Lentivirus production and concentration were performed as previously described.22 Lentiviruses were serially diluted and the titers determined by analyzing the GFP-positive rates of infected 293 T cells. A spinfection protocol was employed to enhance the transduction efficacies of NK cells. Briefly, NK cells were activated for 4 days with cytokines and feeder cells as described above, then mixed with lentivirus at a multiplicity of infection (MOI) of 10 and centrifuged at 1000×g for 2 hours at 32°C. Five days after virus infection, integration of target genes in NK cells was determined by fluorescence-activated cell sorting (FACS).
FACS analysis
Cells were counted, collected and then washed with ice-cold phosphate buffered saline (PBS) containing 2% FBS, after which the cells were incubated with fluorochrome-conjugated antibodies (Biolegend, San Diego, California, USA) or apoptosis detection chemicals (Thermo Fisher Scientific) according to manufacturer’s instructions. Following that, cells were washed three times with ice-cold PBS containing 2% FBS and analyzed using a Canto II or Accuri C6 flow cytometer (Becton Dickinson, Franklin Lakes, New Jersey, USA). For in vivo assays of NK cells, tissues were dispersed and single cell suspensions prepared. Next, samples were stained with fixable viability dye (Invitrogen, Carlsbad, California, USA) and fluorochrome-conjugated anti-human CD45 antibody (Biolegend). The stained samples were then subjected to FACS analysis.
Cytotoxicity assay
Cytotoxic activity of NK cells was determined using a Lactate Dehydrogenase (LDH) Detection Kit (Thermo Fisher Scientific). Tumor cells were incubated with NK cells at various effector to target (E:T) ratios for 6 hours, after which the supernatants were transferred to a new plate, mixed with reaction buffer for 0.5 hour, and subsequently incubated with stop solution. LDH activities were then monitored by measuring optical densities at 490 and 680 nm. Cytotoxic lysis was calculated according to the formula % cytotoxicity = (sample LDH activity-spontaneous LDH activity)/(maximal LDH activity-spontaneous LDH activity)×100.
Cytokine detection
NK cells were incubated with tumor cells at E:T=1 for 24 hours, after which supernatants were collected and cytokine (interferon-γ (IFNγ) and tumor necrosis factor α (TNFα)) secretions determined using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems) according to manufacturer instructions.
Chemotaxis assay
Transwell assays were carried out as previously described.23 Supernatants from tumor cells or media containing CCL5 protein (R&D Systems) were added to the bottom wells. NK cells were added to the 5 µm Transwell chambers (Corning, Corning, New Jersey, USA) and incubated for 1 hour. For the blocking assays, CCL5-neutralizing antibody (R&D Systems) was supplemented to the bottom wells before adding NK cells to the upper chamber. After incubation, cells in the bottom wells were counted to determine the difference in NK cell movement.
Oncolytic virus
Oncolytic vaccinia virus WR (Western Reserve) strain (ATCC) was engineered to express the CCL5 gene; a plasmid containing the CCL5 gene regulated by the viral promoter P7.5 early/late and firefly luciferase genes regulated by the strong synthetic viral promoter SE/L was inserted into the viral thymidine kinase (TK) gene locus by homologous recombination, thus inactivating the TK gene.20 The coding sequence of CCL5 was cloned from the human cDNA library. OV expansion in tumor cells was monitored using bioluminescence or plaque formation assays. Viabilities of tumor cells infected with OV were determined by an MTS cell viability assay for assessing cell survival relative to uninfected controls.
Real-time PCR
NK cells or HCT-116 cells infected with vaccinia virus were collected and washed with PBS. DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), after which real-time PCR was performed to detect the genomic DNA of vaccinia virus. Briefly, 100 ng DNA from NK cells or 5 ng DNA from HCT-116 cells were mixed with primers (0.4 µm for each primer) and TB Green Fast qPCR Mix (TaKaRa, Kusatsu, Japan). Next, 40 cycles of expansion were performed according to the manufacturer’s instruction. Primers were synthesized as previously described24 and the sequences were as follows, forward, 5ʹ-CGGCTAAGAGTTGCACATCCA-3ʹ and reverse, 5ʹ-CTCTGCTCCATTTAGTACCGATTCT-3ʹ. Ct values were used to determine the expansion of viral DNA.
Animal experiments
Female nude mice (NU/J strain) aged 8–10 weeks were obtained from the Jackson Laboratory (Bar Harbor, Maine, USA). HCT-116 cells (1×106) were implanted subcutaneously per mouse, after which OVs (105 or 106 plaque-forming unit (PFU) as required) were intratumorally injected 10 days later. Virus proliferation was monitored for three successive days via bioluminescence assays. Imaging was on an in vivo imaging system (IVIS200; PerkinElmer, Waltham, Maine, USA) after intraperitoneal injection of luciferin substrate. On the fourth day after virus treatment, 5×106 NK cells labeled with Cy5.5 NHS Ester were injected through the tail veins. Two days later, the tumorous accumulation of NK cells was examined using a fluorescence molecular tomography imaging system (FMT2500; Perkin Elmer). In some experiments, tumor homogenates were collected 4 days post virus injection for CCL5 detection. Tumor volumes were calculated twice a week until they reached 1500 mm3.
Statistical analysis
Data are presented as the mean±SD and are representative of at least three independent experiments. Student’s t-test was used to compare two groups of data, while one-way analysis of variance analysis was performed to determine the difference among three or more groups. Kaplan-Meier curves were used to estimate survival and the log-rank test was used to determine the difference in survival. Bonferroni correction was performed when multiple survival curves were compared. P values <0.05 were considered statistically significant.
