Article Text

Abstract
Background The use of checkpoint inhibitors has revolutionized cancer therapy. Unfortunately, these therapies often cause immune-related adverse effects, largely due to a lack of tumor specificity.
Methods We stained human natural killer cells using fusion proteins composed of the extracellular portion of various tumor markers fused to the Fc portion of human IgG1, and identified Nectin4 as a novel TIGIT ligand. Next, we generated a novel Nectin4 blocking antibody and demonstrated its efficacy as a checkpoint inhibitor in killing assays and in vivo.
Results We identify Nectin4 to be a novel ligand of TIGIT. We showed that, as opposed to all other known TIGIT ligands, which bind also additional receptors, Nectin4 interacts only with TIGIT. We show that the TIGIT-Nectin4 interaction inhibits natural killer cell activity, a critical part of the innate immune response. Finally, we developed blocking Nectin4 antibodies and demonstrated that they enhance tumor killing in vitro and in vivo.
Conclusion We discovered that Nectin4 is a novel ligand for TIGIT and demonstrated that specific antibodies against it enhance tumor cell killing in vitro and in vivo. Since Nectin4 is expressed almost exclusively on tumor cells, our Nectin4-blocking antibodies represent a combination of cancer specificity and immune checkpoint activity, which may prove more effective and safe for cancer immunotherapy.
- immunology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Cancer immunotherapy with checkpoint inhibitors has shown great success in treating a wide variety of cancers. Given the immunomodulatory nature of these therapies, many of their side effects stem from immune overactivation. These are so common they have been named ‘immune-related adverse events’. While many are easily managed, some of these autoimmune-like syndromes can prove fatal.1 To help avoid side effects, we searched for immunomodulatory molecules with specificity to tumors.
Natural killer (NK) cells are innate lymphocytes which are best known for their ability to kill virally infected and transformed cells. NK cell activity is regulated by a balance of signals they receive from a variety of activating and inhibitory receptors.2
The inhibitory receptor T-cell immunoreceptor with Ig and ITIM domains (TIGIT) is considered a major new target for cancer immunotherapy.3 It is expressed on most immune cells,3 4 including NK cells and CD8+ tumor infiltrating lymphocytes (TILs) in non‐small cell lung cancer, colon cancer and melanoma.5 Indeed, several anti-TIGIT immunotherapies have already reached phase I or II in clinical trials.5 TIGIT’s known ligands arepoliovirus receptor (PVR), Nectin2 and Nectin3—some members of the Nectin family are overexpressed in many cancers.5–7 Some even serve as diagnostic indicators for several cancers.5 6 8 Interestingly, these ligands are also recognized by the coactivating receptors DNAM1 and CD96 (the latter of which is sometimes considered inhibitory) and by the inhibitory receptor CD112R.7 9
In contrast to the other Nectins, which are found extensively in adult tissues,10 Nectin4 is abundant during fetal development but its expression declines in adult life. Its expression, however, returns specifically in cancers of the breast, bladder, lung and pancreas,11 among others. Nectin4 expression is also associated with poorer prognostic features.10
Here, we report that Nectin4 is a cancer-specific TIGIT ligand, and the only Nectin family member that interacts with TIGIT alone. We have developed a checkpoint blocking mAb against Nectin4, and show its efficacy in vitro and in vivo. Given the properties of Nectin4, our antibody represents a unique synergy between checkpoint inhibition and tumor specificity, which may prove beneficial in clinical use.
Methods
Cells
MDA-MB-453, SK-BR-3, T47D and lymph node carcinoma of the prostate (LNCAP) cells were maintained in Dulbecco’s modified Eagle’s medium and Raji cells were maintained in Roswell park memorial institute culture medium(RPMI) medium, both supplemented with 10% fetal calf serum, 1% Pen/Strep, 1% L-glutamine, 1% MEM Eagle and 1% sodium pyruvate. All cells were incubated in a humidified incubator with 5% CO2 at 37°C.
Fusion proteins
TIGIT-Ig, DNAM1-Ig, CD112-Ig, PVR-Ig, CD96-Ig, Nectin4-Ig ANPEP-Ig, PDGFRa-Ig, HAVcR-1-Ig, CD46-Ig, EFNB2-Ig and murine TIGIT-Ig fusion proteins were cloned into an expression vector containing the mutated Fc portion of human IgG1 (Fc mut pIRESpuro3). The resulting constructs were transfected into HEK-293T cells using the TransIT-LT1 Transfection reagent (Mirus Bio). After 48 hours, transfected cells were subjected to antibiotic selection with 5 μg/mL puromycin (Sigma-Aldrich). Stable pools were analyzed for protein secretion by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS/PAGE). Supernatants were collected and purified on a HiTrap Protein G HP column in the High Pressure Perfusion Chromatography Station, BioCAD (PerSeptive Biosystems) as previously described.12 Nectin4-Ig was generated using these primers: Forward primer: GGGGAATTCGCCGCCACCATGCCCCTGTCCCTGGGA Reverse primer: GGGGGATCCGAGGCTGACACTAGGTCC. HAVcR-Ig was generated using these primers: Forward primer: GGGAATTCGCCGCCACCATGCATCCTCAAGTGGTCA Reverse primer: GGGGATCCCCTTTAGTGGTATTGGCCGT. CD46-Ig was generated using these primers: Forward primer: GGGAATTCGCCGCCACCATGGAGCCTCCCGGCCG Reverse primer: GGAGATCTAAACTGTCAAGTATTCCTTCCTCA. EFNB2-Ig was generated using these primers: Forward primer: GGGCTAGCGCCGCCACCATGGCTGTGAGAAGGGACTCC Reverse primer: GGGGATCCGCAAATAAGGCCACTTCGGAAC. ANPEP-Ig was generated using these primers: Forward primer: AAAACTAGTTACTCCCAGGAGAAGAACAAGA Reverse primer: TTTCTCGAGCTATTTGCTGTTTTCTGTGAACC. PDGFRa-Ig was generated using these primers: Forward primer: AAAGCTAGCGCCGCCACCATGGGGACTTCCCATC Reverse primer: TTTGGATCCGCCACCGTGAGTTC. DNAM1-Ig was generated using these primers: Forward primer: CCGATATCGCCGCCACCATGGATTATCCTACTTTACTTTTG Reverse primer: CGGATCCACAAAGAGGGTATATTGGTTAT. TIGIT-Ig was generated using these primers: Forward primer: ATGCGCTGGTGTCTCCTCCT Reverse primer: TCTTCACAGAGACTGGTTAG. Murine TIGIT-Ig was generated using these primers: Forward primer: CCGAATTCGCCGCCACCATGCATGGCTGGCTGCTCCT. Reverse primer: CGGATCCGGGGCAGTCTGGAACTGAG. CD112R-Ig was generated using these primers: Forward primer: ATGCAGATCCCACAGGCGCC Reverse primer: CCCCCATTCTGCGGGCAGAC.
The fusion proteins used in this work were regularly assayed by SDS protein gels to ensure that the proteins are not degraded. Protein purity of all Ig fusion proteins used approached 100%.
Generation of lentivirus overexpression
Nectin4 was cloned into the pHAGE-DsRED(−)-eGFP(+) lentiviral vector which also contains GFP. Lentiviruses were generated in 293 T cells using a transient three-plasmid transfection protocol as previously described.13 Transduction efficiency into Raji cells was assessed by GFP expression and only cell populations with >90% efficiency were used for experiments. Primers sequences: Human Nectin4 forward: GGGCGGCCGCGCCGCCACCATGCCCCTGTCCCTGGGA, Human Nectin4 reverse: GGACCGGTTCAGACCAGGTGTCCCCGC. Murine Nectin4 forward: GCGGCGGCCGCGCCGCCACCAGCCAATGCCCCTGT, Murine Nectin4 reverse: CGCACCGGTGGCCTGGGTCAGACC.
