Article Text

Abstract
Background Developing novel strategies to overcome the immunosuppressive tumor microenvironment is a critically important area of cancer therapy research. Here, we assess the therapeutic potential of CD244 (2B4/signaling lymphocyte activation molecule family 4), an immunoregulatory receptor found on a variety of immune cells, including exhausted CD8+ T cells, dendritic cells (DCs), and myeloid-derived suppressor cells (MDSCs).
Methods Using de-identified human tumor and blood samples from patients with head and neck squamous cell carcinoma (HNSCC) and HNSCC models in WT and CD244-/- mice, we assessed the therapeutic potential of CD244 using flow cytometry, RT-PCR, Luminex immunoassays and histopathological analyses.
Results Compared with healthy tissues, tumor infiltrating CD8+ T cells from HNSCC patients and a HNSCC mouse model showed significant increased expression of CD244 expression that correlated with PD1 expression. Moreover, CD244 was increased on intratumoral DC and MDSC and high CD244 expression correlated with PD-L1 expression and increased spontaneous expression of immune-suppressive mediators. In addition, CD244 activation inhibited production of proinflammatory cytokines in human DC in vitro. Importantly, CD244-/- mice showed significantly impaired tumor growth of HNSCC and interventional treatment of WT mice with anti-CD244 monoclonal antibody significantly impaired the growth of established HNSCC tumors and increased tumor-infiltrating CD8+ T cells.
Conclusions Together these data suggest that CD244 contributes to the overall immune-suppressive environment and therefore has potential as a new immunotherapy target in the treatment of malignancies.
- immunology
- oncology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Cancers facilitate their growth in part by creating an immunosuppressive tumor microenvironment through the recruitment of suppressive cell types and the promotion of immune cell exhaustion. Several immune cell types have been implicated in the creation and maintenance of the immunosuppressive tumor microenvironment, including regulatory T cells, subsets of dendritic cells (DCs) and myeloid derived suppressor cells (MDSCs), and the presence of such cells is associated with negative clinical outcomes. In addition, exhaustion of effector cells, such as CD8+ T cells, represents a significant factor in the maintenance of the immunosuppressive tumor microenvironment. Exhausted and suppressive immune cells coexpress a variety of immunomodulatory receptors implicated in the initiation and maintenance of the exhausted phenotype. Recently, elucidation of the molecular mechanisms underpinning certain of these receptors on CD8+T cells, such as PD-1 and CTLA-4, has led to the development of clinical checkpoint inhibitors, which provide an effective strategy to ameliorate immune exhaustion1 2 These checkpoint inhibitors have improved patient outcomes in a multitude of solid tumors and hematological malignancies. However, treatment responses remain suboptimal for many patients, demonstrating a need for additional immunotherapy targets.
CD244 (2B4) is an immunomodulatory transmembrane receptor in the signaling lymphocyte activation molecule family that offers a potential target for immunotherapy. CD244 is restricted to hematopoietic cells, including natural killer (NK) cells, a subset of CD8+ αβ T cells, DC and MDSCs.3–8 CD244 binds CD48, a transmembrane receptor ubiquitously expressed on hematopoietic cells.9–11 Its cytoplasmic domain includes four immunoreceptor tyrosine-based switch motifs that interact with at least five distinct adaptor molecules: SAP, EAT-2, SHP1, SHP2 and SHIP-1. It has been shown that intracellular binding of SAP propagates an activating signal, while binding of the SH2 phosphatases propagates an inhibitory signal; binding EAT-2 has been shown to propagate both inhibitory and activating signals.12–14 Factors that determine whether CD244 propagates an activating or inhibitory signal include adaptor molecule expression levels, availability and competitive binding (reviewed in ref. 15). Increased CD244 cell surface density and decreased functional SAP levels have been associated with inhibitory signaling in CD8+ T cells and NK cells.16 17 Inhibitory CD244 signaling has been demonstrated in human cancers and mouse models, and recent research has linked CD244 inhibitory signaling to an exhausted phenotype in CD8+ T cells in chronic infection and cancer.18–28
In the myeloid compartment of the tumor microenvironment, CD244 signaling has been implicated in immunosuppressive phenotypes of both DCs and MDSCs. In DCs, CD244 signaling appears to diminish T cell priming and NK cell activation.4 In MDSCs, CD244 expression correlates with suppression of antigen-specific CD8+ T cells.3 Intratumoral MDSCs are associated with tumor progression, treatment resistance and poor prognosis, yet therapeutic strategies to counter their effects are limited.29–40
In the current study, we explore the expression levels of CD244 in CD8+ T cells, DCs and MDSCs in head and neck squamous cell carcinoma (HNSCC) in mice and humans, and the correlation between these levels and immunosuppressive pathways in each cell type. Additionally, we explore the therapeutic potential of CD244 inhibition in vivo. For these experiments, we use fresh biopsy samples from human (HNSCC) patients and a syngeneic mouse model of HNSCC (MEER).41 We demonstrate that CD244 expression increases in intratumoral CD8+ T cells, MHC II+ CD11c+ DCs and monocytic MDSCs (Mo-MDSCs) compared with peripheral counterparts, and correlates with PD-1 or PD-L1 expression. In addition, increased CD244 expression corresponds to increases in molecules associated with immunosuppressive function in human and mouse intratumoral immune cells. Finally, growth of syngeneic tumors in CD244-/- mice is significantly inhibited compared with WT mice, and most importantly, therapeutic intervention with monoclonal CD244 antibody inhibits the growth of established tumors. These results support the further investigation of CD244 as a potential new immunotherapy target.
