Article Text

Abstract
Background Natural killer and cytotoxic CD8+ T cells are major players during antitumor immunity. They express NKG2D, an activating receptor that promotes tumor elimination through recognition of the MHC class I chain-related proteins A and B (MICA and MICB). Both molecules are overexpressed on a great variety of tumors from different tissues, making them attractive targets for immunotherapy. However, tumors shed MICA and MICB, and the soluble forms of both (sMICA and sMICB) mediate tumor-immune escape. Some reports indicate that anti-MICA antibodies (Ab) can promote the restoration of antitumor immunity through the induction of direct antitumor effects (antibody-dependent cell-mediated cytotoxicity, ADCC) and scavenging of sMICA. Therefore, we reasoned that an active induction of anti-MICA Ab with an immunogenic protein might represent a novel therapeutic and prophylactic alternative to restore antitumor immunity.
Methods We generated a highly immunogenic chimeric protein (BLS-MICA) consisting of human MICA fused to the lumazine synthase from Brucella spp (BLS) and used it to generate anti-MICA polyclonal Ab (pAb) and to investigate if these anti-MICA Ab can reinstate antitumor immunity in mice using two different mouse tumors engineered to express MICA. We also explored the underlying mechanisms of this expected therapeutic effect.
Results Immunization with BLS-MICA and administration of anti-MICA pAb elicited by BLS-MICA significantly delayed the growth of MICA-expressing mouse tumors but not of control tumors. The therapeutic effect of immunization with BLS-MICA included scavenging of sMICA and the anti-MICA Ab-mediated ADCC, promoting heightened intratumoral M1/proinflammatory macrophage and antigen-experienced CD8+ T cell recruitment.
Conclusions Immunization with the chimeric protein BLS-MICA constitutes a useful way to actively induce therapeutic anti-MICA pAb that resulted in a reprogramming of the antitumor immune response towards an antitumoral/proinflammatory phenotype. Hence, the BLS-MICA chimeric protein constitutes a novel antitumor vaccine of potential application in patients with MICA-expressing tumors.
- immunology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
An improved understanding of the cellular and molecular mechanisms that regulate tumor growth in immunocompetent hosts made possible the design of strategies aimed at restoring antitumor immunity, collectively known as immunotherapies.1 However, such strategies must overcome the ability of tumor cells to evade immune cell effector functions, a phenomenon that arises during tumor growth under immunological pressure.2 Therefore, potentially successful approaches to improve clinical success focus on developing and combining novel strategies that improve tumor-specific immunity and at the same time, interfere with tumor escape mechanisms.
Natural killer (NK) cells and cytotoxic CD8+ T lymphocytes (CTL) are major players of antitumor immunity. NK cells recognize and eliminate tumor cells through an array of activating receptors among which CD16 (that recognizes the Fc portion of IgG and is responsible for the antibody-dependent cell-mediated cytotoxicity, ADCC) and NKG2D are the most relevant. In humans, NKG2D recognizes MICA and MICB, as well as members of the UL-16-binding protein (ULBP)/retinoic acid early transcripts family.3 Altogether, these NKG2D ligands (NKG2DL) are expressed on a wide variety of tumors but weakly expressed on healthy cells.3 Although overexpression of NKG2DL may represent a valid strategy to limit tumor progression,4 5 tumors display escape strategies that subvert the biological function of NKG2D.6 7 The underlying mechanisms through which these ‘immunoevasion’ phenotypes resist NKG2D-dependent cytotoxicity involve the proteolytic shedding of MICA and other NKG2DL induced by tumor-secreted metalloproteases.6–8 Released soluble MICA (sMICA) can thereafter bind to NKG2D and induce its downmodulation and degradation. In addition, other mechanisms account for low cell surface expression of MICA and impaired recognition by NKG2D, as we have demonstrated previously.9
It has been shown that Ab-mediated blockade of CTLA4 in mouse models induced a strong antitumor immunity10 and clinical studies with antagonistic anti-CTLA4 Ab demonstrated that it is possible to boost antitumor immunity in patients with melanoma.11 Notably, administration of anti-CTLA4 monoclonal Ab (mAb) induced anti-MICA Ab in some patients that cleared sMICA and interfered with tumor-immune escape.12 13 Additionally, the therapeutic efficacy of mAb-mediated neutralization of sMICA14 or MICA shedding15 has been shown to negatively affect tumor growth in mouse models. In addition, it has been demonstrated that immune complexes formed between a mAb and the α3 domain of MICA can activate human NK cells in a Fc-dependent manner.16 Monoclonal Ab-mediated therapeutic approaches are mostly useful for patients with clinically evident tumors and there are some adjuvant treatment options such as chemotherapy, radiation therapy, hormone therapy, and/or immunotherapy administered to patients with cancer after surgical excision, chemotherapy or radiotherapy of the primary tumor. However, the efficacy of most of these adjuvant therapies is not quite high and many patients remain at risk of tumor recurrence of the primary tumor and/or development of metastases. Therefore, we hypothesized that active induction of anti-MICA Ab may trigger beneficial effects in tumor-bearing hosts and exert a beneficial effect through multiple mechanisms such as clearing of sMICA from serum and promoting ADCC against tumor cells. Taking advantage of an established platform suitable to produce highly immunogenic chimeric proteins17 18 that displays intrinsic adjuvant properties and can induce specific Ab,19 the objectives of this study were to produce a chimeric protein consisting of the ectodomain of MICA fused to BLS to generate an immunogen useful for the induction of anti-MICA Ab in tumor-bearing hosts, to investigate whether these Ab enhance antitumor immunity, and to elucidate the underlying mechanisms.
Methods
Bioinformatic analysis
Data of expression of different NKG2DL in human tumors were obtained from the cBioportal (http://www.cbioportal.org/).20 21
Ab and reagents
The following mAbs were used: unlabeled and phycoerythrin (PE)-labeled anti-human MICA/B (clone 6D4, Biolegend), AlexaFluor488-labeled anti-human MICA (clone 159227, Biotechne), allophycocyanin (APC)-labeled anti-human MICB (clone 236511, Biotechne), PE-labeled anti-F4/80 (clone BM8, Biolegend), PE-labeled, fluorescein isothiocyanate (FITC)-labeled and PerCP/Cy5.5-labeled anti-CD3ε (clone 17A.2; Biolegend), APC-labeled anti-CD8α (clone 53-6.7, Biolegend), FITC-labeled anti-CD4 (clone RM4-5; Biolegend), APC/Cy7-labeled anti-CD11b (clone M1/70, Biolegend), PE/Cy7-labeled anti-CD11c (clone N418, Biolegend), PE/Cy7-labeled anti-Gr-1 (clone RB6.8C5, Biolegend), PerCP/Cy5.5-labeled anti-CD45R/B220 (clone RA3-6B2, Biolegend), FITC-labeled anti-CD86 (clone GL-1, Biolegend), PE/Cy7-labeled anti-CD44 (clone IM7, Biolegend), APC-labeled anti-MHC-II (I-A/E, clone M5/114, Biolegend), PE/Cy7-labeled anti-CD274 (PD-L1, clone 10F.9G2, Biolegend) and PerCP/Cy5.5-labeled anti-CD206 (clone C068C2, Biolegend). Also, a previously obtained anti-MICA/B mAb (D7, IgG2b), an anti-BLS mAb (BI24, IgG2b) and rabbit pAb raised against peptides spanning the α1 or the α2 domain of human MICA were used.22–24 FITC-labeled or PE- labeled anti-mouse IgG (Biolegend) and HRP-labeled anti-mouse or anti-rabbit IgG (Bio-Rad) were also used.
Production of chimeric BLS-MICA protein
Exons 2 to 4 of human MICA*001 (that encode for the full ectodomain), http://www.ebi.ac.uk/ena/data/view/AH008148, were synthesized including an upstream sequence for cloning with NsiI (ATGCAT), a downstream linker (GGTAGC) followed by a downstream sequence for cloning with AflII (CTTAAG), and cloned into the pET11a plasmid containing the open reading frame of Brucella spp lumazine synthase (BLS) as previously reported.17 Of note, as exon 3 contains a NsiI cutting sequence (ATGCAT), this sequence was replaced by the silent substitution ATGCAC. The plasmid was used to transform Escherichia coli BL21 (DE3)-competent cells for expression of the recombinant protein (429 aminoacids; expected size: 48.6 kDa, assessed with the ProtParam tool; http://web.expasy.org/protparam/). The sequences of the ectodomain of MICA*001, the peptide linker and BLS are shown in table 1. Expression of the chimeric recombinant protein was induced with isopropyl-β-d-1-thiogalactopyranoside, and bacteria were lysed with 50 mM Tris–HCl, 5 mM EDTA, 40 µg/mL deoxyribonuclease (DNase), 1 mM phenylmethylsulfonyl fluoride (PMSF), pH 8.0 and sonication. Inclusion bodies were solubilized in 100 mM Tris, 50 mM glycine, 5 mM EDTA, 8 M urea, pH 8.0 at room temperature overnight with agitation. The solubilized proteins were purified by anion exchange chromatography in a Q Sepharose (Pharmacia, GE Healthcare Life Sciences) column using a high performance liquid chromatography (HPLC) equipment (Dionex Ultimate 3000) connected to a UV/vis detector. Elution was performed using a linear gradient between 0 M and 1 M NaCl in a 50 mM Tris–HCl, 8 M urea, 50 mM glycine, pH 8 buffer. The elution fraction containing the recombinant protein, determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), was refolded by sequential dialysis against decreasing concentrations of urea (4 M, 2 M, 1 M and 0 M) in 50 mM Tris–HCl, pH 8, 1 M L-arginine, 0.5 mM EDTA, 0.02% sodium azide buffer. Each dialysis step was of 2 hours. BLS-MICA was stored at 4°C. The fraction containing the recombinant protein was analyzed by SDS-PAGE.