Mouse thymoma (BW) assay
The generation of chimeric TIGIT and DNAM1 and expression in BW cells was previously described in details.14 15 In brief, we expressed chimeric proteins composed of the extracellular portions of TIGIT and DNAM1 fused to the mouse ζ-chain in the BW murine thymoma cell line. The BW assay was performed as previously described. Briefly, BW or BW transfected cells were incubated with irradiated targets (6000 rad) at 1:1 effector to target ratio. After 48 hours, the supernatants were collected and the level of interleukin-2 (IL2) was quantified by sandwich ELISA using anti IL2 mAbs (BioLegend).
Generation of anti-Nectin4 antibodies
The anti-Nectin4 mAbs (clone .01 and clone .05) were generated by immunizing mice with Nectin4-Ig fusion proteins and then by using standard hybridoma techniques. The hybridomas were cultured in RPMI—medium and the antibodies were purified on a protein G column. All antibodies generated are of the IgG1 isotype.
Flow cytometry
Extracellular fluorescence-activated cell sorting (FACS) staining for Nectin4 (MAB2659, R&D Systems), Nectin2 (BLG-337402, BioLegend), PVR (MAB25301, R&D Systems) DNAM1 (BLG-338302, BioLegend) and TIGIT (MAB7898, R&D Systems) was performed with 0.2 μg antibody per 100 000 cells. Binding was detected by a secondary antibody (Alexa Flour 647 anti-mouse IgG, Jackson, cat. 115-606-062), at a dilution of 1:200, incubated for 1 hour on ice. Extracellular FACS staining with TIGIT-Ig, DNAM1-Ig, CD112-Ig, CD96-Ig, PVR-Ig, Nectin4-Ig and murine TIGIT-Ig fusion proteins was performed with 3 μg/well. Binding was detected by a secondary antibody (Allophycocyanin antihuman IgG, Jackson, cat. 709-136-098), at a dilution of 1:200, incubated for 1 hour on ice. For antibody blocking experiments, cells were preincubated with the indicated antibody for 1 hour on ice. All steps were performed in a Phosphate buffered saline (PBS)-based buffer containing 5% bovine serum Albumin (BSA) and 0.05% sodium azide. All staining was assessed using FACS with CellQuest software and analyzed with FCSexpress.
Microscale thermophoresis
The microscale thermophoresis (MST) experiments were performed as previously described.16 Briefly, labeled TIGIT-Ig was mixed with Nectin4-Ig or PVR-Ig (figure 1G) and labeled Nectin4-Ig was mixed with either anti-Nectin4 mAb clone .01 or clone .05 (figure 2E) in standard capillaries at 25°C. Titration of the non-fluorescent antibodies resulted in a gradual change in thermophoresis, which was plotted as a change in the normalized fluorescence (Fnorm), to yield a binding curve.
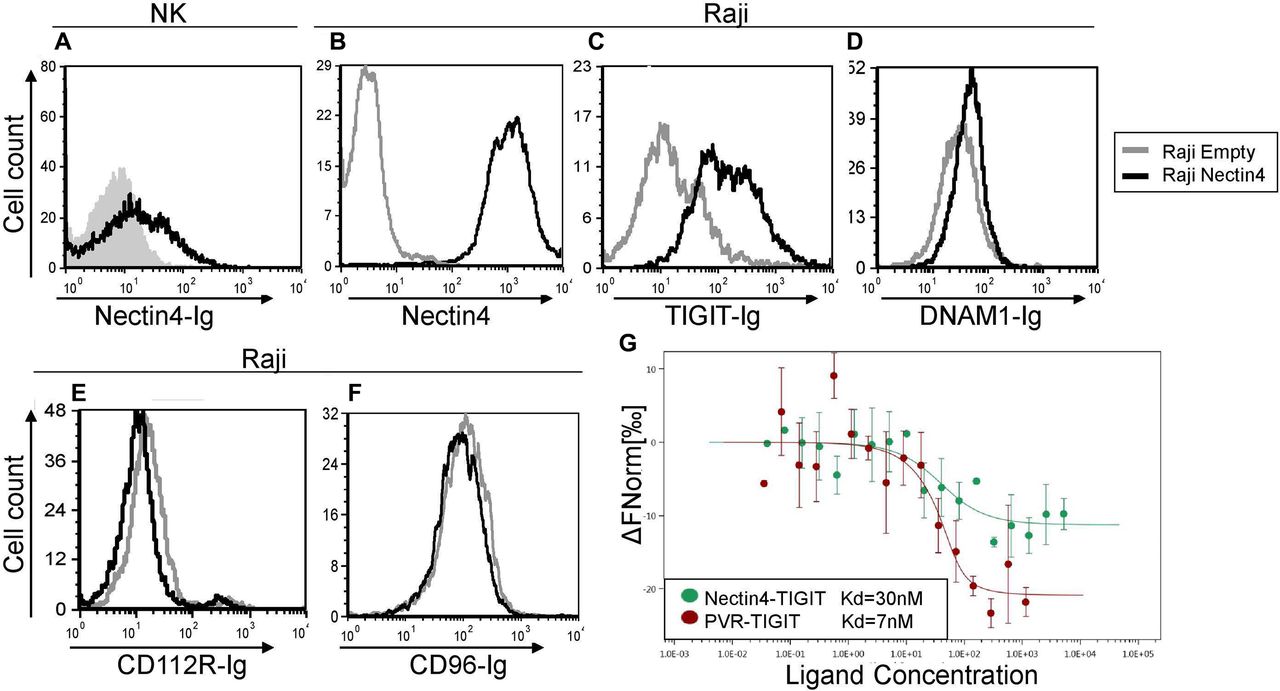
TIGIT but not DNAM1, CD112R or CD96—interacts with Nectin4. (A) FACS staining of IL-2 activated primary NK cells with Nectin4-Ig. Gray filled histogram represents background staining with secondary antibody only; black line histogram represents specific binding as indicated. (B–F) FACS staining of Raji cells transfected either with an empty vector as control (gray histograms) or with Nectin4 (black histograms). Cells were stained with commercial anti-Nectin4 mAb (B), TIGIT-Ig (C), DNAM1-Ig (D), CD112-Ig (E) or CD96-Ig (F). Figures show one representative experiment out of three performed. Graph depicting the mean fluorescence intensity values of the stainings appears in online supplementary figure 2. (G) Direct binding of Nectin4 to TIGIT. The binding of fluorophore-labeled TIGIT-Ig and its ligands PVR-Ig (red) and Nectin4-Ig (green) was determined using MST. Measurements were repeated with at least three independent protein preparations. FACS, fluorescence-activated cell sorting; IL-2, interleukin-2; MST, microscale thermophoresis; NK, natural killer.