Materials and methods
Cell lines, reagents and antibodies
A previously described mouse MEER tumor cell line created by stably transforming mouse oropharyngeal epithelial cells with HPV16 E6 and E7(41) was obtained from Dr. John Morris at University of Cincinnati. All antibodies, ef780 viability dye and brefeldin A were purchased from eBioscience, Biolegend or BD Pharmigen. Surface antibodies included CD45.2 (A20), CD3 (145–2 C11), CD4 (GK1.5), CD8a (53–6.7), NK1.1 (PK136), NKp46 (29A1.4), CD69 (H1.2F3), PD-1 (J43), CD244.2 (m2B4), CD11b (M1/70), CD11c (N148), MHC class II (M5/114.15.2), Ly6G (1A8), Ly6C (HK1.4) and PD-L1 (10F.9G2). Intracellular antibodies included antimouse antibodies to arginase-1 (Met1-Lys322), indolamine-2,3-dioxygenase (2E2/IDO1), interleukin-10 (IL-10) (JES5-16E3), latency-associated peptide (LAP, TW7-16B4) as a marker for transforming growth factor-β1 (TGFβ1), tumor necrosis factor α (TNFα) (MP6-XT22) and IFNγ (XMG1.2). The Invitrogen eBioscience Foxp3/Transcription Factor Staining Buffer Set was used for all intracellular staining. For in vivo experiments, mouse anti-CD244.2 (eBio244F4) monoclonal antibody (mAb) was purchased from ThermoFisher, and IgG2b antibody isotype control (MPC-11) was purchased from BioXcell. Complete IMDM medium (for murine cells) or RPMI 1640 medium (for human cells) was enriched with 10% fetal bovine serum, 2% L-glutamine, 2% penicillin/streptomycin and 50 µM β-mercaptoethanol for all cell-based assays. For freezing human peripheral blood mononuclear cells (PBMCs), Cryostor Cs10 medium was purchased from Stemcell Technologies.
Human blood, spleen and tumor samples
Peripheral blood was drawn from healthy donors and HNSCC patients who provided informed consent. PBMCs were isolated by density gradient centrifugation using Ficoll-Paque and suspended in Cryostor Cs10 before freezing in liquid nitrogen as described in Mallone et al. 42 Fresh tumor tissue was obtained from untreated HNSCC patients who were undergoing tumor resection for clinical indications. The tissue was dissociated using Miltenyi Biotec gentleMACS Tissue Dissociators per manufacturer’s instructions. Tumor cells were immediately stained for flow cytometry. Tumor-associated PBMCs were thawed, stained for flow cytometry and analyzed in groups. Designated healthy donor PBMC controls were taken along with every batch of fresh tumor cells and frozen tumor-associated PBMCs to allow for interexperimental comparison. For human DC assays, deidentified blood samples from healthy donors were obtained from Hoxworth Blood Center. PBMCs were isolated and CD1c (BDCA-1)+ myeloid DCs were isolated using Miltenyi Biotech CD1c (BDCA-1)+ DC Isolation Kit. For human splenic DC, splenic samples were collected through LifeCenter, the major organ procurement center in Cincinnati, at the time organs were harvested for transplantation. Samples identified as being from donors without confounding factors (cancer, or metabolic, infectious, or autoimmune diseases) were processed within a few hours of collection. Spleens were manually disrupted, filtered through a 70 μm strainer, and RBCs were lysed. BDCA1+ DC were then sorted as above. The use of human cells and tissues from patients and healthy donors was approved by the University of Cincinnati Institutional Review Board (IRB# 2014–4755) except for collection of cadaveric spleens, which was exempted on the basis of deidentified samples, and all protocols followed Good Clinical Practice.
Mice
C57BL/6 mice were purchased from The Jackson Laboratory and CD244-/- mice (C57BL/6 background) were a generous gift from Andre Veillette’s laboratory; both strains were bred in-house. Age-matched male mice 5–12 weeks of age were used in all experiments as the MEER model was developed in male C57BL/6 mice and grows more consistently in males. All the animal experiments and handling procedures were approved by the Cincinnati Children’s Institutional Animal Care and Use Committee and performed according to the guidelines. In conducting research using animals, the investigators adhere to the laws of the US and regulations of the Department of Agriculture.
Tumor growth experiments
The MEER model is a syngeneic murine model of HPV-driven HNSCC that expresses oncogenic proteins E6 and E7.41 Tumor cells were injected subcutaneously in mouse flanks bilaterally with MEER cells at 2–3×106 cells per 200 µL PBS using 18-gauge needles. Tumors were measured 2–3 times per week until maximum allowable size was reached (1.7 cm diameter). Tumor volume was calculated using the equation volume=0.5 (length x width2), where length was the longest diameter and width was perpendicular. Mice were euthanized and tumors and spleens were immediately dissected for analysis. For interventional experiments involving in vivo antibody injection, anti-CD244 antibody or isotype antibody (both dosed 50 µg antibody in 200 µL PBS) was injected intravenously via tail vein three times a week for six doses starting after tumors had become measurable (day 9).
Flow cytometry
Single cell suspensions were produced from mouse tumors and spleens by manual dissociation through a 70 μm filter. For experiments measuring production of intracellular molecules in DCs and MDSCs, cells were incubated with brefeldin A at 37°C and 5% CO2 for 5 hours in the absence of any stimulatory agents prior to staining to measure spontaneous production of cytokines/mediators. For experiments measuring production of proinflammatory cytokines, CD8+ T cells were incubated with Brefeldin A at 37°C and 5% CO2 for 5 hours with or without plated CD3 (145–2 C11) antibody. Cells were stained with viability dye and surface antibodies. Intracellular staining was accomplished using eBioscience Foxp3/Transcription Factor Staining Buffer Set according to manufacturer directions. CD244 expression was determined using cells from CD244-/- mice stained in parallel as a negative reference (see online supplementary figure 1). Data were collected using a BD LSRFortessa flow cytometer and analyzed using FlowJo v10. For expression of CD244 in DC subsets, mouse DCs were sorted by Mo-Flo flow cytometry flow sorter into CD8+ DC (live, CD19-, CD3-, NKp46-, CD11c+ MHCII+ CD8+ CD11b-), CD11b+ DC (live, CD19-, CD3-, NKp46-, CD11c+ MHC II+, CD8-, CD11b+) and plasmacytoid DCs (live, CD19-, CD3-, NKp46-, CD11c+, MHC II+, CD8-, CD11b-). Human DCs were sorted into BDCA1+ DC (live, CD19-, CD3-, NKp46-, CD11c+ MHCII+, BDCA3-, BDCA1+) BDCA3+ DC (live, CD19-, CD3-, NKp46-, CD11c+ MHCII+, BDCA3+, BDCA1-), and plasmacytoid DCs (live, CD19-, CD3-, NKp46-, CD11c+, MHCII+, BDCA3-, BDCA1-).
