Article Text

Abstract
Background The generation of antigen-specific cytotoxic T lymphocyte (CTL) responses is required for successful cancer vaccine therapy. In this regard, ligands of Toll-like receptors (TLRs) have been suggested to activate adaptive immune responses by modulating the function of antigen-presenting cells (APCs). Despite their therapeutic potential, the development of TLR ligands for immunotherapy is often hampered due to rapid systemic toxicity. Regarding the safety concerns of currently available TLR ligands, finding a new TLR agonist with potent efficacy and safety is needed.
Methods A unique structural domain (UNE-C1) was identified as a novel TLR2/6 in the catalytic region of human cysteinyl-tRNA synthetase 1 (CARS1) using comprehensive approaches, including RNA sequencing, the human embryonic kidney (HEK)-TLR Blue system, pull-down, and ELISA. The potency of its immunoadjuvant properties was analyzed by assessing antigen-specific antibody and CTL responses. In addition, the efficacy of tumor growth inhibition and the presence of the tumor-infiltrating leukocytes were evaluated using E.G7-OVA and TC-1 mouse models. The combined effect of UNE-C1 with an immune checkpoint inhibitor, anti-CTLA-4 antibody, was also evaluated in vivo. The safety of UNE-C1 immunization was determined by monitoring splenomegaly and cytokine production in the blood.
Results Here, we report that CARS1 can be secreted from cancer cells to activate immune responses via specific interactions with TLR2/6 of APCs. A unique domain (UNE-C1) inserted into the catalytic region of CARS1 was determined to activate dendritic cells, leading to the stimulation of robust humoral and cellular immune responses in vivo. UNE-C1 also showed synergistic efficacy with cancer antigens and checkpoint inhibitors against different cancer models in vivo. Further, the safety assessment of UNE-C1 showed lower systemic cytokine levels than other known TLR agonists.
Conclusions We identified the endogenous TLR2/6 activating domain from human cysteinyl-tRNA synthetase CARS1. This novel TLR2/6 ligand showed potent immune-stimulating activity with little toxicity. Thus, the UNE-C1 domain can be developed as an effective immunoadjuvant with checkpoint inhibitors or cancer antigens to boost antitumor immunity.
- immunology
- oncology
- adjuvants, immunological
- vaccination
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Aminoacyl-tRNA synthetases (ARSs) are essential enzymes involved in protein translation that catalyze the ligation of amino acids to their cognate tRNAs. However, beyond this conventional role in protein synthesis, new functions in inflammation, angiogenesis, and tumorigenesis have been discovered.1–4 These expanded functions are often achieved during evolution by acquiring new domains, some of which are shared by several ARSs or uniquely found in specific ARSs.5 6 Especially, several different cytosolic ARSs exhibit diverse activities in the extracellular space, and their pathophysiological association with immune responses has been reported. For example, the N-terminal extension domain of tryptophanyl-tRNA synthetase 1 (WARS1) stimulates innate immune responses in response to pathogen challenges via Toll-like receptor (TLR)4–myeloid differentiation factor 2 (MD2) complex on macrophages.7 Unique (UNE) domain of asparaginyl-tRNA synthetase 1 elicits chemokine activities, and the endothelial monocyte-activating polypeptide II-like domain of tyrosyl-tRNA synthetase 1 induces proinflammatory cytokine secretion.8 9 These observations suggest that additional or unique domains might be involved in immune responses when secreted from the cell.
Human cysteinyl-tRNA synthetase 1 (CARS1) contains two UNE domains inserted in different locations,10 suggesting that these domains may contribute to the functional expansion of CARS1 beyond protein synthesis. However, the extracellular secretion and activities of CARS1 are not yet understood. Here, we investigated the extracellular activities of human CARS1, especially in immune control, and identified one of the inserted domains responsible for immune-modulating activities. We further determined its therapeutic potential as an effective immunoadjuvant for cancer vaccine and immune checkpoint inhibitor.
Methods
Mice
Ovalbumin (OVA)-specific T-cell receptor transgenic OT-I mice11 and TLR2−/− mice were kindly provided by C-YK, and E K Jo, respectively. Female C57BL/6 mice aged 6–8 weeks were obtained from DooYeolBiotech and maintained in the pathogen-free authorized facility in WOOJUNG BIO CROWISE.
Secretion test
HCT116 cells were cultivated in roswell park memorial institute (RPMI) containing fetal bovine serum (FBS) with 60%–70% confluence. After the cells were incubated in serum-free media for 4 hours, tunicamycin (Sigma-Aldrich) or tumor necrosis factor alpha (TNF-α) (R&D Systems) with different doses was treated for 24 hours. After culture media were centrifuged at 500 g for 10 min, supernatants were centrifuged again at 10 000 g for 30 min to remove further debris. Protein precipitation was conducted using a final concentration of 12% trichloroacetic acid (TCA, Sigma-Aldrich) mixed with supernatant and incubated overnight (O/N) at 4°C. Final samples were obtained by centrifugation at 18 000 g for 15 min, followed by neutralization with 0.1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Sigma-Aldrich), pH 8.0.
Cell-binding assay
THP-1, U937, Daudi, and Jurkat cells were seeded on 9 mm coverslips for immunofluorescence staining. Cells were fixed with 4% paraformaldehyde (Biosesang) for 5 min, followed by a washing step with cold phosphate-buffered saline (PBS). After blocking non-specific binding with CAS-Block (Thermo Fisher Scientific), each cell line was incubated for 1 hour with 30 nM of bovine serum albumin (BSA, GenDEPOT) or CARS1 conjugated with Alexa-Fluor 647 (Invitrogen). Visualization of CARS1 was observed by confocal fluorescence microscopy. For flow cytometry analysis, 30 nM of CARS1 or BSA was incubated for 30 min with different cell types in six-well dishes.
