Article Text

Abstract
Background CD19 chimeric antigen receptor T (CAR-T) cells demonstrate remarkable remission rates in pediatric and adult patients with refractory or relapsed (r/r) acute lymphoblastic leukemia (ALL) and non-Hodgkin's lymphoma (NHL). In 2016, we initiated a clinical trial with in-house produced CD19 CAR-T cells with a CD28 co-stimulatory domain. We analyzed, for the first time, differences in production features and phenotype between ALL and NHL patients.
Methods Non-cryopreserved CAR-T cells were produced from patients’ peripheral blood mononuclear cells within 9 to 10 days. 93 patients with r/r ALL and NHL were enrolled under the same study. CAR-T cells of ALL and NHL patients were produced simultaneously, allowing the head-to-head comparison.
Results All patients were heavily pretreated. Three patients dropped out from the study due to clinical deterioration (n=2) or production failure (n=1). Cells of ALL patients (n=37) expanded significantly better and contained more CAR-T cells than of NHL patients (n=53). Young age had a positive impact on the proliferation capacity. The infusion products from ALL patients contained significantly more naïve CAR-T cells and a significantly higher expression of the chemokine receptor CXCR3. PD-1, LAG-3, TIM-3, and CD28 were equally expressed. 100% of ALL patients and 94% of NHL patients received the target dose of 1×10e6 CAR-T/kg. The overall response rate was 84% (30/36) in ALL and 62% (32/52) in NHL. We further compared CAR-T cell infusion products to tumor infiltrating lymphocytes (TIL), another common type of T cell therapy, mainly clinically effective in solid tumors. CAR-T cells contained significantly more naïve T cells and central memory T cells and significantly less CCR5 compared to TIL infusion products.
Conclusions The in-house production of CAR-T cells is highly efficient and fast. Clinical response rate is high. CAR-T cells can be successfully produced for 99% of patients in just 9 to 10 days. Cells derived from ALL patients demonstrate a higher proliferation rate and contain higher frequencies of CAR-T cells and naïve T cells than of NHL patients. In addition, understanding the differences between CAR-T and TIL infusion products, may provide an angle to develop CAR-T cells for the treatment of solid tumors in the future.
Trial registration number ClinicalTrials.gov; CAR-T: NCT02772198, First posted: May 13, 2016; TIL: NCT00287131, First posted: February 6, 2006.
- tumours
- hematologic neoplasms
- immunotherapy
- t-lymphocytes
- cell engineering
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Adoptive cell therapy (ACT) with autologous CD19 chimeric antigen receptor T cells (CAR-T) has demonstrated remarkable remission rates in patients with relapsed and refractory (r/r) B-cell malignancies. The CAR combines a single chain variable fragment ectodomain of an antibody (scFv) that can target an antigen of choice with an endodomain T cell signaling moiety comprised of the CD3ζ TCR signal and additional co-stimulatory domains. In contrast to the T cell receptor, CAR induces antigen recognition in a major histocompatibility complex (MHC)-independent manner.1 The CAR-T approach was pioneered in the late 1980s2 3 and finally let to its approval by the Food and Drug Administration (FDA) in 2017 and the European Medicines Agency (EMA) in 2018.
Currently two commercial CD19 CAR-T products exist on the market for the treatment of B-cell precursor acute lymphoblastic leukemia (ALL) and various types of non-Hodgkin's lymphoma (NHL). Both CAR-T products, tisagenlecleucel and axicabtagene ciloleucel, were approved for adult patients with r/r B-cell lymphoma including diffuse large B-cell lymphoma (DLBCL), high grade B-cell lymphoma and DLBCL arising from follicular lymphoma.4 In addition, tisagenlecleucel was approved for patients up to 25 years of age with r/r ALL.5–7
ALL is the most common cancer in childhood and young adults. In children the 5 year survival rate approaches 90% with standard treatment, but clinical results for relapsed disease remains poor.8 The majority of adults with ALL who achieve complete response (CR) with standard therapies will relapse after first remission and about 25% will have refractory disease, leading to high mortality.9
Non-Hodgkin's lymphomas are a heterogeneous group of malignancies with distinct prognoses and therapeutic approaches. NHL is the most frequent hematological malignancies in the world with more than 50,000 new cases in USA per year.10 It consists of 40 major subtypes with distinct genetic, morphologic and clinical features.11 DLBCL is the most common subtype, holding 25% to 30% of adult NHL in Western countries,12 however many additional and rare CD19 positive lymphoma types, such as Burkitt lymphoma exist, which do not fall under the approved indications of CD19 CAR-T cells. The same is true for chronic lymphocytic leukemia, the most common type of leukemia in adults, accounting for 30% of cases.13
The addition of CD19 CAR-T cells to the existing treatment repertoire of r/r ALL and various types of NHL was a major breakthrough, however there are still obstacles related to the commercial CAR-T cell products, including high costs and a long turnaround time from leukapheresis to infusion of 21 to 60 days.