Supplemental material
Quantification of cytokine production by human DCs
Flat-bottom 96-well plates were prepared for CD244 crosslinking by coating wells with either RPMI 1640 media alone or antihuman CD244 (C1.7) purified mAb (0.5 mg/mL) and incubating overnight at 4°C. BDCA-1+ DCs, isolated as described above, were resuspended in enriched RPMI 1640 media and washed to remove antibody and isolation buffer. The cells were then plated 1×105 cells/well in the absence or presence of lipopolysaccharide (LPS) from Escherichia coli, Serotype R515 (Re) (Enzo Life Sciences) (100 ng/mL final concentration). The cells were incubated for 24 hours at 37°C and 5% CO2. Following incubation, supernatants were analyzed by Luminex high throughput multiplexing suspension array system per manufacturer’s instructions.
RNA quantification of CD244 in human and mouse DCs
RNA was isolated from healthy human peripheral DCs, human splenic DCs and mouse splenic DCs using Trizol and chloroform, and precipitated using isopropanol. Generation of cDNA was accomplished using a ThermoFisher High Capacity cDNA Reverse Transcription Kit per manufacturer’s instructions. PCR reactions were catalyzed using LightCycler 480 SYBR Green I Master mix. For human DCs, H2B4-A-373+1F (GTATTTGATAAAGTTGAGAAACCCCG) and H2B4−558+1R (GCAGGTATATGTGTGAGTGCCATTA) primers were used. Measurements were normalized against a ubiquitin (7322) housekeeping protein (primers 5- CTACGATCACAGTGGTCTCCAG and 3- CGAGCAATCTCAGGCACTAAAGG). For mouse DCs, 2B4 (1080) 5-GTTGCCACAGCAGACTTTC and 2B4 (1250) 3-TTCCAAC CTCCTCGTACACGGTAC primers were used. Measurements were normalised against an L32 housekeeping protein (primers mL32F: TTAAGCGAAACTGGCGGAACC, mL32R: TTGTTGCTCCCATAACCGATG). Quantifications were done using a LightCycler 480 high- performance, medium-to high-throughput PCR platform.
Microscopy
Mouse tumors were fixed in 4% PFA for 2 hours at room temperature or overnight at 4°C, then embedded in OCT compound on dry ice and stored at −80°C until microtomy. Cryostat sections 5 µm thick from the midpoint of each tumor were affixed to glass slides. H&E staining was performed using a well-established protocol.43 Digital microscopy images were obtained using Leica Aperio ImageScope, V.12.4.0.7018. Using the same software, the geographical necrosis of each tumor was outlined and areas of the total tumor and the geographical necrosis were calculated. Percentage area necrosis was calculated using Microsoft Excel.
Statistics
All experiments were done with biological replicates greater than or equal to four. Data are presented as proportions (percentages) or as the mean±SEM, as indicated in the figure legends. Statistics were performed using paired and unpaired Student’s t-tests as appropriate for each set of experimental conditions using GraphPad Prism V.8, and p<0.05 was considered significant.
Results
Tumor growth is impaired in CD244-/- mice
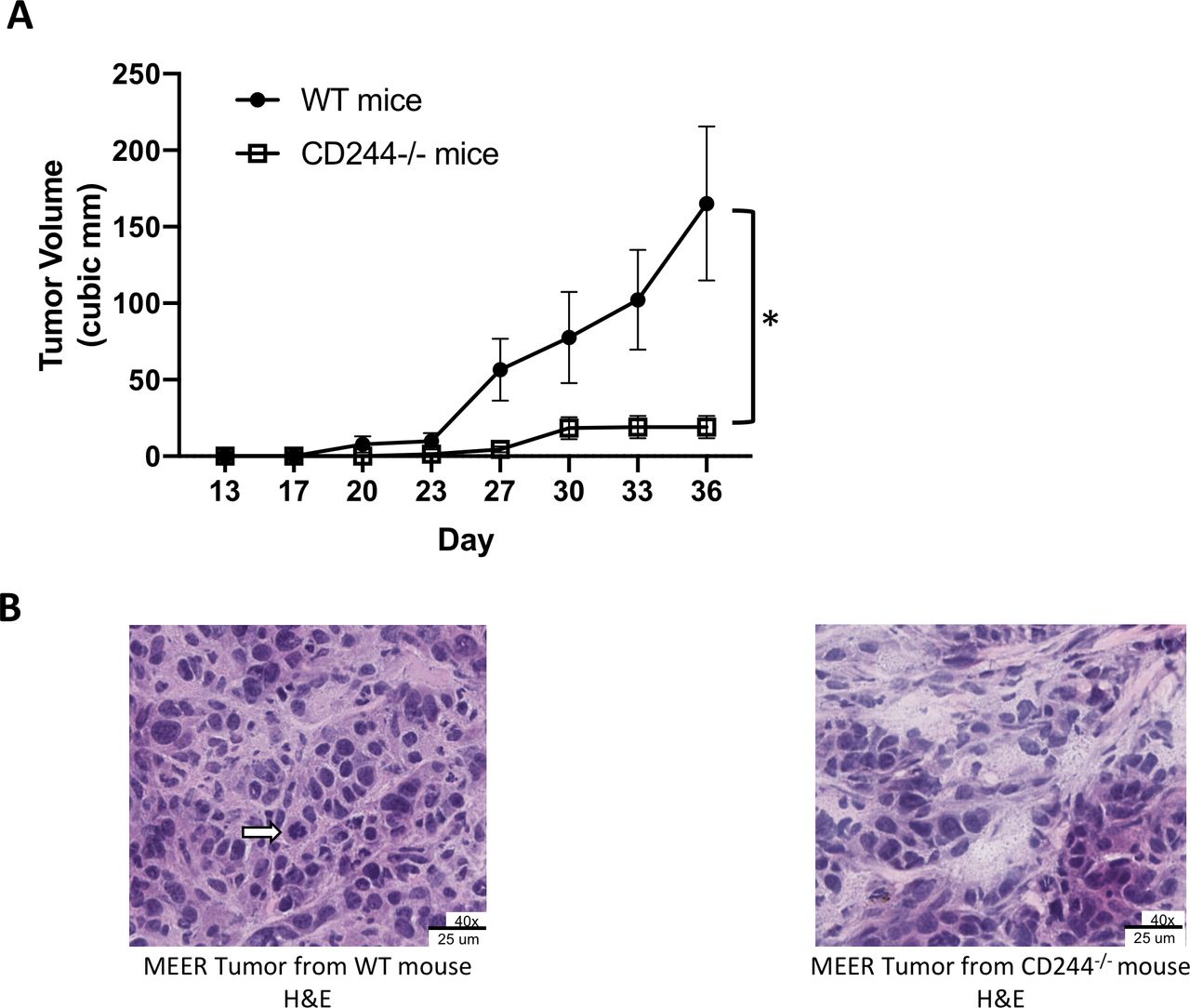
To evaluate whether CD244 expression on host immune cells affects tumor growth, we compared the growth of MEER tumors in CD244-/- and WT mice. Using syngeneic MEER tumors grown subcutaneously in WT C57BL/6 and CD244-/- C57BL/6 mice, we observed in repeated experiments that spontaneous tumor growth in the CD244-/- mice was significantly impaired compared with growth in the WT mice (figure 1A and online supplementary figure 2). We proceeded to investigate the immunological mechanisms underlying the significant difference in tumor growth. Since CD244 expression is restricted to immune cells, we first examined tumor morphology and infiltration of T cells by microscopy. H&E staining of MEER tumors from both WT and CD244-/- mice showed a fundamentally patternless cellular arrangement with delicate stroma in the background. Hyperchromatic round tumor nuclei in were in the moderate size range (frequently two to fourfold), occasionally molding, with a small to moderate amount of cytoplasm. Occasional mitoses and karyorrhexis were noted. As expected due to relative size, the larger tumors from the WT mice showed greater percentages of area necrosis in a geographical (ischemic) distribution than the tumors from the CD244-/- mice (data not shown). Intriguingly, tumors from CD244-/- mice demonstrated viable portions similar to those from WT mice, but with focal to spotty degenerative features, tumor cell dropout, and rare small acellular devitalized foci, as shown here (figure 1B). The profound effect of CD244 absence warrants further study into the biology of CD244 in immune cells of the tumor microenvironment.