Supplemental material
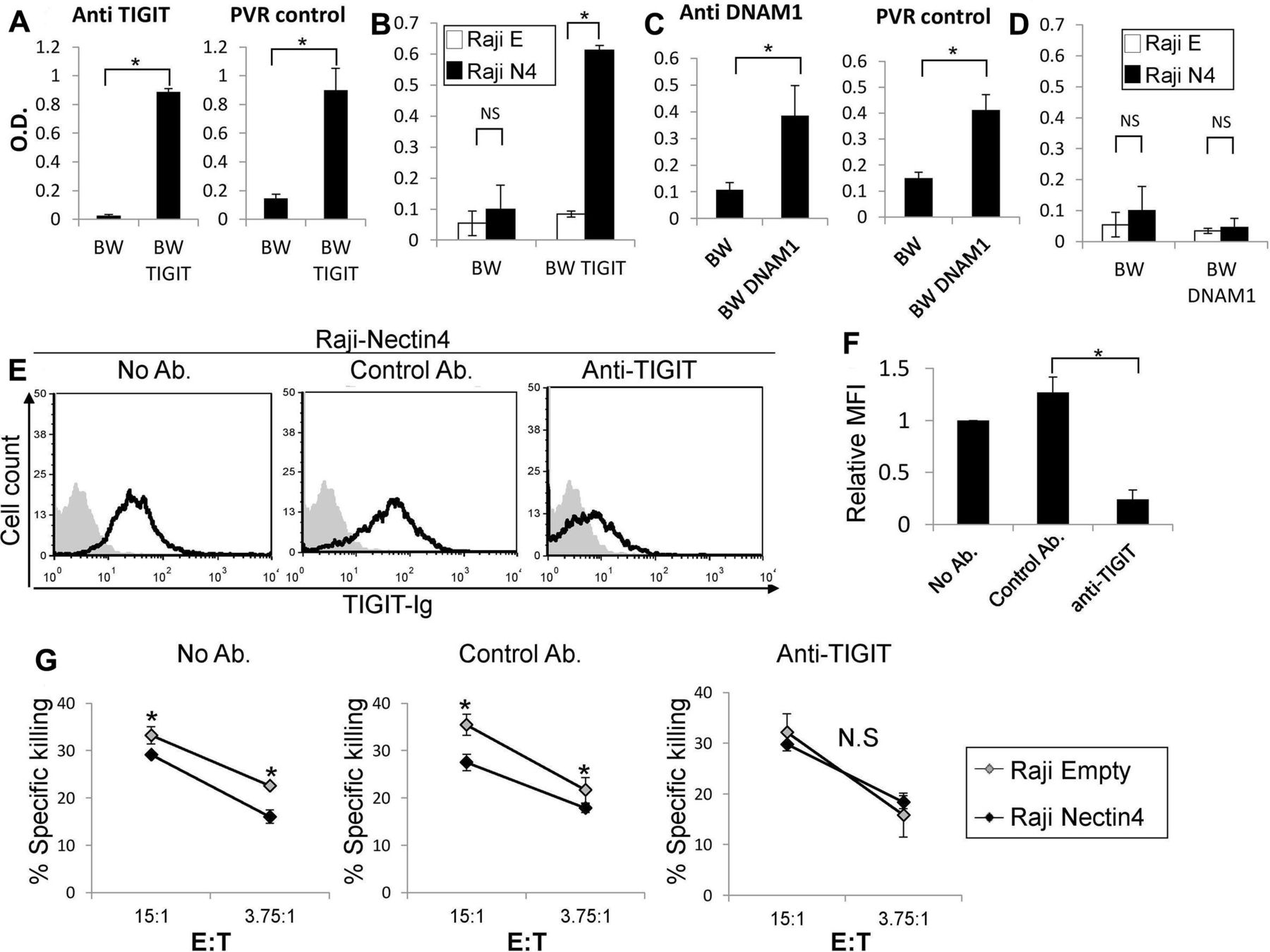
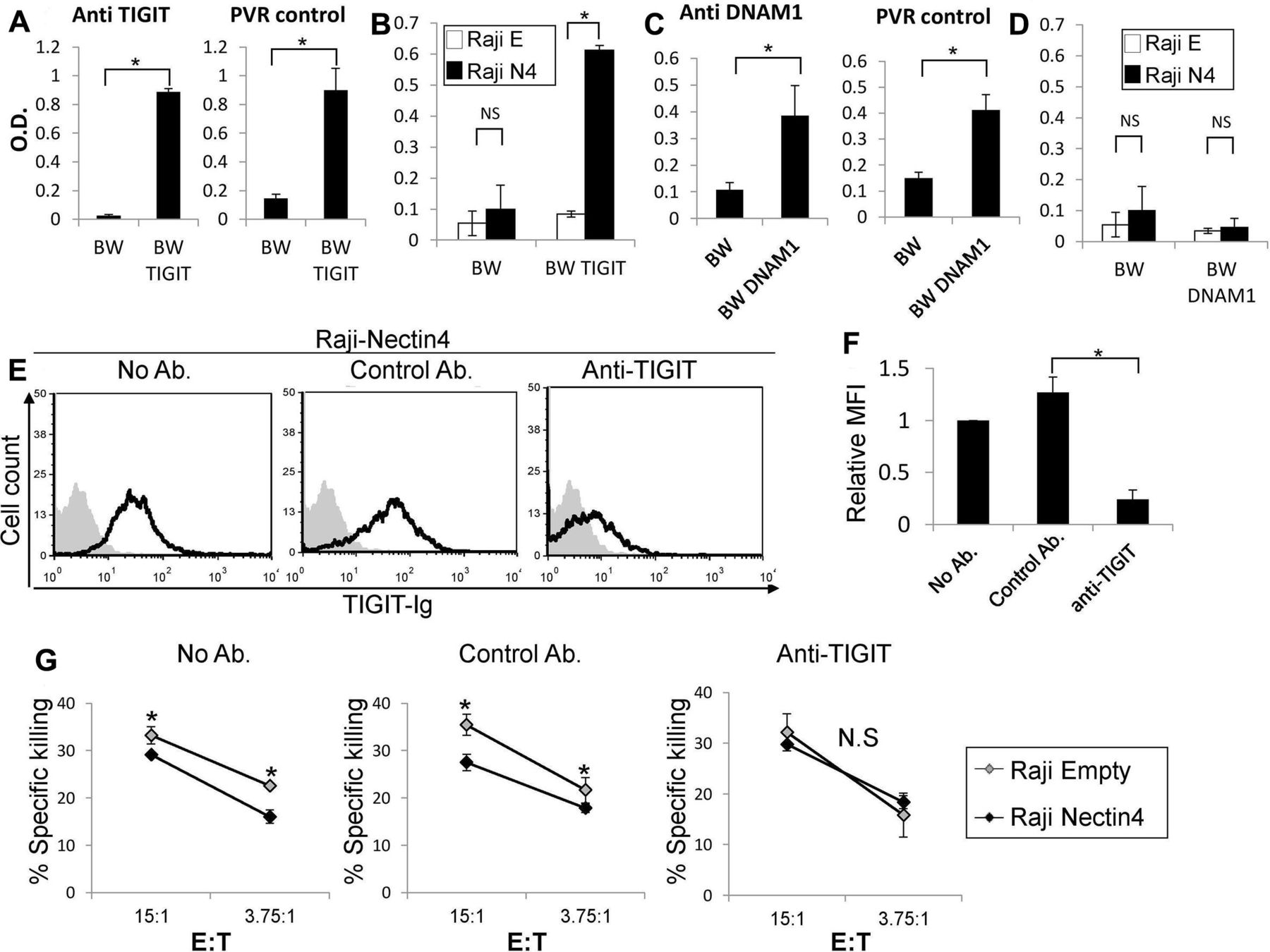
.Nectin4 inhibits NK cytotoxicity via TIGIT. (A, C) IL2 secretion by parental BW (A, C), (A) BW-TIGIT and (C) BW-DNAM1 cells. IL2 secretion was determined by ELISA (od 650 nm) following incubation with control anti-TIGIT or anti-DNAM1 antibodies (left in a and C) or with PVR expressing cells (right in a and C). (B, D) IL2 secretion of parental BW (B, D), (B) BW-TIGIT and (D) BW-DNAM1 cells. IL2 secretion was determined by ELISA (od 650 nm) following incubation with Raji cells transfected either with an empty vector (Raji E) as a control, or with Nectin4 (Raji N4). Figure shows one representative experiment out of 3 performed. *P<0.05. (E) FACS staining of Raji cells overexpressing Nectin4 with TIGIT-Ig. TIGIT-Ig was preincubated with no antibody (left), with a control mAb (anti-CD99 mAb clone 12E7, middle) or with anti-TIGIT blocking antibody (mAb #4 generated as described previously,14 right). Black line histograms represent TIGIT-Ig binding. Gray filled histograms represent background staining of the secondary antibody only. (F) Mean fluorescence intensity (MFI) values of the TIGIT-Ig staining shown in (E) relative to NO antibody staining, *p<2×10-4. (G) [35S] methionine-labeled Raji cells transfected either with an empty vector as control (Raji empty—gray) or with Nectin4 (Raji Nectin4—black), were incubated for 5 hours with NK cells. NK cells were preincubated with no antibody (left), with a control antibody (anti-CD99 mAb clone 12E7, middle) or with an anti-TIGIT antibody (mAb #4 generated as described previously,14 right). The effector to target (E:T) ratios are indicated on the x-axis. Figure shows one representative experiment out of three performed. Shown is the relative average killing ±SD, *p<0.05. FACS, fluorescence-activated cell sorting; IL2, interleukin-2; NK, natural killer; NS, not significant.