In-house production of CAR-T cells can overcome most of these obstacles, leading to a raising number of clinical centers generating their own CAR-T products today. In 2016, the Sheba Medical Center, Israel, initiated a phase 1b/2 study with in-house produced CD19 CAR-T cells for the treatment of CD19 positive B-cell malignancies. Patients undergo a single leukapheresis procedure. Peripheral blood mononuclear cells (PBMC) are isolated, activated and transduced with a gamma retrovirus encoding for the anti CD19 scFv, a CD28 co-stimulatory domain and a CD3-ζ signaling domain. Following expansion, fresh, non-cryopreserved CAR-T cells are administered to the patients after a turnaround time of only 9 to 10 days. In contrast to the commercial products, also patients with off-label CD19 positive B-cell malignancies, such as adults with r/r ALL and Burkitt lymphoma were enrolled to the study and CAR-T cells were manufactured in just 10 days. Clinical results of the first 21 patients with r/r ALL and one patient with Burkitt lymphoma were previously published.14 15
Here, we describe the manufacturing process of 91 in-house produced CAR-T products. Patients with r/r ALL and NHL were enrolled under the same study and CAR-T were produced in parallel, allowing true head-to-head comparison of CAR-T cell production and characterization in ALL and NHL.
Material and methods
Patients
This study was designed as a phase 1b/2 trial, approved by the Israeli Ministry of Health and registered at ClinicalTrials.gov. All enrolled patients signed an informed consent. Inclusion criteria were age between 1 and 50 years, failure of at least two prior therapeutic protocols, a CD3 count greater than 250/µl blood, absence of clinical signs of graft-versus-host disease and no immunosuppressive treatment. Patients above age 50 were permitted to the study after approval by the Sheba Medical Center Institutional Review Board (IRB). Depending on age, the minimal performance score was 50 on a Lansky scale or on a Karnofsky scale. Patients with prior CD19 directed therapies were eligible for the study. Lympho-depleting conditioning was inducted by fludarabine 25 mg/m2 for 3 days (2 to 4 days before infusion) and cyclophosphamide 900 mg/m2 for 1 day (2 days before infusion), followed by infusion of 1×10e6 transduced CAR-T cells per kilogram weight. Primary endpoints of the study were production feasibility, patient safety and best overall response rates, documented 1 to 2 months after infusion.
CD19 CAR retroviral vector
The retroviral supernatant was generated from the CD19 CAR producer line PG13-CD19-CAR-H3 kindly provided by Dr. Steven Rosenberg, National Cancer Institute (NCI). Vector construction and master cell bank production were conducted at the NCI.16 17 In short, a plasmid encoding the CD19 CAR containing of the mouse stem-cell virus gamma-retroviral backbone engineered to a scFv derived from the mouse anti-CD19 hybridoma, FMC63,18 fused to intracellular domains from human CD28 and CD3-ζ, was used for viral vector production. Retroviral vector supernatant was harvested in accordance with current good manufacturing practices at the Indiana University Viral Production Facility.
CAR-T production
If not otherwise indicated, a fresh leukapheresis product was used as starting material for CD19 CAR-T cell production. PBMC were isolated from the apheresis product by density gradient with Ficoll-Hypaque (Lymphocyte Separation Medium, Axis-Shield Diagnostics, Scotland). 400×10e6 PBMC were re-suspended at the concentration of 1×10e6 cells per ml in complete medium (CM), containing 10% human AB serum (Valley, Virginia, USA), 2 mM L-Glutamine (Biological Industries, Israel), Pen/Strep (Biological Industries, Israel) and 300 IU/mL IL-2 (Chiron Novartis, New Jersey, USA) in AIM-V medium (Invitrogen, California, USA). 50 ng/mL anti-CD3 monoclonal antibody OKT-3 (Miltenyi Biotec, Bergisch Gladbach, Germany) were added to the medium on the day of initiation. After 2 days around 60×10e6 cells were transduced with the CD19 CAR retroviral vector and the rest of the cells discarded. For this purpose, non-tissue culture treated 6-well plates were coated with 10 µg/mL RetroNectin (Takara Bio Inc, Otsu, Japan) in phosphote-buffered saline (PBS) for 2 hours at room temperature or overnight at 4°C, followed by 30 min blocking with 2.5% human albumin (Bio Products Laboratory, Zenalb20) in PBS and washed. Retroviral supernatant was thawed and diluted 1:1 in AIM-V medium with 5% human AB serum. Four ml of the diluted vector were added per well of the retronectin-coated plates and centrifuged at 2000xg for 2 hours at 32°C. The supernatant was aspirated and cells were re-suspended in CM medium with IL-2. 2.5×10e6 cells were added to each well (total of 60×10e6 cells in four 6-well plates), centrifuged for 15 min at 1000xg and incubated at 37°C overnight. On day 3 the cells were transferred to T175 or GRex100 (Wilson Wolf, Minnesota, USA)19 culture flasks and maintained at a concentration of 0.5 to 2.0×10e6 cells/mL in T175 flasks or 0.5 to 8.0×10e6 cells/mL in GRex100 in CM medium with IL-2 until day 9 or 10. When using GRex100, flasks were topped up to 450 ml with IL-2 containing CM medium on day 6. On the day of infusion, cells were washed, counted and 1×10e6 CD19 CAR expressing cells/kg were re-suspended in 100 mL 0.9% sodium chloride (Baxter,) containing 2.5% human albumin and 300 IU/mL IL-2. The cell product was delivered to the patient for immediate infusion. CAR-T cells for ALL and NHL patients were produced in parallel and by the same laboratory staff.