Aminoacid sequences of the ectodomain (α1–α3 domains) of the MICA*001 allele, of BLS, the linker and the chimeric protein BLS-MICA
Production of mouse cell lines stably expressing MICA
The mouse lymphoma cell line EL4 (ATCC TIB-39) and the bladder carcinoma MB49,25 both of C57BL/6 background, were transduced to express the MICA*008 allele on their cell surface. For expression of human MICA, cells were infected with retroviruses encoding this MICA allele and also the puromycin resistance gene. For retrovirus production, the packaging cell line PT67 was transfected with viral DNA (pMSCV and pMSCV/MICA*008) and the packaging vectors (pCMVgag-pol and pMD2.G) using FUGENE HD Transfection Reagent (Promega). pMSCV (negative control) and pMSCV/MICA*008 (encoding the MICA*008 allele) viral DNA were kindly provided by Dr Alessandra Zingoni and Dr Angela Santoni from the Laboratory of Molecular Immunology and Immunopathology, Department of Molecular Medicine, Sapienza University of Rome, Italy. After 48 hours, virus-containing supernatants were harvested, filtered, and used for infection as follows: 1 mL of viral supernatants containing polybrene (8 mg/mL) was used to infect 5×105 EL4 and MB49 cells for 2 hours. Cells were cultured in the presence of puromycin (Enzo Life Sciences). Cells expressing high amounts of MICA were screened by flow cytometry (FC) with the anti-MICA/B mAb 6D4 labeled with PE (Biolegend). Positive cells for MICA were further selected by cell sorting (FACS) with the anti-MICA/B mAb 6D4 labeled with PE, cultured and stored until use. Thereafter, another round of infection, selection and sorting was performed. After these two rounds of infection, cells were reanalyzed by FC with the anti-MICA/B mAb 6D4, the anti-MICA mAb 159 227 or the anti-MICB mAb 236 511 and acquired in a FACSCanto II FC (BD). Cell sorting was performed in a FACSAria II plus cell sorter (BD).
Immunization of mice and collection of sera for passive immunization
C57BL/6 mice were obtained from the animal facility of the School of Veterinary, University of La Plata (Argentina) and housed at the animal facility of the Institute of Biology and Experimental Medicine according to NIH guidelines. Mice (6–10-weeks old) were immunized with BLS or BLS-MICA by intraperitoneal inoculation (10 or 20 µg/mouse, respectively, in order to compensate for the different molecular mass of each immunogen), with or without complete Freund’s adjuvant (CFA). Mice received one immunization every 2 weeks and 1 week after the last immunization, blood samples were obtained by venipuncture from the facial vein, and sera were obtained and analyzed for the presence of anti-MICA pAb by ELISA. Sera from mice with high titers of anti-MICA pAb were collected and stored at −20°C for passive transfer studies. As titration of the sera by ELISA with rMICA as coating antigen showed that mice immunized with BLS-MICA with or without CFA developed a similar titer of anti-MICA pAb, subsequent immunizations aimed at obtaining sera with anti-MICA pAb were performed without CFA. All studies with mice have been approved by the institutional review committee for animal care.
ELISA
Polystyrene plates (High binding, Costar) were coated with 1 µg/mL of rMICA*001 produced and used as previously described26 or BLS-MICA. After blocking with 5% non-fat dry milk in phosphate buffered saline (PBS) for 1 hour at 37°C, plates were incubated with an anti-MICA or an anti-BLS mAb, with rabbit anti-MICA pAb or with serial dilutions of sera from the different mice for 1 hour at 37°C, washed with PBS-Tween 20 0.05%, incubated with horseradish peroxidase (HRP)-labeled anti-mouse or anti-rabbit IgG for 1 hour at 37°C, washed and developed with H2O2 and 3,3′,5,5′-tetramethylbenzidine. Optical density was determined in a Thermo Multiscan FC ELISA reader at 450 nm.
Flow cytometry
Expression of cell surface receptors and markers was analyzed by FC using fluorochrome-labeled mAb as previously described.27 28 Data were acquired in a MACSQuant10 (Miltenyi Biotec) or a FACSCanto II (BD) flow cytometers, analyzed using FlowJo X software (Tree Star) and expressed as median fluorescence intensity (MFI) or as percentage of positive cells.
Mice and tumor challenge experiments
C57BL/6 mice (6–10 weeks) were used (males and females for experiments with EL4 cells and males for experiments with MB49 cells). Studies have been approved by the institutional ethics review committee. Control or MICA-expressing EL4 or MB49 tumor cells were inoculated by subcutaneous route in one flank of the animals (2×105 cells per mouse), tumor size was measured using a caliper and tumor volume was calculated by the modified ellipsoidal formula (length × width2)/2 as described.9 For active prophylactic immunization experiments mice were immunized four times by intraperitoneal route with BLS or with BLS-MICA (10 or 20 µg/mouse, respectively) without adjuvants every 9–10 days. Three weeks after the last immunization, mice were challenged with MB49-MICA*008 tumor cells, and tumor growth was assessed. For passive therapeutic immunization experiments, once tumors reached a palpable/measurable size (between 7 and 10 days after inoculation), mice were passively immunized by intraperitoneal route with 200 µL of a 1/4 dilution of a pool of sera from mice previously immunized with BLS (control pAb, Ctrl pAb) or with a similar volume of a pool of sera from mice previously immunized with BLS-MICA (anti-MICA pAb) every 2 days until the euthanasia of each animal. In some experiments, mice were euthanized after 12 days of tumor inoculation and tumors were collected to analyze the composition of the cell infiltrate. For active therapeutic immunization experiments, 1 day after tumor challenge, mice were actively immunized by intraperitoneal route with BLS or with BLS-MICA (10 or 20 µg/mouse, respectively) without adjuvants every 7 days until the euthanasia of each animal. Additionally, adjuvant treatment simulation was assessed, for which mice were challenged with control or MICA-expressing MB49 tumor cells, tumors were removed by surgery when they became palpable (about 1 week after the inoculation) and mice were thereafter treated with Ctrl pAb or anti-MICA pAb as described above. For these experiments, mice received tramadol (40 mg/kg) in drinking water just before and after surgery. To mitigate risk of infections, surgery wound was cleaned with thimerosal and mice received enrofloxacin (50 mg/kg) in drinking water for 2 days before surgery and up to 2 days after finishing the experiment. Also, in vivo ADCC was assessed using tumor cells that were incubated with sera from mice immunized with BLS-MICA or with BLS alone as described below, were injected in naive C57BL/6 mice by subcutaneous route and tumor growth was assessed. In all cases, tumor growth was assessed as described and animals were euthanized when tumors reach a size of 1000 mm3. Also, experiments with MB49 cells were terminated earlier as these tumors tend to ulcerate and due to ethical reasons, mice were euthanized if their tumors ulcerated.
ADCC in vitro
Control or MICA-expressing tumor cells were labeled with eFluorDye670 (eBiosciences) for 15 min at 37°C, washed and incubated with sera from mice immunized with BLS-MICA or with BLS alone for 30 min. The excess of pAb was washed, and tumor cells were incubated with mouse NK cells isolated from spleens from healthy C57BL/6 mice using a positive selection reagent (Miltenyi) for 5–6 hours. Thereafter, viability in the eFluorDye670 population (target cells) was assessed using ZombieAcqua (Biolegend) and FC using a MACSQuant10 flow cytometer. Data were analyzed using FlowJo X software, and results were expressed as percentage of cytotoxicity calculated as the percentage of ZombieAcqua+ cells in the eFluorDye670+ cell population.
ADCC in vivo
Control or MICA-expressing tumor cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) or eFluorDye670 (eBiosciences), respectively, for 15 min at 37°C, washed, mixed in equal proportions and incubated with sera from mice immunized with BLS-MICA or with BLS alone for 1 hour. Then, 4×105 tumor cells were injected in naive C57BL/6 mice by subcutaneous route. After 72 hours, mice were euthanized, cells were recovered from the inoculation site, and the relative abundance of CFSE+ and eFluorDye670+ cells was analyzed by FC using a MACSQuant10 flow cytometer and expressed as percentage of recovered cells. Data were analyzed using FlowJo X software. In other experiments, unlabeled control or MICA-expressing tumor cells were incubated with sera from mice immunized with BLS-MICA or with BLS alone as described before, injected in naive C57BL/6 mice by subcutaneous route and tumor growth was assessed as described.