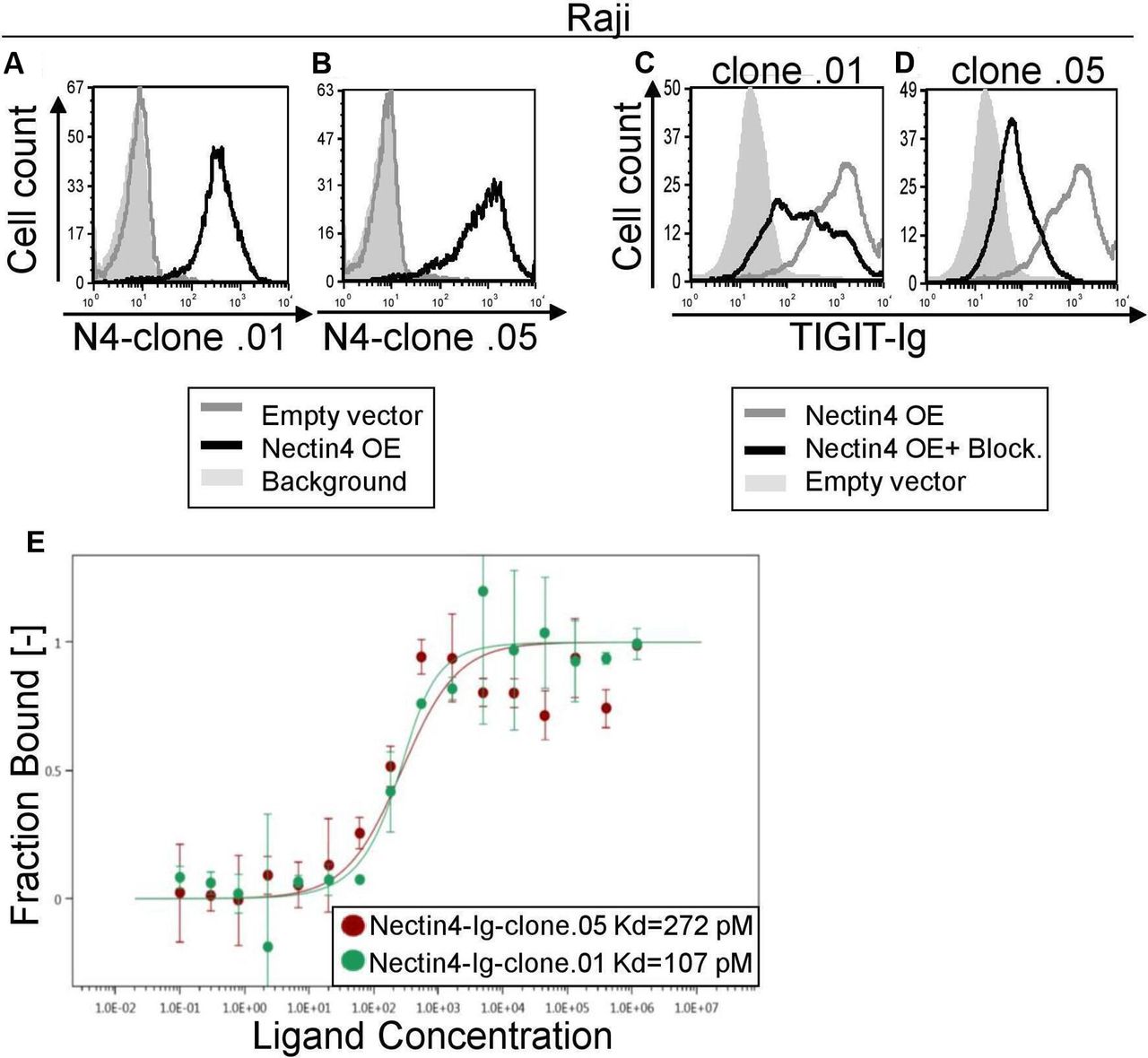
Novel checkpoint inhibitor anti-Nectin4 antibodies block TIGIT binding. (A, B) FACS staining of Raji cells transfected either with an empty vector (gray histograms) or with Nectin4 over-expression (black histograms). Gray filled histograms represent background staining of secondary antibody only. Cells were stained with anti-Nectin4 mAb clone .01 (A) or clone .05 (B). (C, D) FACS staining with TIGIT-Ig of Raji overexpressing Nectin4 with (black histograms) or without (gray histograms) preincubation with anti-Nectin4 mAb clone .01 (C) or clone .05 (D). Gray, filled histograms represent background staining. Figures show one representative experiment out of three performed. Graph depicting the mean fluorescence intensity values of the stainings appears in online supplementary figure 3a–d. (E) Avidity quantification between Nectin4-Ig and anti-Nectin4 mAb clone .01 and clone .05 using MST. Measurements were repeated with at least three independent protein preparations. FACS, fluorescence-activated cell sorting; MST, microscale thermophoresis
NK cell killing assay
To evaluate NK cell cytotoxic activity against target cells, [35S] release assays were performed as described previously.17 For antibody blocking, cells were preincubated with the indicated antibodies for 1 hour on ice. The reaction mixtures were incubated for 5 hours. Percentages of killing were calculated as follows: [counts per minute (cpm) (sample) − cpm (spontaneous release)]/[cpm (total release) − cpm (spontaneous release)]×100.
Western blot analysis
Lysates of the cells were prepared and SDS gel electrophoresis was performed. Proteins were transferred onto a nitrocellulose membrane with the tank blot procedure and specific protein bands were detected using antibodies detecting murine Nectin4 (MAB3116, R&D Systems (1:200)) or Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (SC-32233, Santa Cruz (1:1000)) as loading control. Antibodies were diluted in 5% BSA in PBS. Chemiluminescence from detection antibody-linked horseradish peroxidase (Jackson ImmunoResearch, Pennsylvania, USA) was detected.
Mouse tumor development
All experiments were performed using 6–8 weeks severe combined immunodeficiency disease (SCID)-beige female mice. All mice were housed under Specific pathogen-free (SPF) conditions, in normal light/dark cycles at 22°C±2°C. All experiments were performed in accordance with the guidelines of the ethics committee of the Hebrew University Medical School (Ein-Kerem, Jerusalem). Every group of mice contained seven females (n=7). Xenografts were generated by administering the indicated cells subcutaneously into the left flank region. Injections of 75 ug anti-Nectin4 clone .05 or control (anti-CD3, InVivoMAb-clone 17A2) antibodies were administrated intraperitoneally twice a week, starting from the time point when the tumor could be seen as a white dot under the skin. The mice were monitored daily, and sacrificed at any indication of illness such as bristled fur, difficult breathing, or tremor, among others. When tumors reached a maximal volume of 1000 mm3, all mice were sacrificed. No differences were observed between the various groups in their general health at baseline.
