Article Text

Abstract
Background Phosphorylated peptides presented by MHC molecules represent a new class of neoantigens expressed on cancer cells and recognized by CD8 T-cells. These peptides are promising targets for cancer immunotherapy. Previous work identified an HLA-A*0201-restricted phosphopeptide from insulin receptor substrate 2 (pIRS2) as one such target. The purpose of this study was to characterize a second phosphopeptide, from breast cancer antiestrogen resistance 3 (BCAR3), and to evaluate safety and immunogenicity of a novel immunotherapic vaccine comprising either or both of these phosphorylated peptides.
Methods Phosphorylated BCAR3 protein was evaluated in melanoma and breast cancer cell lines by Western blot, and recognition by T-cells specific for HLA-A*0201-restricted phosphorylated BCAR3 peptide (pBCAR3126-134) was determined by 51Cr release assay and intracellular cytokine staining. Human tumor explants were also evaluated by mass spectrometry for presentation of pIRS2 and pBCAR3 peptides. For the clinical trial, participants with resected stage IIA–IV melanoma were vaccinated 6 times over 12 weeks with one or both peptides in incomplete Freund’s adjuvant and Hiltonol (poly-ICLC). Adverse events (AEs) were coded based on National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) V.4.03, with provision for early study termination if dose-limiting toxicity (DLT) rates exceeded 33%. The enrollment target was 12 participants evaluable for immune response to each peptide. T-cell responses were assessed by interferon-γ ELISpot assay.
Results pBCAR3 peptides were immunogenic in vivo in mice, and in vitro in normal human donors, and T-cells specific for pBCAR3126-134 controlled outgrowth of a tumor xenograft. The pIRS21097-1105 peptide was identified by mass spectrometry from human hepatocellular carcinoma tumors. In the clinical trial, 15 participants were enrolled. All had grade 1 or 2 treatment-related AEs, but there were no grade 3–4 AEs, DLTs or deaths on study. T-cell responses were induced to the pIRS21097-1105 peptide in 5/12 patients (42%, 90% CI 18% to 68%) and to the pBCAR3126-134 peptide in 2/12 patients (17%, 90% CI 3% to 44%).
Conclusion This study supports the safety and immunogenicity of vaccines containing the cancer-associated phosphopeptides pBCAR3126-134 and pIRS21097-1105, and the data support continued development of immune therapy targeting phosphopeptides. Future studies will define ways to further enhance the magnitude and durability of phosphopeptide-specific immune responses.
Trial registration number NCT01846143
- immunogenicity
- vaccine
- immunotherapy
- melanoma
- vaccination
- antigens
- neoplasm
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The development of therapeutic cancer vaccines based on peptides presented by MHC-I molecules to CD8 T-cells has been pursued for many years. Studies of these types of vaccines have recently been revitalized by the successes of checkpoint blockade and adoptive T-cell immunotherapies and by interest in patient-specific mutated antigens as vaccine targets. Peptide antigens tested in cancer vaccines have commonly been derived from source proteins in one of three categories: tissue-specific differentiation proteins, cancer-germ cell proteins or mutated proteins. The immunogenicity of antigens derived from non-mutated proteins may be compromised by mechanisms of self-tolerance.1 While antigens carrying tumor-specific mutations may avoid some tolerance concerns, identification of relevant antigens in each patient and preparation of an appropriate vaccine are technically challenging and resource-intensive undertakings,2–4 and their therapeutic value is likely to vary widely. Importantly, only a small number of previously-identified antigens in any of these categories are derived from proteins that are objectively linked to mechanisms of cellular growth control, survival or metastasis. Such antigens are particularly appealing as immunotherapeutic targets for cancer control because their alteration as a means of immune escape may compromise one or more aspects of the malignant phenotype. We have identified peptide antigens of this type that are modified by intracellular phosphorylation, naturally processed and presented by MHC-I molecules on cancer cells and specifically recognized by CD8 T-cells.5–7 Phosphorylation is associated with a variety of cellular control processes, some of which are dysregulated in cancer cells. The source proteins for MHC-I-associated phosphopeptides identified to date are in large part known phosphoproteins,6 supporting the idea that the peptides are processed from folded proteins participating in signaling pathways.
One such protein is insulin receptor substrate 2 (IRS2). IRS proteins are adapters that link signaling from upstream activators to multiple downstream effectors to modulate growth, metabolism, survival and differentiation.8 9 IRS2 overexpression at the gene or protein level is evident in many cancer types and has been demonstrated to cause tumorigenesis and enhanced metastasis in vivo.8–10 IRS2 phosphorylated at Ser1100 is detectable in multiple cancers, and the resulting phosphopeptide (pIRS21097-1105) is endogenously processed and presented by the MHC-I molecule HLA-A*0201 at levels that allow strong immune recognition (7 and unpublished). pIRS21097-1105-specific CD8 T-cells can be generated from HLA-A2 transgenic mice by immunization with the phosphopeptide.6 7 These T-cells are capable of recognizing and killing human melanoma and breast tumors in vitro and controlling tumor growth in a xenograft tumor model system.7
A second cancer-relevant HLA-A*0201 restricted phosphopeptide is derived from breast cancer antiestrogen resistance 3 (BCAR3). BCAR3 activates the PI3K/Akt pathway and mediates both cellular migration and acquisition of resistance to therapeutic antiestrogens in breast cancer cells, processes that are associated with malignancy.11 12 A BCAR3-derived phosphopeptide (pBCAR3126-134) was initially shown by us to be presented by HLA-A*0201 on melanoma cells.6 Here, we show that it also is immunogenic in HLA-A2 transgenic mice and that the resulting CD8 T-cells can directly recognize melanoma and breast cancer tumor cell lines.
Several reports, including our own, have established the safety and immunogenicity of multipeptide vaccines derived from non-mutated tissue-specific proteins.13–18 In the clinical trial reported here, we now evaluate the safety and immunogenicity of a vaccine consisting of these two oncologically and immunologically relevant phosphorylated peptides, pBCAR3126-134 and pIRS21097-1105.
Methods
Preclinical studies
Mice
Mice expressing a chimeric class I MHC molecule composed of the α1 and α2 domains of human HLA-A2 and the α3 domain of murine H-2Dd (AAD) have been described.19 NOD/SCID/interleukin-2 (IL-2)Rγc-/- (NSG) mice were purchased from Jackson Immunoresearch Laboratories (Bar Harbor, Massachusetts, USA).
Tumor cell lines
All tumor cell lines were of human origin. Melanoma line SLM2 was obtained from the American Type Culture Collection (Bethesda, Maryland, USA). Melanoma line DM331 was established from a patient whose tumor was resected at Duke University. VMM12 and VMM18 lines were established from metastatic melanoma resected from patients at the University of Virginia. VMM18 also has been deposited with the ATCC. Melanoma line 1363Mel was obtained from Suzanne Topalian (Johns Hopkins University). Breast cancer lines BT-549, MCF7, MDAMB231, MDAMB468 and T47D were obtained from Sarah Parsons (University of Virginia). Melanoma lines were grown at 37°C with 5% CO2 in RPMI-1640 containing 10% fetal bovine serum, 15 mM HEPES, 2 mM L-glutamine and 50 µg/mL gentamycin. Breast cancer lines were grown at 37°C with 8% CO2 in DMEM +10% FBS, 1 mM sodium pyruvate, 15 mM HEPES, 2 mM L-glutamine and 50 µg/mL gentamycin.