Results
CCR5-engineered NK cells show enhanced migrating ability but not cytotoxic function
We first analyzed CCR5 expression on NK cells purified from patients with cancer and healthy donors. The purities of expanded NK cells ranged from 85% to 90% (online supplementary figure S1A). As shown in figure 1A, CCR5 was expressed at low levels on both freshly isolated and expanded NK cells (Less than 30% of NK cells were CCR5+). Due to the limited expression of CCR5, NK cells would not efficiently respond to endogenous or virus-delivered CCL5, and thus remain in the peripheral blood. Therefore, we attempted to overexpress CCR5 in NK cells with lentiviruses; NK cells were successfully engineered and CCR5 expression was significantly increased after lentivirus infection (online supplementary figure S1B and figure 1B). The transduction efficacies with NK cells from different donors ranged from 30% to 70%. Next, we examined the proliferation, cytotoxicity and directional movement of these engineered NK cells. During expansion, untransduced (UTD), GFP- (NK-GFP) and CCR5-transduced NK cells (NK-CCR5) showed similar proliferating curves (online supplementary figure S1C), indicating that transgenic modifications have limited impact on NK cell expansion. When coincubated with tumor cells at various E:T ratios, different NK cells demonstrated similar cytotoxicity (figure 1C). To further dissect the effects of CCR5 modification on NK cell cytotoxicity, the apoptotic death of tumor cells and the secretion of effector cytokines were tested at an E:T ratio of 1:1. As shown in figure 1D, tumor cell apoptosis was efficiently induced by NK cells 6 hours post incubation. The percentages of dying tumor cells induced by different NK cells were not significantly different (p>0.05; figure 1D and online supplementary figure S2). Consistent with this, the IFNγ and TNFα concentrations in supernatants after 24 hours of incubation exhibited minor differences among the three groups (p>0.05; figure 1E). These findings indicate that lentivirus-mediated CCR5 overexpression does not affect cytotoxic cytokine production and the direct tumor-killing activity of NK cells.
Supplemental material
Supplemental material

Functional assays of transgenic NK cells. (A) NK cells from peripheral blood of patients with cancer or healthy donors were purified and expanded. CCR5 was determined using flow cytometer on day 0 (resting) and day 15 (expanded). (B) GFP and CCR5 expression in NK cells. NK cells were infected with GFP or CCR5-expressing lentivirus, and the GFP+ ratios and mean fluorescent intensities (MFI) of CCR5 levels were determined in whole NK cells after 5 days. As a control, GFP and CCR5 expressions were analyzed in UTD NK cells. (C) Different NK cells were incubated with tumor cells at indicated E:T ratios for 6 hours, then NK cell cytotoxicity was determined. (D) Tumor cells were mixed with NK cells at E:T=1:1 for 6 hours, then tumor cell apoptosis was detected. (E) Tumor cells were coincubated with NK cells at E:T=1:1 for 24 hours, after which the supernatants were collected and ELISA was performed to determine IFNγ and TNFα secretion. (F) Transwell assay of NK cells. Media containing 1000 pg/mL of recombinant CCL5 or culture supernatants from HCT-116 cells were supplemented with bovine serum albumin (BSA) or CCL5-neutralizing antibody (anti-CCL5, 0.2 µg/mL) and then added to the bottom wells. Next, NK cells were plated in the upper chambers, and 1 hour later, the cells at the bottom wells were counted. UTD NK cells in wells with BSA were used as control to calculate the migration indices. (G) NK cells were labeled with proliferation-indicative eFluor670 dye and cocultured with HCT-116 cells at E:T=1:1 for 48 hours. Proliferation was determined using flow cytometry. Data are presented as the means±SD and are representative of independent experiments with samples from at least five healthy donors. One-way ANOVA analysis was performed to determine the statistical significances among different groups. *P<0.05, **P<0.01, ***P<0.005. ANOVA, analysis of variance ; E:T, effector to target; GFP, green fluorescent protein; IFNγ, interferon-γ; NK, natural killer; TNFα, tumor necrosis factor α; UTD, untransduced.
We next analyzed the migratory ability of modified NK cells. Transwell assays showed that NK cells rely on the CCR5-CCL5 axis for directional migration and that CCL5 deprivation impaired NK cell movement (figure 1F). Compared with the UTD and GFP-transgenic counterparts, NK-CCR5 cells exhibited increased migration ability in response to CCL5 protein (p<0.005) or tumor cell culture supernatant (p<0.005; figure 1F). When CCL5 was neutralized, the migration of NK-CCR5 cells was completely attenuated (p<0.005; figure 1F), indicating that the CCR5-CCL5 axis enhances NK cell migration. Furthermore, NK cells and their modified counterparts showed similar proliferation rates after coincubation with target cells (figure 1G), indicating that CCR5 modification has limited impact on the proliferation of activated NK cells.
CCR5-overexpressing NK cells show enhanced antitumor effects in vivo
GFP-expressing and CCR5-expressing NK cells were labelled with fluorescent dyes and injected through the tail veins of mice. After 48 hours, we analyzed the tumorous accumulation of NK cells via FMT imaging. As shown in figure 2A and B, tumor infiltration of NK cells was enhanced by the CCR5 transgene (p<0.05). In agreement, NK-CCR5 cells markedly delayed tumor progression and improved mouse survival (p<0.005), although NK-GFP cell infusion led to tumor suppression and prolonged survival as well (p<0.01;figure 2C,D). Together with our in vitro data, the results indicate that CCR5 modification enhances tumor infiltration and subsequently the cytotoxic effects of NK cells.