Statistical analysis
All p values were determined using Student’s t-test, except the Kaplan-Meier curves p values, which were generated from the X2 score with 1 df.
Results
Nectin4 interacts with the immune checkpoint receptor TIGIT
In testing whether certain tumor markers interact with NK cells, we generated fusion proteins composed of the extracellular portion of various tumor markers, including Nectin4, fused to the Fc portion of Human IgG1 (Nectin4-Ig). We next stained human NK cells and observed staining only with Nectin4-Ig (figure 1A, online supplementary figure 1a–e). Since other Nectin family members interact with TIGIT, DNAM1, CD112R and CD96,7 we next tested whether Nectin4 interacts with these receptors. We overexpressed Nectin4 on Raji cells, which do not normally express any ligands of these receptors, including Nectin4 (figure 1B, online supplementary figure 1f–h). We stained the transfected cells with TIGIT-Ig, DNAM1-Ig, CD112R-Ig and CD96-Ig. Interestingly, Nectin4 overexpression led to increased binding of the inhibitory receptor TIGIT-Ig alone (figure 1C–F), uniquely allowing for a purely inhibitory effect.
To demonstrate that Nectin4 interacts directly with TIGIT, we used MST. We found that TIGIT binds Nectin4 with relatively high affinity, similar to that of PVR-TIGIT binding (figure 1G). Collectively, we concluded that Nectin4 is not only a TIGIT ligand, but also the only Nectin family member that interacts exclusively with TIGIT, and not with DNAM1, CD96 or CD112R.
Nectin4 is a functional ligand for TIGIT
To further determine the binding of Nectin4 to TIGIT, we used a cell-based reporter assay. We expressed a chimeric protein composed of TIGIT fused to mouse ζ-chain in the mouse BW thymoma cell line and named the cells BW TIGIT (online supplementary figure 1i). In this system, BW cells secrete IL2 on engagement of the chimeric TIGIT receptor by its ligand (present on target cells), thus reporting for binding. We determine the amount of IL2 in the supernatants using ELISA. To test that the reporter system is functional, we initially triggered the chimeric TIGIT protein with antibody or incubated the BW TIGIT cells with cells expressing PVR, a known TIGIT ligand. In both cases, IL2 secretion was observed when BW TIGIT cells were used, while little or no IL2 secretion was detected when parental BW cells were used (figure 2A). We then incubated BW parental and BW TIGIT cells with RAJI cells transfected either with Nectin4 (RAJI N4) or empty vector (RAJI E). Little or no IL2 was observed when the parental BW cells were incubated with RAJI cells with or without Nectin4 expression (figure 2B). In contrast, when BW TIGIT cells were incubated with RAJI Nectin4 cells, significantly more IL2 was secreted as compared with RAJI Empty cells (figure 2B), indicating, using an isolated reporter system, that TIGIT interacts with Nectin4.
As a negative control, we used DNAM1 that is also known to interact with PVR.5 BW expressing DNAM1 cells were generated, in a similar manner to the BW TIGIT cells described above, and expression of DNAM1 was verified (online supplementary fiugre 1j). We verified that the BW DNAM1 reporter cells function by stimulating with anti-DNAM1 mAb or with cells expressing PVR (figure 2C). In agreement with the lack of Nectin4 recognition by DNAM1-Ig, no significant IL2 expression was observed on BW DNAM1 incubation with either Raji Empty vector or Raji Nectin4 cells (figure 2D).
We next sought to address the impact of TIGIT-Nectin4 interaction using primary human NK cells. We first tested whether a blocking anti-TIGIT mAb14 impairs this interaction. On preincubation of the anti-TIGIT mAb with TIGIT-Ig, binding of TIGIT-Ig to cells overexpressing Nectin4 was reduced (figure 2E, summarized in figure 2F). We also performed NK cytotoxicity assays with activated primary human NK cells that express TIGIT (online supplementary figure 1k). As expected, expression of Nectin4 on Raji cells led to reduced NK cell cytotoxicity (figure 2G; see No Ab and Control Ab). Preincubation of NK cells with the blocking anti-TIGIT mAb abrogated this difference (figure 2G). These results demonstrate that Nectin4 is indeed a functional ligand for TIGIT.
Generation and characterization of Nectin4 blocking antibodies
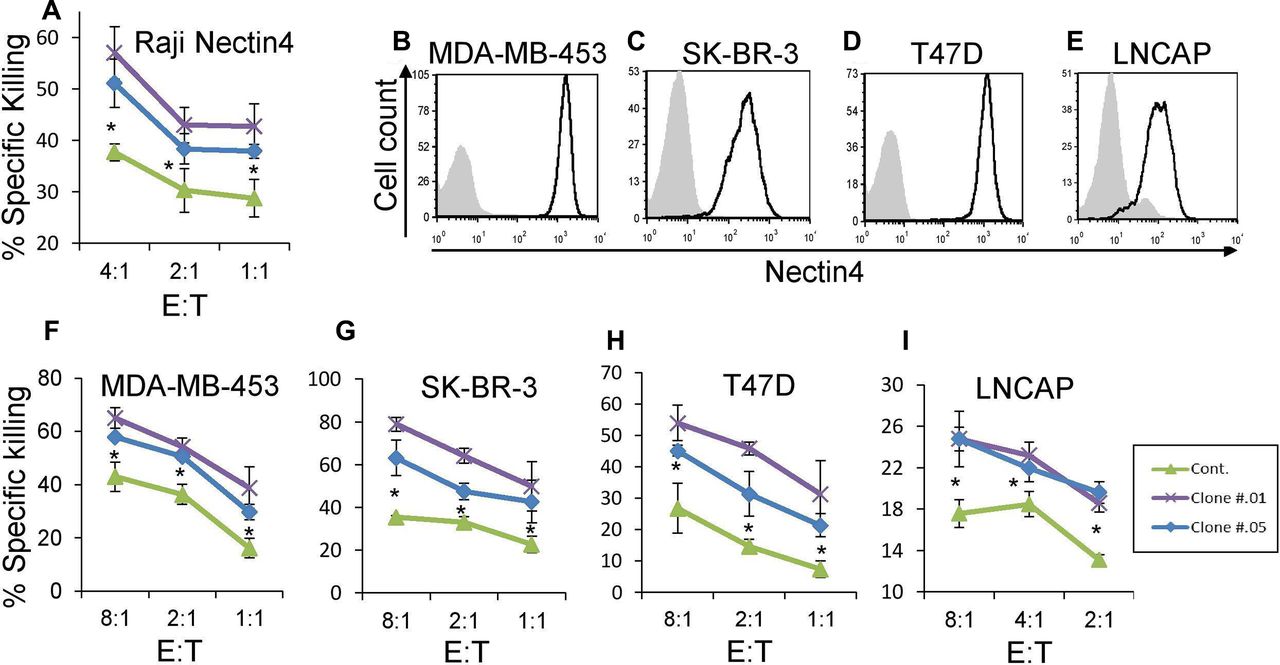
Nectin4 is a well-known tumor marker, as it is overexpressed on various tumors including breast, bladder, lung and pancreas.11 Therefore, we wanted to generate checkpoint blocking antibodies against Nectin4. We immunized mice against Nectin4-Ig in order to generate hybridoma cell lines and select those that secrete blocking antibodies. Two of them, clones .01 and .05, are shown in figures 2 and 3. First, we confirmed specific binding of these antibodies to Nectin4 by staining Raji cells overexpressing Nectin4 (figure 3A,B). We then assessed the ability of these antibodies to block TIGIT-Ig binding (figure 3C,D). Both antibodies effectively block Nectin4-TIGIT interaction. Using MST, we determined the affinities of the mAbs to Nectin4, which are in the pM range (figure 3E). To evaluate functional ability of the anti-Nectin4 mAbs to block TIGIT-mediated inhibition, we used Raji cells overexpressing Nectin4. Preincubation of target cells with either antibody led to increased killing by NK cells (figure 4A).