TIL production
Tumor infiltrating lymphocytes (TIL) were isolated from tumor biopsies of metastatic melanoma patients, enrolled to a phase II TIL ACT trial at the Sheba Medical Center. The generation of TIL was conducted precisely as described before.20 21 In short, fragmentation, enzymatic digestion and tissue remnant culture techniques were used to isolate TIL from surgically resected metastatic melanoma lesions. Cells were cultured in CM medium with IL-2 (3000 IU/mL) and gave rise to TIL cultures within 2 to 4 weeks. Next, TIL were expanded in a rapid expansion procedure (REP) using anti-CD3 monoclonal antibody OKT-3 (30 ng/mL), IL-2 (3000 IU/mL) and 50 Gy irradiated feeder cells of healthy donors. Within 2 weeks, cultures expanded by about 1000-fold. On day 14, TIL were harvested, washed, re-suspended in 400 mL 0.9% sodium chloride containing 2.5% human albumin and 300 IU/mL IL-2 and immediately intravenously administered to the patient.
In vitro reactivity
In order to demonstrate in vitro anti-tumor reactivity, IFNγ secretion was measured following co-incubation of CAR-T cells with target cells.17 22–25 Untransduced T cells served as negative control. The following CD19-expressing target cell lines were used: NALM-6 (acute lymphoid leukemia); Toledo (B-cell diffuse large cell lymphoma). The CD19-negative cell line CCRF-CEM (T cell leukemia) was used as negative control. All tumor lines were kindly provided by Dr. Steven Rosenberg, NCI.
The co-culture was performed with an effector to target ratio of 1:1 (1×105 each) in a total of 200 µl medium overnight at 37°C. Supernatant was collected, if necessary diluted and IFNγ secretion was determined by ELISA (Human IFN- ELISA MAX Deluxe Set, BioLegend, San Diego, California). Measurements were performed in triplicates.
Flow cytometry
The following antibodies were used: CD3 (VioBlue; Miltenyi Biotech or Pacific blue and PE; BioLegend), CD4 (FITC or APC-Cy7; BioLegend), CD8 (PE-Cy7; BioLegend), CD3/CD19 antibodies (FITC/PE; BD), CD28 (PerCP-Cy5.5; eBioscience), PD-1 (FITC; clone: EH12.2H7; BioLegend), TIM-3 (APC-Cy7; BioLegend), LAG-3 (VioBlue; Miltenyi Biotech), CD45RA (APC-Vio770; Miltenyi Biotec or Brilliant Violet; BioLegend), CCR7 (PerCP-Vio770; Miltenyi Biotec or PerCP; BioLegend), CCR2 (APC; Biolegend), CCR4 (PE; Biolegend), CCR5 (Alexa Influenza 488; Biolegend), CXCR2 (PE-Cy7; Biolegend) and CXCR3 (FITC; Biolegend).
Transduction efficacy was determined on day 6 and day 9 of culture by labeling CAR-T cells with biotin-labeled polyclonal goat anti-mouse F(ab)2 antibody (anti-Fab, Jackson Immunoresearch, West Grove, Pennsylvania) and streptavidin (APC conjugated; BioLegend). CD3+F(ab)2+ cells were defined as CAR-T cells. Isotype labeled cells (Jackson Immunoresearch) and untransduced cells served as negative controls. For further characterization, cells were stained with antibodies mentioned above. Cells were washed and re-suspended in cell staining buffer (BioLegend), incubated for 30 min with the antibodies on ice, washed and measured using MACSQuant FACS cytometer (Miltenyi Biotec). Samples were analyzed by FlowJo software (FlowJo LLC, Ashland, Oregon).
Statistics
Significance of variation between groups was evaluated using a non-parametric two-tailed Student's t-test. The differences between proportions were tested using two-sided Fisher's exact test. Analysis of covariance (ANCOVA) was used to exam the differences in the mean values of patients’ characteristics on variable production parameters.
Results
Patients’ characteristics and clinical response
Between June 2016 and August 2019, 93 patients with r/r B-cell malignancies were enrolled to the trial. All patients were heavily pretreated. Three enrolled patients (3%) dropped out from the study due to clinical deterioration (n=2) or failure to produce CAR-T cells (n=1; absence of CAR-T cells in the infusion product). One patient was treated twice. Of the treated patients, 37 patients had r/r ALL and 53 patient's r/r NHL, including DLBCL (n=36), Burkitt lymphoma (n=3), PMBCL (n=7), follicular lymphoma (n=4), gray zone lymphoma (n=1), mediastinal lymphoma (n=1) and high-grade lymphoma (n=1).