Immunoprecipitation
His-tagged CARS1 and UNE-C1 proteins were constructed in the pET-28a vector and purified as described previously. TLR2 and TLR4 were purified from human embryonic kidney (HEK) 293 cells transfected with pCMV3-TLR2-flag, and pCMV3-TLR4-flag, respectively (Sino Biological). Two micrograms of anti-His (Santa Cruz Biotechnology) or anti-Flag antibody (Thermo Fisher Scientific) was incubated with protein G agarose (Invitrogen) for 1 hour. After incubating TLR2 or TLR4 with his-tagged proteins for 4 hours mixtures were incubated with antibody-bound protein G complex for an additional 1 hour. Three times of washing with tris-buffered saline with tween 20 (TBS-T) were performed and subjected to immunoblotting. Anti-His and anti-FLAG antibodies were used for detecting His or Flag-tagged proteins.
HEK blue detection
HEK cells were cultured in DMEM containing 10% FBS, 1% streptomycin, and 100 µg/mL normocin. Different doses of CARS1 and UNE-C1 were added in a flat-bottom 96-well plate. Then, 50 000 cells of hTLR2, hTLR4, hTLR2/TLR6, and hTLR1/TLR2 HEK-Blue cells (Invivogen) were added per well. The plates were then incubated for 24 hours at 37°C and supernatants were collected. QUANTI-Blue solution (Invivogen) was incubated with collected supernatant at 37°C. Activities were observed through measuring optical density (OD) value at 620 nm.
In vivo antigen presentation and activation of dendritic cells (DCs)
OVA was purchased from Sigma-Aldrich. Mice were immunized subcutaneously with OVA alone or OVA plus UNE-C1. A day after, draining lymph nodes (dLNs) were harvested for measuring antigen presentation and activation of DCs. Lymph nodes were passed through 70 µm strainer and red blood cells (RBCs) were removed. Single cells were stained with CD8α, B220, CD11c, SIINFEKL-MHCI, and CD86 (Biolegend) to identify CD8+ DCs (CD11c+, CD8α+, and B220−), CD8− DCs (CD11c+, CD8α−, and B220−), and pDCs (CD11c+ and B220+) as previously reported.12
In vitro CD8+ T-cell priming
C57BL/6 mice were immunized with OVA with or without UNE-C1. After 24 hours, DCs from lymph nodes and spleen were collected using Pan-DC cell isolation kit (Miltenyibiotec). Meanwhile, CD8+ T cells from OT-1 transgenic mice were purified using a CD8+ T-cell isolation kit (Miltenyibiotec). Cells of DCs (5×105) and cells of CD8+ T cells (5×105) were then cocultured in RPMI complete media for 24 hours. The supernatant was collected to check interferon gamma (IFN-γ) levels, and collected cells were stained with CD3, CD8, and CD69 antibodies (Biolegend) to analyze CD69 expression on OT-1 T cells.
In vivo CD8+ T-cell priming
Female C57BL/6 mice of 6–8 weeks of were subcutaneously injected with 10 µg OVA alone or OVA plus 100 µg of UNE-C1 total of two injections once per week. Splenocytes and dLN cells were harvested 7 days after the final injection. After RBC lysis, single cells were stained with h-2kb OVA (257-264) tetramer and CD8 to analyze the frequency of OVA-specific CD8+ T cells.
In vivo cytotoxicity killing assay
C57BL/6 mice were injected subcutaneously with 10 µg of OVA with or without UNE-C1 once a week. After 7 days of second injections, splenocytes from naïve C57BL/6 mice were harvested. Half of the splenocytes were pulsed with SIINFEKL peptide, and half remained unpulsed. After 2 hours, pulsed and unpulsed groups were labeled with 2.5 and 0.25 µM of carboxyfluorescein succinimidyl ester (CFSE, Invitrogen), respectively, for 15 min and the remaining dyes were quenched with serum media. Equal numbers of pulsed and unpulsed cells were mixed evenly and injected intravenously into the immunized mice. After 24 hours, splenocytes were collected, and CFSEhigh and CFSElow cells were determined and specific target cell killing was calculated with the following formula: [1–(% of CFSEhigh in immunized sample/% of CFSElow in immunized sample)/(% of CFSEhigh in unimmunized sample/% of CFSElow in unimmunized sample)]×100.
Anti-OVA IgG detection
After 2 weeks of immunization, serum from immunized mice was obtained and subjected to ELISA. OVA (10 µg/mL) was coated on Maxisorp plates (Nunc) O/N at 4°C in sodium carbonate buffer. After washing three times with PBS-T, the plates were blocked with 5% BSA for 1 hour and washed with PBS-T three times, and the diluted samples were incubated for 2 hours. Anti-mouse IgG, IgG1, and IgG2c conjugated with HRP (Thermo Fisher Scientific) were added for 1 hour. After seven times of washing, 3,3’,5,5’-tetramethylbenzidine (TMB) substrate was added for development, and absorbance was measured at 450 nm.
Therapeutic mouse model
For the therapeutic cancer vaccine model, 1×106 cells of E.G7-OVA were injected subcutaneously at the right flank. On days 3 and 10, 10 µg of OVA or OVA plus 100 µg of UNE-C1 were injected at the opposite dorsal side. For the depletion model, 200 µg of anti-CD4, anti-CD8, and anti-NK1.1 antibodies (BioXcell) was injected intraperitoneally on days 2, 5, 8, and 11. For testing the synergy effect of anti-CTLA-4 antibody and UNE-C1, 200 µg of CTLA-4 antibody (BioXcell) in saline was injected intraperitoneally on days 3, 6, 9, and 12. For the TC-1 mouse model tumor, mice were inoculated with 1×105 cells of TC-1. On days 6 and 13, 20 µg of E743–62 (GQAEPDRAHYNIVTFCCKCD) or E743–62 plus 100 µg of UNE-C1 was injected. Tumors were measured with a digital caliper and calculated using the formula 0.52×length×width.2 When the tumor reached higher than 1500 mm3, the mice were euthanized.
Analysis of tumor-infiltrating immune cells
Tumor cells from immunized mice were dissociated using tumor dissociation kit (Miltenyibiotec) under the manufacturer's protocol. After lysing the red blood cells, tumor cells were stained with CD8, CD3, OVA (H-2Kb-SL8) tetramer and CD45 antibodies for 30 min at 4°C. For staining IFN-γ, tumor-infiltrating immune cells were restimulated with OVA257−264 (SIINFEKL) or E749–57 (RAHYNIVTF) for 6 hours. Peptides were synthesized from GenScript. Fixation/permeabilization solution kits (BD) were used for staining intracellular IFN-γ, and the frequency of IFN-γ+ cells were analyzed from CD3+ CD8+ cells.