Antitumor effects of NK cells in vivo. A total of 1×106 HCT-116 cells were inoculated subcutaneously per nude mouse. Ten days later, PBS containing NK-GFP or NK-CCR5 cells (5×106 per mouse) prelabeled with Cy5.5 dyes, or the same volumes of PBS, were injected through the tail veins. (A, B) FMT was used to determine tumor accumulation of NK cells 2 days after injection. n=4 mice per group. (C, D) Tumor growth (C) and mouse survival (D) were monitored after receiving different treatments. The median survivals were 19.5, 30 and 40. 5 days, respectively, in indicated groups. n=6 mice per group. A t-test was used to compare NK cell accumulation within tumors; Kaplan-Meier curves and log-rank tests were used for survival curves. *P<0.05, **P<0.01, ***P<0.005. FMT, fluorescence molecular tomographic; NK, natural killer; PBS, phosphate buffered saline.
CCL5-expressing oncolytic vaccinia virus retains its oncolytic capacity
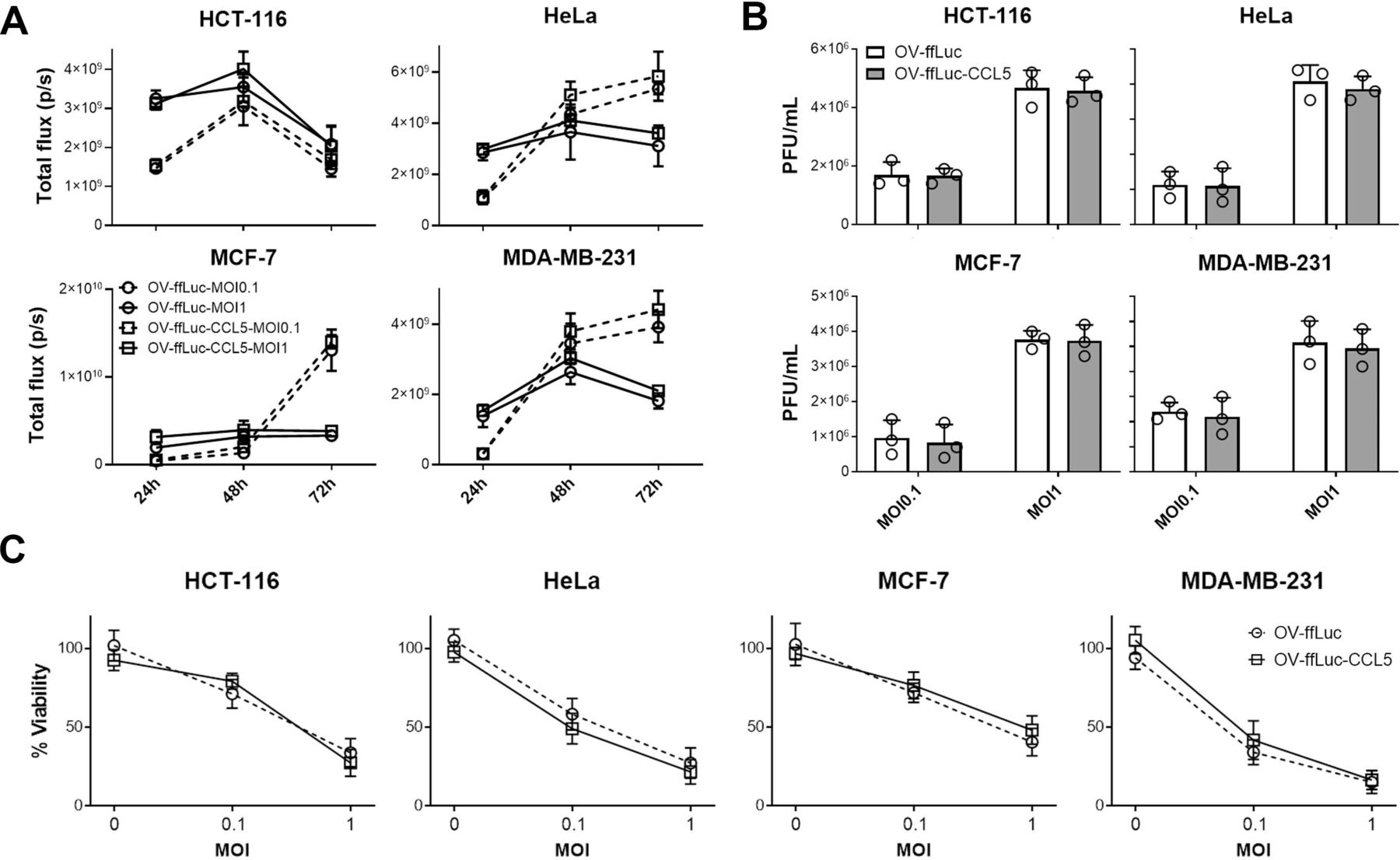
CCR5-expressing NK cells delayed tumor growth. However, none of the treated mice reached complete regression (figure 2C and D). One possible explanation is the insufficient expression of the CCR5 ligand CCL5 in tumors, which largely decides the infiltrating intensities of CCR5-expressing immune cells. We, thus, explored whether the intratumorous production of CCL5 by the OV increases NK cell trafficking and tumor cell elimination. First, we constructed a human CCL5-producing oncolytic vector (OV-ffLuc-CCL5) based on the TK deficient vaccinia virus (OV-ffLuc); TK deficiency limits viral replication to tumor cells. Next, human cancer cell lines were infected with OV-ffLuc or OV-ffLuc-CCL5 at an MOI=0.1 or 1. Bioluminescent imaging (BLI) demonstrated that both the prototype and CCL5-producing vaccinia virus replicated efficiently in malignant cells (figure 3A). Plaque formation assays also supported that CCL5 insertion had limited effect on OV replication (p>0.05; figure 3B). Furthermore, we analyzed the viabilities of infected cells and found that the proliferation rates of all four cancer cell lines were similarly inhibited by OV-ffLuc and the derivate OV-ffLuc-CCL5 (p>0.05; figure 3C). These data indicate that the CCL5-modified vaccinia virus has similar infectivity and replicating potency to the prototype OV.