Analysis of soluble MICA
Analysis of sMICA in sera of mice was performed by an in-house developed system. Briefly, avidin-coated microspheres (SPHERO Avidin Coated Particles, Spherotech) were coated with biotinylated anti-MICA/B mAb 6D4 and used to capture sMICA from sera of different groups of mice for 16 hours. Microspheres were washed and bound sMICA was detected with AlexaFluor488-labeled anti-MICA mAb 159 227 by FC using a MACSQuant10 flow cytometer. Data were analyzed using FlowJo X software and results were expressed as MFI. The analysis was performed using sera of mice that were euthanized after 15 days of tumor challenge.
Statistical analysis
Two-way analysis of variance (ANOVA) with Sidak’s posthoc tests were used for comparison of tumor growth. For comparison of two experimental groups, unpaired two-tailed Student’s t-tests were used. For Kaplan-Meier analysis, log-rank Mantel-Cox test was used. Data were analyzed using GrapPad Prism V.6.0 software.
Results
MICA is overexpressed in tumors including those that display the highest number of deaths and incidence
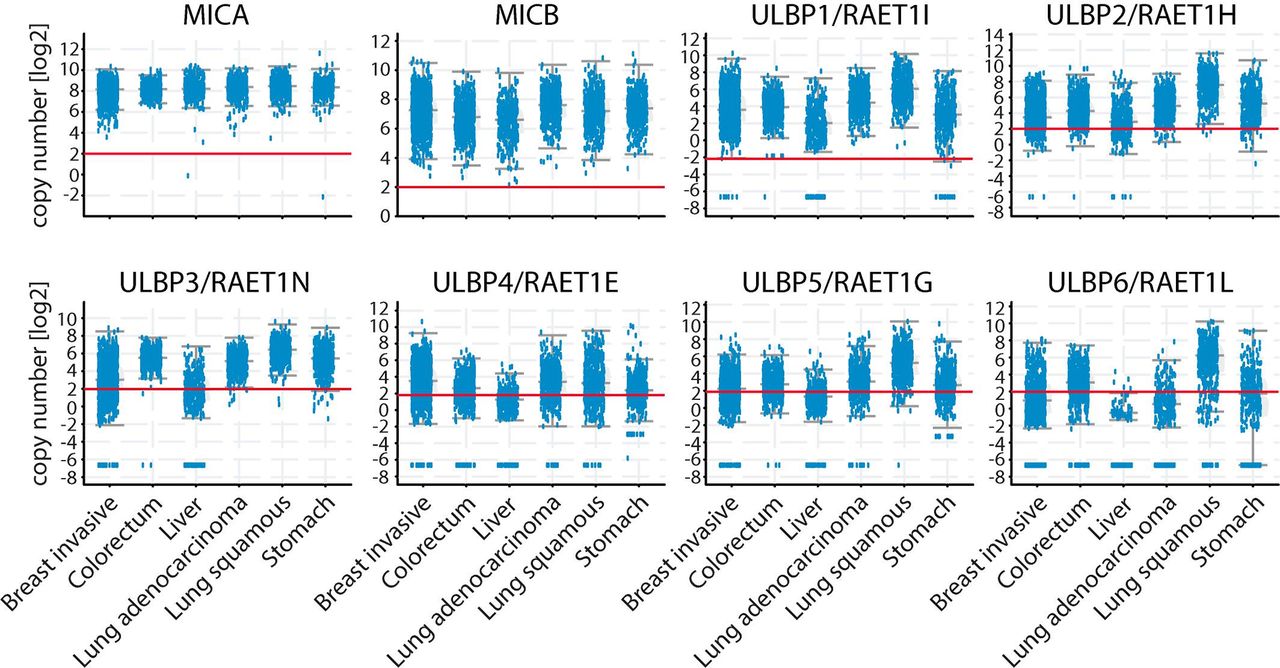
According to GLOBOCAN (https://gco.iarc.fr/), lung, colorectal, stomach, liver and breast cancers exhibit the highest number of deaths. To explore the expression of NKG2DL in these types of tumors, we performed a bioinformatic analysis of their expression. The analysis of RNA sequencing (seq) data in the cBioportal database (http://www.cbioportal.org/)20 21 showed that all these tumors expressed MICA, MICB and the 6 ULBP (figure 1). However, MICA was the NKG2DL that exhibited the most homogeneous expression within each of these tumors and among them, MICA was the NKG2DL that exhibited the highest expression. Therefore, we focused our study on MICA, as its overexpression in human tumors that exhibit a high mortality suggests a potential role for MICA as target for an Ab-mediated therapy.

MICA RNA is overexpressed in the human tumors that exhibit high number of deaths compared with matched normal tissues. RNAseq data of expression of different NKG2DL (indicated at the top of each panel) in the indicated human tumors. The red horizontal line in each graph indicates the cut-off value to consider upregulated expression. Data presented were obtained from the cBioPortal for cancer genomics (http://www.cbioportal.org/). The graph corresponds to 1084 breast tumors, 594 colorectal tumors, 372 liver tumors, 566 lung adenocarcinomas, 487 lung squamous cell carcinomas and 440 stomach carcinomas. MICA, MHC class I chain-related protein A; MICB, MHC class I chain-related protein B; NKG2DL, ligand of NKG2D.
Immunization with the chimeric BLS-MICA protein generates high titer of anti-MICA Ab that recognize the cell surface expressed form of MICA
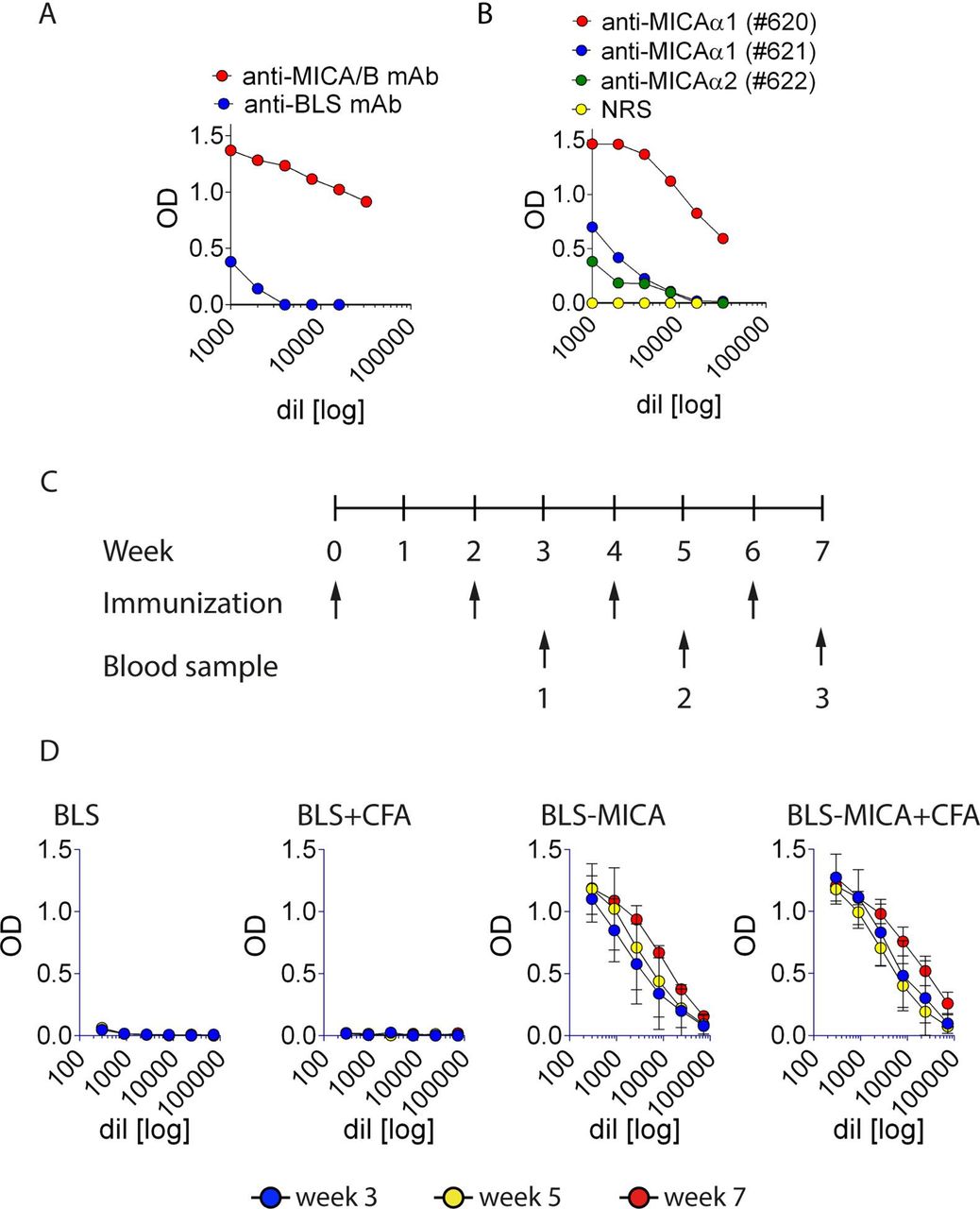
Reasoning that MICA constitutes a potential target for Ab-mediated antitumor therapy, we devised a strategy to generate pAb against this human NKG2DL using a chimeric protein. First, we produced this chimeric/immunogenic protein (named BLS-MICA) that combines the immunogenic scaffold provided by BLS coupled to the ectodomain of MICA (table 1). This protein was produced in bacteria as inclusion bodies. After solubilization, refolding and removal of the urea used during solubilization, this protein displayed the expected size (~49 kDa) by SDS-PAGE analysis (not shown). Moreover, BLS-MICA was recognized by an anti-MICA/B mAb D7 (previously produced in our laboratory22), by an anti-BLS mAb (BI24, figure 2A), and by rabbit pAb produced against peptides of the α1 or the α2 domains of MICA23 (figure 2B). Next, this chimeric protein was used to immunize C57BL/6 mice with or without the use of CFA following a standard immunization schedule (outlined in figure 2C). Only mice immunized with BLS-MICA (with or without CFA) developed anti-MICA pAb (figure 2D). Therefore, subsequent immunizations were performed without CFA. Sera with high titer of anti-MICA pAb were pooled and used for further experiments.