Toxicity test
C57BL/6 mice were injected subcutaneously with OVA alone or OVA plus an excess amount of UNE-C1 at 25 mpk (28.6 nmol) and 100 mpk (114.4 nmol). As control, Pam2CSK4 (114.4 nmol), Pam3CSK4 (114.4 nmol), and CpG-ODN 1826 (28.6 nmol) from Invivogen were injected. At the indicated time, serum cytokines were measured using mouse interleukin (IL)-6, TNF-α, and IL-12 p40 ELISA set (BD). Body weights were measured 24 and 48 hours after injection. Spleen mass was divided by the body weight of the mouse to decide on splenomegaly.13
Statistics
Data were analyzed with GraphPad Prism V.7.0, and results are presented as mean±SD or SEM. Statistical significance was analyzed with Student’s t-tests, and p<0.05 was considered to be statistically significant for differences between the two groups. For the survival curve, statistical analysis was performed using Kaplan-Meier with the log-rank (Mantel-Cox) test.
Results
Effect of secreted CARS1 on TNF-α secretion from macrophages
To explore the novel function of CARS1, we first tested whether it is secreted in response to various stimuli. We used colon cancer cells since the secretion of many ARSs has been previously reported from colon cancers and CARS1 was also identified among the secreted ARSs by liquid chromatography–tandem mass spectrometry.14–16 We found that CARS1 is secreted from HCT116 cells under endoplasmic reticulum and inflammatory stresses, but not by growth factor and cytokine treatments (figure 1A). To confirm the secretion of CARS1, we incubated HCT116 cells with TNF-α or tunicamycin and found that CARS1 secretion was gradually increased in dose-dependent and time-dependent manners (figure 1B,C). Moreover, the secretion of tubulin was not observed in media, suggesting that the CARS1 secretion did not result from cell rupture. Altogether, these data indicate that CARS1 would be secreted during stress conditions that tumors often encounter in their local microenvironment.17 Next, to understand the activities of the secreted CARS1, we checked its effect on immune cells since some of the secreted ARSs were previously reported to play roles in immune responses.7 14

Effect of secreted CARS1 on TNF-α secretion from macrophages (A) CARS1 secretion was tested by incubating HCT116 cells under different conditions, including SF, TNF-α (10 ng/mL), tunicamycin (1 µg/mL), arsenite (12.5 µM), CoCl2 (100 µM), Wnt3a (200 ng/mL), IL-2 (10 ng/mL), IL-4 (10 ng/mL), VEGF (20 ng/mL), BMP4 (50 ng/mL), PDGF (100 ng/mL), EGF (100 ng/mL) FGF-2 (50 ng/mL), FGF-4 (50 ng/mL), IGF (50 ng/mL), and glutamine-free conditions for 24 hours. The amount of CARS1 secreted in the medium and WCL was detected. (B) HCT116 cells were treated with tunicamycin (Tunica) in dose-dependent and time-dependent manners. Proteins in the medium were precipitated and detected by an antibody against CARS1. (C) HCT116 cells were treated with TNF-α in dose-dependent and time-dependent manners to detect secreted CARS1. (D,E) CARS1 and BSA were conjugated with Alexa-Fluor 647 and treated for 30 min to different cell types. CARS1-bound cells were visualized and analyzed by confocal microscopy (D) and flow cytometry (E), respectively. (F) RAW264.7 cells were treated with either 100 nM of CARS1 or KARS1 for 4 hour. Boiled CARS1 and KARS1 were used for negative controls. (G) The production of TNF-α was measured by ELISA after treating 100 nM of CARS1 on PMA-differentiated THP-1 cells. CARS1 was preincubated with proteinase K (50 µg/mL) or boiled for 1 hour. Before adding CARS1, some cells were preincubated with polymyxin B (10 µg/mL) for 1 hour. Data are representative of three independent experiments. Results are presented as mean±SD, and statistical significance was analyzed with Student’s t-test (***p<0.001). BSA, bovine serum albumin; BMP4, bone morphogenetic protein 4; CARS1, cysteinyl-tRNA synthetase 1; IL, interleukin; LPS, lipopolysaccharide; PDGF, platelet-derived growth factor; PMA, phorbol 12-myristate 13-acetate; SF, serum-free; TNF-α, tumor necrosis factor alpha; VEGF, vascular endothelial growth factor; WCL, whole-cell lysate.
We first determined the binding specificity of CARS1 to different immune cells using immunofluorescence and flow cytometry analyses and found preferential binding of CARS1 to macrophage (RAW264.7) and monocyte (THP-1) cells, but not to B (Daudi) or T (Jurkat) cells (figure 1D,E), suggesting that CARS1 would primarily function on monocyte and macrophages. We thus treated macrophage RAW264.7 cells with purified recombinant CARS1 and lysyl-tRNA synthetase 1 (KARS1) (below 0.04 EU/mg from limulus amebocyte lysate (LAL) test) for 4 hours and compared TNF-α secretion.18 Results showed that CARS1 and KARS1 stimulate TNF-α secretion with comparable efficacy (figure 1F). Next, to exclude the possibility of lipopolysaccharide (LPS) contamination, we incubated CARS1 at high temperature or with protease K and observed that both treatments resulted in loss of TNF-α secretion, suggesting that the activity would result from CARS1. CARS1-induced TNF-α secretion was also observed from phorbol 12-myristate 13-acetate (PMA)-differentiated THP-1 cells, and this activity was not affected by the LPS inhibitor, polymyxin B (figure 1G), excluding the possibility of LPS contamination. Based on these results, human CARS1 appears to be secreted from cancer cells in stressful microenvironment and activate antigen-presenting cells (APCs) such as macrophages.