Patients’ demographic and clinical characteristics are shown in table 1. As expected more pediatric patients were in the ALL population, resulting in a significantly lower age (17±14 years) and weight (44±21 kg) compared with NHL patients (44±15 years and 75±20 kg; p values ≤0.001). Both, ALL and NHL patients received an average of three prior lines of therapy. Thirty-two of 90 patients (36%) received a hematopoietic stem cell transplantation prior CAR-T therapy, including 17 allogenic or haloidentical stem cell transplantations in patients with ALL (n=15) and NHL (n=2). Ten of 37 (27%) ALL patients received prior therapy directed against CD19, such as blinatumomab. ALL patients had significantly more CNS involvement (p ≤0.001).
Baseline characteristics of treated patients
Following lympho-depleting preconditioning, fresh, non-cryopreserved CAR-T cells were intravenously infused to the patients. Clinical response was evaluated 1 to 2 months after CAR-T cell administration. One ALL patient died of sepsis before evaluation and one NHL patient is still awaiting his evaluation. Of 36 evaluated ALL patients, 24 (67%) achieved minimal residual disease (MRD) negative CR, 6 (17%) MRD positive CR and 5 patients (14%) progressed. One ALL patient with an initial response was treated a second time with CAR-T, but did not respond. Of 52 evaluated NHL patients, 32 (62%) achieved an objective response, including 16 complete remissions and 16 partial responses. Twenty (38%) patients had disease progression.
Leukapheresis, stimulation and expansion
PBMC were collected by leukapheresis. The average CD3 T cell blood count before leukapheresis was 1,712±762 CD3 cells/µl, with no statistical difference between ALL and NHL patients (p=0.871) (table 2). Three ALL patients and one NHL patient with CD3 count below 250 cells/µl were permitted to enroll to the study after approval by the Sheba Medical Center IRB due to rapid disease progression with no alternative treatment options. Ninety-one full manufacture processes were performed, including 38 productions for ALL patients (37 patients and 1 patient treated twice) and 53 NHL patients.
Leukapheresis, production and infusion characteristics
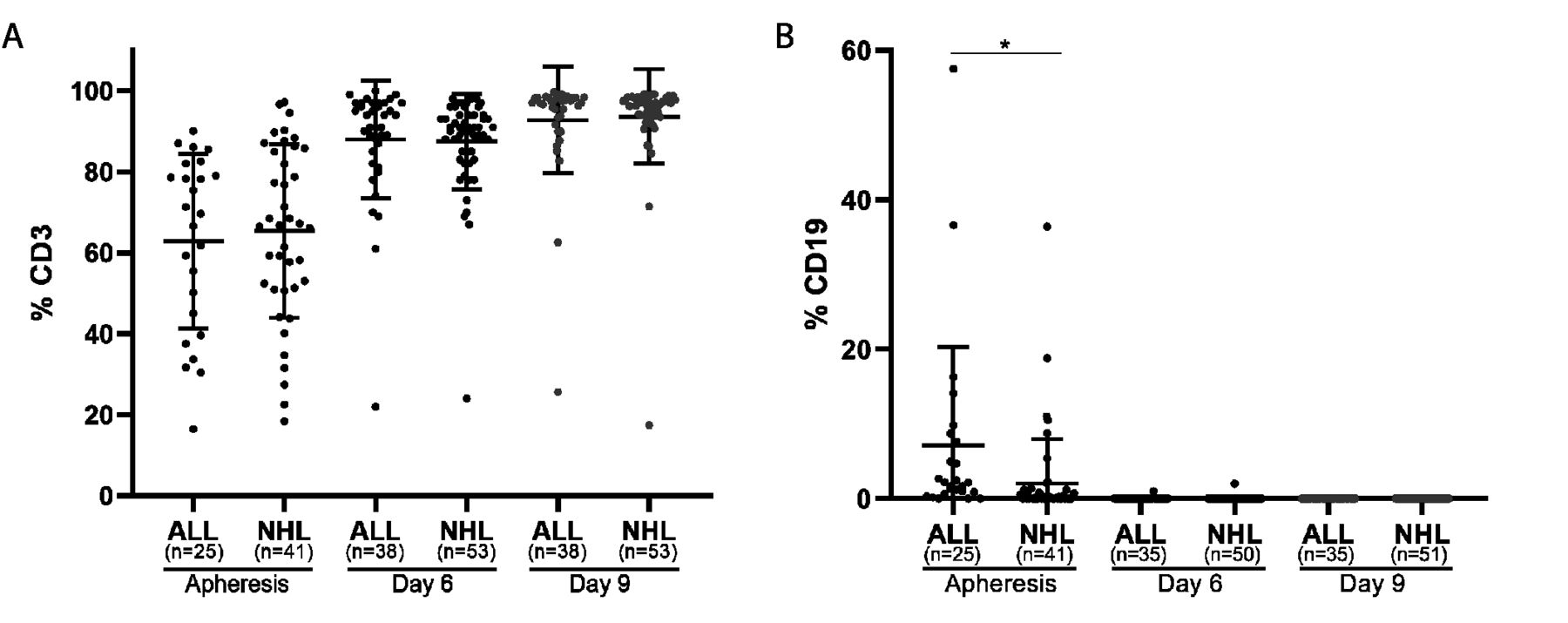
Time of apheresis procedure varied between 2 to 5 hours, depending on the weight and CD3 T cell blood count of the patient. The average number of mononuclear cells obtained by leukapheresis was 3.8±2.9×10e9 (ALL patients, 2.7×109±2.7×109; NHL patients 4.5±3.0×109; p≤0.002). PBMC were purified by ficoll-hypaque density gradient and CD3 T cell and CD19 B-cell contents were determined by FACS analysis. The frequency of CD3 T cell in ALL and NHL patients was 62.9%±21.1% (range 16% to 90%, n=25) and 65.2%±21.1% (range 18% to 97%, n=41), respectively, (p=0.640) and of CD19 B-cells 7.2%±12.9% and 1.6%±3.8% respectively (p=0.013) (figure 1, ‘apheresis’). There was a strong correlation (r=0.600) between the CD3 blood count and the CD3 frequency after ficol purification in the ALL population and a moderate correlation in the NHL population (r=0.572). Additional phenotype analysis on four apheresis products demonstrated the presence of monocytes (26.4%±14.0%) and NK cells (13.4%±6.0%).