Immunization of mice with BLS-MICA generates high titers of anti-MICA pAb. (A) ELISA with plates coated with BLS-MICA and incubated with serial dilutions of an anti-MICA/B (D7, red) or an anti-BLS (BI24, blue) mAb. (B) ELISA with plates coated with BLS-MICA and incubated with serial dilutions of rabbit pAb raised against peptides spanning the α1 (#620 and #621) or the α2 (#622) domains of MICA. (C) Outline of the immunization schedule used to immunize C57BL/6 mice. After 3, 5 and 7 weeks of starting the protocol, blood samples were collected from the animals (indicated by numbers 1, 2 and 3, respectively) to analyze the presence of anti-MICA pAb. (D) ELISA with plates coated with rMICA and incubated with serial dilutions of sera from mice collected after 3 (blue), 5 (yellow) and 7 (red) weeks after starting the immunization protocol. Graphs show representative results of one immunization plan performed with five animals per group immunized with BLS alone, BLS and CFA, BLS-MICA alone or BLS-MICA and CFA. Mean±SEM is shown. Ab, antibody; BLS, Brucella lumazine synthase; BLS-MICA, BLS coupled to MICA; CFA, complete Freund’s adjuvant; mAb, monoclonal antibody; MICA, MHC class I chain-related protein A; MICB, MHC class I chain-related protein B; NRS, normal rabbit serum; pAb, polyclonal Ab.
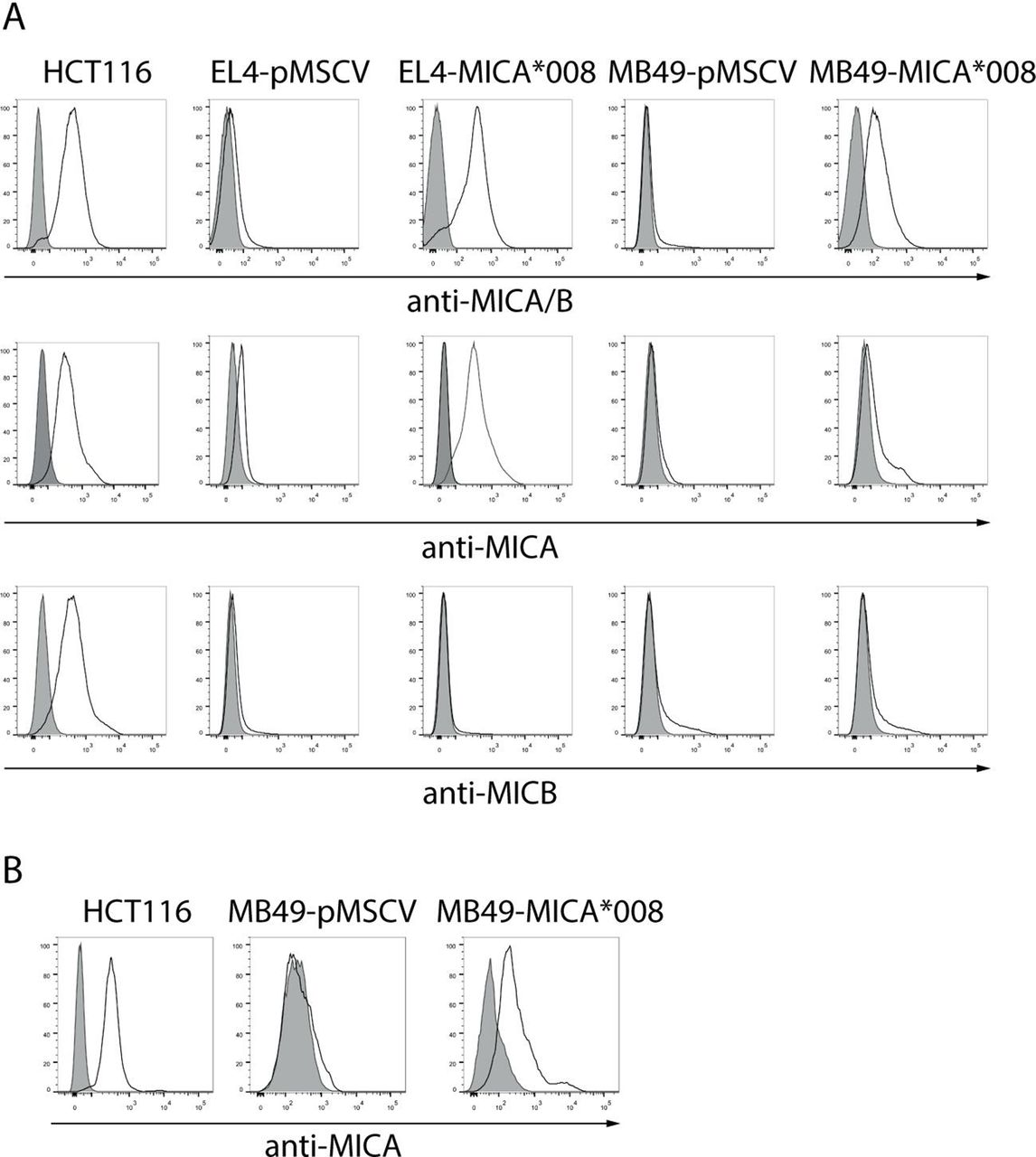
A critical step to evaluate our hypothesis was the in vivo testing of the therapeutic efficacy of the anti-MICA pAb in mouse models. As mice do not have the MICA gene and consequently do not express MICA, we generated two mouse cell lines (EL4 and MB49) engineered to express the most frequent allele of MICA (MICA*008, www.allelefrequencies.net) on their cell surface using retroviral vectors. After two rounds of infection, we obtained control cell lines (EL4-pMSCV and MB49-pMSCV), and cell lines expressing MICA*008 (EL4-MICA*008 and MB49-MICA*008) on their cell surface (figure 3A) that did not display differences in their in vitro growth rate (not shown). Moreover, anti-MICA pAb generated with BLS-MICA bound to MB49-MICA*008 cells, suggesting that BLS-MICA elicits Ab activity against native MICA (figure 3B). Although figure 1 shows data from tumors that exhibit the highest number of deaths and considering that we were interested in MICA as promising target for immuno-oncology, we selected the EL4 and MB49 mouse tumor models due to their C57BL/6 background, their availability and the fact that they constitute one model of liquid tumor and one model of solid tumor, both of which could be enforced to express MICA.

Mouse cell lines transduced with retroviral vectors encoding MICA express high amounts of this NKG2DL on their cell surface that is recognized by sera of mice immunized with BLS-MICA. (A) EL4 and MB49 cells were infected with control (empty) retroviruses (pMSCV) or with retroviruses encoding MICA*008 to generate the EL4-pMSCV, EL4-MICA*008, MB49-pMSCV and MB49-MICA*008 cell lines, and expression of MICA was assessed with the 6D4 mAb (anti-MICA/B), the anti-MICA mAb 159 227 or the anti-MICB mAb 236 511 by FC. Gray: IC mAb; black lines: anti-MICA/B, anti-MICA or anti-MICB mAb. HCT116 cells: positive control for MICA and MICB expression. (B) MB49-pMSCV, MB49-MICA*008 and HCT116 cell lines were labeled using a pool of sera of mice immunized with BLS-MICA (‘anti-MICA’, black line histogram) or with normal mouse serum (preimmune, gray histogram) and analyzed by FC. Ab, antibody; BLS, Brucella lumazine synthase; FC, flow cytometry; IC, isotype control; BLS-MICA, BLS coupled to MICA; mAb, monoclonal antibody; MICA, MHC class I chain-related protein A; MICB, MHC class I chain-related protein B; NKG2DL, ligand of NKG2D.