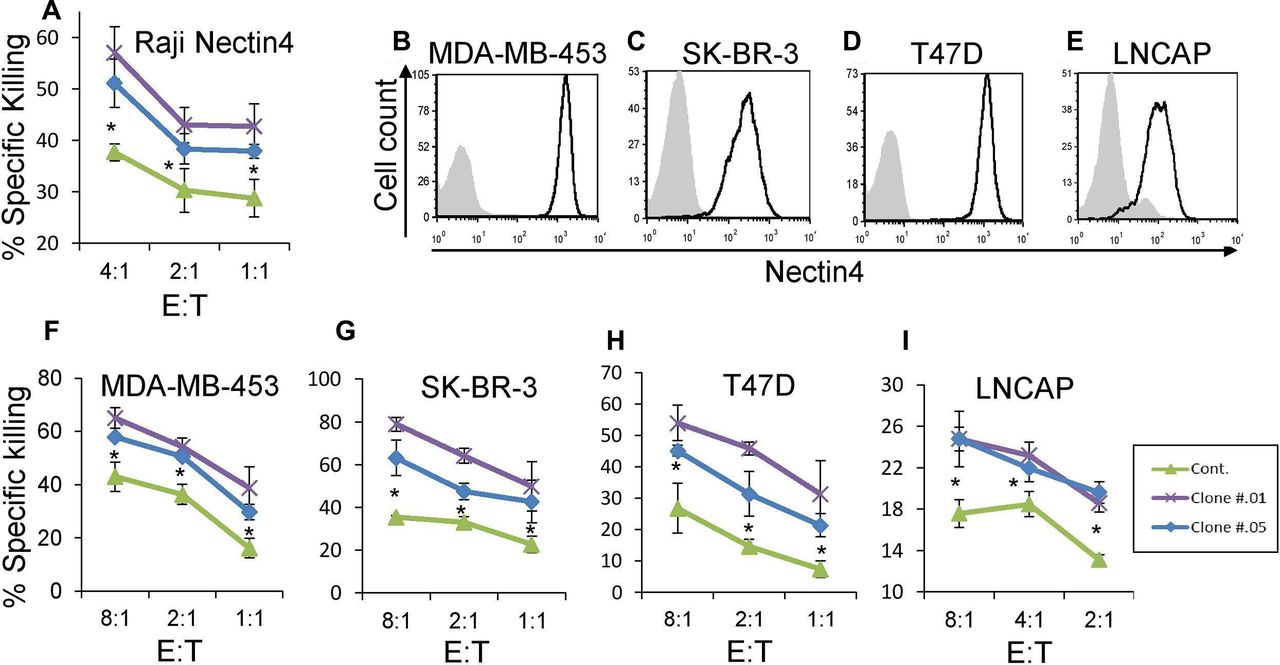
Anti-Nectin4 antibodies increase NK cytotoxicity. (A) [35S] methionine-labeled Raji cells transfected with Nectin4 were incubated with 1 µg/well of either mouse IgG1 as control Ab (green) or with clone .01 (purple) or clone .05 (blue) for 1 hour and then incubated with activated human NK cells for 5 hours. The effector to target (E:T) ratios are indicated on the X-axis. Figure shows one representative experiment out of three performed. Shown is the relative average killing ±SD., *p<0.05 (significance between the clones’ blocking and control antibody blocking). (B–E) FACS staining of cell lines stained with commercial anti-Nectin4 antibody (black line histograms). Gray filled histograms are background staining with secondary antibody only. Cell lines used are MDA-MB-453 (B), SK-BR-3 (C), T47D (D) and LNCaP (E). Figures show one representative experiment out of three performed. Graph depicting the mean fluorescence intensity values of the stainings appears in online supplementary figure 3e–h. (F–I) [35S] methionine-labeled MDA-MB-453 (F), SK-BR-3 (G), T47D (H), and LNCaP (I) cells were incubated with either mouse IgG1 as control mAb (green) or with clone .01 (purple) or clone .05 (blue) for 1 hour and then incubated with activated human NK cell cultures for 5 hours. The E:T ratios are indicated on the X-axis. Figures show one representative experiment out of three performed. Shown is the relative average killing ±SD., *p<0.05 (significance between the clones’ blocking and the control antibody). FACS, fluorescence-activated cell sorting; NK, natural killer.
To establish that endogenous Nectin4 is a central ligand for TIGIT, even in the context of other TIGIT ligands, we assayed various cell lines (breast cancer cell lines MDA-MB-453, SK-BR-3, T47D and prostate cancer LNCAP) that naturally express Nectin4 (figure 4B–E) along with other TIGIT ligands. We performed NK cytotoxicity assays on these cells as well. Preincubation of all cell lines with either anti-Nectin4 antibody led to increased killing by primary NK cells (figure 4F–I). Accordingly, we concluded that Nectin4 is a new immune checkpoint ligand, and that our antibodies can functionally block its inhibitory effect.
Anti-Nectin4 antibody can attenuate tumor growth in vivo
Before moving to in vivo models, we first investigated whether murine Nectin4 interacts with murine TIGIT. We overexpressed murine Nectin4 in Raji cells (figure 5A) and stained these cells with the murine TIGIT-Ig. As no binding was detected (figure 5B), we concluded that the orthologous system in mice is irrelevant.
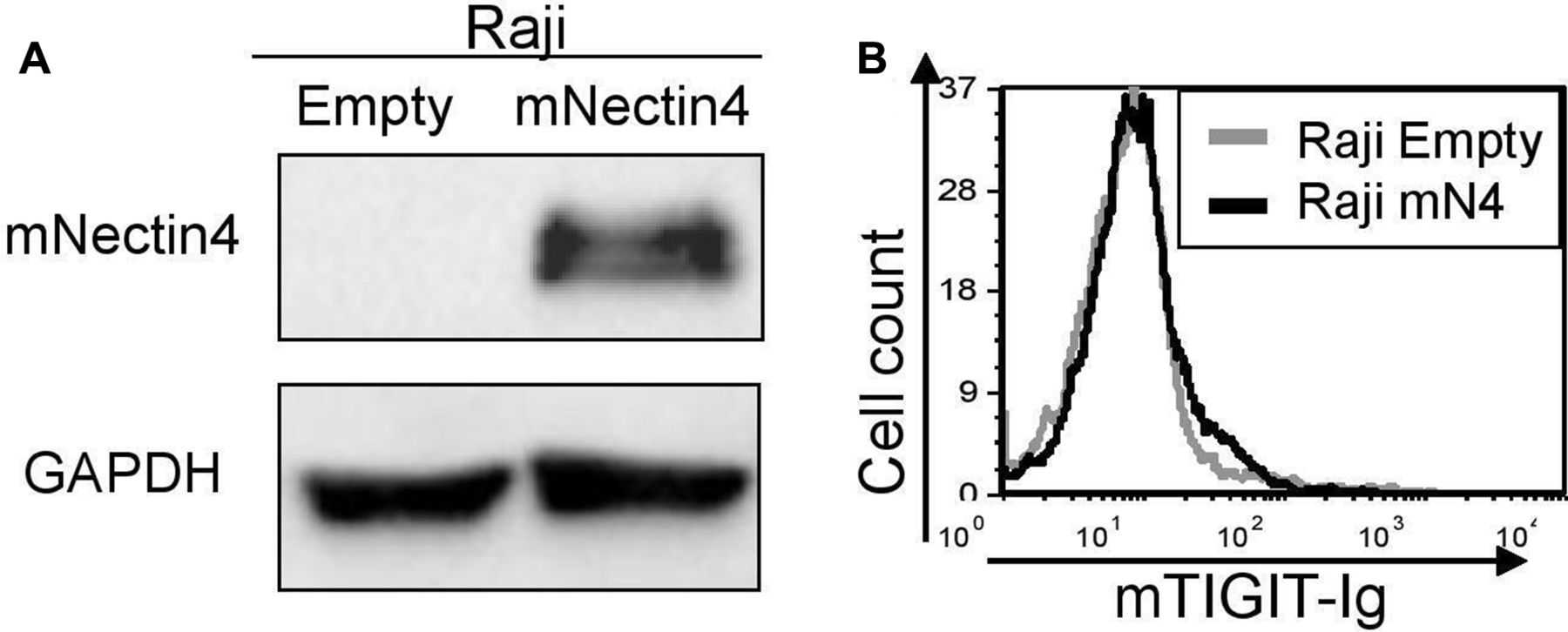
Murine Nectin4 does not bind murine TIGIT. (A) Overexpression of murine Nectin4 (indicated as mNectin4) on Raji cells. Western blots were performed with antimurine Nectin4 AB and expression was compared with Raji cells expressing empty vector (indicated as empty). Staining for GAPDH was used as a loading control. (B) FACS staining of Raji cells transfected either with an empty vector as control (gray histograms), or with murine Nectin4 (black histograms). Cells were stained with murine TIGIT-Ig. Figures show one representative experiment out of three performed. Graph depicting the mean fluorescence intensity values of the stainings appears in online supplementary figure 4 FACS, fluorescence-activated cell sorting.