Supplemental material

Tumor growth is inhibited in CD244-/- mice compared with WT mice. (A) WT (n=7) and CD244-/- (n=6) male mice received 2×106 MEER cells s.c. in each flank. Tumors were measured twice weekly until maximum allowable size was reached. Significant (*) (p=0.0137) inhibition of tumor growth was seen in the CD244-/- mice (unpaired Student’s t-test). The experiment was repeated three times with consistent results. Data are expressed as mean±SEM. (B) H&E staining of MEER tumors grown in WT and CD244-/- mice (40x magnification). Tumor from the WT mice demonstrated a patternless cellular arrangement with delicate stroma in the background. Occasional mitoses (arrow) and karyorrhexis were noted. Tumors from the CD244-/- mice demonstrated viable portions fundamentally similar to those from WT mice, but with focal to spotty degenerative features with tumor cell drop out.
CD244 expression is increased in intratumoral CD8+ T cells in mice and humans and correlates with PD-1 expression
Induction of CD8+ T cell exhaustion is a well-established strategy by which cancers escape immune surveillance and killing. Tumor-associated exhausted CD8+ T cells are known to coexpress multiple inhibitory receptors. We investigated patterns of CD244 expression on CD8+ T cells in MEER tumor-bearing mice and HNSCC patients using multichannel flow cytometry and compared the expression of CD244 to the well-established immune checkpoint receptor PD-1 (see online supplementary figure 3 for gating strategy). In the spleens of tumor-bearing WT mice, nearly all of the CD8+ T cells were negative for CD244 and PD-1. In contrast, approximately half CD8+ T cells in the tumor were double positive for CD244+ PD-1+ (figure 2A). The concomitant increase in CD244 and PD-1 expression on tumor-infiltrating CD8+ T cells was seen early, on day 12 after tumor inoculation, persisted through the end of the experiment, and was independent of tumor size. The increase in CD244+ PD-1+ co-expression in CD8+ tumor-infiltrating lymphocytes (TIL) compared with splenic CD8+ T cells was significant at both time points (p=0.0049 and p=0.0007, respectively).
Supplemental material

CD244 expression in mice and humans increases in CD8+ tumor-infiltrating lymphocytes and correlates with PD-1 expression. MEER tumor cells were injected subcutaneously into the flanks of WT mice. Spleens and tumors were harvested on indicated days and analyzed by flow cytometry. (A) Data shown are gated on live CD45+CD8+ cells. Representative flow data and means with SD are shown. (B) Fresh human HNSCC tumor samples were stained for flow cytometry along with healthy donor PBMC controls. CD244 and PD-1 expression is significantly increased on intratumoral CD8+ T cells compared with healthy PBMCs and compared with HNSCC patient PBMCs. HNSCC patient PBMCs were frozen and stained separately. Graphs show means with standard errors and individual samples; p values were calculated using unpaired Student’ t-tests. HNSCC, neck squamous cell carcinoma; PBMC, peripheral blood mononuclear cell.
Using PBMCs from healthy donors and HNSCC patients, and fresh, untreated human HNSCC tumor samples, we compared the expression of CD244 and PD-1 on CD8+ T cells from each source. We found that the proportion of CD244+ PD-1+ CD8+ T cells was significantly lower in the PBMCs of healthy donors than in the PBMCs of HNSCC patients (p=0.0356) and, furthermore, that the proportion of CD244+ PD-1+ CD8+ T cells in the PBMCs of HNSCC patients was lower than in the HNSCC tumors themselves (p=0.0410) (figure 2B). Thus, the frequency of CD244 expressing CD8+ T cells increases in the peripheral blood of HNSCC patients, but is greatest in the CD8+ T cells within the tumor itself. The significant increase in CD244 expression on CD8+ TILs concomitant with increased PD-1 expression in human HNSCC and mouse MEER tumors implicates CD244 signaling in CD8+ TIL exhaustion. Interestingly, across mouse and human tumor samples, the dominant proportion of tumor infiltrating CD8+ T cells were almost always CD244+ PD-1+, followed by the proportion of PD1+ CD244- CD8+ TILs. However, almost all CD244+ CD8+ TILs coexpressed PD-1. These trends suggest that PD-1 expression and CD244 expression may be induced by different environmental triggers and affect CD8+ TILs in different ways, warranting further study.