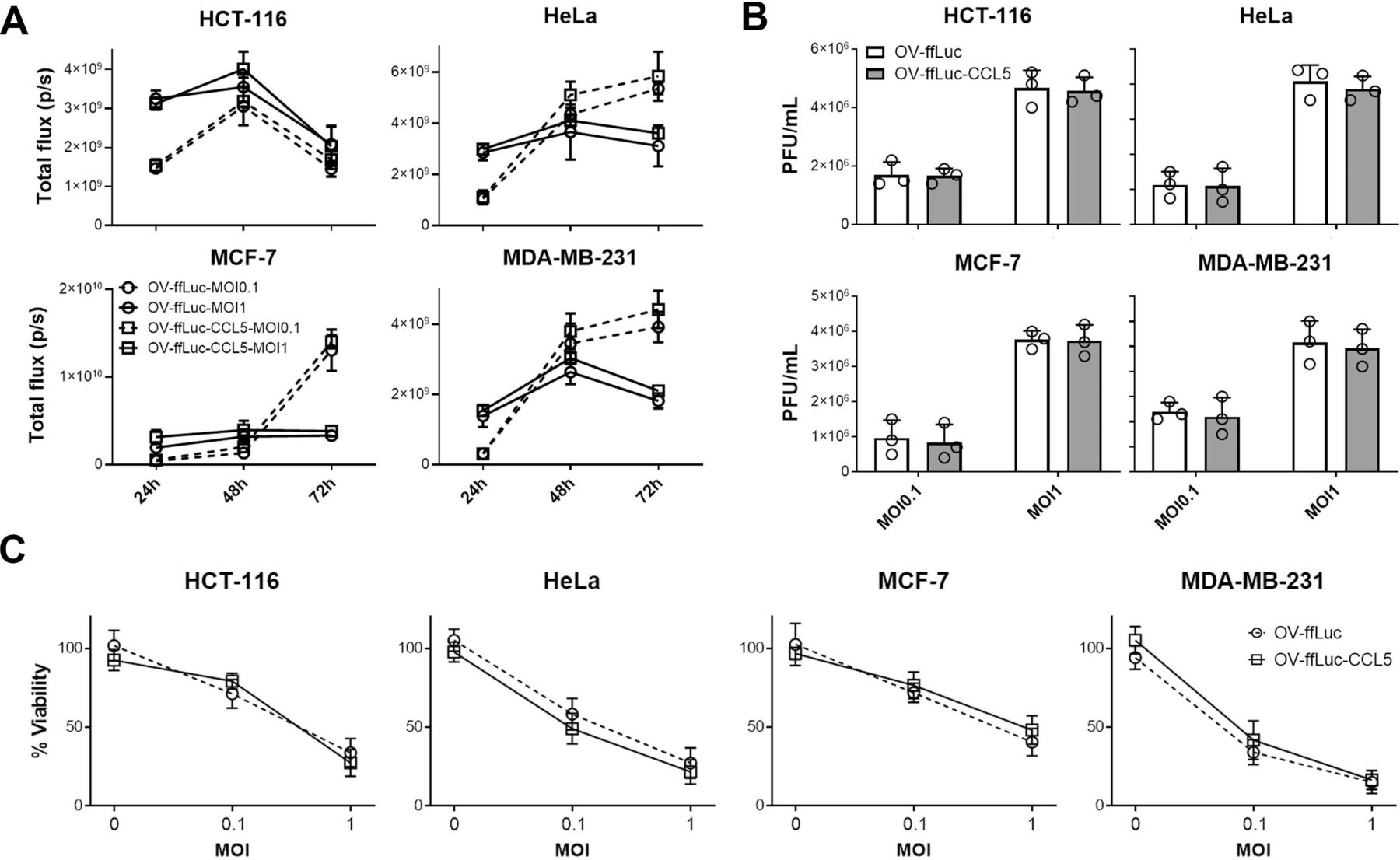
Oncolytic effects of vaccinia virus. (A, B) Tumor cells were infected with OV-ffLuc or the derivate OV-ffLuc-CCL5 virus at MOI=0.1 or MOI=1. The IVIS imaging was used to determine virus expansion within tumor cells (A), while the resultant virus amounts were determined by plaque formation assays (B). (C) LDH assay for determining tumor cell viability 48 hours after infection with OV-ffLuc or OV-ffLuc-CCL5 virus. Data are presented as the means±SD of three independent experiments. Statistical analysis was performed via t-test and one-way ANOVA. ANOVA, analysis of variance; IVIS, in vivo imaging system; LDH, lactate dehydrogenase; MOI, multiplicity of infection; OV, oncolytic viruse; PFU, plaque-forming unit.