Determination of UNE-C1 as the activity determinant
The two unique domains were suggested to be embedded in human CARS1 based on the previous report and multiple sequence alignments10 (figure 2A), and they were defined as UNE-C1 (residues 106–228) and UNE-C2 (residues 571–748). To determine whether these domains are responsible for the induction of TNF-α secretion by CARS1, we prepared the peptides spanning these two domains and CARS1 peptides lacking each UNE-Cs (figure 2B). Interestingly, all four CARS1 fragments were stably expressed and could be homogeneously purified (figure 2C). We then compared them with full-length CARS1 with respect to TNF-α secretion from PMA-differentiated THP-1 cells. TNF-α secretion was significantly increased by CARS1, ΔUNE-C2 (residues 1–566), and UNE-C1 (figure 2D), but not by UNE-C2 and ΔUNE-C1, suggesting that UNE-C1 is responsible for the activity. To ensure that the activity mediated by the isolated UNE-C1 would result from its native structure, we checked whether its activity would be affected by heat, protease, and polymyxin B treatment. The activity was not affected by polymyxin B treatment, suggesting that it was not due to potential LPS contamination. The activity of UNE-C1 was retained even after the peptide was boiled, whereas it was eradicated by protease treatment (figure 2E). Circular dichroism (CD) spectrum analysis of UNE-C1 showed that the peptide is mainly composed of alpha-helices and has a Tm around 60°C, and the CD spectrum of UNE-C1 was restored when cooled after boiling (online supplementary figure S1A), suggesting its unusual thermostability and potential as an independent functional domain. After characterizing CARS1 activity on innate immune cells is mediated by UNE-C1, we next checked the involvement of UNE-C1 in the catalytic activity of CARS1. The activity was observed from full-length CARS1, but not from the other constructs, including ΔUNE-C1, ΔUNE-C2, and UNE-C1 (online supplementary figure S1B). These findings indicate that UNE-C1 is not only involved in immune stimulation but also protein translation.
Supplemental material

Determination of UNE-C1 as the activity determinant. (A) Multiple sequence alignment comparing UNE-C1 sequences from different species. (B) Schematic demonstration of CARS1 fragments containing the full-length, two unique domains (UNE-C1 and UNE-C2), and the full-length without UNE-C1 and UNE-C2 (ΔUNE-C1 and ΔUNE-C2). (C) Purified CARS1 and its fragments CARS1 were stained by Coomassie blue. (D) PMA-differentiated THP-1 cells were treated with CARS1 and its fragments for 4 hours. The secretion level of TNF-α was quantified from the collected supernatants. (E) PMA-differentiated THP-1 cells were treated with UNE-C1, which have been untreated, pretreated with proteinase K or boiled. Some cells were preincubated with polymyxin B before treatment. Data are representative of three independent experiments. The results are presented as mean±SD, and statistical significance was analyzed with Student’s t-test (***p<0.001). CARS1, cysteinyl-tRNA synthetase 1; LPS, lipopolysaccharide; PMA, phorbol 12-myristate 13-acetate; TNF-α, tumour necrosis factor alpha.
We also treated bone marrow-derived dendritic cells (BMDCs) with each of the four CARS1 fragments for 24 hours and determined expression levels of the costimulatory molecules, CD40, CD80, and CD86, which are known as essential molecules for stimulating adaptive immune responses.19 Consistently, these molecules were upregulated by CARS1, ΔUNE-C2, and UNE-C1 (figure 3A and online supplementary figure S2A). Likewise, these three fragments significantly increased secretion of the proinflammatory molecules, IL-6, IL-12p70, as well as TNF-α (figure 3A and online supplementary figure S2A), and also stimulated costimulation molecules and cytokine production from bone marrow-derived macrophages (online supplementary figure S2B,C). Altogether, UNE-C1 appears to work as an independent entity even if it is separated from CARS1.

UNE-C1-mediated activation of APCs via TLR2/6. (A) BMDCs were incubated with different CARS1 fragments for 24 hours. Costimulatory molecules were analyzed from the gated CD11c+ population. CD86 expression was evaluated by flow cytometry, and IL-6 and IL-12p70 secretion in supernatants was quantified by ELISA. (B) hTLR2 and hTLR4 HEK-Blue cells, expressing SEAP reporter gene in response to NF-Kβ activity, were treated with CARS1 or UNE-C1 in a dose-dependent manner. HEK-Blue TLR2 and TLR4 activation was evaluated by measuring SEAP secretion in culture media. (C) PMA-differentiated THP-1 cells were preincubated with the indicated amount of anti-human TLR2 or anti-human TLR4 for 1 hour and treated with CARS1 or UNE-C1 for an additional 4 hours. TNF-α from supernatants of PMA-differentiated THP-1 was measured by ELISA. (D) His-tagged CARS1 or UNE-C1 were incubated with TLR2-Flag or TLR4-flag proteins. His-ab or Mock-ab bound protein G agarose was used for immunoprecipitating his-tagged proteins. (E) Reciprocal immunoprecipitation was performed using Flag-ab or Mock-ab bound protein-G agarose. His-CARS1 or -UNE-C1 was incubated with TLR2-Flag or TLR4-flag. Interactions were determined by immunoblotting (F) BMDCs from naïve and TLR2−/− mice were treated with CARS1 or UNE-C1 for 24 hours. IL-6 and IL-12p70 levels in supernatants were quantified. (G) CARS1 and UNE-C1 were treated on hTLR2/6 and hTLR1/2. SEAP activities were measured at OD 620 nm. Data are representative of three independent experiments. Results are presented as mean±SD, and statistical significance was analyzed with Student’s t-test (***p<0.001). APC, antigen-presenting cell; BMDC, bone marrow-derived dendritic cell; CARS1, cysteinyl-tRNA synthetase 1; IL, interleukin; LPS, lipopolysaccharide; NF-Kβ, nuclear factor kappa-light-chain-enhancer of activated B cells; NS, not significant; OD, optical density; PMA, phorbol 12-myristate 13-acetate; SEAP, secreted embryonic alkaline phosphatase.