Increased CD244 in intratumoral MHC II+ CD11c+ DCs correlates with PD-L1 expression and corresponds to increased expression of suppressor molecules in vitro
CD244 expression on CD11c+ DCs in mice has been previously described.44 DCs play a crucial role in the induction and maintenance of antitumor immunity by cross-presenting tumor antigens and cross-priming tumor-specific T cells. However, recent studies have demonstrated that DCs can play an activating or inhibitory role in the tumor microenvironment depending on DC subset, maturation status and presence of (co)stimulatory and inhibitory receptors and cytokines.45 Using RT-PCR and flow cytometry, we first evaluated whether CD244 expression is comparable between mouse and human DC subsets. We measured levels of CD244 mRNA and CD244 protein within DC subsets from naïve mouse spleens, healthy human peripheral blood and cadaveric human spleens (without history of cancer). For both mRNA levels and protein expression, CD244 demonstrated similar patterns of expression between human and mice in each subset, with highest expression on conventional DC2 (cDC2) populations (CD11b+ DCs in mice and BDCA1+ DCs in humans), intermediate on cDC1 populations (CD8+ DC in mice and BDCA3+ DC in humans), and low to intermediate on plasmacytoid DCs (figure 3A).

CD244 expression is increased in intratumoral DCs and corresponds to increased production of suppressor molecules. (A) DCs were sorted from human PBMCs, human splenocytes and mouse splenocytes. CD244 mRNA levels were measured by RT-PCR and expressed in relative units (RUs). In parallel, CD244 expression was assessed by flow cytometry. By both measures, CD244 expression shows similar patterns between mouse and human in each subset. (B) Separately, 7 MEER tumors and spleens grown in WT mice were dissociated, incubated with GolgiPlug for 5 hours at 37°C 5% CO2 and stained for flow cytometry. CD244 versus PD-L1 expression on live CD45+CD11c+MHCII+ DCs in tumor and spleen is shown as representative flow shows and bar graphs (means
±
SEM). (C) Bar graphs (means
±
SEM) show MFI of 4 suppressor molecules spontaneously produced by intratumoral CD11c+ MHCII+ DCs in vitro. DCs with high CD244 expression spontaneously produced more arginase-1, IDO, IL-10, and TGF
β
1 (as measured by LAP) compared with DCs with low CD244 expression.(*) denotes significance. (D) DCs with high PD-L1 expression spontaneously produced more IL-10 and TGF
β
1 compared with DCs with low CD244 expression, with no significant difference in arginase-1 and IDO production. (E) CD11c+ DCs were sorted from healthy donor PBMCs (n=3) and stimulated with LPS in the presence or absence of CD244 activation with a cross-linking CD244 monoclonal antibody. Production of cytokines TNF IL-12p70, IL-8 was measured using Luminex. Data were normalized and unpaired Student’s t-tests were performed. DC, dendritic cell; IL-10, interleukin-10; LAP, latency-associated peptide; LPS, lipopolysaccharide; MFI, mean fluorescent intensity; PBMCs, peripheral blood mononuclear cells; TNFα, tumor necrosis factor α.
IL-12p70, IL-8 was measured using Luminex. Data were normalized and unpaired Student’s t-tests were performed. DC, dendritic cell; IL-10, interleukin-10; LAP, latency-associated peptide; LPS, lipopolysaccharide; MFI, mean fluorescent intensity; PBMCs, peripheral blood mononuclear cells; TNFα, tumor necrosis factor α.
Using MEER tumor-bearing WT mice, we analyzed the expression of CD244 in intratumoral MHC II+ CD11c+ DCs (see online supplementary figure 4A and B for gating strategy). In parallel with CD8+ T cells, the level of CD244 expression was higher in intratumoral MHC II+ CD11c+ DCs than in their splenic counterparts, although this trend fell just shy of statistical significance (p=0.0506) (figure 3B). CD244 expression in tumor and splenic DCs using a syngeneic B16-OVA melanoma model showed the same pattern with statistical significance (see online supplementary figure 4C and D) CD244 expression in MHC II+ CD11c+ DCs also correlated with PD-L1 expression in both spleen and tumor; cells with the highest levels of CD244 also expressed the highest levels of PD-L1. We next evaluated the relationship between the level of CD244 expression on intratumoral DCs and spontaneous production of suppressor molecules arginase-1, IDO, IL-10 and TGF β 1 (measured by LAP) using intracellular flow cytometry. For all four suppressor molecules, spontaneous production by MHC II+ CD11c+ DCs in vitro was significantly higher in CD244hi DCs than CD244lo DCs in the tumor (figure 3C).
Supplemental material
Since CD244 expression correlates with PD-L1 expression on DCs, we assessed whether production of these suppressor molecules could be related to PD-L1 rather than CD244. PD-L1hi DCs did produce higher mean fluorescent intensity (MFI) of IL-10 than PD-L1lo DCs (p=0.0467). However, in contrast to the differences seen with the level of CD244 expression, there were no significant differences in the spontaneous production of arginase-1, IDO, or TGF β 1 (LAP) by PD-L1hi vs PD-L1lo DCs (figure 3D), suggesting that increased CD244 expression on intratumoral DCs is associated with increased production of suppressor molecules. As PD-1/PD-L1 interactions have been established in the promotion of suppressive immune cell function in the tumor microenvironment, the association between increased levels of CD244 on DCs and increased spontaneous production of arginase-1, IDO and TGF β 1 not correlated with PD-L1 expression suggests that the mechanism by which CD244 may contribute to an immunosuppressive or tolerogenic phenotype in DCs likely differs from that of PD-1/PD-L1.