Immunization with the chimeric BLS-MICA protein generates a therapeutic effect against MICA-expressing tumors that is mediated by anti-MICA pAb
Active immunization with BLS-MICA delays growth of tumors that express MICA
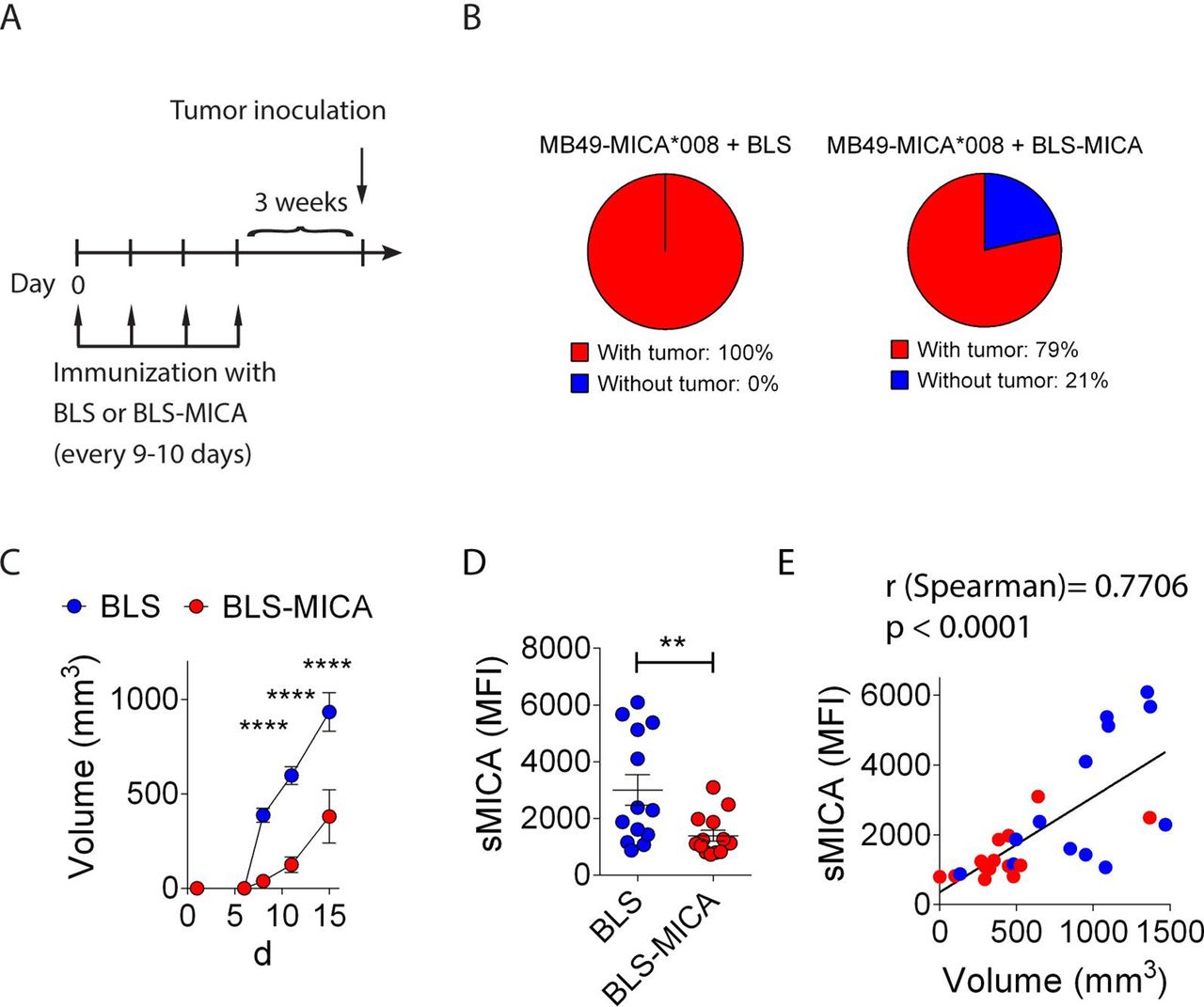
To assess the effect of the immunization with BLS-MICA on tumor growth, mice were challenged with MB49-MICA*008 cells and 1 day later, mice were immunized with BLS-MICA or with BLS alone. However, we did not observe differences in the tumor growth between both groups (not shown), likely due to the rapid tumor growth which is much faster than the time required to elicit an effective anti-MICA immune response and a hypothetical therapeutic effect. Therefore, we set-up a prophylactic immunization protocol in which naive mice were immunized four times with BLS or with BLS-MICA, and after 3 weeks, mice were challenged with MB49-MICA*008 cells as outlined in figure 4A. We selected MB49 cells as this tumor grows slower in vivo, providing a more suitable experimental approach to test our hypothesis. We observed that 100% of the mice immunized with BLS developed palpable tumors at day 8 after challenge but that only 79% of mice immunized with BLS-MICA developed palpable tumors at day 8 after challenge (figure 4B). Although mice without tumor did eventually develop tumors, the analysis of tumor growth revealed that active immunization with BLS-MICA induced a statistically significant slower tumor growth of MICA-expressing tumors than immunization with BLS (figure 4C). In addition, mice immunized with BLS-MICA exhibited lower amounts of sMICA in serum than mice immunized with BLS (figure 4D). In the control group (mice immunized with BLS), we observed that 5/13 mice displayed high sMICA and 8/13 displayed low sMICA. Although in these experiments we did not analyze if this difference was due to an endogenous immune response against MICA; additional data indicate that mice challenged with MICA-expressing tumors can develop endogenous/spontaneous anti-MICA Ab (online supplementary figure 1). Therefore, the difference between these two groups of mice challenged with MICA+ tumors might be due to a different endogenous response against MICA. Moreover, there was a positive correlation between sMICA and tumor volume (figure 4E). Therefore, prophylactic immunization with BLS-MICA appears to trigger a protective immune response that promotes scavenging of sMICA likely mediated by pAb against MICA.
Supplemental material

Immunization with BLS-MICA delays growth of tumors that express MICA. (A) Outline of the active immunization and tumor challenge schedule. Mice were immunized with BLS or BLS-MICA as indicated (four immunizations). Then, mice were rested for 3 weeks, challenged with MB49-MICA*008 tumors, and tumor growth was assessed. (B) Pie charts showing the percentage of mice immunized with BLS (left) or BLS-MICA (right) that developed palpable tumors at day 8 after challenge. (C) Tumor growth curves in mice immunized with BLS (blue) or with BLS-MICA (red). Mean±SEM is shown. Statistically significant differences between groups started at day 8 after tumor challenge. ****p<0.0001, two-way ANOVA with Sidak’s posthoc test. (D) Amounts of sMICA (expressed as MFI) detected in sera of mice immunized with BLS (blue dots) or BLS-MICA (red dots) and challenged with MB49-MICA*008 tumor cells at the time of euthanasia (15 days after tumor challenge). Each dot corresponds to one mouse (**p=0.008, unpaired two-tailed Student’s t-test. (E) Correlation analysis between the amount of sMICA and tumor volume (R Spearman=0.7706, p<0.0001). Data from mice immunized with BLS (blue) or with BLS-MICA (red) were plotted together. Data shown in the graph correspond to cumulative results from two independent experiments. n=14 mice per group (B–D) and n=28 (E). ANOVA, analysis of variance; BLS, Brucella lumazine synthase; BLS-MICA, BLS coupled to MICA; MFI, median fluorescence intensity; MICA, MHC class I chain-related protein A; MICB, MHC class I chain-related protein B; sMICA, soluble MICA.