UNE-C1-mediated activation of APCs via TLR2/TLR6
To understand the mechanism underlying the stimulation of APCs by UNE-C1, we performed RNA sequencing of BMDCs after UNE-C1 treatment for 24 hours. Consistent with the aforementioned findings, genes related to proinflammation, such as il-6, nos2, il-12, cd40, cd80, and cd86, were upregulated by UNE-C1 (online supplementary figure S3A). Especially, TLR-related genes, such as il-6, irf7, cxcl9, ccl5, cxcl11, and cxcl10, were among the top 100 upregulated genes, and their expression was upregulated by at least 20-fold when compared with control levels (online supplementary figure S3A). We also evaluated Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways through the DAVID software20 using the top 100 upregulated genes induced by UNE-C1. UNE-C1 was strongly connected with the TLR signaling pathway with the lowest p value in the KEGG pathway analysis (online supplementary figure S3B). To identify the potential receptor of UNE-C1-induced TLR signaling, we focused on TLR2 and TLR4 since they are well-studied extracellular receptors known to activate related innate immune responses.21 22 We treated TLR2-expressing and TLR4-expressing HEK-Blue cells with CARS1 or UNE-C1 and assessed the resulting responses. Both CARS1 and UNE-C1 activated TLR2-expressing, but not TLR4-expressing, HEK-Blue cells in a dose-dependent manner (figure 3B). To confirm TLR2 specificity, we preincubated PMA-differentiated THP-1 cells with TLR2 or TLR4 neutralizing antibodies. TNF-α secretion from PMA-differentiated THP-1 cells was significantly decreased by the anti-TLR2 antibody but was unaffected by the anti-TLR4 antibody, suggesting that UNE-C1-mediated TNF-α secretion occurs specifically through TLR2 (figure 3C). Further, we checked the interaction between UNE-C1 and TLR2 using immunoprecipitation assays. After purifying TLR2 and TLR4 from transfected HEK-293 cells, CARS1 or UNE-C1 were incubated together with each and their interactions were determined by coimmunoprecipitation. Both CARS1 and UNE-C1 specifically interacted with TLR2 but not with TLR4 (figure 3D,E). Then, we treated BMDCs from a TLR2−/− mouse with CARS1 to investigate the dependency of TLR2 in CARS1-mediated stimulation. As expected, UNE-C1-mediated cytokine production was abrogated when BMDCs from a TLR2−/− mouse were treated with UNE-C1, confirming the TLR2 specificity of UNE-C1 (figure 3F). Since TLR2 forms heterodimers with either TLR1 or TLR6 during activation,23 we treated HEK-Blue cells expressing TLR1/TLR2, or TLR2/TLR6 with UNE-C1. Pam3CSK4 (Pam3) and Pam2CSK4 (Pam2) were used as positive controls for TLR1/TLR2 and TLR2/TLR6 activation, respectively.24 25 Both CARS1 and UNE-C1 preferentially activated TLR2/TLR6 cells (figure 3G).
UNE-C1-dependent stimulation of humoral and cellular immune responses in vivo
DCs are known to play a crucial role in the adaptive immune system. In this regard, TLR agonists are often tested as vaccine adjuvants whether they can boost antigen-specific adaptive immunity through DC activation. Since UNE-C1 stimulates DCs via TLR2/TLR6 in vitro, we further assessed whether it could activate DCs in vivo. After injecting OVA and UNE-C1 into mice, we analyzed the maturation of DC subsets in dLNs by flow cytometry since different types of DCs exist and particular DCs are potent for cross-presentation in secondary lymphoid organs.26 UNE-C1 increased antigen presentation on H-2Kb and CD86 on CD11c+, CD8+ and CD8− DCs, but not on plasmacytoid dendritic cells (pDCs) (figure 4A), suggesting the specificity of UNE-C1 activity among DC subsets. These results are consistent with the previous findings that CD8+ DCs are activated by TLR2 agonists and pDCs express low levels of TLR2 and TLR6 on their plasma membranes.27 28 We next assessed the priming of CD8+ T cells using a coculture system. Mice were injected with OVA antigen and UNE-C1 for 24 hours, and DCs were collected from the dLNs and spleens. Then, CD8+ T cells of OT-1 mice were cocultured with collected DCs for 24 hours, and CD69 expression on CD8+ T cells was analyzed. In the OVA+UNE-C1 group, the expression of CD69 on OVA-specific CD8+ T cells was higher than that in the OVA group. Also, the secretion of IFN-γ from the OVA+UNE-C1 group was significantly increased when compared with the OVA-alone group (figure 4B). These data suggest that UNE-C1 activates the cross-presentation of DCs and primes T cells. Moreover, we investigated the priming of CD8+ T cells by checking the presence of OVA-specific CD8+ T cells in vivo. Seven days after the last immunization, SIINFEKL tetramer+ cells were counted from CD8+ T cells obtained from spleens and dLNs. Mice immunized with OVA and UNE-C1 showed higher or comparable OVA-specific CD8+ T-cell priming in both spleens and dLNs, compared with those immunized with OVA+Pam3 or OVA+CpG-ODN 1826 (CpG) (figure 4C). To investigate whether generated OVA-specific CD8+ T cells are functional, we checked their activity through in vivo cytotoxicity tests. Most SIINEFKL peptide-pulsed splenocytes were eliminated in the OVA+UNE-C1 injected mice, whereas mice immunized with OVA alone did not show significant killings (figure 4D). Compared with other TLR2 agonists, UNE-C1 appears to show superior antigen-specific cytotoxic T lymphocyte (CTL) responses in vivo. To explore UNE-C1-mediated humoral responses in vivo, we immunized the mice with OVA plus UNE-C1. One week after the last vaccination, the level of OVA-specific antibodies in the serum was evaluated. We found that UNE-C1+OVA immunization produced more OVA-specific antibodies than immunization with OVA alone. Notably, the level of Th1-related IgG2c was higher in the UNE-C1 group than that in the Pam3 group. Meanwhile, the levels of total IgG and Th2-related IgG1 from the UNE-C1 group were lower than those in the Pam3 group, implying that UNE-C1 would mediate Th1 skewing adaptive immune response (figure 4E). Altogether, these data demonstrate that immunizing mice with an antigen combined with UNE-C1 produces specific antibodies against the antigen and induces functional antigen-specific CD8+ T cells through DC maturation.