CD244 cross-linking decreases proinflammatory cytokine production in healthy human DCs in vitro
We next investigated the functional correlation of CD244 expression and cytokine repertoire in healthy human DCs. BDCA1+ DCs were sorted from healthy human donor PBMCs and stimulated with LPS in the presence or absence of a CD244 cross-linking antibody. After 24 hours, cytokine production was measured with a Luminex assay. CD244 cross-linking significantly inhibited the LPS-stimulated production of several proinflammatory cytokines in human DCs, including TNF α (p=0.0002), IL-12p70 (p=0.0087) and IL-8 (p=0.0001) (figure 3E). Interestingly, production of anti-inflammatory cytokines such as IL-10 was not altered on cross-linking CD244 in healthy peripheral DCs. The inhibitory CD244 signaling in DCs demonstrated by our functional in vitro studies and the increased CD244 expression on tumor-infiltrating DCs in mice and humans suggest that CD244 may also contribute to the immunosuppression of DCs in the tumor microenvironment.
CD244 expression on intratumoral MDSCs corresponds to increased spontaneous production of intracellular suppressor molecules
Increased numbers of MDSCs have been associated with tumor progression and poor prognosis in a wide variety of cancer types. Two morphologically distinct subtypes of MDSCs are found in both mice and humans: Mo-MDSC and granulocytic MDSC (Gr-MDSC). Using flow cytometry, we identified Mo-MDSC (live CD45.2+ CD11b+ Ly6Chi cells) and Gr-MDSC (live CD45.2+ CD11b+ Ly6Cint Ly6G+ cells) populations among tumor-infiltrating immune cells from MEER tumors grown in WT mice (figure 4A). We then examined the levels of CD244 expression on each MDSC population and the relationship between CD244 expression and spontaneous production of suppressor molecules (see online supplementary figure 5 for gating strategy.) Consistently, Mo-MDSC demonstrated higher levels of CD244 expression than Gr-MDSC, and this correlated with higher frequencies of suppressor molecule expression in Mo-MDSC (figure 4B). Quantitatively, CD244 expression was significantly higher among Mo-MDSCs than Gr-MDSCs (p=0.0253), and Mo-MDSC produced significantly more arginase-1 (p=0.0004), IL-10 (p=0.0271) and TGFβ1 (p=0.0003) than Gr-MDSC (figure 4C). The difference in IDO production between the two subsets was not significant (p=0.1623).
Supplemental material

CD244 expression corresponds to increased spontaneous production of intracellular suppressor molecules in intratumoral MDSCs. Seven MEER tumors grown in WT mice were dissociated, incubated with GolgiPlug for 5 hours at 37°C 5% CO2, and stained for flow cytometry. (A) Mo-MDSC were identified as live CD45.2 CD11b+ Ly6Chi cells; Gr-MDSC were identified as live CD45.2 CD11b+ Ly6Cint Ly6G+ cells. (B) Frequencies of CD244hi Mo-MDSC (black line) versus CD244hi Gr-MDSC (grey) are shown, followed by frequencies of intracellular suppressor molecules for arginase, IDO, IL-10, and TGF β 1 (as measured by LAP). (C) Bar graphs show mean fluorescence intensity (MFI) of these four suppressor molecules as expressed as means ± SEM. (*) denotes significance. (D) Mo-MDSC were divided into subsets based on low vs high CD244 expression. High CD244 expression corresponded to increased spontaneous production of arginase-1, IL-10 and TGF β 1 compared with low CD244 expression. (E) Mo-MDSC was divided into subsets based on low versus high PD-L1 expression. Subsets were compared using paired Student’s t-tests. Gr-MDSC,granulocytic MDSC; IL-10, interleukin-10; LAP, latency-associated peptide; MDSCs, myeloid derived suppressor cells; Mo-MDSC, monocytic MDSC; TGFβ, transforming growth factor-β.
We next investigated the relationship between CD244 expression levels on Mo-MDSCs and spontaneous production of these four suppressor molecules. Spontaneous production was significantly higher among the CD244hi Mo-MDSC compared with CD244lo Mo-MDSC for arginase-1 (p=0.0237), IL-10 (p=0.0125) and TGFβ1 (p=0.0252) (figure 4D). IDO production did not correlate with the frequency of CD244 expression in Mo-MDSCs. In contrast to CD244, there was no significant difference in the production of IDO, IL-10 or TGFβ1 between PD-L1hi and PD-L1lo MDSC populations (figure 4E), indicating that the positive correlation between CD244 expression and spontaneous production of IL-10 or TGFβ1 by Mo-MDSCs is not confounded by PD-L1 signaling. Arginase-1 did show a significant increase in PD-L1hi Mo-MDSC compared with PD-L1lo MDSC, consistent with previously published findings in human carcinoma-associated MDSC.46 Taken together, these data associate high CD244 expression with increased immunosuppressive function in intratumoral Mo-MDSCs.