CCL5-producing OV enhances NK cell migration in vitro
As the CCL5 transgene had limited impact on OV infectivity, we asked whether the transgenic CCL5 can be efficiently expressed in host cells. To test this, tumor cells were infected with different viruses and the supernatants collected. ELISA showed that CCL5 levels were increased in supernatants derived from OV-ffLuc-CCL5-infected tumor cells (p<0.005; figure 4A). CCL5 concentrations from tumor cells treated with OV-ffLuc were mildly increased compared with those of PBS-treated samples (figure 4A), indicating that enhanced CCL5 levels result from the translation of the CCL5 transgene in the virus and are not induced by virus infection. We next performed Transwell assays and interestingly found that supernatants from tumor cells infected with CCL5-expressing OV improved the directional movement of NK-CCR5 cells (p<0.005) but not NK-GFP cells (p>0.05; figure 4B). To assess whether the enhanced movement of NK cells was mediated by CCL5 and not other chemokines induced after virus infection, CCL5-specific neutralizing antibody was added to the supernatants. We, thus, found that the directional movement of both NK-GFP and NK-CCR5 cells was inhibited (figure 4C), indicating that the production of CCL5 by OV specifically promotes NK-CCR5 cell migration. Of note, CCL5 was present in supernatants from chemokine-expressing OV-infected cells in markedly high levels (figure 4A). We, thus, asked whether NK cells expressing different levels of CCR5 exhibit inconsistent responses to CCL5. To test this hypothesis, media containing various concentrations of CCL5 were prepared. As shown in figure 4D, CCL5 enhanced the movement of CCR5-overexpressed NK cells in a concentration-dependent manner; however, NK-GFP cell migration showed limited improvement when CCL5 levels were over 1000 pg/mL (figure 4D). These findings indicate that CCR5-expressing NK cells respond better to CCL5 at higher concentration ranges. Therefore, we further analyzed the possibility of combining NK-CCR5 cells with OVs in the following experiments.

CCL5 production by vaccinia virus in infected tumor cells. (A) Tumor cells were infected with indicated virus at MOI=0.1, after which CCL5 concentration in the supernatant was determined 48 hours later. Data are representative of three independent experiments. (B) Transwell assays of NK-GFP or NK-CCR5 cell migration to supernatants from tumor cells infected with different viruses. NK cells were collected from five healthy donors. (C) Migration of NK cells after addition of anti-CCL5 antibody (0.2 µg/mL). (D) Media were supplemented with various concentrations of recombinant CCL5, after which the directional movement of NK-GFP or NK-CCR5 cells was monitored. The chemokine production tests in tumor cells were repeated thrice. Transwell assay was performed using NK cells from five donors and the representative results are presented. One-way ANOVA was performed to compare the difference among samples. Data are presented as the means±SD. *P<0.05, **P<0.01, ***P<0.005. ANOVA, analysis of variance; GFP, green fluorescent protein; MOI, multiplicity of infection; NK, natural killer; OV, oncolytic viruse; PBS, phophate buffered saline.
OV does not replicate effectively in NK cells
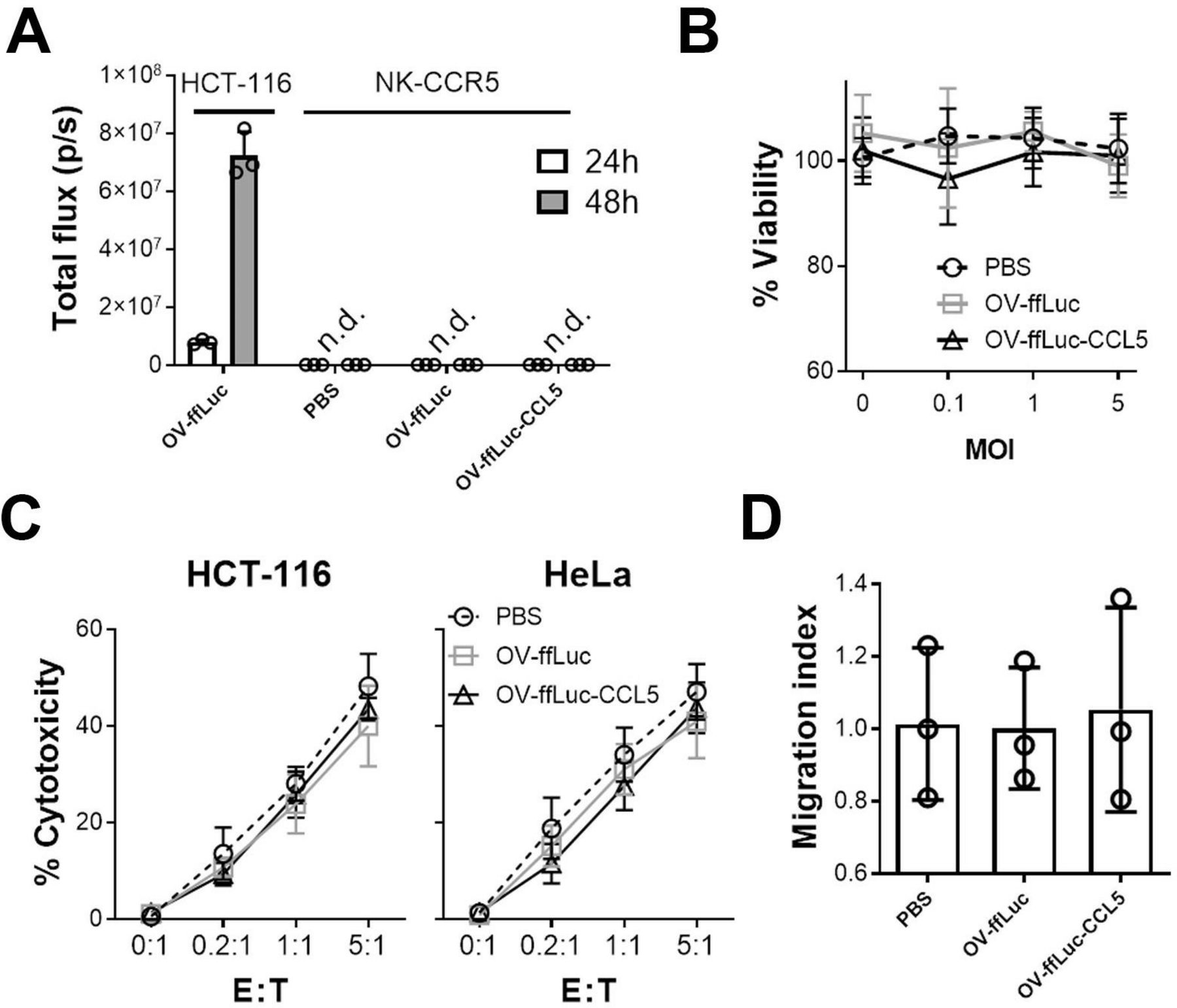
One concern of combining OVs with NK cells is whether the immune cells would be infected and consequently destroyed. To address this, we performed the following assays; NK-CCR5 were incubated with OV-ffLuc or OV-ffLuc-CCL5 at an MOI=1. Real-time PCR showed that small amounts of OV infected NK cells but the virus did not expand in those cytotoxic cells (online supplementary figure S3A). Consistently, BLI demonstrated that OV genome transcription was hampered in NK cells (figure 5A). These findings suggest that both the infection and replication of OV were inefficient in NK cells. Furthermore, MTS assays demonstrated that NK cell viability was not compromized after inoculation with OV, even when the MOI was increased to 5 (p>0.05; figure 5B). Next, we examined the cytotoxic activity of NK cells; following coincubation with OV for 24 hours, NK cells were washed with fresh media and then mixed with tumor cells for 6 hours (to detect cytolysis of tumor cells) or 24 hours (to detect cytokine secretion of NK cells). Compared with the counterparts pretreated with PBS, NK cells preincubated with OV-ffLuc or OV-ffLuc-CCL5 induced similar levels of tumor cell death (figure 5C and online supplementary figure S3B). Moreover, ELISA confirmed that inflammatory cytokine secretion was not affected in NK cells previously incubated with OV (online supplementary figure S3C). Meanwhile, the trafficking ability of NK cells responding to CCL5 was not altered by OV pretreatment (figure 5D). Our results indicate that NK cells are resistant to OV infection and retain their potent cytolytic activity after encountering OVs.