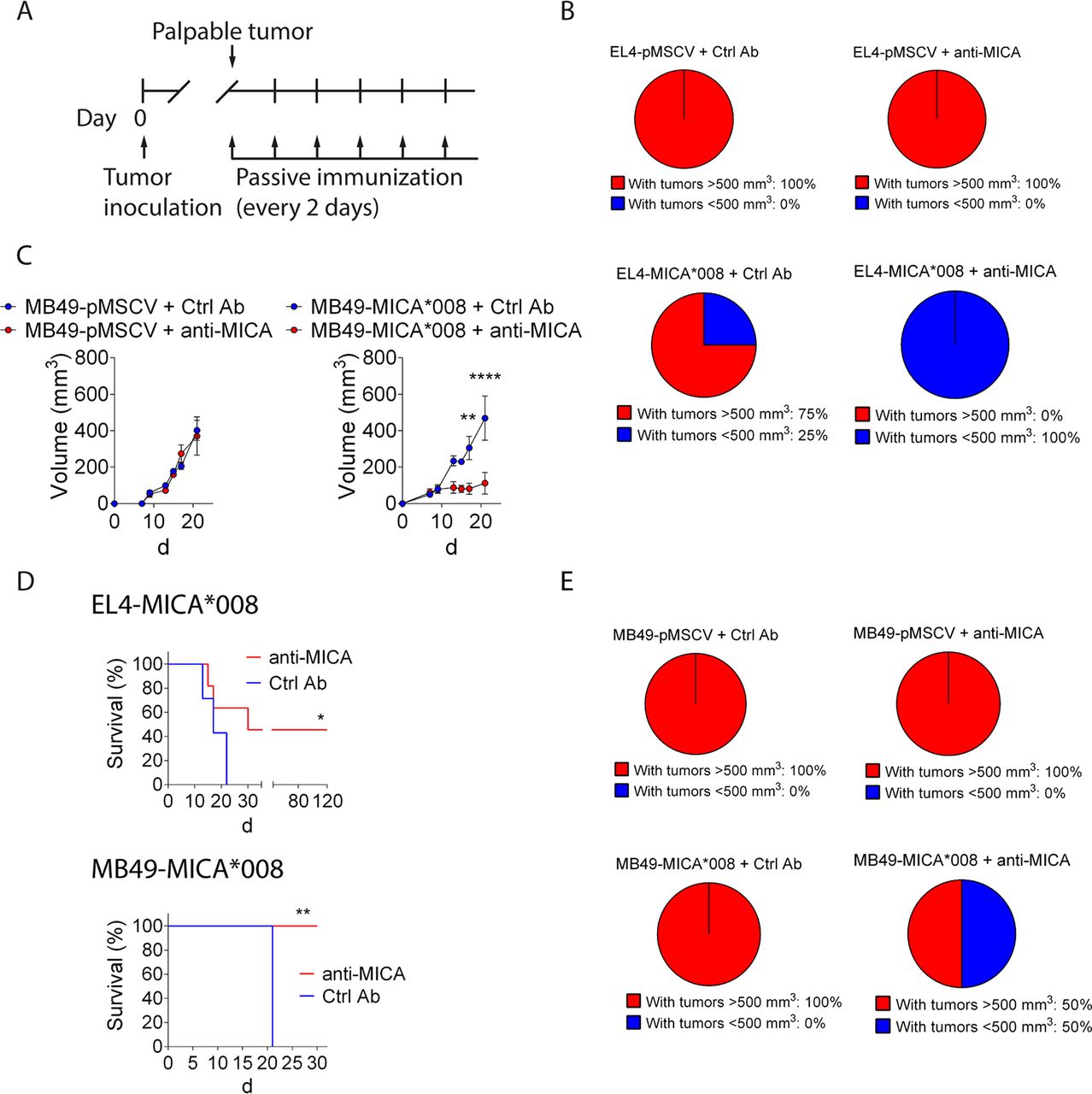
Passive immunization of tumor-bearing mice with anti-MICA pAb produced with BLS-MICA delay growth of tumors that express MICA and reshape the tumor microenvironment
To confirm this hypothesis, we challenged C57BL/6 mice with EL4-pMSCV, EL4-MICA*008, MB49-pMSCV or MB49-MICA*008 cells and, once tumors were palpable, mice were passively immunized with a pool of sera from mice immunized with BLS (Ctrl pAb) or with BLS-MICA (anti-MICA pAb), as outlined in figure 5A. In the case of EL4 tumors, when we analyzed the proportion of mice that developed tumors greater than 500 mm3 at day 11 after challenge, we observed that 100% of mice challenged with control tumors experienced tumor development regardless of the treatment, that 75% of mice challenged with MICA-expressing tumors and treated with Ctrl pAb developed tumors but that none of the mice challenged with MICA-expressing tumors and treated with anti-MICA pAb developed tumors (figure 5B). Moreover, in the case of MB49 tumors, we observed that the anti-MICA pAb induced a significant delay in tumor growth in mice challenged with MICA-expressing tumors, compared with mice treated with Ctrl pAb and challenged with MICA-expressing tumors or to mice treated with Ctrl pAb or anti-MICA pAb and challenged with control tumors (figure 5C). The therapeutic effect of the anti-MICA pAb on MICA expressing tumors resulted in an extended survival, as indicated by the Kaplan-Meier analysis of both models, EL4 and MB49 tumors (figure 5D). Remarkably, some mice challenged with MB49-MICA*008 and treated with anti-MICA pAb displayed complete tumor regression and these mice (n=3/8) remained tumor-free for more than 2 months, even without continuous passive administration of anti-MICA pAb (which was stopped at day 19 after tumor challenge), indicating that the robust therapeutic effect was long-lasting. Moreover, to investigate if anti-MICA pAb can affect tumor recurrence, mice were challenged with MB49-pMSCV or MB49-MICA*008 cells, tumors were removed by surgery once they became palpable and then the animals were passively immunized with Ctrl pAb or anti-MICA pAb and monitored for tumor reappearance (figure 5E). Notably, all mice challenged with MB49-pMSCV and treated with Ctrl pAb or with anti-MICA pAb, as well as mice challenged with MB49-MICA*008 tumors and treated with Ctrl pAb developed palpable tumors at day 42 after surgery, while only a 50% of mice challenged with MB49-MICA*008 tumors and treated with anti-MICA pAb developed palpable tumors at that time. These results indicate that the anti-MICA Ab delay re-emergence of tumors if they express this NKG2DL.


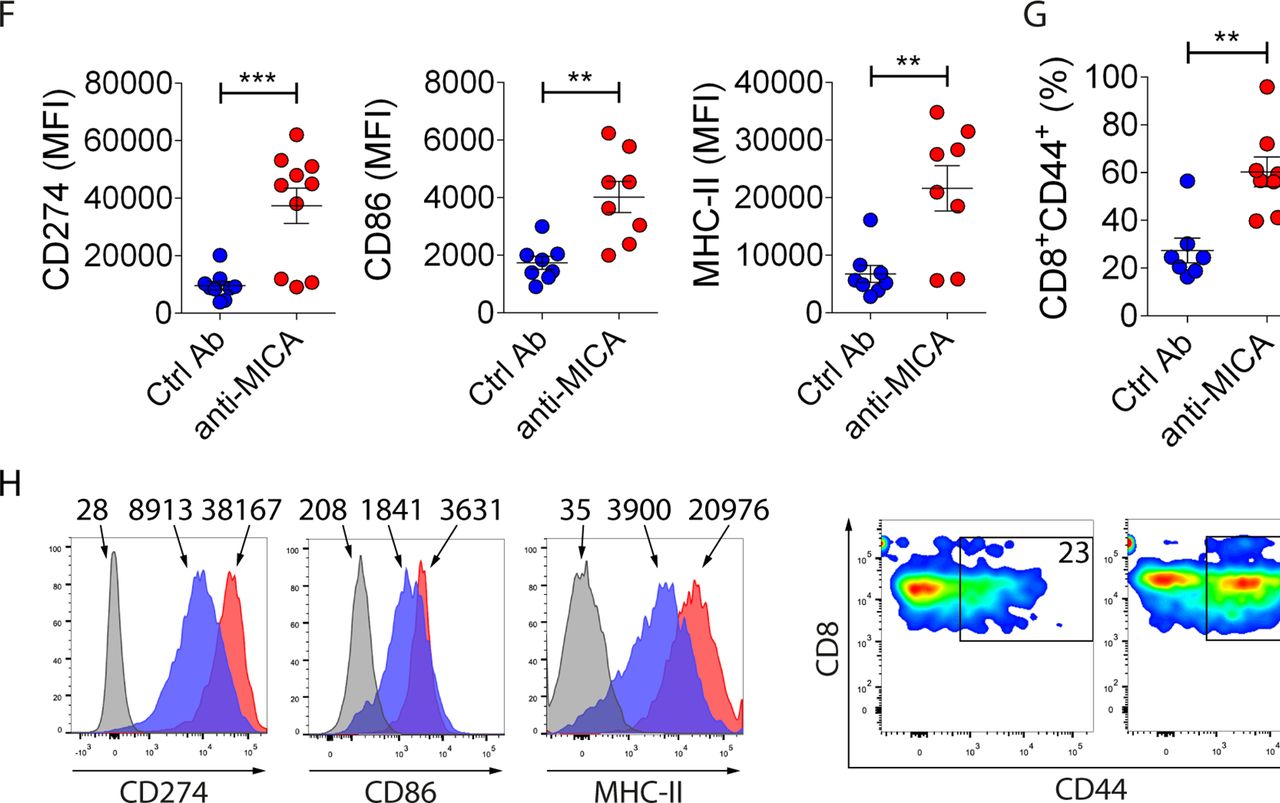
Anti-MICA pAb passively administered to tumor-bearing mice delay growth of tumors that express MICA associated with an increased accumulation of tumor infiltrating M1/proinflammatory macrophages and antigen-experienced CD8+ T cells. (A) Outline of the tumor challenge and passive immunization schedule. Mice were challenged with control (EL4-pMSCV or MB49-pMSCV) or MICA-expressing (EL4-MICA*008 or MB49-MICA*008) tumors at day 0. When tumors reached a palpable size, mice were passively immunized with sera from mice previously immunized with BLS (Ctrl pAb) or with BLS-MICA (anti-MICA) every 2 days, and tumor growth was assessed. (B) Pie charts showing the percentage of mice challenged with EL4-pMSCV (upper charts) or EL4-MICA*008 (lower charts) cells and immunized with Ctrl pAb (left) or anti-MICA (right) that developed tumors greater than 500 mm3 at day 11 after challenge. The tumor volume of 500 mm3 was selected because in our experience tumors of that size invariably progress. (C) Tumor growth curves of MB49-pMSCV (left) or MB49-MICA*008 (right) cells, in mice treated with Ctrl pAb or with anti-MICA pAb. **p<0.01, ****p<0.0001, two-way ANOVA with Sidak’s posthoc test. (D) Kaplan-Meier curves corresponding to mice challenged with EL4-MICA*008 cells (upper graph) or MB49-MICA*008 cells (lower graph) and treated with Ctrl pAb or with anti-MICA pAb. Differences between the two groups were significant (*p=0.0223 for EL4-MICA*008 cells and **p=0.016 for MB49-MICA*008 cells, log-rank Mantel-Cox test). Of note, it was not possible to extend the experiments up to 120 days with MB49 cells as these tumors tend to ulcerate and due to ethical reasons, mice were euthanized. (E) Pie charts showing the percentage of mice challenged with MB49-pMSCV (upper charts) or MB49-MICA*008 (lower charts) cells that underwent surgery to remove the tumor and that were then immunized with Ctrl pAb (left) or anti-MICA (right) that developed palpable tumors at day 42 after surgery (tumors of more than 60 mm3). n=4 per group (B–E). Data shown in B to E correspond to one representative experiment which was performed twice with similar results. (F–H) Mice were challenged with MB49-MICA*008 tumor cells and passively immunized with control pAb (Ctrl Ab, blue dots) or with anti-MICA pAb (anti-MICA, red dots) every 2 days starting when tumor became palpable as indicated in panel A. After 12 days, mice were euthanized, and the composition of the tumor infiltrate was analyzed by FC (outlined in online supplementary figure 2). (F) MFI of CD274, CD86 and MHC-II in F4/80+ cells (macrophages). (G) Percentage of CD44+CD8+ cells. (H) Representative histograms (numbers correspond to MFI) of CD274, CD86 and MHC-II in F4/80+ cells (macrophages) and pseudocolor plots (numbers correspond to percentages) of CD44+CD8+ cells. Each dot in F and G represents the data from one mouse. Data shown in the graphs correspond to cumulative results from two independent experiments. **p<0.01, ***p<0.001. An unpaired two-tailed Student’s t-test was used in F and G. CD274: n=9 (Ctrl Ab), n=10 (anti-MICA Ab), p=0.0007. CD86: n=8 (Ctrl Ab and anti-MICA Ab), p=0.0016. MHC-II: n=8 (Ctrl Ab and anti-MICA Ab), p=0.0032. CD44+: n=7 (Ctrl Ab), n=8 (anti-MICA Ab), p=0.0016. Ab, antibody; ANOVA, analysis of variance; BLS, Brucella lumazine synthase; BLS-MICA, BLS coupled to MICA; Ctrl Ab, control Ab; FC, flow cytometry; MFI, median fluorescence intensity; MICA, MHC class I chain-related protein A; MICB, MHC class I chain-related protein B pAb, polyclonal Ab.