T cell and B-cell content during production. Frequency of CD3 T cells (A) and CD19 B cells (B) at initiation (apheresis), during cell expansion (day 6), and before infusion (day 9) in ALL and NHL patients. *p≤0.05.
CAR-T production was typically initiated with 400×10e6 PBMC, obtained from fresh (n=84) or cryopreserved (n=7) apheresis products. Only four collections of pediatric ALL patients under age 9 yielded less than 400×10e6 PBMC and were initiated with less cells (322×10e6 on average) and one production was initiated with 600×10e6 PBMC. PBMC were stimulated by addition of soluble anti CD3 antibody and IL-2. Since this condition stimulates CD3 T cells, but less other cell types, a drop in total cell number was typically observed after 2 days.
The cell count 2 days after stimulation dropped by approximately 30% and was 304±171×10e6 in ALL patients and 251±205×10e6 in NHL patients (p=0.203) (figure 2A, table 2). There was no correlation between the drop in cell number and the frequency of CD3 cells in the purified apheresis product (R=0.292). Transduction was typically performed with 60×10e6 cells (range 48 to 72×10e6 cells) on retronectin coated plates overnight and the remaining cells were discarded. On day 3, cells were transferred to T175 or GRex100 flasks for expansion. Cell counts were performed on days 3, 6 and on day 10, the day of infusion. Figure 2A demonstrates the total cell number throughout the production and figure 2B the fold expansion compared with day 2.

Comparison of the expansion capacity and transduction efficacy of CAR-T cells from ALL (n=38) and NHL (n=53) patients. (A) Total viable cell numbers (x10e6) ± SE. On day 2 only part of the cells were transduced and further expanded. (B) Fold expansion ± SE in comparison to the number of cells taken for transduction on day 2. (C) Fold expansion of total viable cells according to age. (D) Transduction efficacy displayed as percent of CD3+F(ab)2+ CAR-T cells on day 6 and day 9. *p≤0.01; **p≤0.001. ALL, acute lymphoblastic leukemia;CAR-T, chimeric antigen receptor T cells; NHL, non-Hodgkin's lymphoma.
As shown in figure 2A and B, cells of ALL patients expanded significantly better than cells of NHL patients. The fold expansion on day 6 compared with day 2 was 8.6±6.3 in ALL patients and 5.7±4.9 in NHL patients (p=0.016) and on day 10, 28.7±24.7 in ALL patients and 17.8±13.5 in NHL patients (p=0.009). Independent of the disease type, CAR-T cells of younger patients expanded significantly better, than of patients above age 21 (age ≤20, 32.4±27.5; age >20, 17.0±11.2; p=0.0003) (figure 2C).
On day 6, the frequency of CD3 T cells was 87% in both patient populations and increased to 93%±12% on day 10 (figure 1A). CD3 negative cells were mostly NK cells. CD19 + B-cell frequency was below 0.5% on day 6 and 0% on day 10 (figure 1B). Interestingly, transduction efficacy, measured by CD3+F(ab)2+ expression, was significantly higher in ALL patients than in NHL patients on day 6 (ALL, 64.1%±15.5%; NHL 54.8%±17.6%; p=0.012) and on day 9/10 (ALL, 73.5%±16.3%; NHL, 62.6%±19.6%, p=0.007) (table 2, figure 2D).
Quality control testing required a minimum of 10% of CAR-T cells on day 6 and of 15% on day 9 or 10. All 91 CAR-T products passed these criteria.
As a consequence of the significantly higher fold expansion and transduction efficacy in ALL patients, the total number of CAR-T cells on day 10 was almost double in ALL patients and reached 1,174±901×10e6 cells compared with 688±635×10e6 CAR-T cells in NHL patients (p=0.004).