Supplemental material

Vaccinia virus has limited impact on NK cells. (A) NK-CCR5 cells were incubated with OV-ffLuc or OV-ffLuc-CCL5 at MOI=1. Virus proliferation was monitored by the IVIS imaging 24 and 48 hours later. HCT-116 cells infected with OV-ffLuc at MOI=0.1 were used as positive control; PBS-treated NK cells were used as negative control. (B) NK-CCR5 cell viability was determined 48 hours after infection with virus at different MOI values. (C) After incubation with virus at MOI=1 for 48 hours, NK-CCR5 cells were mixed with tumor cells at the indicated E:T ratios for 6 hours and NK cell cytotoxicity determined. (D) The impact of oncolytic vaccinia virus on NK cells was determined; NK cells were pretreated with OV for 48 hours, following which the Transwell assay was performed. After counting the number of migrated cells, the migrating indices were determined relative to NK cells treated with PBS. NK cells were collected from five healthy donors. Data are presented as the means±SD. Statistical analysis was performed with t-tests and one-way ANOVA. ANOVA, analysis of variance; n.d., not detected; E:T, effector to target; IVIS, in vivo imaging system; MOI, multiplicity of infection; NK, natural killer; OV, oncolytic viruse; PBS, phosphate buffered saline.
CCL5-expressing OV enhances NK cell accumulation within tumors in vivo
As depicted in figure 6A, we explored whether CCL5-producing OV can improve NK cell infiltration and antitumor activity. Immunodeficient mice engrafted with HCT-116 were inoculated with different doses of OV (105 or 106 PFU) intratumorally. Next, the expanding potentials of OV were monitored by BLI. OV-ffLuc and OV-ffLuc-CCL5 showed similar replicating kinetics in vivo (figure 6B and C); virus replication peaked 48 hours after infection and then decreased (figure 6C and online supplementary figure S4). On day 10, the virus was hardly detected in vivo (online supplementary figure S4). Further analysis showed that the replication and persistence of OV in vivo were not affected by the infusion of NK cells (online supplementary figure S4). Compared with OV-ffLuc, CCL5-transgenic OV increased the specific chemokine concentrations in a dose-dependent manner within tumors (figure 6D). Four days after virus injection, when virus titers had decreased but CCL5 levels were significantly increased, NK-CCR5 cells were administered (figure 6A). Two days after NK cell injection, the tumorous accumulation of engineered immune cells was markedly improved by chemokine-expressing OV, especially at a high dose of OV (figure 6E,F). Consistent with this, FACS analysis showed that the directional movement of NK-CCR5 cells was enhanced by CCL5-expressing OV, especially by a high dose of specific OV on day 6 (p<0.0005; online supplementary figure S5B). In agreement with these findings, enhanced accumulation of NK cells was noted within tumor lesions from OV-ffLuc-CCL5-treated mice on day 9 and 14 (p<0.05; online supplementary figure S5B). On day 24, there were still more NK cells within CCL5-expressing OV-treated tumors than in those treated with NK alone or in combination with OV-ffLuc; however, the difference was not statistically significant (online supplementary figure S5B). Similarly, NK cell accumulation was enhanced by OV-ffLuc-CCL5, especially by a high dose of CCL5-producing OV, in HeLa cell-xenografted models (online supplementary figure S6). In addition, transient retention of NK-CCR5 cells was observed within normal tissues, including the liver, lung and spleen (online supplementary figure S5B); these accumulations were comparable between mice treated with NK alone or in combination with OVs (online supplementary figure S5B). These findings suggest that CCL5-expressing OV can enhance the tumor-targeted movement of NK cells in vivo.