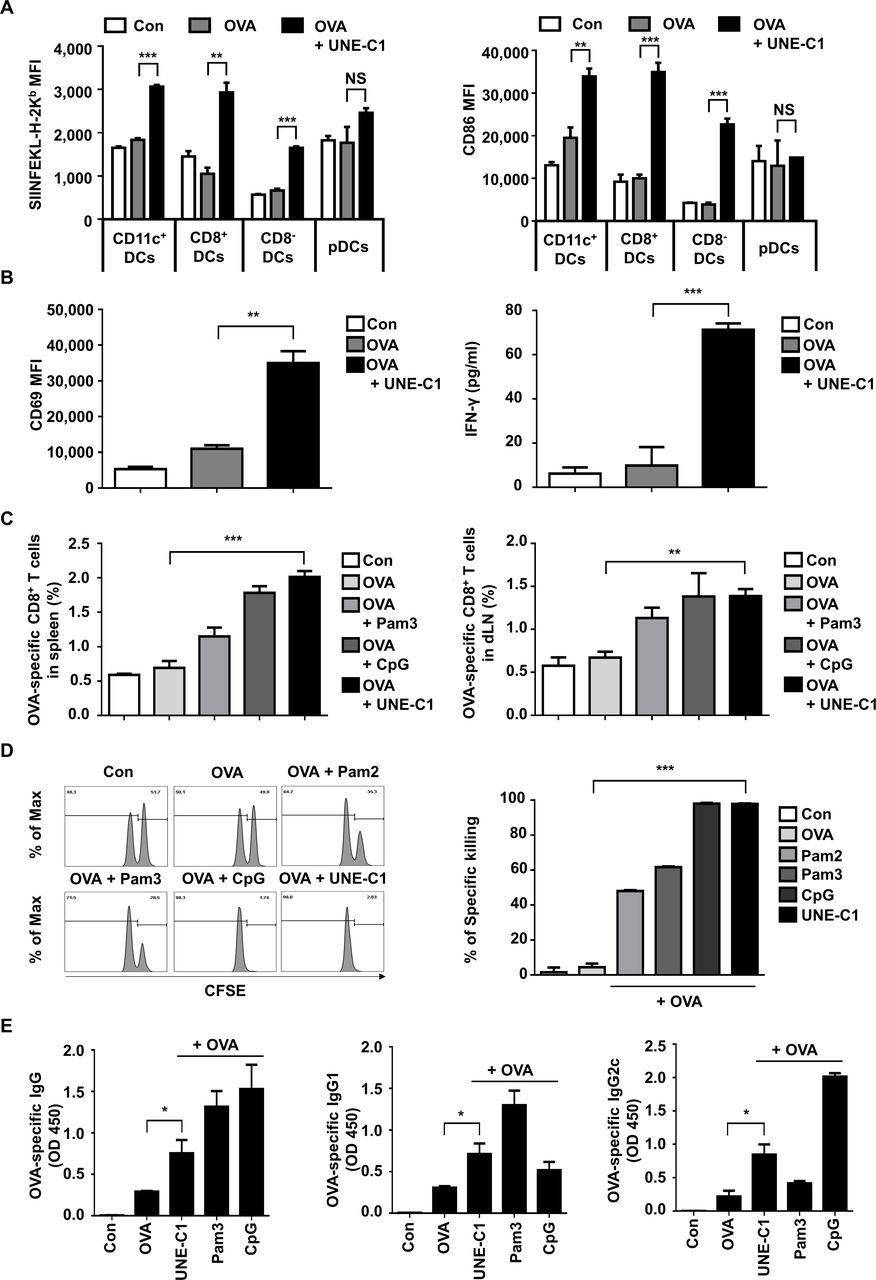
UNE-C1-dependent stimulation of humoral and cellular immune responses in vivo. (A) C57BL/6 mice (n=3) were injected subcutaneously with indicated reagents. A day after, dLNs were collected, and antigen presentation on h-2kb and CD86 in a different subset of DCs was quantified. (B) After immunizing mice (n=3), pan-DCs of each group were collected from its dLNs and spleens. Collected DCs were cocultured with CD8+ T cells from OT-1 transgenic mice for 24 hours. Expression levels of CD69 on OT-1 T cells were quantified and the production of IFN-γ in the supernatants was measured by ELISA. (C) Indicated reagents were used for mice (n=5) immunization on days 0 and 7. Seven days after the last immunization, spleens and dLNs were collected from immunized mice. Percentages of OVA-specific CD8+ T cells in the spleens and dLNs were measured. (D) Mice (n=3) were immunized on days 0 and 7. On day 14, mice were injected intravenously with SIINFEKL peptide-pulsed and unpulsed splenocytes labeled with a high or low concentration of CFSE, respectively. After 24 hours, the percentage of antigen-specific killing was measured by flow cytometry. Representative flow cytometry plots showing remained pulsed and unpulsed cells from immunized mice, and bar diagram with quantitative comparison. (E) Seven days after the final immunization, the serum was collected from each group of mice (n=5), and OVA-specific total IgG, IgG1, and IgG2c were measured by ELISA. Data are representative of three independent experiments. Results are presented as mean±SEM, and statistical significance was analyzed with Student’s t-test (*p<0.05, **p<0.01, ***p<0.001). CFSE, carboxyfluorescein succinimidyl ester; DC, dendritic cell; dLN, draining lymph node; IFN-γ, interferon gamma; MFI, mean fluorescence intensity; OVA, ovalbumin; pDC, plasmacytoid dendritic cell.
Effect of UNE-C1 on immune responses against tumor growth in vivo
Since UNE-C1 stimulates potent humoral and cellular responses, we tested the efficacy of UNE-C1 as an adjuvant for cancer vaccines using an E.G7-OVA cancer model.11 29 After implanting E.G7-OVA to the right flanks of C57BL/6 mice, we injected UNE-C1 and 10 µg of OVA to the opposite side of the tumor injection site on day 3. After 7 days, the mice received a booster immunization, and tumor volumes were recorded (figure 5A). Compared with mice immunized with saline or OVA antigen alone, UNE-C1+OVA injection induced profound tumor inhibition (figure 5A). To investigate the cellular mechanism of anticancer effect, we analyzed the tumor-infiltrating immune cells from tumor tissues. The UNE-C1+OVA-treated group showed enhanced CD8+ T-cell infiltration, antigen-specific CD8+ and IFN-γ+ CD8+ T cells in the tumor, compared with the saline or OVA-treated group (figure 5B). Then, we extended our study on HPV16-E6/E7 tumor (TC-1) syngeneic mouse model using synthetic long peptides. Six days after TC-1 inoculation, mice received E7 peptide and UNE-C1, followed by a booster immunization (figure 5C). Results demonstrated that the UNE-C1+E7 peptide more significantly inhibited tumor growth than saline or the peptide alone (figure 5C). Consistently, the UNE-C1+E7 peptide showed enhanced infiltration of antigen-specific CD8+ and IFN-γ+ CD8+ T cells into the tumor microenvironment (figure 5D), indicating that UNE-C1 would effectively work in both whole protein and long peptide antigen models. Next, to examine the immune cells required for UNE-C1-mediated anticancer effects, immune cells were depleted using CD4−, NK−, and CD8-specific antibodies. The anticancer effect of UNE-C1 was abolished by the CD8 antibody, but not by the CD4 and NK1.1 antibodies (figure 6A). These data indicate that CD8+ T cells are required for UNE-C1-mediated antitumor activity.