The target dose of CAR-T cells for infusion in this study was 1×10e6 CAR-T cells per kg body weight. Taking into account an average body weight of 44±21 kg and an average total number of 1,174±901×10e6 CAR-T cells in ALL patients, the in-house production yielded an average of 27-times more CAR-T cells than required. For NHL patients, an average of 9-times more CAR-T cells were obtained (688±635×10e6 CAR-T cells and 75 kg ±20 kg weight). The target dose of 1×10e6 CAR-T cells was reached in 97% (88 of 91) of patients. In three NHL patients the CAR-T cell number on day 10 was 0.3×10e6, 0.6×10e6 and 0.8×10e6 CAR-T per kg. Following the approval by the Sheba Medical Center IRB, also these three patients received CAR-T therapy.
All 91 CAR-T products passed the required microbiological tests for sterility, endotoxin and mycoplasma, and no replication competent retrovirus was detected. The viability in all infusion products was above 90%.
Impact of age and gender
As described before, CAR-T cells of patients below age 20 demonstrated a significantly increased fold expansion compared with older patients (p=0.0003, figure 2D). To further investigate the impact of age on production characteristics, ALL patients were divided into two age groups; age 1 to 19 years (10.4±5.1 years; n=28) and age 20 to 59 years (35.8±13.31 years; n=10). This analysis was not relevant for NHL patients, as only 3 of 53 patients were below the age of 20 years. As expected, the average number of mononuclear cells collected by leukapheresis (p=0.029) and the weight (p≤0.00001) was significantly lower in younger ALL patients. The fold expansion by day 10 was 33.4±27.6 in ALL patients below age 20, and 15.7±6.17 above age 20 (p=0.053). Young ALL patients had a significantly higher number of CAR-T cells on the day of infusion (patient age ≤19 years, 1357±986×10e6 cells; patient age ≥20 years, 664±345×10e6 cells; p=0.037). The age had no impact on % CD3 T cells frequency in the ficol-purified apheresis product (p=0.634), the transduction efficacy (p=0.625) or the CD4/CD8 content in the infusion product (p=0.960).
Fifteen of 38 patients with ALL had a prior allogeneic or haloidentical hematopoietic stem cell transplant. When accounting for donor age in these patients, representing the biological age of the lymphocytes, results were similar, with significantly increased fold expansion (donor age ≤19 years, 35.3±27.7; donor age ≥20 years, 14.5±6.6; p=0.015) and a significantly higher number of CAR-T cells by day 10 (donor age ≤19 years, 1436±979×10e6 cells; donor age ≥20 years, 609±345×10e6 cells; p=0.008) in younger patients. Of note, only two patients below age 19 received an allogeneic transplant from patients above age 20.
Gender had no impact on any of the parameters.
ANCOVA analysis adjusted by age was applied to all ALL and NHL patients and confirmed that differences between CAR-T frequency in the infusion product were disease dependent (ALL vs NHL, p=0.031), whereas cell count or fold expansion at day 10 were not disease dependent (p=0.420), but rather effected by the age.
Anti-tumor reactivity
The potency of the CAR-T cells was evaluated after co-culture with CD19 expressing tumor cell lines (NALM-6, Toledo) followed by IFNγ ELISA. In all cases, the IFNγ levels after co-incubation with CD19 positive target cells were far above the required limit of 200 pg/mL IFNγ. Following co-culture with CD19 positive NALM-6 and Toledo, cells from ALL patients secreted an average of 4.5±4.8 mg/mL and 6.3±7.9 mg/mL IFNγ, respectively; and from NHL patients 8.5±12.7 mg/mL and 10.5±12.5 mg /mL IFNγ respectively (p values >0.084). There was no significant difference between the two patient populations. Untransduced T cells, or transduced T cells co-incubated with CD19 negative target line CCRF-CEM, resulted in an approximately 1000-times lower secretion. The results of this assay demonstrate the highly specific anti-tumor reactivity and potency of CAR-T cells.
Phenotype analysis of CAR-T cells
The differentiation status of CAR-T cells and expression of co-inhibitory molecules have previously been related to clinical response.26–29 Therefore, we analyzed head-to-head the phenotype of ALL and NHL-derived CAR-T cells infusion products. CD8/CD4 subpopulation analysis of 91 infusion products did not reveal any significant differences between the two patient populations (CD8: ALL 67%±19%, NHL 74%±17%, p=0.071; CD4: ALL 33%±19%, NHL 26%±17%, p=0.070). Additional phenotype analysis was performed on 24 infusion samples of 12 ALL patients (seven MRD negative complete responders and five non-responders) and 12 NHL patients (six objective responders and six non-responders). The cells were analyzed for their differentiation status, determined by CCR7/CD45RA co-expression, expression of co-inhibitory receptors (PD-1, TIM-3, LAG-3), the co-stimulatory receptor CD28, the chemokine receptors CCR2, CCR4, CCR5, CXCR2, CXCR3 and the presence of gamma-delta T cells. Table 3 demonstrates the results gated on CAR-T cell of ALL versus NHL patients and online supplementary table 1 of clinical responders versus non-responders.