Supplemental material
Supplemental material
Supplemental material

CCL5-expressing vaccinia virus enhances NK cell infiltration. (A) Schematic description of animal experiments. Ten days after HCT-116 cell inoculation, 106 or 105 PFU of oncolytic virus were injected intratumorally. The viral replications were monitored for 3 days by IVIS imaging. Next, 5×106 NK-CCR5 cells were injected 4 days after virus injection. The accumulation of NK-CCR5 cells was examines by FMT assay. (B, C) Vaccinia virus expansion was not affected by CCL5 transgene in vivo (n=5). (D) Four days after virus treatment, tumors were isolated and homogenized (n=3). CCL5 concentrations were determined using ELISA. (E, F) NK cells labelled with Cy5.5 dye were injected into mice pretreated with PBS or indicated virus (n=4), after 48 hours, the tumorous infiltration of NK cells was determined. Data are presented as the means±SD. *P<0.05, **P<0.01, ***P<0.005. FMT, fluorescence molecular tomographic; IVIS, in vivo imaging system; i.t., intratumorous; i.v., intravenous; NK, natural killer; PFU, plaque-forming unit; S.C., subcutaneous; OV, oncolytic viruse; PBS, phosphate buffered saline.
Synergistic administration of OV and NK in vivo
After chemokine-transgenic OV was found to improve NK cell filtration into tumors, we monitored the long-term antitumor efficacy of combined therapies. As expected, NK cells or OV alone delayed tumor growth rather than completely eradicating the malignant cells (figure 7A,B). In mice receiving both NK cells and OV-ffLuc treatment, tumor growth was slower than that of single treatment due to sequential injections of cytolytic virus and cells (figure 7A,B); nevertheless, no complete responses were noted after such therapy. In contrast, the combination of OV-ffLuc-CCL5 with CCR5-NK cells led to better prognoses, especially in the group injected with a high dose of CCL5-producing OV, with more than half of mice showing a complete response (figure 7). These in vivo results indicate that CCL5-induction within tumors can improve NK cell accumulation and tumor regression.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CCL5-expressing vaccinia virus augmented NK-CCR5 cell therapy. As depicted in figure 6, mice inoculated with HCT-116 cells received the indicated treatments. (A, B) Tumor growth (A) and mouse survival (B) were monitored separately. The number and median survival time (days) of mice from different groups are indicated in corresponding plots. Kaplan-Meier curves and log-rank tests were used to estimate and compare survival, respectively, followed by Bonferroni correction. *P<0.05, **P<0.01, ***P<0.005. n.s. indicates no significant difference; NK, natural killer; OV, oncolytic viruse; PFU, plaque-forming unit.
Discussion
OVs have been extensively tested for enhancing the efficacy of cancer immunotherapy.25–28 Oncolytic vaccinia virus demonstrates very limited infectivity of NK cells29 30 and can boost the antitumor effects of immune cells instead of impairing their cytotoxic function.31 32 More importantly, vaccinia virus has been shown not to hinder immune cells in humans in a clinical setting.33 Within the lesions infected by vaccinia virus, NK cells are often increased.33 This work also confirmed that NK cells were resistant to vaccinia virus mediated killing (figure 5 and online supplementary figure S3). Oncolytic vaccinia virus, therefore, represents a potential tool to combine with engineered NK cells.
One important potential use of such a combination could be to enhance the tumor-directed movement of the infused cytotoxic cells. As the chemokine–chemokine receptor system is critical in determining the tissue-specific distribution of immune cells,11 much efforts have been made to alter the expressions of specific chemokines within the tumor microenvironment. In previous studies on immunocompetent mice,31 34 35 we found that the intratumorous production of certain chemokines by OVs can enhance the recruitment of endogenous immune cells, including NK cells. Based on these findings, we speculated whether OVs can improve the directional movement of human NK cells by adjusting chemokine expression patterns in the tumor, especially in an in vivo setting. Although xenograft models cannot completely model the situation in a human body, immunodeficient mice are frequently used to evaluate the in vivo antitumor effects of human T and NK cells.36–39 Hence, we employed immunodeficient mice in the present study to test the combination of OV with human NK cells. As expected, OVs expressing CCL5 increased the intratumorous expression of this chemokine and so could be used to enhance NK cell recruitment in vivo (figure 6), indicating the combination strategy may be applicable to humans.