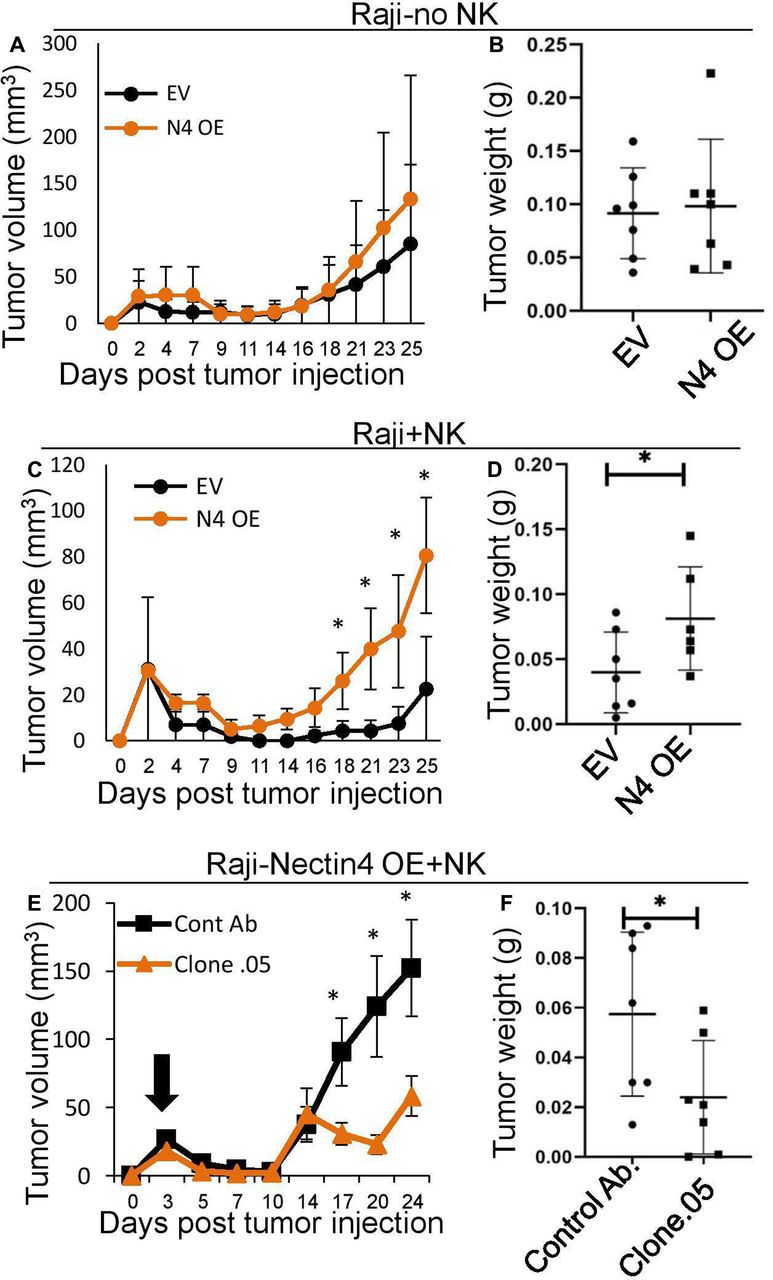
Therefore, to demonstrate the functionality of the interaction of Nectin4 and TIGIT in vivo, SCID-beige mice were subcutaneously implanted with Raji cells with either human Nectin4 overexpression or empty vector, with or without the addition of human NK cells. In the absence of NK cells, Nectin4 overexpression did not lead to any detectable difference in tumor size and weight (figure 6A,B). In contrast, when NK cells were included, tumor size was significantly larger in Raji tumors overexpressing Nectin4 (figure 6C,D). Therefore, we show that Nectin4 is an immune checkpoint ligand in vivo.