Effect of UNE-C1 on immune responses against tumor growth in vivo. (A) After the tumor implantation, mice (n=5) were immunized on days 3 and 10. Schematic illustration of UNE-C1 treatment schedule and the recorded tumor volumes are shown. (B) E.G7-OVA tumor-bearing mice (n=5) were treated as described. Tumors were harvested on day 17, and CD8+ T cells in the tumor were analyzed. The frequencies of tumor-infiltrating CD8+ T cells, OVA-specific CD8+ T cells, and IFN+ CD8+ T cells were measured. (C) C57BL/6 mice were injected subcutaneaously with 1×105 of TC-1 cells. On days 6 and 13, mice (n=5) were injected subcutaneaously with the E7 peptide (20 µg) and UNE-C1 (100 µg). (D) TC-1 tumor-bearing mice (n=5) were treated as described. Tumors were harvested on day 19, and CD8+ T cells in the tumor were analyzed. The frequencies of CD8+ T cells, E7-specific CD8+ T cells, and IFN+ CD8+ T cells were measured. Data are representative of three independent experiments. Results are presented as mean±SEM, and statistical significance was analyzed with Student’s t-test (*p<0.05, **p<0.01, ***p<0.001). IFN-γ, interferon gamma; OVA, ovalbumin.
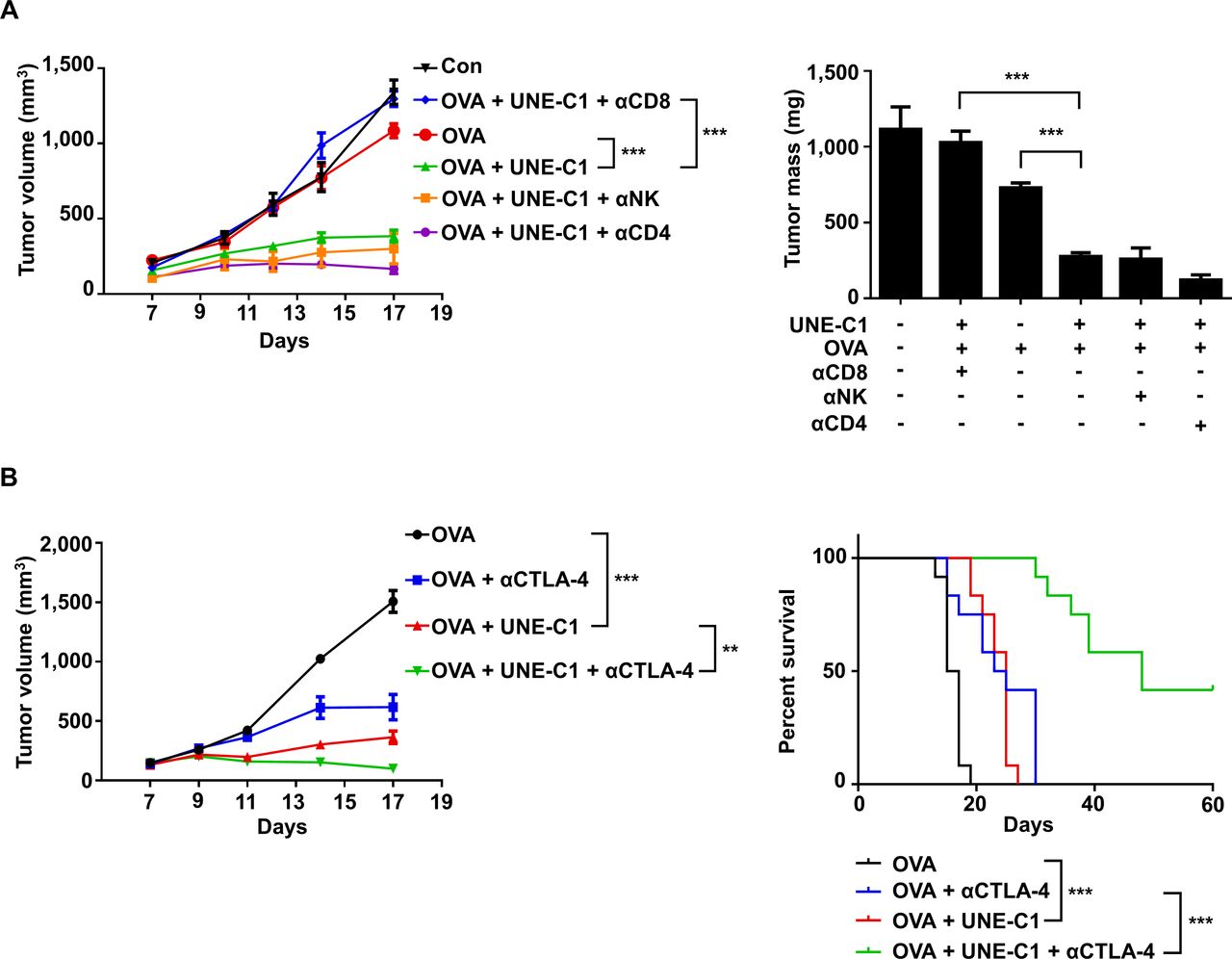
UNE-C1 induces CD8+ T-cell-mediated antitumor immune responses and shows synergy with anti-CTLA-4 antibody. (A) After E.G7-OVA tumor-bearing mice (n=5) were immunized on days 3 and 10, the mice were injected intraperitoneally with anti-CD8, anti-CD4, and anti-NK1.1 antibodies on days 2, 5, 8, and 11. Tumor volumes were monitored and tumors were collected on day 17 to determine tumor weights. (B) C57BL/6 mice (n=12) were implanted with 1×106 E.G7-OVA cells. OVA plus UNE-C1 was injected subcutaneously on days 3 and 10. Anti-CTLA-4 was injected intraperitoneally on days 3, 6, 9, 12, and 15 to check the synergy effect. Tumor volumes were measured and the percent of tumor survival is shown. Data are representative of three independent experiments. All results are presented as mean±SEM. P values of tumor volumes were calculated by Student’s t-test, and survival days were analyzed by the Mantel-Cox test (**p<0.01, ***p<0.001). OVA, ovalbumin.