Western blot
Cell lysates were generated as described.7 Each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and proteins were transferred onto polyvinylidene fluoride membranes (Immobilon-FL, 0.45 µm, Millipore, Bedford, Massachusetts, USA). Blots were probed with specific primary antibodies overnight at 4°C and then with horseradish peroxidase-conjugated secondary antibodies. Pixel densities were determined using the AlphaEaseFC software and the densities of BCAR3 and of BCAR3 phosphorylated at T130 (phosphoT130-BCAR3) were normalized to those from glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Antihuman BCAR3 was from Bethyl labs (Montgomery, Texas, USA). Anti-GAPDH was from Santa Cruz (Santa Cruz, California, USA). A polyclonal antibody recognizing T130 phosphorylated BCAR3 protein was generated by AbboMax (San Jose, California, USA) by immunization of a rabbit with a 13mer phosphopeptide (CRHIMDR[pT]PEKLK), and sequential affinity purification on columns derivatized with the 13mer and its non-phosphorylated counterpart. Specificity was established by Western analysis with or without competing 1.5 µg of phosphorylated or unphosphorylated 13mer BCAR3 peptides per 5 µg of antibody (online supplementary figure 1A). Protein lysates from cells in which expression of BCAR3 had been knocked down by RNA interference and used in a Western blot verified that the antibody recognized a band of ~90 kDa corresponding to BCAR3 (online supplementary figure 1B), and detection of this band was abrogated by treatment with lambda phosphatase (online supplementary figure 1C). However, this antibody also detected an unidentified phosphoprotein with a molecular weight larger than 102 kDa (online supplementary figure 1B), precluding its use in immunohistochemical analyzes.
Supplemental material
Phosphopeptide detection by mass spectrometry
Tumor and normal adjacent tissues from HLA-A*0201+ hepatocellular carcinoma patients were processed and phospholigandome analysis performed as described.20 Peptides were isolated from tissue extracts using the pan-human MHC class I antibody W6/32, desalted and concentrated via STop And Go Extraction tips, and resuspended in formic acid. Phosphopeptides were enriched using iron-III-iminodiacetic acid immobilized metal affinity chromatography and analyzed using high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. Data analysis was performed using Xcalibur software (Thermo Electron Corporation) and raw data files were searched using Byonic (Protein Metrics) against the Swissprot human protein database. Phosphopeptide sequences were confirmed by accurate mass measurement and manual interpretation of MS2 spectra. Relative abundances of detected phosphopeptides were calculated based on internal phosphorylated and nonphosphorylated peptide standards present at fixed concentrations within each sample.
Peptides for preclinical studies and HLA-A*0201 binding assays
pBCAR3126-134 (IMDR[pT]PEKL), pBCAR3(V)126-134 (IMDR[pT]PEKV), pS33-βcat30-39 (YLD[pS]GIHSGA), and their nonphosphorylated counterparts were synthesized by GenScript (Piscataway, New Jersey, USA). Binding of peptides to purified HLA-A*0201 was determined in an equilibrium assay by competition with a radiolabeled indicator peptide as described.21 22
In vitro culture of human T-cell and cytotoxicity assay
Autologous mature dendritic cells were pulsed with phosphopeptides of interest for 2 hours at room temperature and then irradiated at 3500 rad. T-cells were enriched from donor peripheral blood mononuclear cells (PBMCs) using a human Pan T-cell isolation kit II (Miltenyi, Bergisch Gladbach, Germany) and then incubated with phosphopeptide-pulsed dendritic cells in microwells at 37°C with 5% CO2 for 2 weeks in medium with 20 ng/mL IL-7, 10 ng/mL IL-15, 1 ng/mL IL-10 and 20 U/mL IL-2. The cultures were restimulated with phosphopeptide-pulsed irradiated autologous PBMCs every 7 days. Two weeks after in vitro culture, the T-cells were incubated with phosphopeptide-pulsed 51Cr-labeled targets at a ratio of 60:1 for 4 hours. Radioactivity was measured on a Wallac Wizard 1470 Automatic Gamma Counter. Normalized percent lysis was calculated as: ((radioactivity values for pulsed targets – background values for unpulsed targets)/values for positive control) ×100.
Immune responses of AAD mice to phosphopeptides
Bone marrow derived dendritic cells (BMDC) from AAD mice were prepared as described.5 23 BMDC were pulsed with 10 µg/mL phosphopeptide and 10 µg/mL β2-microglobulin for 2–3 hours, irradiated (3000 cGy), washed and injected intravenous (1.5–2.5×105) into AAD mice. Mice received intravenous booster immunizations 6 days later with phosphopeptide, CpG (type C, ODN 2395, InvivoGen (San Diego, California, USA)), and antimouse CD40 (FGK45, gift of Stephen Schoenberger, La Jolla Institute of Allergy and Immunology). CD8α+ T-cells were enriched from spleens and lymph nodes 5 d later using magnetic beads (Miltenyi) and incubated for 5 hours with peptide-pulsed stimulators (C1R-AAD) in medium containing 0.5 µg/mL anti-CD107a, 5 µg/mL Brefelden A (Sigma-Aldrich) and 1 µM monensin (eBioscience). Cells were stained for CD8α (eBioscience), permeabilized and fixed with Cytoperm/Cytofix (BD Bioscience), and stained for intracellular interferon-γ (IFNγ) (eBioscience). Samples were analyzed on a FACSCanto I or II (BD Bioscience) using FlowJo software (Tree Star, Ashland, Oregon, USA).
Tumor control using pBCAR3126-134-specific T-cell lines
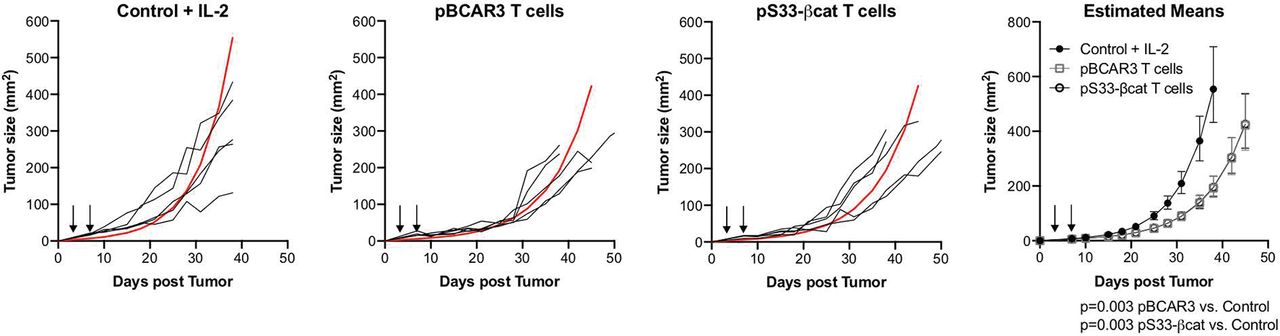
pBCAR3126-134-specific T-cell lines were established using enriched CD8 cells harvested from mice immunized as described above and repetitively recultured with pBCAR3(V)126-134 peptide-pulsed irradiated AAD+ splenocytes in media supplemented with IL-2. Lines were established within 3–4 weeks, as documented by 70%–85% of the cells responding to peptide pulsed stimulators in the effector function assays described above. For tumor control experiments, male NSG mice aged 7–8 weeks were inoculated s.c. with 1.4×106 SLM2 human melanoma cells. Three days later, mice were injected intravenous with 6×106 pBCAR3126-134- or pS33-βcat30-39-specific T-cells. Four days later, the mice received an additional 0.3×106 T-cells. 1500U IL-2 was administered i.p. every other day for 10 days beginning on the day of T-cell transfer. Tumor size was measured with a digital caliper and calculated as L x W. Tumor growth was analyzed using repeated measure models after transforming to the log scale assuming an AR1 covariance structure. Specific comparisons between pBCAR3- or pS33-βcat-treated and control mice were made with F-tests based on contrasts and estimates were transformed back for plotting.
Clinical trial
Patients
Patients with resected AJCC stage IIA–IV (V.7) melanoma24 were eligible, as well as patients with disease who failed other approved therapies. However, all who enrolled were clinically free of disease. Inclusion criteria included: HLA-A2 expression, ages 18 years and above, Eastern Cooperative Oncology Group performance status 0–1, adequate liver and renal function, and ability to give informed consent. Exclusion criteria included: pregnancy; cytotoxic chemotherapy, IFN, immunomodulatory therapies, or radiation within the preceding month; known or suspected allergies to vaccine components; multiple brain metastases; use of steroids; Class III–IV heart disease; systemic autoimmune disease with visceral involvement or uncontrolled diabetes (hemoglobin A1C ≥7%).