Treatment with anti-CD244 mAb causes inhibited tumor growth and increased proportions of tumor-infiltrating CD8+ T cells
Significantly impaired tumor growth in CD244-/- mice compared with WT, and correlations between CD244 expression and signs of increased immunosuppressive function in CD8+ T cells, DCs, and MDSCs, spurred us to investigate CD244 as a target for immunotherapy. For a therapeutic trial with CD244 mAb, MEER tumors were established in WT mice, which then received intravenous treatments of either CD244 mAb or the corresponding isotype control. Treatments were given three times a week for a total of 6 doses. Tumors were measured throughout the experiment and harvested 3 days after the final dose. CD244 mAb treatment significantly inhibited tumor growth compared with isotype control (p=0.0264) (figure 5A), recapitulating the phenotype seen in MEER tumors grown in CD244-/- mice (figure 1A). The experiment was repeated three times with similar results each time.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Treatment with CD244 monoclonal antibody inhibits tumor growth and increases proportions of tumor-infiltrating CD8+ T cells. WT mice were injected subcutaneously in bilateral flanks with MEER tumor cells (2×106 cells/injection). After tumors became measurable, intravenous injections of either anti-CD244 monoclonal antibody (CD244 mAb) or the corresponding isotype were started on day 10 and administered three times a week for six doses. Tumor growth was measured twice weekly. Tumors were harvested 3 days after the final dose (day 24). (A) Tumor growth curves for isotype group (n=17) and CD244 mAb treatment group (n=20) show significantly impaired tumor growth in the CD244 mAb group. Arrows denote treatment days. (B) Tumors were analysed by flow cytometry. The graph shows a significantly increased proportion of infiltrating CD8+ T cells in the tumors treated with CD244 mAb compared with isotype-treated controls. (C) Tumors treated with isotype or with CD244 mAb were stained with H&E stained. Slides were scanned and image analysis was performed to measure necrotic area relative to total tumor area (mid tumor). Necrosis in all tumors was consistent with ischemic geographical necrosis (not shown). There was no significant difference in per cent area necrosis between tumors treated with isotype versus CD244 mAb. (D) Compared with WT CD8+ TILs, CD244-/- CD8+ TILs encompassed a smaller proportion of PD1+ cells. PD-1 expression intensity (MFI) was similar between PD-1+ CD244-/- CD8+ TILs and PD-1+ WT CD8+ TILs. (E) Spontaneous production of proinflammatory cytokines IFNγ and TNFα by intratumoral CD8+ T cells from untreated CD244-/- mice was increased compared with those from untreated WT mice. Representative flow plots are shown. (F) Graphs show significantly increased percentages of IFNγ-producing intratumoral CD8+ T cells from untreated CD244-/- mice compared with WT, under both unstimulated and CD3-stimulated conditions ex vivo. (G) Graphs show increased percentages of TNFα-producing intratumoral CD8+ T cells from untreated CD244-/- mice compared with WT, under both unstimulated and CD3-stimulated conditions ex vivo. Cohorts were compared using one-tailed Student’s t-tests. IFNγ, interferon-γ; mAb, monoclonal antibody; MFI, mean fluorescent intensity; TIL, tumor-infiltrating lymphocytes; TNFα, tumor necrosis factor α.
Tumors from the treatment experiments were analysed by flow cytometry. Intriguingly, tumors treated with CD244 mAb showed a significantly increased proportion of infiltrating CD8+ T cells compared with isotype treated controls (p=0.0017) (figure 5B). Tumors from each cohort in each experiment were fixed and H&E stained. Tumors from both groups showed the histologic patterns noted in untreated tumors from WT mice, as shown in figure 1B, including areas of central geographic necrosis (data not shown). The percentage area of necrosis was not significantly different between treatment groups, suggesting that the mechanism by which CD244 mAb treatment impairs tumor growth is not related to increased ischemia (figure 5C).
These intriguing findings raised questions about the effect of CD244 signaling on intratumoral CD8+ T cell phenotype and functionality. We evaluated the phenotypic composition of CD8+ TILs in terms of CD244 and PD-1 expression in WT mice. Compared with WT CD8+ TILs, CD244-/- CD8+ TILs encompassed a smaller proportion of PD1+ cells (figure 5D). Notably, PD-1 expression intensity (MFI) was similar between PD-1+ CD244-/- CD8+ TILs and PD-1+ WT CD8+ TILs (figure 5D). We next examined the difference in proinflammatory cytokine production from intratumoral CD8 +T cells grown in CD244-/- versus WT mice using flow cytometry. Tumors were processed into single cell suspensions and incubated for 5 hours in the absence of presence of plate-bound CD3 to assess spontaneous and stimulated cytokine production. After incubation, a significantly higher percentage of CD244-/- CD8+ TILs produced IFNγ than did WT CD8+ TILs spontaneously and with CD3 stimulation (figure 5E and F) without change in PD-1 expression intensity. Additionally, a significantly higher percentage of CD244-/- CD8+ TILs produced TNFα compared with WT CD8+ TILs under both conditions (figure 5E,G; online supplementary figure 6). These findings suggest that CD244 signaling in the tumor microenvironment may contribute to an immunosuppressive phenotype in CD8+ TILs. This in conjunction with the significantly inhibited tumor growth and corresponding significant increase in tumor-infiltrating CD8+ T cells in CD244 mAb treated MEER tumors supports further investigations into CD244-directed immunotherapy.
Supplemental material
Discussion
The observation that syngeneic tumors grown in CD244-/- male mice demonstrate significantly inhibited growth compared with those grown in male WT mice suggests a role for inhibitory CD244 signaling in tumor-associated immunosuppression. Our experiments demonstrate a consistent pattern of increased CD244 expression in intratumoral CD8+ T cells, DCs, and Mo-MDSCs compared with their peripheral counterparts, and a corresponding increase in immunosuppressive function not explained by concomitant increases in PD-1 or PD-L1. Consistent with previous studies,8 we found CD244 to be ubiquitously expressed on NK cells in tumors and spleens. Furthermore, the proportion of NK cells within the intratumoral infiltrate did not vary between MEER tumors grown in WT versus CD244-/- mice, and only a minority (<20%) expressed activation marker CD69 (see online supplementary figure 7). Given the relative preponderance of research into CD244 in NK cells, compared with other cell types important to tumor immunity, we chose to focus our studies on CD244 in CD8+ T cells, DCs and Mo-MDSCs. The significant increase in CD244 expression on intratumoral CD8+ T cells associated with increased PD-1 expression in human HNSCC patients and tumor-bearing mice is consistent with previous studies implicating CD244 in the development of CD8+ T cell exhaustion in chronic viral illness23 24 and in cancers such as melanoma and multiple myeloma.26 27
Supplemental material
Within human and mouse tumors, we observed predominant populations of CD244+ PD-1+ CD8+TILs, but also distinct populations of PD-1+ CD244- CD8+ TILs. Previous studies of CD8+ T cell exhaustion in chronic viral illness have shown concomitant increases in PD-1 and CD244 expression, followed by a slow decline in CD244 expression but persistence of PD-1.47 Thus, CD244 expression may be a biomarker for earlier stages of CD8+ T cell exhaustion, while its loss in the setting of persistent PD-1 may be a marker of more severe exhaustion moving toward apoptosis. That is, concomitant CD244+ PD-1+ expression on CD8+ T cells may predict improved response to exhausting checkpoint inhibitors compared with PD-1 expression alone. Importantly, comparisons in cytokine production by CD8+ TILs from WT and CD244-/- mice suggest that CD244-/- PD-1+ CD8+ TILs, but not PD-1+ CD244+ CD8+ TILs produce proinflammatory cytokines IFNγ and TNFα. While we cannot exclude that absence of CD244 on other cell types in the tumor microenvironment contributed to the improved CD8+ TIL functionality, these data provide support for our hypothesis that CD244 blockade could provide therapeutic benefit. Furthermore, in related experiments done in our laboratory, we see the same trends in CD244 expression in multiple syngeneic mouse models, including B16 melanoma, EL-4 lymphoma and M3-9-M embryonal rhabdomyosarcoma, suggesting that CD244 may be a useful biomarker across a broad variety of cancers.