Phenotype analysis of CD19 CAR-T cells infusion products of ALL and NHL patients
As shown in table 3 and figure 3A, infusion products from ALL patients contained significantly more CD45RA+CCR7+naïve (TN) CAR-T cells (ALL 60.5%±17.3%, n=12; NHL 41.2%±22.4%, n=12; p=0.027) and significantly less CD45RA-CCR7+ central memory (TCM) CAR-T cells (ALL 15.8%±11.8%, n=12; NHL 27.2%±14.8%, n=12; p=0.048). The co-inhibitory molecules PD-1, LAG-3 and TIM-3 and the co-stimulatory molecule CD28 were equally expressed on ALL and NHL-derived CAR-T cells. The average expression of TIM-3 was with 63.3%±13.7% (n=24) almost double than of PD-1 (32.4%±11.6%, n=24) and LAG-3 (31.0%±12.1%, n=24). Also chemokine receptors were mostly equally expressed on CAR-T cells of ALL and NHL CAR-T, with the exception of CXCR3, which was significantly higher expressed in ALL patients (ALL 87.4%±8.2%, n=12; NHL 79.1%±11.1%, n=12; p=0.047). In general, the expression level of CXCR3 (83.2%±9.6%, n=24) was found to be the highest compared with other tested chemokine receptors (figure 3B).

{kind=link}
{kind=link}
{kind=link}
Phenotype analysis of CAR-T infusion products derived from ALL (n=12) and NHL (n=12) patients. Cells were gated on CD3+ F(ab)2+ CAR-T cells. (A) Differentiation status. TN (naïve T cells), CD3+CD45RA+CCR7+; TCM (central memory T cells), CD3+CD45RA−CCR7+; TEM (effector memory T cells), CD3+CD45RA−CCR7−; TEMRA (effector T cells), CD3+CD45RA+CCR7−. (B) Chemokine receptor profile. *p<0.01. SD are presented. ALL, acute lymphoblastic leukemia; CAR-T,chimeric antigen receptor T cells; NHL, non-Hodgkin's lymphoma.
We did not find any marker which was significantly different between responders and non-responders, besides the significantly higher expression of TIM-3 on CD8+ CAR-T cells of responding ALL patients (responders 38.8%±12.4%, n=7; non-responders 25.2%±5.4%, n=5; p=0.046).
CAR-T infusion products compared to TIL infusion products
Another type of adoptive T cell therapy is the administration of tumor infiltrating lymphocytes. In comparison to CAR-T cells, TIL are not genetically-engineered and occur in lesions of solid tumor. TIL have a natural capability to recognize intracellular tumor antigens, which are presented as peptides on MHC molecules of the tumor cell. These tumor peptides may be shared between patients or derive from unique tumor mutations.30–32 Adoptive TIL therapy has mostly been described for patients with r/r metastatic melanoma and yields objective response rates of 30% to 50% in a highly advanced patient population.20 33–36 TIL T cells are isolated from tumor lesions, grown for about 1 month to generate a pure lymphocyte culture and further expanded in a rapid expansion procedure (REP).21 24 REP uses similar reagents as the expansion of CAR-T cells; however, the addition of irradiated donor feeder cells at REP initiation induces an average fold expansion of about 1000-fold, compared with approximately 20-fold for CAR-T cells. Over the last 15 years we have manufactured over 100 TIL infusion products for melanoma patients. Although the production process of TIL and CAR-T cells largely differs, CAR-T and TIL are the two most common types of T cell therapy to date. We therefore compared the phenotype of CAR-T infusion products (n=24) with TIL infusion products (n=40–88). As shown in table 4, TIL and CAR products had a similar content of CD4 and CD8 T cells, as well as PD1 and CD28 expressing T cells. CAR-T cells demonstrated a significantly higher expression of LAG-3 (CAR-T, 25%±11%; TIL, 10%±4%; p≤0.001) and significantly lower expression of TIM-3 (CAR-T 50%±15%; TIL, 61%±14%; p=0.004). As expected, TIL demonstrate a more differentiated phenotype and were almost exclusively (97%) effector memory T cells, whereas 49%±18% and 17%±14% of CAR-T cells were naive or central memory T cells, respectively. The chemokine receptor CCR5 was significantly higher expressed on TIL (CAR-T, 12%±13%; TIL, 80%±24%; p≤0.001).
Comparison of CAR-T and TIL infusion products
Discussion
CD19 CAR-T cells have demonstrated high effectiveness for the treatment of r/r ALL and NHL patients, leading to FDA approval of two commercial products in 2017. Although highly potent, the commercial CD19 CAR-T products have the disadvantage of being very costly. In addition, as a consequence of the high turnaround time of 1 to 2 months from leukapheresis to infusion, many rapidly progressive patients clinically deteriorate during the waiting period. The JULIET and ELIANA phase 3 trials with tisagenlecleucel reported an intent-to-treat analysis from screening to treatment of only 47% and 70%, respectively.5 37 As a result, a rising number of clinical centers produce their own CAR-T cells in-house. To date, the Sheba Medical Center, Israel, has conducted one of the highest numbers of treatments with in-house produced CD19 CAR-T cells and TIL worldwide. All patients were heavily pretreated and CAR-T cells were mostly given as salvage therapy. We demonstrated that the short turnaround time of only 9 to 10 days, enabled the treatment of almost all enrolled patients. Only 2 of 93 (2%) patients dropped out due to clinical deterioration. The production success was 99% (91 of 92).