Vaccine preparation and administration
pBCAR3126-134 (IMDR(pT)PEKL) and pIRS21097-1105 (RVA(pS)PTSGV) were synthesized and purified (>95%) by PolyPeptide Laboratories (San Diego, California, USA), solubilized in aqueous solution, sterile-filtered, vialed in sterile single-use vials, lyophilized and stored at −80°C. The tetanus helper peptide AQYIKANSKFIGITEL (P2830-844, modified by adding alanine to the N-terminus),25 26 was prepared, sterile filtered, vialed and lyophilized under Good Manufacturing Practice conditions as described.17 25 Vials were submitted for quality-assurance studies including sterility, identity, purity, concentration, general safety, pyrogenicity and stability in accordance with Code of Federal Regulations guidelines and BB-IND #15 134. Polyinosinic-polycytidylic acid stabilized with poly-L-lysine and carboxymethylcellulose (poly-ICLC, Hiltonol; Oncovir, Washington, D.C., USA) was provided by the Ludwig Institute (New York, New York, USA) as a clinical grade reagent. Each vaccination consisted of 100 µg of either or both phosphopeptides and 200 µg of the tetanus peptide in a water-in-oil emulsion with an equal volume of an incomplete Freund’s adjuvant (IFA) (Montanide ISA-51 VG adjuvant, Seppic, Paris, France). Vaccines were administered subcutaneously (0.5 mL) and intradermally (0.5 mL) at one skin puncture site. One mg of poly-ICLC was administered immediately thereafter by a separate 0.5 mL injection (0.25 mL subcutaneously and 0.25 mL intradermally) into the precise sites where the peptide emulsion was given. Vaccine sites were rotated to different skin regions on each vaccination date, using a minimum of two separate extremities. Vaccines were administered in two treatment cycles (figure 1).

Design for clinical trial to evaluate safety and immunogenicity of phosphopeptide-based vaccine in melanoma patients. Participants were eligible for all arms with stage IIIB-IV melanoma, with measurable disease, or after surgical resection. Eligibility for arm C was extended to stage II–IIIA patients, after surgical resection. The study was also opened to participants with other solid tumors, but only participants with melanoma enrolled. IFA, incomplete Freund’s adjuvant; PBMC, peripheral blood mononuclear cell; pIRS2, phosphopeptide from insulin receptor substrate 2; tet, tetanus helper peptide.
Clinical trial design
This was an open-label, pilot, proof-of-concept study to assess phosphopeptide vaccine safety and immunogenicity. It was designed with three arms to assess each phosphopeptide individually before assessing the combination: Arm A, pBCAR3126-134 only; Arm B, pIRS21097-1105 only; and Arm C, pBCAR3126-134 + pIRS21097-1105. Arms A and B were restricted to patients with stage IIIB–IV melanoma (at original presentation or at recurrence, with or without measurable disease). Once safety was evident for each peptide in these higher-risk patients, Arm C was open for stage IIIB–IV patients and also for patients with lower risk (stage IIA–IIIA) melanoma rendered clinically free of disease by surgery, other therapy or spontaneous remission. Accrual to arm C occurred only after continuation was deemed appropriate in the initial safety phase of arms A and B. Maximum target accrual was set at 21 eligible participants who received a vaccination. Provided safety rules were met, immunogenicity would be assessed for each phosphopeptide in up to 12 evaluable participants total.
Toxicity assessment
The study was monitored continuously for treatment-related adverse events. Adverse events were described and coded based on NCI CTCAE v4.03. A dose-limiting toxicity (DLT) was defined as any unexpected adverse event that is possibly, probably or definitely related and: (1) ocular ≥grade 1, (2) non-hematologic/non-metabolic ≥Grade 3, or (3) hematologic/metabolic ≥Grade 3. Toxicities with known or likely autoimmune features and affecting vital organs, including colitis, hepatitis, hypophysitis, uveitis and adrenal insufficiency, were recorded and assessed with special interest. These were considered DLTs if ≥grade 2 and probably or definitely related. Grade 2 autoimmune toxicities involving these organs/tissues were not considered DLTs if only possibly related. Vitiligo was not considered a DLT, but was recorded. Hyperthyroidism or hypothyroidism was considered a DLT if ≥grade 3. All vaccinated participants were followed for a minimum of 4 weeks for assessment of DLTs.
Safety assessment
The number of vaccinated participants who experienced DLTs guided decisions about safety. In the initial safety phase for arm A or Arm B, one participant was accrued. If that subject did not experience a DLT within 4 weeks, 2 additional participants were accrued. If 0/3 DLTs were observed, then the initial safety criteria for the arm were satisfied; if ≥2/3 participants experienced a DLT, then accrual to that arm would be halted permanently; otherwise three additional participants would be accrued to the arm to assess safety. Accrual to arm C occurred only after the initial safety criteria for arms A and B were satisfied. A maximum of nine participants were accrued in sequential cohorts of 3. Accrual continued as long as no more than 33% of participants experienced a DLT.
ELISpot analysis of T-cell response to phosphopeptides
Blood (100–140 mL) was drawn on days indicated in figure 1. PBMCs were isolated from 80 mL (except 120 mL week 0) using Ficoll gradient centrifugation and cryopreserved in 10% dimethyl sulfoxide/90% serum. Thawed PBMCs were incubated 2 hours at 37°C with 40 µg/mL each of pBCAR3126-134, pIRS21097-1105 and pS33-βcat30-39 (control), washed to remove unbound peptide, and resuspended in medium containing 10 ng/mL recombinant human IL-7 (Peprotech, Rocky Hill, New Jersey, USA) for 3 days. New medium containing recombinant human IL-7 plus 10 ng/mL IL-15 (Peprotech) was added on days 3, 7 and 10. Cells were assayed on day 14 for IFNγ production by ELISpot assay using methods and criteria described previously.14 15 Briefly, T2-B7 cells were pulsed with each of the three phosphopeptides for each assay date. Negative controls included no peptide, irrelevant peptide (HIV gag peptide restricted by HLA-A2; Atlantic Peptides, LLC, Lewisburg, Pennsylvania, USA), and positive controls included PMA-ionomycin, PHA. Responses were corrected for the fraction of CD8 T-cells in PBMC as determined by flow cytometry of individual samples at culture initiation. T-cell responses were calculated using the following definitions: Nvax=number T-cells responding to vaccine peptide; Nneg=number T-cells responding to maximum negative control; Rvax=Nvax/Nneg. A patient was considered to have a T-cell response to vaccination (binary yes/no), only if all of the following criteria were met: (1) Nvax exceeded Nneg by at least 100 spots/100 000 CD8 cells, (2) (Nvax – 1 SD) ≥ (Nneg +1 SD) and (3) Rvax after vaccination ≥2 x Rvax prevaccine. Prevaccine Rvax values less than one (eg, control counts exceed number of responding T-cells) were set equal to one to indicate no response and to prevent overinflating adjusted fold-increases due to prevaccine ratios less than one or division by zero. Interassay coefficients of variation (CVs) were calculated for the response of 2 normal donors to a mixture of viral peptides (CEF peptide pool, Proimmune, Sarasota, Florida, USA): for the high responder, mean number of spots per 1 00 000 cells was 309, and CV was 29%.
Statistical assessment of immune response
Since this was a first-in-humans clinical trial with phosphopeptide antigens for cancer, a first goal was to assess safety with a small study. The biological primary endpoint was evidence of immunogenicity. The prespecified level of interest was two or more patients with T-cell responses. Beyond that, CI for the observed proportions would determine how much the data supported immunogenicity. Assuming that the response to one phosphopeptide is not affected by the presence of the other for those in arm C, the immunogenicity of each phosphopeptide was based on the total number of participants vaccinated with the specific phosphopeptide, alone or in combination with the other (ie, Arm A+Arm C for pBCAR3126-134; Arm B+Arm C for pIRS21097-1105). Based on the upper limit of a one-sided 90% CI, if the upper limit of the observed bound was >35% to any single phosphopeptide, we would conclude that the phosphopeptides was immunogenic and worthy of further study. To define the CD8 T-cell response rate with higher precision would require a much larger sample size, which was not indicated in this first-in-humans trial.
Analysis of exploratory endpoint
All participants were clinically free of disease at enrollment. Disease-free survival (DFS) is defined as the time from on-study until the earliest documentation of a new metastasis or death from any cause, or censored at the date of last follow-up for those without a documented event. Overall survival is defined as the time from on-study to death from any cause, or censored at the date of last follow-up for those still alive.