CCL5 is overexpressed in a variety of solid tumors and directs the migration of CCR5-expressing cytotoxic cells including NK and T cells.14–16 18 The direct effects of CCL5 on tumor cells are controversial,40 41 but the majority of the evidence indicates that CCL5 is required for increasing antitumor immunity and benefits tumor suppression.13 15 42 43 In a previous report, CCL5 has been exploited to enhance the tumor infiltration of T cells.44 Additionally, the CCR5–CCL5 interaction is important for NK cell infiltration of tumor.16 Nevertheless, CCR5 expression is typically low in human NK cells.45 In agreement, our results showed that CCL5 could induce NK cell migration but CCR5 was expressed only in a small fraction of NK cells (figure 1), which would limit the movement of NK cells towards tumor lesions even when CCL5 is expressed. Thus, we selected the CCL5-CCR5 axis to manipulate the migration of NK cells in this study.
Previous studies have shown that the enhanced expression of either chemokines or chemokine receptors can promote the tumor-specific migration of human cytotoxic cells.19 31 46 47 For NK cells, the enforced expression of specific chemokine receptors can improve their accumulation with tumors.19 Consistent with this, we showed that CCR5 overexpression enhances the directional movement of NK cells (figures 1 and 2). However, the chemotactic movement of immune cells relies on the pairing of chemokines with their cognate receptors.48 In the clinic, such strategy would not be effective if the patients have low levels of expressions of specific chemokines or chemokine receptors. In our study, it was noticed that the movement of natural NK cells, which had low levels of CCR5, could not be obviously improved by elevated CCL5 (figure 4). This phenomenon was also noted in previous reports,19 44 46 47 in which the cytotoxic cells having native chemokine receptors are not efficiently recruited even by the upregulated chemokines within tumors. Similarly, the cytotoxic cells are not efficiently moved into malignant lesions when the specific chemokines have low expressions in tumors.31 32 35 Together, it is suggested that the separate expression of chemokine or chemokine receptor may fail to induce the movement of immune cells if either component is suppressed. Hence, a more potent and reliable strategy is to concomitantly overexpress the matched chemokine and receptor. Our work confirmed that CCR5-overexpressing NK cells were more significantly attracted by various concentrations of CCL5 (figure 4). As well the in vivo data showed higher concentrations of CCL5 could better recruit CCR5-overexpressing NK cells, resulting in better suppression of tumor progression (figure 6).
The oncolytic vaccinia virus can be delivered via intraperitoneal, intravenous or intratumorous injections.20 31 49 We wanted to explore the effects of CCL5 concentration on NK cell migration, so intratumorous injection was used in order to more reliably regulate the production of CCL5 through giving different doses of the vaccinia virus. However, in a clinical setting intravenous delivery of the virus might be used. Indeed, CCL5 production from the OV was found to be viral dose dependent in vivo (figure 6). As a result, more NK cells were recruited into tumor lesions (online supplementary figure S5) and tumors were markedly suppressed in mice receiving high doses of OV-ffLuc-CCL5 (figure 7). This strategy of regulating tumorous expression of specific genes by viruses via intratumorous injection is also likely to be more efficient and has been used in previous studies.20 25 44
Although our study demonstrates that the simultaneous upregulation of CCR5 and CCL5 efficiently improves NK cell-based therapy, there remains several issues to overcome. First, the route of administration may be important for effective OV therapy. In the clinic, systemic infusion of OV is preferred. Although oncolytic vaccinia virus has demonstrated systemic delivery both in mouse models49 and in a clinical setting, it may require careful control of the doses and timing to combine OV and NK cells when intravenous infusion is performed. Additionally, the antitumor effects of the combining strategy were only tested in HCT-116 cell xenograft models in the present work. CCL5–CCR5 interaction represents a general mechanism to move NK cells towards tumor and it is believed mouse and human cross-react.50 We did find that CCL5 upregulation enhanced the tumor infiltration of NK cells in different tumor models (online supplementary figures S5 and S6). However, many factors including virus replication efficacy, blood vessel formation, presence of immunosuppressive ligands and other elements may affect the antitumor efficacy of the OV and NK cell combination therapy. In particular, it would be of interest to examine the effects of this combination in the context of a fully intact immune system, such as in a humanized PDX tumor model. Although NK cells and OV were cytotoxic against different cancer lines in vitro, it is not clear whether such a combination is as efficient in other tumors as it is in the HCT-116 tumor model. Our future studies will focus on those questions and determine the effects of the above-mentioned factors on the combination of OV and NK cells.
Conclusion
Adoptive transfer therapy faces many challenges in solid tumors. A critical challenge is how to direct cytotoxic cells into the tumors as effectively as possible. Therefore, efforts should be made to improve the distribution of infused cells in vivo. Our present study investigated the possibility of upregulating matched chemokines and chemokine receptors in tumor and NK cells, respectively, to enhance tumor infiltration. We found that this approach can improve NK cell-based therapies, warranting further study in additional cancer models.
Supplemental material
Acknowledgments
We would like to thank the flow facility and animal facility for their assistance.
References
Footnotes
FL and YS are joint first authors.
FL and YS contributed equally.
Contributors YZ, ST and FL conceived of and designed the study; FL, YS, WH, PS and DB developed the methodology; FL, YS, WH, PS and DB acquired the data; FL, YS and YZ analyzed and interpreted the data; FL, YS, ST and YZ wrote/reviewed the manuscript; ST and YZ provided administrative, technical or material support and supervized the study. All authors read and approved the final manuscript.
Funding This work received funds from the National Key Research and Development Program (2018YFC1313400); the National Natural Science Foundation of China (81502689, 81502628) and the provincial foundations of Henan Province (192102310035, SB201903004).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Blood collection and processing from heathy and cancer patients were performed according to the protocols (protocol number: research-2015-LW-511) approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University. Animal experiments abided by the protocol approved by the University of Pittsburgh Institutional Animal Care and Use Committee (Protocol Number: 11312848).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.