Supplemental material
The therapeutic effect of anti-MICA pAb generated with BLS-MICA include a reprogramming of the tumor microenvironment and stimulation of ADCC
To analyze the effect of the anti-MICA pAb on the characteristics of the tumor cell infiltrate, mice bearing MB49-MICA*008 tumor cells were passively immunized with anti-MICA pAb or with control pAb by intraperitoneal route following the previously described scheme (figure 5A), euthanized at day 12, and tumors were collected to analyze the composition of the infiltrate as outlined in online supplementary figure 2. Significant differences in the profile of intratumoral macrophages (CD11b+F4/80+ cells) and antigen-experienced CD8+ T cells were observed in mice that received anti-MICA pAb. Intratumoral macrophages of these mice displayed higher expression of CD274, CD86 and MHC-II than intratumoral macrophages of mice that received Ctrl pAb, indicating that they display markers associated with a M1/proinflammatory phenotype (figure 5F), concomitant with a higher percentage of tumor-infiltrating CD8+CD44+ T cells (antigen-experienced CD8+ T cells, figure 5G; representative results are shown in figure 5H). No differences were observed between groups in terms of granulocytic and monocytic myeloid-derived suppressor cells (G-MDSC or CD11b+Gr1high cells and M-MDSC or CD11b+Gr1low cells, respectively; online supplementary figure 3). These results indicate that anti-MICA pAb can reprogram the tumor-infiltrating cells toward an antitumoral/proinflammatory phenotype.
Supplemental material
Next, to explore the mechanisms that account for the therapeutic effect, we assessed whether anti-MICA pAb generated with BLS-MICA can promote ADCC, and its effect on tumor growth (figure 6). We observed that these pAb indeed triggered ADCC in vitro (figure 6A). Moreover, less MICA-expressing tumor cells than control tumor cells were recovered after 72 hours of in vivo growth if they were preincubated with anti-MICA pAb. In addition, similar MICA-expressing tumor cells and control tumor cells were recovered after 72 hours of in vivo growth if they were preincubated with Ctrl pAb (figure 6B). This effect resulted in a strong inhibition of the tumor growth as we observed that MICA-expressing tumor cells preincubated with anti-MICA pAb displayed a very slow tumor growth compared with MICA-expressing tumor cells pre-incubated with Ctrl pAb and to control tumor cells preincubated with control or anti-MICA pAb before injection (figure 6C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Anti-MICA pAb passively administered to tumor-bearing mice promote ADCC. (A) In vitro ADCC of mouse NK cells against MB49-pMSCV or MB49-MICA*008 cells preincubated with control pAb (Ctrl Ab) or anti-MICA pAb (anti-MICA) assessed at 2 E:T ratios (indicated in the x-axis). YAC-1 cells were used as positive control of NK cell-mediated cytotoxicity. (B) In vivo ADCC against MB49-pMSCV (yellow dots) or MB49-MICA*008 tumor cells (green dots) preincubated with control pAb (Ctrl Ab) or anti-MICA pAb (anti-MICA). The relative abundance of CFSE+ and eFluorDye670+ cells (each tumor cell) recovered after 72 hours of subcutaneous challenge of naive mice with tumor cells preincubated with control or anti-MICA pAb was analyzed by FC. An unpaired two-tailed Student’s t-test was used. ****p<0.0001. (C) Tumor growth of tumor cells preincubated with control or anti-MICA pAb injected in naive mice. MB49-pMSCV (blue and green dots) or MB49-MICA*008 tumor cells (yellow and red dots) were incubated in vitro with control pAb (Ctrl Ab, blue and yellow dots) or anti-MICA Ab (green and red dots), injected in naive mice and tumor growth was assessed. Each dot in B and C correspond to one mouse. n=4 per group (B, C). Data shown correspond to one representative experiment which was performed twice with similar results. ****p<0.0001, two-way ANOVA with Sidak’s posthoc test. Ab, antibody; ADCC, antibody-dependent cell-mediated cytotoxicity; ANOVA, analysis of variance; CFSE, carboxyfluorescein succinimidyl ester; Ctrl Ab, control Ab; FC, flow cytometry; MFI, median fluorescence intensity; MICA, MHC class I chain-related protein A; pAb, polyclonal Ab.
Overall, these results indicate that immunization with BLS-MICA generates a protective effect against MICA-expressing tumors that involves the generation of anti-MICA pAb, scavenging of sMICA and stimulation of ADCC.
Discussion
During the last years, MICA emerged as an attractive target for Ab-mediated immunotherapy strategies in patients with cancer due to its overexpression in a wide variety of human tumors. The possibility of MICA being a target for Ab in human patients was first demonstrated in individuals with solid transplant rejection.26 Later, a significant correlation between anti-MICA Ab and graft loss was described.29 In a tumor setting, it has been demonstrated that anti-MICA Ab spontaneously arise in patients treated with an anti-CTLA4 mAb and vaccinated with lethally irradiated, autologous tumor cells engineered to secrete granulocyte–macrophage colony-stimulating factor. Moreover, such spontaneously induced anti-MICA Ab promoted scavenging of sMICA and interfered with tumor-immune escape, resulting in a recovery of NK and CD8+ T cell-mediated cytotoxicity and a better course of the disease.12 13 These results highlight the therapeutic potential of anti-MICA Ab for patients with cancer. Accordingly, some mAb against MICA that demonstrated therapeutic efficacy against tumors in mouse models through the promotion of mAb-mediated neutralization of sMICA14 or inhibition of MICA/B shedding and stabilization of cell surface expression of MICA/B15 have been developed.