Results
phosphoT130-BCAR3 is expressed in cancer cell lines but not normal melanocytes
We previously found that pIRS21097-1105 is naturally presented by HLA-A*0201 on cancer cells with high levels of protein, and that it serves as a target for immune mediated tumor control.7 Using mass spectrometry, we identified another HLA-A*0201-associated peptide on melanoma cells comprising residues 126–134 of the BCAR3 protein, which was phosphorylated at T130 (pBCAR3126-134).6 We evaluated additional melanoma and breast cancer cell lines for expression of the phosphorylated source protein using a commercially available polyclonal BCAR3 antibody and a validated phosphoT130-BCAR3 antibody (described in Methods and online supplementary figure 1). BCAR3 protein expression was quite heterogenous in both melanoma and breast cancer cells (figure 2A,B). Nonetheless, phosphoT130-BCAR3 was detected in most of the melanoma and breast cancer cell lines (figure 2A–C). Because of a strong cross-reactive band evident in Western blots, the phosphoT130-BCAR3 antibody could not be used for immunohistochemical analysis of human tumor specimens. A survey by mass spectrometry of presented peptides extracted from several HLA-A*0201+ primary tumor samples failed to identify pBCAR3126-134 (Dennis Underwood, Agenus, personal communication). However, the pIRS21097-1105 peptide was directly identified by mass spectrometry in naturally presented peptides extracted from 4/5 hepatocellular carcinoma (HCC) samples at 1–5 (two tumors) and 6–20 (two tumors) copies per cell (table 1). It was found at less than one copy per cell on 1/4 normal adjacent tissue samples. In addition, a phosphoproteome analysis identified phosphoT130-BCAR3 in 3/105 primary breast cancer samples, while 22/105 of these same samples expressed phosphoS1100-IRS2.27 28 PhosphoS1100-IRS2 has also been identified by phosphoproteome analysis in an unspecified fraction of high-grade serous ovarian28 and non-small-cell lung carcinomas,29 2/4 chronic lymphocytic leukemia isolates and 1/1 primary mantle cell lymphoma.30 These results suggest that the expression of pBCAR3126-134 on HLA-A*0201+ tumors may be limited, although a more comprehensive survey of additional tumor types is warranted, while demonstrating that pIRS21097-1105 is expressed broadly and frequently on multiple tumor types.
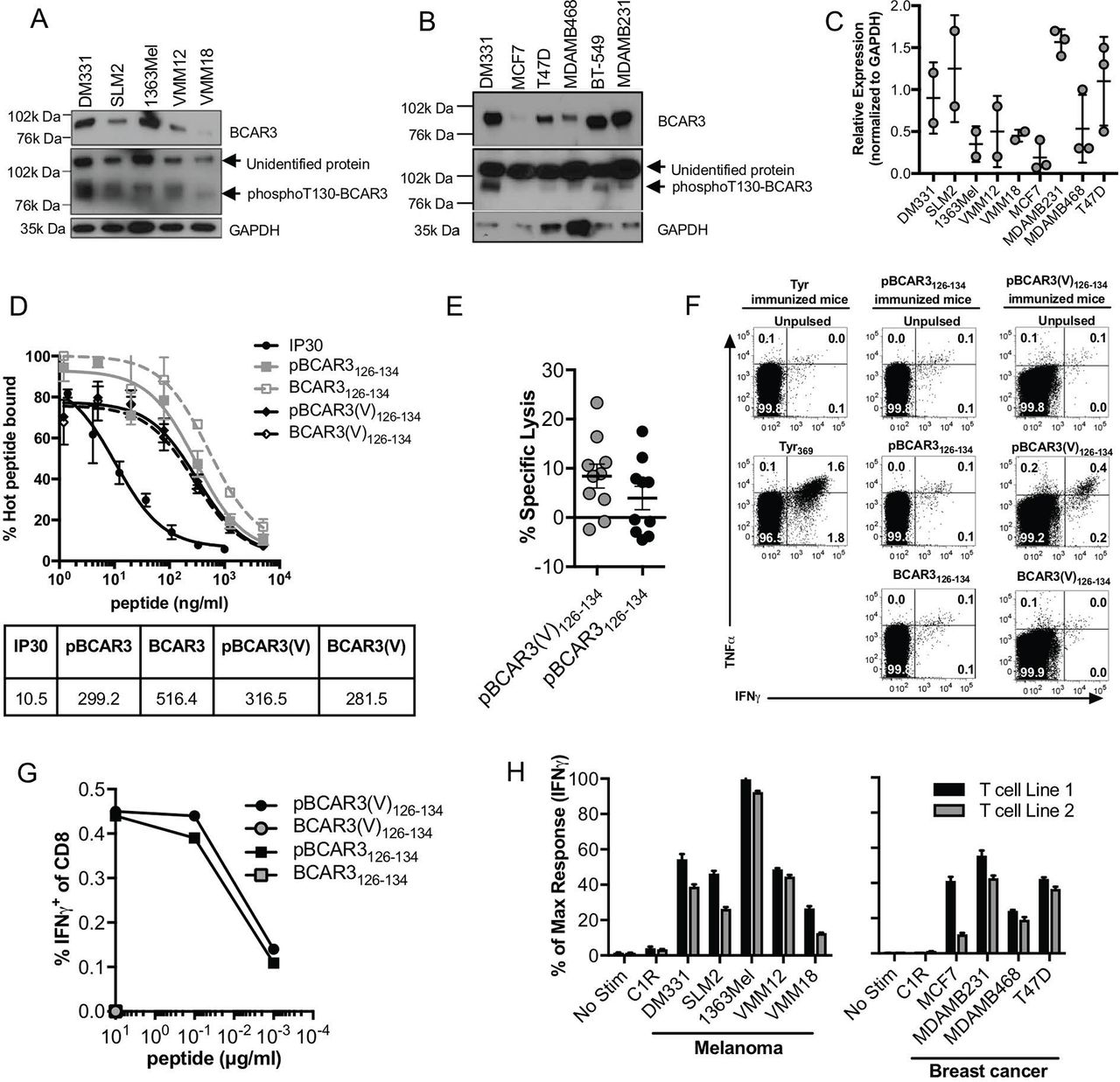
Expression of phosphoT130-BCAR3 protein and recognition of pBCAR3126-134 phosphopeptide in melanoma and breast cancer cells. (A–C) Expression of phosphoT130-BCAR3 and BCAR3 in melanoma (A) and breast cancer (B) cells was evaluated by Western blot using the validated phosphoT130-BCAR3 antibody and a commercially available BCAR3 antibody. GAPDH expression served as a loading control, and DM331 melanoma cells were included in both panels for comparison. Data are representative of two (melanoma) and three (breast) independent experiments. (C) Relative expression of phosphoT130-BCAR3 in melanoma and breast cancer cells normalized to the respective GAPDH levels. (D) pBCAR3126-134, pBCAR3(V)126-134, the non-phosphorylated forms and a control peptide (IP30) were tested for their ability to compete with a radiolabeled peptide derived from hepatitis B core protein for binding to purified HLA-A*0201. The dose required to reduce binding of the radiolabeled peptide by 50% (dotted line) corresponds to the IC50 value. Data are representative of three independent experiments. (E) Human T-cells from one donor were cultured in 10 microwells containing irradiated stimulators pulsed with pBCAR3126-134 or pBCAR3(V)126-134 and evaluated for pBCAR3- or pBCAR3(V)-specific responses in a 51Cr-release assay after 2 weeks using targets pulsed with either the phosphorylated or unphosphorylated forms of the peptide. Data points represent individual microcultures from one donor. (F) AAD transgenic mice were immunized with BMDC pulsed with pBCAR3126-134 or pBCAR3(V)126-134. Six days later, mice received a boost of phosphopeptide, CpG, and anti-CD40. T-cells were harvested from the spleens and lymph nodes and evaluated for cytokine production after 5 hour incubation with pulsed stimulators. Dot plots are gated on CD8 cells. (G) Mice were immunized with pBCAR3(V)126-134, and the percentages of CD8 T-cells isolated from them that recognized the immunogen or the parental pBCAR3126-134 were determined by intracellular staining for IFNγ. Data are representative of two experiments. (H) Melanoma and breast cancer cells expressing endogenous phosphoT130-BCAR3 were incubated separately with two independently derived murine pBCAR3126-134 specific T-cell lines and the fraction that produced IFNγ quantified by flow cytometry. Data are normalized to % of maximal response that was quantified using stimulators pulsed with 10 µg/mL of pBCAR3126-134 (range of maximal response was 70%–85%). Data are representative of three independent experiments (F–H).