In this study, the CAR-T production platform was identical for ALL and NHL patients, allowing true comparison between cells from different patient populations. Although the average CD3 blood counts were very similar in ALL and NHL patients (p=0.871) at the time of leukapheresis, CAR-T cells from ALL patients expanded significantly better than of NHL patients (17.8±13.5 fold) and reached a fold expansion of 28.7±24.7 compared with day 2 (p=0.009). This difference was explained by the variance of age rather than the disease type. The transduction efficacy was higher in ALL derived infusion products than in NHL (p=0.007), independent of age. The target CAR-T cell number in this trial was 1×10e6 CAR-T cells per kilogram weight, since this dose was found effective and relatively safe in adults with NHL38 and children and young adults with ALL.39
As a consequence of the younger age, the lower weight, the better expansion rate and the higher transduction efficacy, an average of 27-times more CAR-T cells were produced for ALL patients than required. For NHL patients 9-times more CAR-T cells were manufactured on average. Three NHL patients received a dose below 1×10e6/kg, compared with none of the ALL patients.
The produced CAR-T cells were highly potent and secreted IFNγ in the range of milligrams per ml after co-culture with CD19 expressing tumor lines. Specificity was confirmed by at least a 1000-times lower secretion of IFNγ, when co-incubated with a CD19 negative tumor line.
The infused cell products were clinically effective, leading to remission in 84% of the ALL patients and 62% of evaluated NHL patients.
The differentiation status analysis of ALL derived CAR-T cells revealed a higher content of naïve T cells, whereas CAR-T cells of NHL patients had a higher frequency of central memory T cells. Both, naïve and central memory T cells were shown to enhance the potency and durability of CAR-T cells.26 40 However, naïve CD4 T cells were reported to secrete more cytokines than TCM.26 Other than that, ALL and NHL-derived CAR-T had a very similar phenotype, except for the chemokine receptor CXCR3, which was significantly higher expressed on CAR-T cells derived from ALL patients. Due to alternative splicing of the CXCR3 mRNA, three variants have been reported, CXCR3-A, CXCR3-B, and CXCR3-Alt, which exhibit opposing cellular effects.41 To understand the potential implication of a higher expression level of CXCR3 in CAR-T products derived from ALL patients, further investigation will be required.
CAR-T therapy has emerged as a promising approach for hematological malignancies, but is yet less effective in solid tumors. Challenges which need to be overcome in solid tumors, include tumor target selection, the immunosuppressive tumor-microenvironment, tumor heterogeneity and inadequate intratumoral T cell trafficking.42–44 Adoptive T cell therapy with TIL on the other hand, has demonstrated high objective response rates in melanoma and other solid tumors.20 33–36 Analyzing differences of TIL and CAR-T cell infusion products, might provide a way to gain insight and improve CAR-T cell therapies for solid tumors. Interestingly, FACS analysis of CAR-T and TIL infusion products revealed a more differentiated phenotype in TIL (97% effector memory T cells) and higher expression of CCR5. This may indicate a lower persistence of TIL, but also a higher infiltration capacity. Further analysis will be required. Of note, it was shown that CCL3, CCL4, and CCL5, the corresponding ligands of CCR5, are a major determinant of immune cellular infiltration in tumors.45 Since active trafficking of T cells into tumor mass partially depends on the interaction between chemokines in tumor and chemokine receptors presented on T cells, new CAR-T cell therapies armed with chemokine receptors are currently under development.46 47
Conclusions
The in-house production of CAR-T cells is highly efficient. CAR-T cells were successfully produced for 99% of patients in just 9 to 10 days, which allowed the treatment of 97% of enrolled patients. CAR-T cells of ALL patients demonstrated increased proliferation and transduction capacity compared with NHL-derived CAR-T cells, which may explain improved clinical remission in these patients.
Acknowledgments
The authors would like to thank Mrs Rina Sareli and Mrs Bella Baturov from the leukapheresis units and Ran Avrahami for performing the ANCOVA analysis. The authors would also like to thank the Lemelbaum family for their generous support.
References
Footnotes
Contributors MJB, OI, and EJ designed the study. EJ, AT, AA, ANagler, GM, and JS recruited and treated the patients, and are investigators. OI, ANissani, ML, AK, KB, HB, LZ, HV, and MR collected and analyzed the data. OI, ANissani, ML, AK, KB, HB, LZ, and MJB led the production and quality control of the in-house CAR-T cell and TIL program. OI, ANissani, and MJB wrote the paper. All authors read and approved the final manuscript.
Funding The clinical trial was funded by the Sheba Medical Center and a donation of the Lemelbaum family.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study has been performed in accordance with the Declaration of Helsinki and was approved by the Israeli Ministry of Health Approval numbers: 20 161 748 (CAR-T) and SMC356616 (TIL). An informed consent to participate in the study was obtained from all participants or their legal guardian for minors.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.