In this work, we hypothesized that active induction of anti-MICA pAb could represent a novel opportunity for some unmet medical needs. On one hand, used as primary treatment, alone or combined with other strategies, such pAb may reinstate antitumor immunity in patients with tumors that express MICA. But on the other hand, an active induction of anti-MICA pAb may represent a novel prophylactic approach useful for patients with cancer as adjuvant therapy after surgical excision, chemotherapy or radiotherapy of the primary tumor in order to reduce the risk of tumor recurrence of the primary tumor and/or development of metastases. To generate an immunogen useful to induce anti-MICA pAb, we used an established platform suitable to produce highly immunogenic chimeric proteins based on the use of BLS, a lumazine synthase from Brucella spp.17–19 We generated a chimeric protein in which we coupled the ectodomain of MICA to BLS. This chimeric protein was highly immunogenic in mice and elicited high titers of anti-MICA pAb, even without the use of additional adjuvants, and recognized MICA naturally expressed on the cell surface of human tumor cells and on mouse tumor cells engineered to express MICA. Prophylactic active immunization with BLS-MICA was sufficient to significantly delay the growth of MICA-expressing tumors in part due to the ability to promote scavenging of sMICA from mouse sera. Moreover, there was a linear association between sMICA levels and tumor size. These results support our working hypothesis and suggest that BLS-MICA might function as an antitumor vaccine. Remarkably, we used one allele to produce the chimeric protein (MICA*001) and another allele for the transduction of the cell lines (MICA*008) in order to develop a strategy that is independent of allele-specific pAb. In this way, we mitigated the risk of eliciting antitumor immunity that is mediated by allele-specific pAb and generated pAb with the chimeric protein that can trigger antitumor immunity even against tumors that express another allele (MICA*008) which, on the other hand, is the most frequent allele in the population.
To further explore the mechanism underlying this antitumor effect and using a passive immunization approach, we demonstrated that such effect was mediated by anti-MICA pAb induced by the chimeric protein. This robust antitumor effect was observed with two different mouse tumors (EL4 lymphoma and MB49 bladder carcinoma) engineered to express MICA*008, with no effects on control tumor cells that do not express this NKG2DL, suggesting that the approach of using anti-MICA pAb as immunotherapy might be a strategy applicable to tumors of different histotypes. This is reinforced by the bioinformatic analysis performed here which showed MICA overexpression in a variety of tumors, including those that exhibit high number of deaths. Of note, in some animals challenged with MICA-expressing tumors and treated with anti-MICA pAb, we observed complete tumor regression. We also established that the anti-MICA pAb elicited with BLS-MICA tilted the balance of tumor-infiltrating cells toward an antitumoral/proinflammatory phenotype characterized by an increased expression of markers associated with proinflammatory macrophages in intratumoral macrophages, and an increased percentage of antigen-experienced CD8+ T cells. The underlying mechanisms for this therapeutic efficacy include the stimulation of ADCC by the anti-MICA pAb that resulted in a delayed tumor growth, as we demonstrated in vitro and in vivo.
Therefore, the use of BLS-MICA as vaccine to induce therapeutic anti-MICA pAb constitutes a ‘two-in-one’ strategy as such pAb may promote tumor elimination through ADCC and simultaneously interfere with a tumor-immune escape mechanism through scavenging of sMICA. Essentially, this is because the ectodomain of MICA that is the target for ADCC-mediated therapeutic effects is almost identical to the soluble form of MICA. Actually, it is possible to target MICA with pAb or mAb. In this work, we explored the strategy of eliciting pAb against this NKG2DL using an immunogenic chimeric protein. However, anti-MICA mAb also are showing promising results.14 15 Regardless of the strategy (mAb or pAb), it is convenient that Ab that target MICA-expressing tumors should trigger ADCC and scavenging of sMICA, as such Ab will likely display therapeutic activity in patients with tumors that express MICA on their cell surface, tumors that shed significant amounts of sMICA or both, and such therapeutic efficacy in last instance will depend on the relative amounts of cell surface versus soluble MICA expressed by any particular tumor type. Moreover, as ADCC and scavenging of immune complexes depend on FcR, Ab with FcR-binding activity will promote efficient tumor regression through one or both mechanisms. In this regard, a dimorphism in MICA (position 129) is associated with significantly higher amounts of sMICA and progression of multiple myeloma,30 highlighting the relevance of targeting both, membrane and soluble MICA with Ab as immunotherapy. It is important to mention that shed MICA has been also reported to be associated with exosomes instead of soluble molecule released by metalloproteases, and that such exosome-associated MICA also promotes immune suppression.31 Besides, anti-MICA Ab (either pAb or mAb), probably can also target this form of MICA and contribute to the restoration of tumor immunity. Also, as some chemotherapeutic drugs have been shown to stimulate MICA shedding,32 strategies that combine chemotherapeutic regimes with anti-MICA Ab appear as promising approaches.
At this point, we can imagine some limitations of our approach. The first one is that we immunized mice with a human protein (MICA). The high titer of anti-MICA Ab that we generated might be in part because in this setting, it is not necessary to break tolerance. Although this introduces a potential limitation in the translation to human patients, the fact that some tumor-bearing patients spontaneously developed anti-MICA Ab12 13 indicates that it is possible to break tolerance to a self, human protein. Other limitations could be antigen down-modulation by the tumors and/or the selection of resistant tumor cells that lost MICA expression (as a consequence of a selective killing of tumor cells that express the target), and the occurrence of off target and side/unwanted effects during clinical trials in human beings. However, regarding the chimeric protein itself, we consider that the use of BLS as scaffold for the immunogen is safe as this protein is naturally expressed by Brucella spp, the bacteria that causes human brucellosis, there are no reports of infected individuals that exhibit infection-triggered autoimmunity and pAb against BLS arise naturally during such infections but their titer decreases after successful treatment of the patients with antibiotics. Therefore, anti-BLS pAb do not seem to cause unwanted effects in human patients and BLS-MICA appears as a promising approach to break tolerance against MICA in human patients.
We believe that vaccination with BLS-MICA may constitute a groundbreaking twist in the field of immuno-oncology as this vaccination approach elicits a polyclonal response with only a limited number of doses that could be therapeutically effective in patients bearing a broad range of neoplastic diseases as MICA is widely expressed by different tumors.6 9 33–44 Remarkably, this approach might also be suitable to cover an unmet medical need such as the prophylactic treatment of oncologic patients that had their primary tumor removed by surgery, or after chemotherapy or radiotherapy, as the presence of prophylactic anti-MICA pAb may reduce the risk of tumor recurrence and/or development of metastases. Passive treatment with pAb against MICA elicited with BLS-MICA is an unlikely option for the near future due to the complication in their obtention after vaccination with BLS-MICA. However, considering the recent development of mAb against MICA for passive treatment,14 15 we consider a promising approach the active immunization (vaccination) of tumor-bearing patients with BLS-MICA (use of BLS-MICA as primary treatment option) and/or the vaccination of patients whose primary tumor was removed by surgery or treated with chemotherapy or immunotherapeutic agents (use of BLS-MICA as secondary treatment option to induce anti-MICA pAb that may prevent or delay recurrence and/or metastasis). In addition, combination of these approaches (chemotherapy or immunotherapy and active immunization with BLS-MICA, either simultaneous or sequentially), especially if the chemotherapeutic agent promotes MICA/B upregulation as has been described for some drugs45 also emerge as promising future therapeutic strategies.
Conclusions
As demonstrated in this work, we developed a chimeric, highly immunogenic protein consisting of MICA fused to BLS that was effective as tumor vaccine through the induction of anti-MICA pAb. The immunization with this chimeric protein delayed the growth of MICA-expressing tumors, promoted scavenging of sMICA, induced ADCC against MICA-expressing tumor cells and led to a reprogramming of the tumor microenvironment toward an antitumoral/proinflammatory phenotype. Therefore, this chimeric protein constitutes an innovative antitumor vaccine of potential application in patients with cancer who bear tumors that express MICA that might be used as monotherapy or as combination therapy for therapeutic or prophylactic purposes.
Acknowledgments
We would like to thank to Dr Alessandra Zingoni and Dr Angela Santoni from the Laboratory of Molecular Immunology and Immunopathology, Department of Molecular Medicine, Sapienza University of Rome, Italy for providing vectors for transduction of mouse cell lines, to Dr Rosana Felice from GSK Argentina for her help during this project, and to Dr Gabriel A Rabinovich for providing unlimited access to the FACSCanto flow cytometer.
References
Footnotes
Contributors NT performed and designed most of the experiments and analyzed the data. MVR, FS, ADF, RGS, XLRI, SYN, JMS, AZ and MCS contributed experimentally to the data presented in figures 3–6 and supplementary figures. NG contributed with the in vivo experiments of figures 4 and 5. EA, CL and RP contributed to the production of the chimeric protein. CID, MBF, KPM, KWH and ISG contributed with critical discussions and design of experiments. VZ and FAG contributed to the design of the chimeric protein. NWZ conceived, designed and supervised the study and wrote the manuscript. All the authors reviewed the manuscript.
Funding This work was supported by grants from the National Agency for Promotion of Science and Technology from Argentina (ANPCYT), the National Research Council of Argentina (CONICET), the Trust in Science Program from GlaxoSmithKline (GSK), the International CentreCenter for Genetic Engineering and Technology (ICGEB, Trieste, Italy) and the University of Buenos Aires (UBA), all to NWZ. We also thank Fundación Williams and Fundación René Barón for providing financial assistance (donations) to our laboratory.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All studies with mice have been approved by the institutional review committee for animal care.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. Data presented in figure 1 are available in a public, open access repository. Data presented in the manuscript were generated locally and included in the article.