Detection of pIRS21097-1105 in human hepatocellular carcinoma tissues by mass spectrometry of MHC-associated peptide extracts
pBCAR3126-134 is immunogenic and enables immune control of tumor outgrowth in a preclinical model
The unphosphorylated BCAR3126-134 peptide is predicted to bind weakly to HLA-A*0201 by the artificial neural network (ANN) algorithm31 and the stabilized matrix method32 (IEDB analysis resource, http://www.immuneepitope.org), and this was confirmed by direct binding assays (figure 2D). pBCAR3126-134 bound to HLA-A*0201 somewhat better than the unphosphorylated peptide, in keeping with the role of phosphate as a surface anchor residue,22 but still bound with a significantly lower affinity than the IP30 peptide that we routinely use as a high affinity positive control21 (figure 2D). A pBCAR3 peptide modified by substitution of V for L at residue 134, the P9 anchor position, (pBCAR3(V)126-134) was predicted to have higher affinity for HLA-A*0201 by ANN analysis, but showed no difference in direct binding assays. Both peptides induced antigen specific human T-cells in vitro (figure 2E), while only pBCAR3(V)126-134 was immunogenic in vivo in HLA-A2 transgenic mice (figure 2F). Nonetheless, all T-cells generated by immunization with pBCAR3(V)126-134 showed complete cross-reactivity with pBCAR3126-134 as assessed by quantifying the fraction of IFNγ+ T-cells after a 5-hour incubation with stimulators pulsed with either the modified or the natural phosphopeptides, and this was maintained over graded doses of the phosphopeptides (figure 2G).
We used two T-cell lines generated from pBCAR3(V)126-134–immunized mice to assess the expression of pBCAR3126-134 on human cancer cell lines. Melanoma and breast cancer cell lines that express the phosphoT130-BCAR3 protein by Western blot were recognized well by both T-cell lines, and the extent of T-cell recognition varied among the cells, suggesting that the epitope is displayed at varied levels on the tumor cells (figure 2H). However, the level of T-cell recognition did not correlate with phosphoT130-BCAR3 protein levels by Western blot, suggesting variability in the efficiency of Ag processing and presentation, MHC levels or expression of molecules supporting T-cell recognition might vary among these cells. We also evaluated the ability of pBCAR3126-134-specific T-cell line 1, and murine T-cells specific for another phosphopeptide derived from β-catenin, pS33-βcat30-39, to control growth of SLM2 tumors in a xenograft model. Both T-cell populations reduced tumor growth in vivo (figure 3). These results supported the evaluation of both pBCAR3126-134 and pS33-βcat30-39 as candidates for cancer immunotherapy. Food and Drug Administration (FDA) concerns about possible expression of pS33-βcat30-39 in normal tissues precluded inclusion in a human clinical trial. However, the FDA approved evaluation of both pIRS21097-1105 and pBCAR3126-134 in humans.
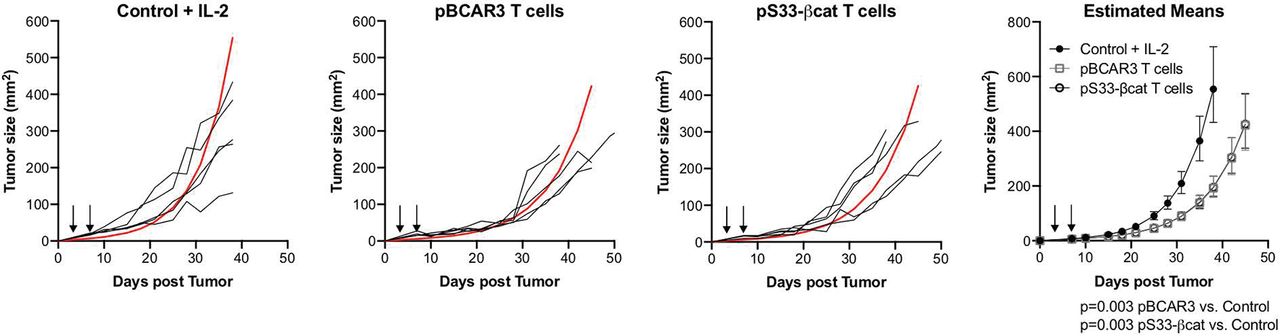
Phosphopeptide specific T-cells mediate tumor control. Control of subcutaneous SLM2 tumors growing in NOD/SCID-IL-2Rγc-/- mice by pBCAR3126-134– or pS33βcat30-39-specific murine T-cells was determined as outlined in Methods. T-cells were injected as indicated by arrows. Estimated mean for each group is shown in red. Data are representative of two independent experiments (n=5 per group per experiment).
Clinical presentation of patients vaccinated with phosphopeptides
Having identified pIRS21097-1105 and pBCAR3126-134 phosphopeptides as attractive targets for cancer immunotherapy, we designed a clinical trial to test their safety and immunogenicity in cancer patients (figure 1). Vaccines were formulated as phosphopeptides together with a tetanus helper peptide emulsified in IFA (Montanide ISA-51) and coadministered with poly-ICLC. To avoid risk of off-target immune responses, the native pBCAR3126-134 phosphopeptide sequence was used. While awaiting FDA approval to accrue to pIRS21097-1105 peptide (Arm B), the first participant was accrued to pBCAR3126-134 (Arm A), after which accrual was randomly assigned 1:1 to arm A and Arm B until cohort conditions were satisfied. Sequential accrual to Arm C occurred after the initial safety criteria for Arms A and B were satisfied and no additional safety bound was crossed. Fifteen eligible patients were enrolled, each of whom had undergone surgical resection of stage II, III or IV melanoma, with three patients each in Arms A and B, and nine in Arm C. Patient demographics and clinical features are summarized in table 2.
Patient demographics by treatment group
Clinical toxicities
Treatment-related adverse events are detailed for all 15 patients in table 3. There were no treatment-related grade 3–4 toxicities, no deaths on study and no DLTs. All patients had grade 1 and 2 adverse events, usually limited to 24–48 hours after each vaccine. All patients developed grade 2 vaccine injection site reactions, which were more persistent and included induration in 13 patients and skin ulceration in 2 patients. Other common treatment-related grade 1 and 2 toxicities were fatigue, chills, headache, myalgias, arthralgias, autoimmune disorders, fever, nausea and diarrhea. Meaningful differences in adverse events among study arms were not evident. Autoimmune toxicities were observed in three patients: two in arm A and one in arm B, and all were grade 1. These were all treatment-associated asymptomatic elevations of serum antinuclear antibody (ANA), and were identified in patients 1A (day 183), 3B (day 85) and 4A (day 85). Three patients had pretreatment elevations of rheumatoid factor (10C) or ANA (7C, 15C). No patients developed treatment-related vitiligo.
Treatment-related adverse events
Immune responses to phosphopeptides in vaccinated patients
All 15 eligible patients were evaluable for immune responses in PBMC. Initial ex vivo ELISpot assays to detect IFNγ production in response to peptide antigen, performed on four patients representing arms A (2A), B (3B) and C (7C, 8C), were negative. Thus, subsequent ELISpot assays were performed after one in vitro sensitization for all patients.