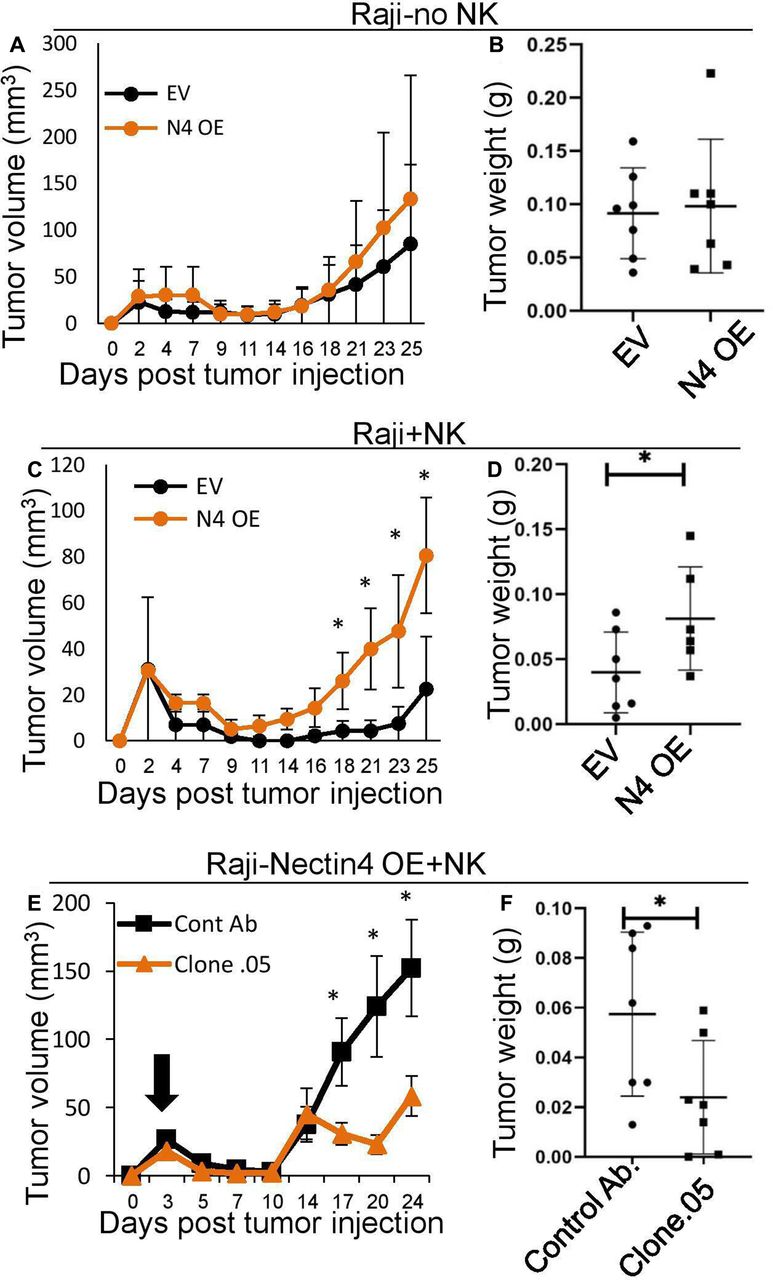
In vivo effect of anti-Nectin4 mAb on tumors overexpressing Nectin4. (A–D) SCID-beige mice were subcutaneously implanted with 5×106 Raji cells either overexpressing Nectin4 (N4 oe) or empty vector (EV) either alone (A, B) or with 1×106 NK cells (C, D). (E, F) SCID-beige mice were subcutaneously implanted with 5×106 Raji cells overexpressing Nectin4 (N4 oe) and 1×106 NK cells. Mice were then treated with either anti-Nectin4 clone .05 mAb (clone .05) and or a control Ab. (A, C, E) Tumor growth was followed with standard caliper. Starting day of antibody treatment is marked by a black arrow. Tumor volumes were calculated by the formula: length×width2×0.5.21 (B, D, F) Tumors ware harvested and weighed on day 21 (B, D) or 27 (F) post tumor injection. For all murine experiments n=7. *p<0.05. NK, natural killer.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The checkpoint inhibitor anti-Nectin4 mAb enhances NK killing of tumors expressing all ligands of TIGIT in vivo. (A–D) SCID-beige mice were subcutaneously implanted with 5×106 MDA-MB-453 cells either alone (A, B) or with 7×105 NK cells (C, D). Half of each group was treated with anti-Nectin4 clone .05 mAb (clone.05) or a control Ab. (A, C) Tumor growth was followed with standard caliper. starting day of antibody treatment is marked by a black arrow. Tumor volumes were calculated by the formula: length×width2×0.5.21 (B, D) Tumors ware harvested and weighed on day 23 post-tumor injection. For all murine experiments n=7. *P<0.05. (E) Kaplan-Meier curves of lung adenocarcinoma patients (left, Geo dataset ID: GSE36471) and colon cancer patients (right, Geo dataset ID: GSE17538) after stratification by Nectin4 expression level. *P<0.05. Data were obtained from DRUGSURV.22 23 NK, natural killer
In order to test the efficacy of our antibodies in vivo, we used them along with human NK cells to treat mice with Raji tumors overexpressing Nectin4. We compared tumor growth with injection of either a control antibody or anti-Nectin4 clone .05 mAb (selected due to greater blocking as shown in figure 2C,D). We found that clone .05 successfully suppresses tumor growth (figure 6E,F). Next, we wanted to assess the mAb’s effect in the context of other TIGIT ligands when Nectin4 is expressed endogenously. To this end, we subcutaneously injected MDA-MB-453 cells (which express Nectin4 and other ligands from the Nectin family, online supplementary figure 1l–n) into SCID-beige mice with or without human NK cells. Mice were treated with clone .05 or a control Ab twice a week, while tumors were visible (indicated by arrows in figures 6,7). As shown, tumor weight was similar in the presence or absence of the antibody, when no NK cells were injected, indicating that clone .05 by itself does not affect tumor growth (figure 7A,B). When NK cells were injected along with clone .05, however, tumor growth was suppressed (figure 7C,D).
Finally, we show that high expression of Nectin4 is associated with poor overall survival in cancer patients (figure 7E) and marks a poorer overall prognosis in several cancer types.18 Therefore, further investigation of our antibody as a drug for these patients is of great clinical value.
Discussion
Checkpoint inhibitors such as anti-CTLA-4 and anti-PD-1 have revolutionized cancer therapy, but they can carry many side effects. Much of this is attributed to off-target immune activation.1 Anti TIGIT blocking mAbs have also been shown to induce immune-related adverse events.19
Here, we report that Nectin4 is a previously undescribed TIGIT ligand and characterize a Nectin4 blocking antibody we generated. As Nectin4 exhibits tumor specificity,11 our antibody aims to induce an immune response specifically against the tumor, thereby minimizing side effects. This is in contrast to other checkpoint inhibitors, which may have a more general effect on the immune system and lead to autoimmunity. As we have shown, unlike other ligands in its family, Nectin4 binds only to TIGIT. TIGIT is a powerful immune checkpoint as it is expressed on TILs and is associated with immune cell exhaustion.3 4 TIGIT binds several ligands, including Nectin1, Nectin2 and Nectin3, in order to allow close monitoring and fine tuning of this immune checkpoint. All known TIGIT ligands are also bound by other immune receptors such as the inhibitory receptors CD112R and CD96 (that sometimes is considered as activating20) and the coactivating receptor DNAM1, which physically influence ligand availability for TIGIT binding.2 It is, therefore, hard to predict how blocking of TIGIT will influence tumor growth as it might affect other receptors in the system. In contrast, here, we show in vitro and in vivo that blocking Nectin4 which, as we show, interacts with TIGIT only, leads to increased tumor killing. Therefore, blocking Nectin4–TIGIT interaction using anti-Nectin4 mAb, may lead to significant and specific induction of the immune system against tumors.
The synergy of a purely inhibitory effect, tumor-restricted expression, and its status as an immune checkpoint ligand makes Nectin4 a unique candidate for cancer immunotherapy. Our blocking antibody is, therefore, the founding member of a new generation of tumor-specific checkpoint inhibitors which can enhance tumor killing in a specific manner with minimal side effects.
Conclusions
In this study, we introduce Nectin4 as a novel major ligand for TIGIT, an inhibitory checkpoint receptor. We further generate anti Nectin4 antibodies that block its interaction with TIGIT and demonstrate that they increase tumor killing in vitro and in vivo. We expect these new anti Nectin4 antibodies to become a new form of cancer specific, autoimmune side effect free, cancer checkpoint immunotherapy.
Acknowledgments
The authors would like to thank Daphna Burowski for her excellent advice, as well as Karmela Miklic and Suzana Malic for technical support.
References
Footnotes
SJ and OM contributed equally.
Contributors AR conducted experiments, analysed the data and wrote the manuscript. YO initiated the project. IK conducted part of the initial screening. PT and BS advised. BI and YCA contributed to the in vivo studies. NS edited the manuscript. PKB and TL generated blocking antibodies against Nectin4. SJ provided reagents and supervised the project. OM supervised the project and wrote the manuscript.
Funding AR is supported by the Einstein Kaye scholarship. This work was supported by the Israel Innovation Authority (Kamin grant), the Israel Science Foundation (Moked grant), the GIF Foundation, the ICRF professorship grant, the ISF Israel-China grant, and the Ministry of Science and Technology grant (all to O.M.). SJ was supported by the grant 'Strengthening the capacity of CerVirVac for research in virus immunology and vaccinology', KK.01.1.1.01.0006, awarded to the Scientific Centre of Excellence for Virus Immunology and Vaccines.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The research protocol was reviewed and approved by the Ethics Committee of the Hebrew University (Ethical approval number: MD-17-15165-4).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplementary information. Data are available in a public, open access repository.