In the myeloid compartment, the significant, positive correlation between CD244 expression on DCs and Mo-MDSCs and spontaneous production of immunosuppressive mediators suggests that inhibitory CD244 signaling in these myeloid cells may directly contribute to immunosuppression in the tumor microenvironment. In addition, the finding that CD244 cross-linking in healthy human DCs decreases the LPS-stimulated production of certain proinflammatory cytokines build on previously published studies demonstrating that CD244-/- DCs produce increased levels of proinflammatory cytokines and increased activation of NK cells.4 Our studies were limited by the inability to isolate sufficient DCs from our human tumor samples to test the hypothesis that CD244 signaling in intratumoral DCs likewise decreases proinflammatory cytokine production. However, it is notable that genome-wide expression profiling has shown that human DCs express specific adaptors implicated in CD244 signaling that promotes suppressive function in these cells, such as SHP-2, but not SAP, which facilitates activating CD244 signaling in NK cells.48 These phenotypic and genotypic characterizations suggest that CD244 signaling in DCs predominantly promotes tolerogenicity and may contribute to DC-mediated immunosuppression in the tumor microenvironment.
Treatment of male mice growing established syngeneic tumors with CD244 mAb significantly inhibited tumor growth compared with isotype treated controls without depleting CD8+ T cells in tumors or spleens; in fact, anti-CD244 treatment led to increased proportions of tumor-infiltrating CD8+ T cells compared with controls. The limited tumor growth seen in the CD244 treatment group recapitulated the spontaneously inhibited tumor growth observed in CD244-/- mice, reflecting an inhibitory effect of the CD244 antibody. To the best of our knowledge, these results represent the first evidence that CD244 inhibition may provide a therapeutic effect in a solid tumor model. Previous leukaemia studies, however, have suggested a therapeutic role for CD244 in leukemia: CD244 deletion has been shown to result in impaired leukemogenesis in human leukemia cell lines and in a mouse model of acute myeloid leukemia.49 These observations provide a framework for future studies of anti-CD244 therapy in both solid tumors and hematological malignancies.
Given the established clinical success of PD-1 inhibition for many cancer types, we have chosen to emphasise the relationship between CD244 and PD-1/PD-L1 expression in this study. However, increased expression of CD244 has also been associated with increased expression of other receptors implicated in tumor-associated immunosuppression, including CLTA-4, TIM-3, and LAG-3 on CD8+ T cells through the VHL/HIFα pathway.50 This common underlying mechanism underscores the likelihood of redundant immunosuppressive pathways and the potential for synergistic coinhibition, or alternatively, upstream targeting to prevent upregulation of these coinhibitory receptors altogether. Likewise, the increase in immunosuppressive mediators by CD244hi myeloid cells provides a basis for interplay among multiple immunosuppressive pathways. Inhibition of CD244 may, therefore, offer synergy with some of the host of therapeutics currently in development targeting the arginine, IDO and anti-inflammatory cytokine pathways.
This study builds on past work establishing CD244 as a marker of CD8+ T cell exhaustion, demonstrating similar increases in CD244 expression in intratumoral DCs and Mo-MDSCs that correspond to signs of increased immunosuppressive function, and importantly, demonstrating that therapeutic CD244 inhibition significantly impairs tumor growth in established syngeneic mouse tumors. The consistent correlation between CD244 and PD-1/PD-L1 also suggests the hypothesis that CD244 and PD-1 inhibition may synergize. Investigations exploring this hypothesis are ongoing. Questions to be addressed in future studies regard the signaling mechanisms of CD244 in CD8+ T cells, DCs and MDSCs, which likely differ from those elucidated in NK cells due to variance in endogenous levels of relevant specific adaptor molecules, and therapeutic implications of targeting CD244 in cancer.
Acknowledgments
We would like to acknowledge Cincinnati Children’s Hospital Medical Center Research Flow Cytometry Core and Pathology Research Core for technical support on this project.
References
Footnotes
Contributors LA performed experiments, analysed and interpreted data, and was the primary contributor in writing the manuscript. ML, KL, RC and CH performed experiments and provided laboratory support. SS provided expertise in the pathology analysis of tumor specimens. TW-D and LC provided expertise on human HNSCC and clinical immunotherapy. KH provided expertise in tumor immunology. EMJ supervised the design, analysis and interpretation of all experiments, provided expertise in tumor immunology, and was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
Funding This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Peer Reviewed Cancer Research Program, under Award No. W81XWH-17-1-0374 (EMJ, TW-D and LC). The US Amy Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick MD 21702-5014 is the awarding and administering acquisition office. Additional funding was provided by the National Cancer Institute (CA201918-01; EMJ and CA95286; LC) and the National Institute on Aging (AG053498; EMJ).
Disclaimer Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The use of human cells and tissues from patients and healthy donors was approved by the University of Cincinnati Institutional Review Board (IRB# 2014-4755) except for collection of cadaveric spleens, which was exempted on the basis of de-identified samples, and all protocols followed Good Clinical Practice. All the animal experiments and handling procedures were approved by the Cincinnati Children’s Institutional Animal Care and Use Committee (IACUC) and performed according to the guidelines.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.