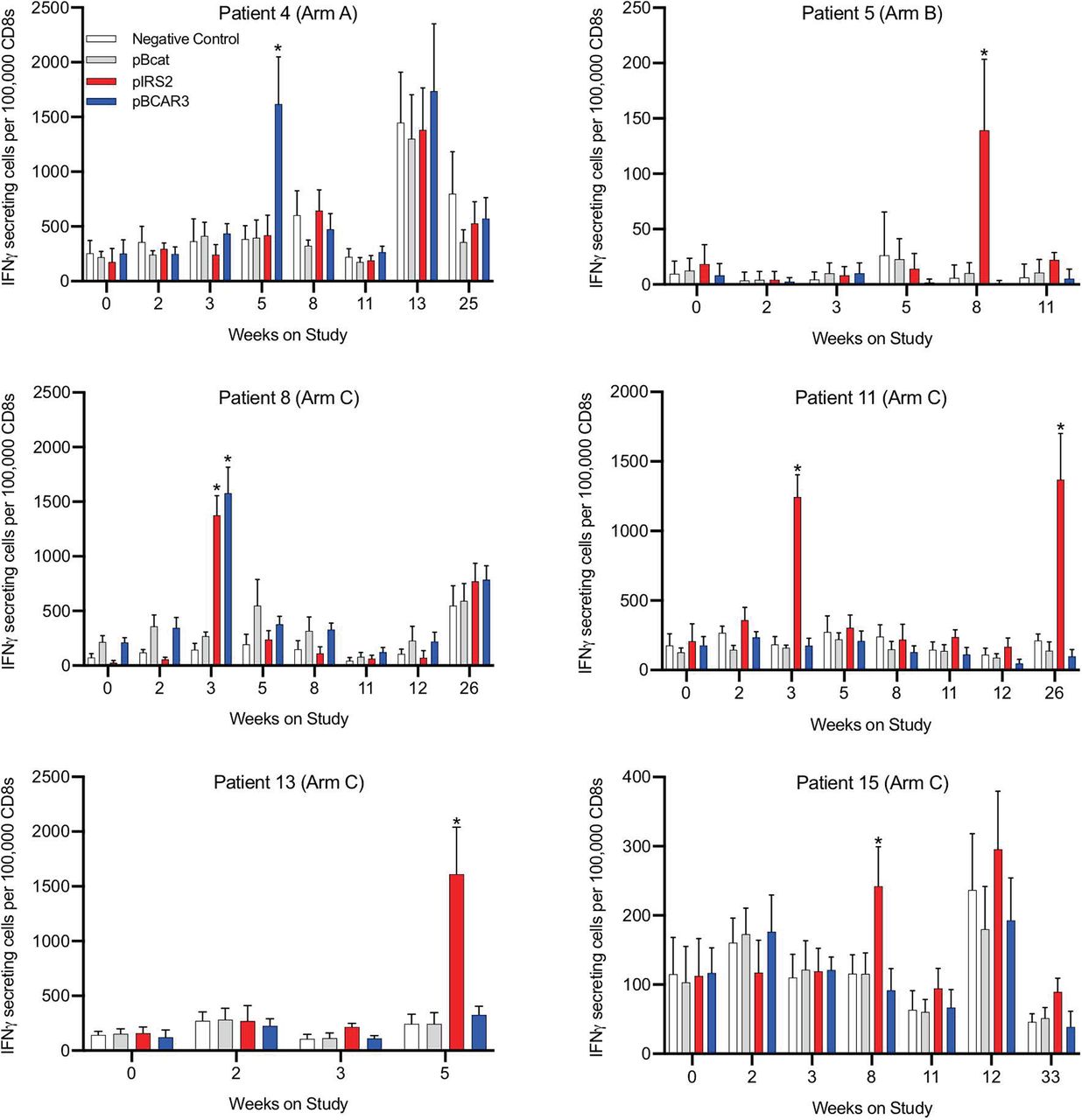
Two patients had evidence of pre-existing immune responses to phosphopeptides based on a greater than twofold increase, and increase of at least 100 spots/100 000 CD8 cells, compared with the maximum negative control at baseline. Specifically, the ratios and spot counts per 100 000 CD8 cells at baseline were as follows: patient 7, for pIRS2 (2.8x and 373 spots difference); patient 8, for pBCAR3 (2.8x and 137 spots difference) and for pbeta-catenin (which was not in the vaccine, 2.9x and 139 spots difference). Responses to vaccine required increases compared with any pre-existing responses (table 4, figure 4 and online supplementary figure 2). T-cell responses to vaccine peptides were identified against pBCAR3126-134 in 2/12 patients (17%, Arms A+C) and against pIRS21097-1105 in 5/12 patients (42%, Arms B+C) (figure 4, table 4). Overall 6 of 15 patients (40%) had a T-cell response to either or both of these peptides. The majority of immune responses were detected at a single time point, typically at weeks 3, 5 or 8. One patient (#11) also showed an additional immune response at week 26. No responses against the negative control phosphopeptide, pS33-βcat30-39, were observed at any time. In addition, responses in arm A and B patients were specific for the vaccinating peptide only. Also, there were indications of subthreshold responses in several patients at other time points that were specific for the same peptide (4A, 8C, 15C). Associations of immune response with autoimmune toxicities were not evident (table 4). These results give confidence that the significant responses observed are a consequence of vaccination, despite their transience. The study design set a goal for immunogenicity based on an upper limit of a 90% CI for the observed immune response rate of 35%. The values calculated for pBCAR3126-134 and pIRS21097-1105 were 44% and 68%, respectively, for each peptide (table 4). These both exceeded the protocol specified upper limit immunogenicity criteria target of >35% supporting further study and development. The prespecified target for immune response rate coincided with 2 or more responses in 12 evaluable patients for each peptide, with greater interest in further development with higher immune response rates.
Supplemental material
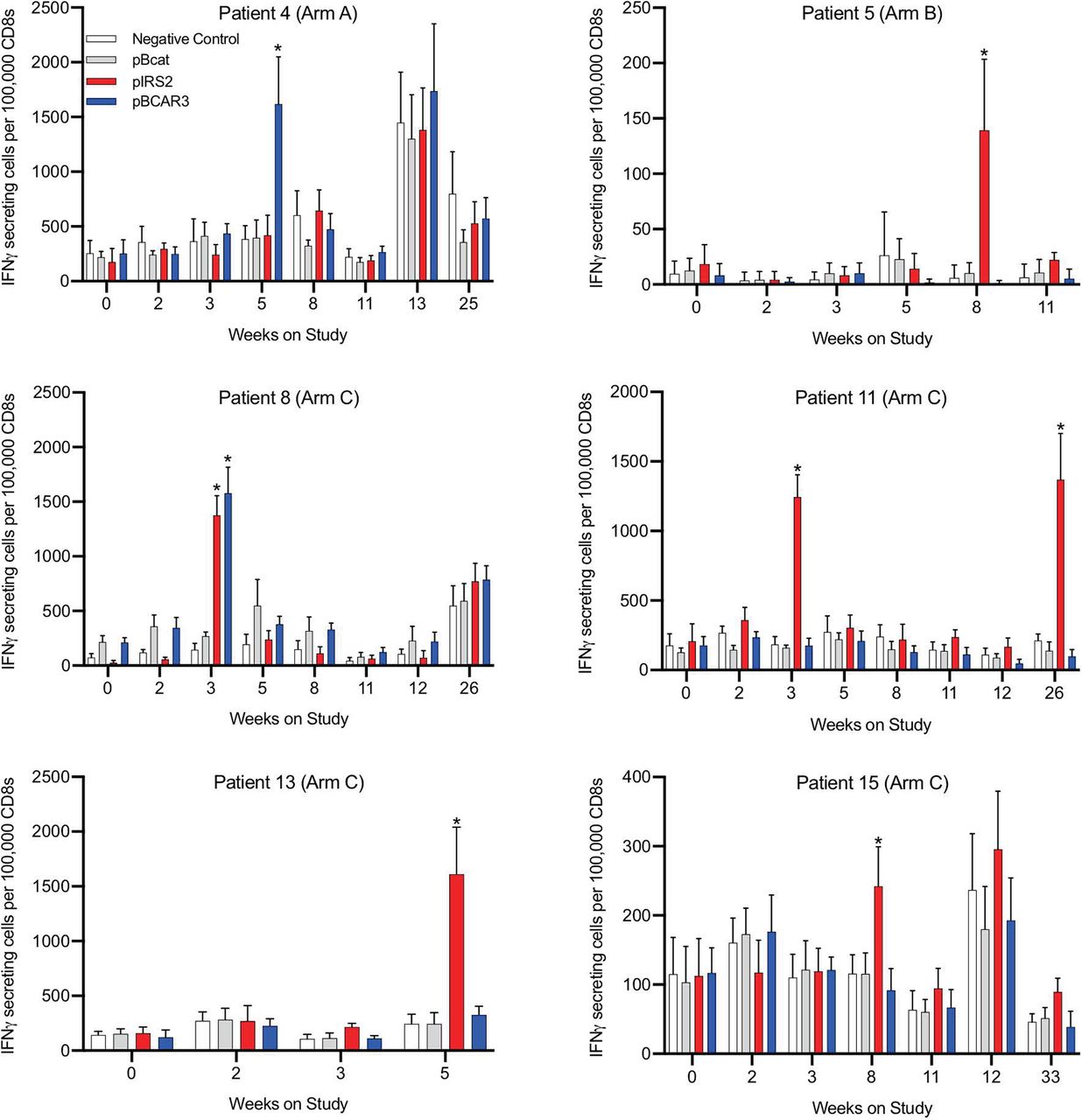
Vaccine-associated increases in immune reactivity to phosphopeptides. Immune responses to phosphopeptides were determined by ELISpot assay of 14 days in vitro stimulated peripheral blood mononuclear cells as outlined in the Methods section. Values are shown for the six patients with immune responses to either or both peptides. Time points with data that meet criteria for an immune response to pBCAR3 or pIRS2 are denoted by asterisks.
IFN-γ T-cell responses, autoimmune toxicities and clinical outcomes by patient and in aggregate
Clinical outcomes
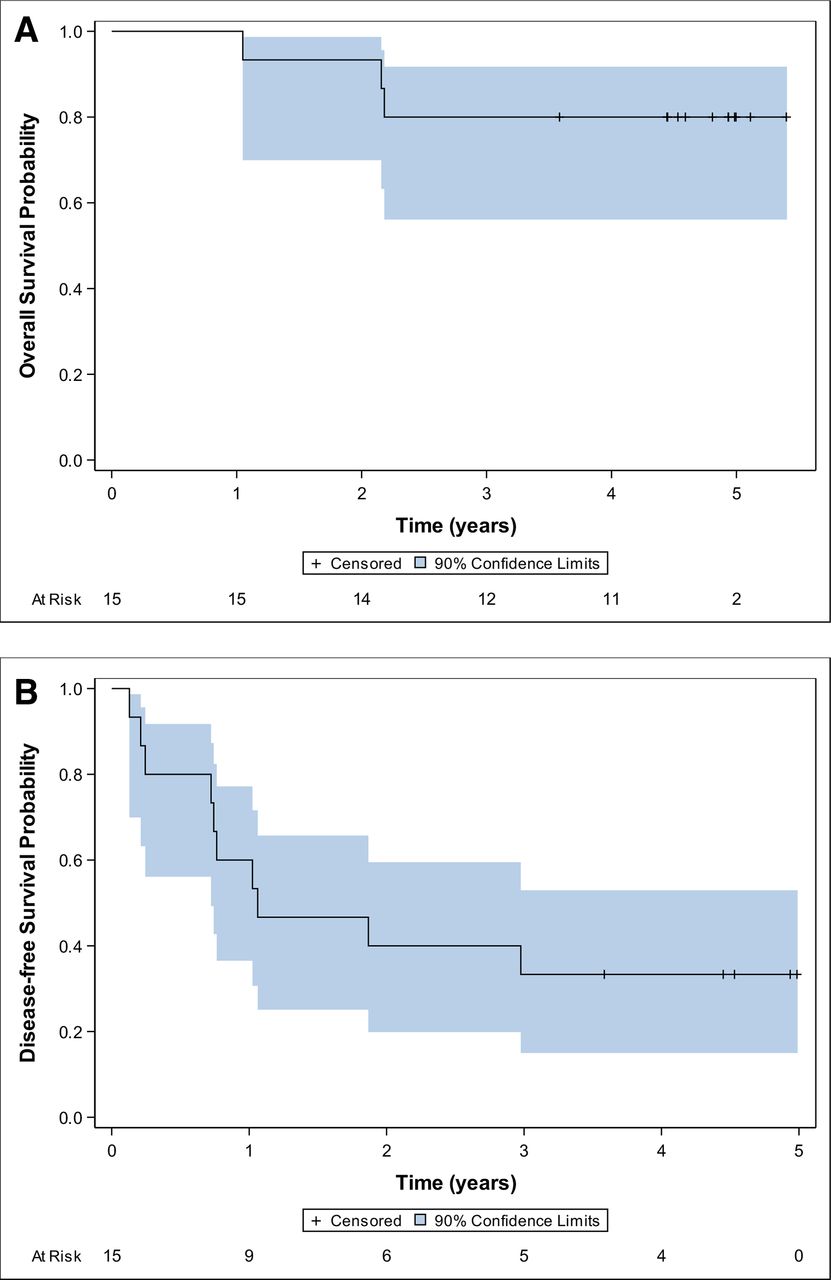
Only three patients have died from melanoma with a median follow-up of 4.9 years for those still alive. Estimated survival and disease-free survival curves are displayed in figure 5A. Four-year survival is estimated at 80%. New melanoma metastases were identified in 10 patients, with a median disease-free survival of just over 1.0 year and 4 years disease-free survival of 33% (95% CI 15% to 53%) (figure 5B). The early phase study was not powered to assess association of immune response with clinical outcome with reliable inference, as is reflected by the width of the confidence bounds. Participant specific immune responses and outcomes are noted in table 4 with no clear associations noted in this small study.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical outcomes of vaccinated patients. Kaplan-Meier estimates of overall survival (A) and disease-free survival (B) outcomes are shown for all 15 patients as of October 2019.
Discussion
Phosphopeptides function as therapeutic targets for T-cell antitumor immunity, and those that arise from cancer-associated protein phosphorylation represent an entirely new category of cancer neoantigens. We have identified hundreds of phosphopeptides from a wide range of human cancers that are collectively presented by numerous common HLA alleles. An important feature of phosphopeptides is that the phosphate can enhance binding of peptides that would otherwise bind poorly to an MHC-I molecule.22 In addition, the phosphate creates an antigenic form that is quite distinct from that of the unphosphorylated peptide,5–7 based on the presence of the phosphate in the T-cell receptor contact zone and the induction of a distinct MHC-bound peptide conformation.22 33 As a consequence, there is negligible cross-reactivity by phosphopeptide-specific T-cells on the unphosphorylated homolog.
A first challenge in selecting phosphopeptides for cancer vaccines is to define those that are uniquely presented on the surface of cancer cells, but not on normal cells. We have previously identified pIRS21097-1105 as a promising target based on its breadth of expression,7 and the identification of IRS2 as an oncogene that is overexpressed in multiple human cancers.8–10 The data presented here establish its prevalence in hepatocellular carcinoma. We have also provided new data on another promising phosphopeptide target, pBCAR3126-134. BCAR3 activates the PI3K/Akt pathway and mediates both cellular migration and acquisition of resistance to therapeutic antiestrogens in breast cancer cells, processes that are associated with malignancy.11 12 Here, we showed that BCAR3 phosphorylated at T130 is expressed in a number of malignanT-cell lines from melanoma as well as breast, that the pBCAR3126-134 peptide is immunogenic in humans and mice, and that pBCAR3126-134 specific T-cells were able to reduce the outgrowth of of human tumor xenografts. While pBCAR3126-134/BCAR3 phosphorylated at T130 was expressed in a high percentage of tumor cell lines, it was found infrequently among tumors surveyed to date, suggesting limited therapeutic potential. However, it but may be more prevalent on other kinds of tumors that remain to be evaluated. Based on the fact that both peptides are also presented by the widely expressed HLA-A2 molecule, we prioritized them for evaluation as cancer immunotherapeutic candidates.
Peptide vaccines have generally been found to be very safe,29 but the novelty of phosphopeptides, and the lack of reagents to test for their expression across all normal human tissues, makes it important to assess their safety in humans. Thus, we have performed a first-in-humans evaluation of a vaccine preparation comprised of these two phosphopeptides. Our findings demonstrate safety of vaccination with pIRS21097-1105 and pBCAR3126-134 phosphopeptides administered with IFA plus polyICLC. Adverse events were mild and comparable to those observed with other vaccines containing IFA. T-cell responses to both peptides met criteria for immunogenicity worthy of further investigation. Limitations of this pBCAR3/pIRS2 vaccine were that the immune responses were not detected ex vivo and they were usually transient, most being detected only at a single time point. Cancer vaccines with other antigens have also induced transient immune responses,34 but a goal of vaccines is to induce more durable responses. We designed the trial such that immune response rates with the upper bound of the 90% CI exceeded 35%. The fraction of patients responding to pIRS21097-1105 was 42% (90% CI 18% to 68%), but the fraction responding to pBCAR3126-134 was lower at 17% (90% CI 3% to 44%). In two earlier melanoma vaccine trials testing the immunogenicity of 12 peptides that had been previously identified as valid T-cell targets, the range of immune response rates based on ex vivo ELISpot assays for each peptide ranged from 0% to 78% and 7% to 50%, respectively, with the median immune response rates across the 12 peptides of 21% and 13%, respectively.15 17 In two trials with data based on ELIspot assays after one in vitro stimulation, those rates ranged from 0% to 75% (median 62%) and 0% to 90% (median 68%), respectively.14 15 In other work, we have established that both pIRS21097-1105 and pBCAR3126-134 can be targets of pre-existing immune memory in normal individuals, but the frequency of responders to pIRS21097-1105 is higher (Lulu, Cummings and Engelhard, unpublished). That prior finding is supported by evidence of pre-existing immunity to those same peptides in one patient each, as well as pre-existing immunity to a third peptide (not included in the vaccine), from phospho-beta-catenin. While the natural exposure leading to pre-existing immune memory to phosphopeptides is not known, these results could indicate that pIRS21097-1105 is inherently more immunogenic. On the other hand, the magnitude of vaccine-induced responses to pBCAR3126-134 was comparable to those induced to pIRS21097-1105. This raises the possibility that patient-specific factors as well as differences in inherent immunogenicity may be factors in limited responses to pBCAR3126-134.
An important goal for future investigation is to identify vaccination strategies that enhance the magnitude and persistence of the immune responses. We have found that vaccination with cocktails of peptides can enhance overall immune response rate to 100% even though individual peptides may have lower immune response rates.14 Thus, one strategy to enhance the immunologic activity of phosphopeptide vaccines is to create cocktails with additional phosphopeptide antigens, and to vaccinate with peptides modified to enhance MHC binding (eg, pBCAR3(V)126-134). Another approach is to optimize the vaccine adjuvants. The TLR3 agonist poly-ICLC has enhanced immune responses to other peptide vaccines, especially when combined with IFA.35 In the present trial, the poly-ICLC was not incorporated in the peptide/IFA emulsion, but was administered by separate injection into the same site as the emulsion. In other experience where poly-ICLC enhanced immunogenicity of peptide vaccines containing IFA, poly-ICLC was incorporated in the emulsion.35 36 Thus, other approaches to enhance the immune responses to phosphopeptides include incorporation of TLR agonists directly in the vaccine emulsion or vaccination in combination with other immune adjuvants, such as agonistic CD40 antibody. The pBCAR3126-134 peptide may also be rendered more immunogenic if the C-terminal residue is modified to valine, as per the preliminary data. It is also important to establish whether the transience of responses can be mitigated by the inclusion of checkpoint blockade inhibitors, or other modalities that target immunosuppressive mechanisms.
While phosphopeptides represent a new class of tumor neoantigens, the vaccine strategies that employ them are also worthy of further investigation. It would be difficult to rely on endogenous kinase activity to phosphorylate proteins expressed via a genetically based vaccine, and at present we expect that phosphopeptide vaccines will remain based on synthetic peptides. It would be useful to explore whether such vaccines might benefit from the use of longer peptides, which have shown promise in other studies,37–39 or by coupling them to various lipids or adjuvants.37–39 While we presently have no evidence that the phosphate moiety is removed by plasma phosphatases, we have found that MHC binding stabilizes the phosphate against removal by phosphatases in vitro (Obeng et al, unpublished). Thus, some consideration to the use of non-hydrolyzable phosphophate analogs is reasonable, although these have frequently been shown to be antigenically distinct.40 The availability of preclinical models employing HLA transgenic mice is of particular value in testing these approaches on peptides of human origin prior to incorporating them into clinical trials.
Conclusions
The findings from this clinical trial highlight the safety of vaccination with phosphopeptides arising from oncogenic phosphoproteins. The pIRS21097-1105 and pBCAR3126-134 phosphopeptides are expressed not only by melanomas, but also by several additional kinds of cancer cells. Thus, our results open the door to vaccines using these phosphopeptides for an array of other cancers. Enhancing phosphopeptide immunogenicity with other adjuvant strategies is expected to elevate the promise for this approach, and to increase the prospect for clinical benefit. Phosphopeptides are presented by most MHC-I alleles so far examined, enabling the approach to be exploited to a broad range of patients. In addition, individuals without a clinical diagnosis of cancer often have T-cells that recognize some of these phosphopeptides30 (Lulu, Cummings and Engelhard, unpublished). An intriguing possibility is that these T-cell responses may arise as a result of successful immune surveillance of early cancers, and it is notable that patients who develop clinical cancers typically have diminished T-cell responses to phosphopeptides.30 The presence of T-cell responses to phosphopeptides in otherwise healthy individuals also provides strong evidence that T-cells reactive to those peptides are safe and that induction of such responses in cancer patients will not induce significant autoimmunity to normal tissues. The present findings provide rationale for next generation phosphopeptide vaccines targeting a larger number of antigens presented by a range of MHC-I alleles, and also provide a rationale for adoptive cell therapies targeting these peptides.
Acknowledgments
We acknowledge assistance of Donna Deacon for vaccine preparation and Carmel Nail and Patrice Neese for assistance in vaccine administration. We acknowledge Erin Jeffrey, Paisley Myers, Dennis Underwood and Andy Hurwitz, all of Agenus, Inc., for sharing information on expression of pBCAR3126-134 and pIRS21097-1105 on tumor samples.
References
Footnotes
VHE, RCO, KLC, GRP, ALA and KAC-B contributed equally.
Contributors RCO, KLC and AA performed the preclinical cellular immunological studies. NB and MC procured hepatocellular carcinoma samples and performed peptide extraction. PM, KM, JS and DFH conducted mass spectrometry and analyzed the data. AA, KAC-B, GRP and CLS designed the clinical trial. KH, GW, EG and CLS recruited patients, administered vaccines and assessed adverse events. EA was the primary clinical research coordinator, who also participated in patient enrollment, safety assessments, and data management and reporting. KTS, KLC, AL and NV analyzed patient immune responses. GRP and MS conducted statistical analyses of preclinical and clinical immune response data. VE, RCO, KLC, GRP and CLS summarized immunological and clinical outcomes and participated in writing the manuscript. All authors have reviewed the manuscript.
Funding This study was supported by USPHS grants R01 CA134060 and CA190665 (VE), R01 AI033993 (DFH), P30 CA44579 (University of Virginia Cancer Center: Biorepository and Tissue Research Facility, Biostatistics), the Commonwealth Foundation for Cancer Research, and the Ludwig Institute for Cancer Research. RCO was supported by USPHS grant F31 CA119954, AA was a recipient of a Sidney Kimmel Scholar award, and AL was supported by USPHS grant T32 CA009109.
Competing interests VE, RCO, KLC, AL, PM, JS and DFH have ownership interest in Agenus, which has licensed phosphopeptide technology and associated patent applications from the University of Virginia Licensing and Ventures Group. PM is now an employee of Agenus. VE is a consultant and the recipient of a Sponsored Research Agreement from Agenus. CLS is the recipient of research grants from GlaxoSmithKline, Merck, and Celldex, and is an inventor for patents for other peptides used in cancer vaccines, which are held by the UVA Licencing and Ventures Group; and is (or has been) a consultant/advisory board member for Immatics. Curevac, and Polynoma. MC is now an employee of AstraZeneca.
Patient consent for publication Not required.
Ethics approval Patients were studied following informed consent, and with University of Virginia Health Sciences Institutional Review Board (IRB#15832) and FDA (BB-IND #15134) approval. Animals were housed and bred in pathogen-free facilities and used between 6 and 22 weeks of age. The University of Virginia Animal Care and Use Committee approved all procedures. Experiments were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplementary information. All relevant data generated or analyzed during this study are included in this published article (and its online supplementary information files). Additional data generated from the clinical trial during the current study are available from the corresponding author on reasonable request.