Synergistic effect of UNE-C1 with anti-CTLA-4 treatment on tumor regression and survival in vivo
We also tested the effect of UNE-C1 in combination with an immune checkpoint inhibitor against tumor growth. Since the immunization of UNE-C1+antigens stimulated antigen-specific CD8+ T cells, we expected that blocking inhibitory signaling on T cells would further boost antitumor immunity. To test the possible synergistic effect, we injected E.G7-OVA tumor-bearing mice with UNE-C1+OVA, as shown in figure 5A. Meanwhile, anti-CTLA-4 antibody was injected intraperitoneally to prolong antitumor immunity. In the E.G7-OVA mouse model, the OVA+anti-CTLA-4 antibody or the UNE-C1-treated group slowed the tumor growth but failed to completely eradicate tumors (figure 6B and online supplementary figure S4A). In addition, survival time was increased in the OVA+anti-CTLA-4 antibody-treated or the UNE-C1-treated group compared with the antigen-alone group, but all mice died 30 days after tumor implantation (figure 6B). However, the combination of OVA+UNE-C1 with anti-CTLA-4 treatment significantly improved survival of tumor-bearing mice. Out of the 12 mice, five were free of tumors until the last day of our observation, indicating that UNE-C1 would show synergistic effects with the anti-CTLA-4 antibody against tumor growth (figure 6B).
Low systemic toxicity of UNE-C1
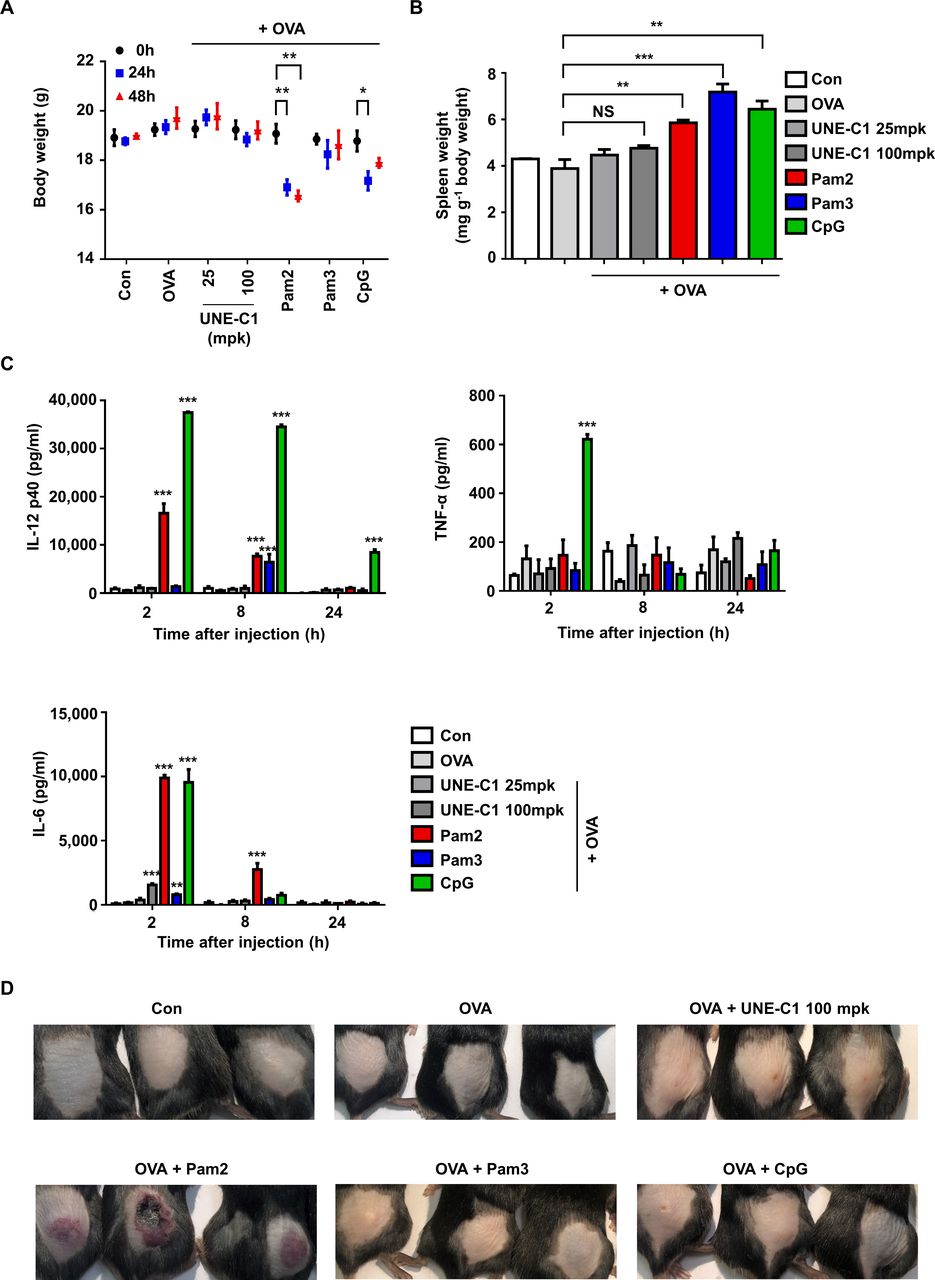
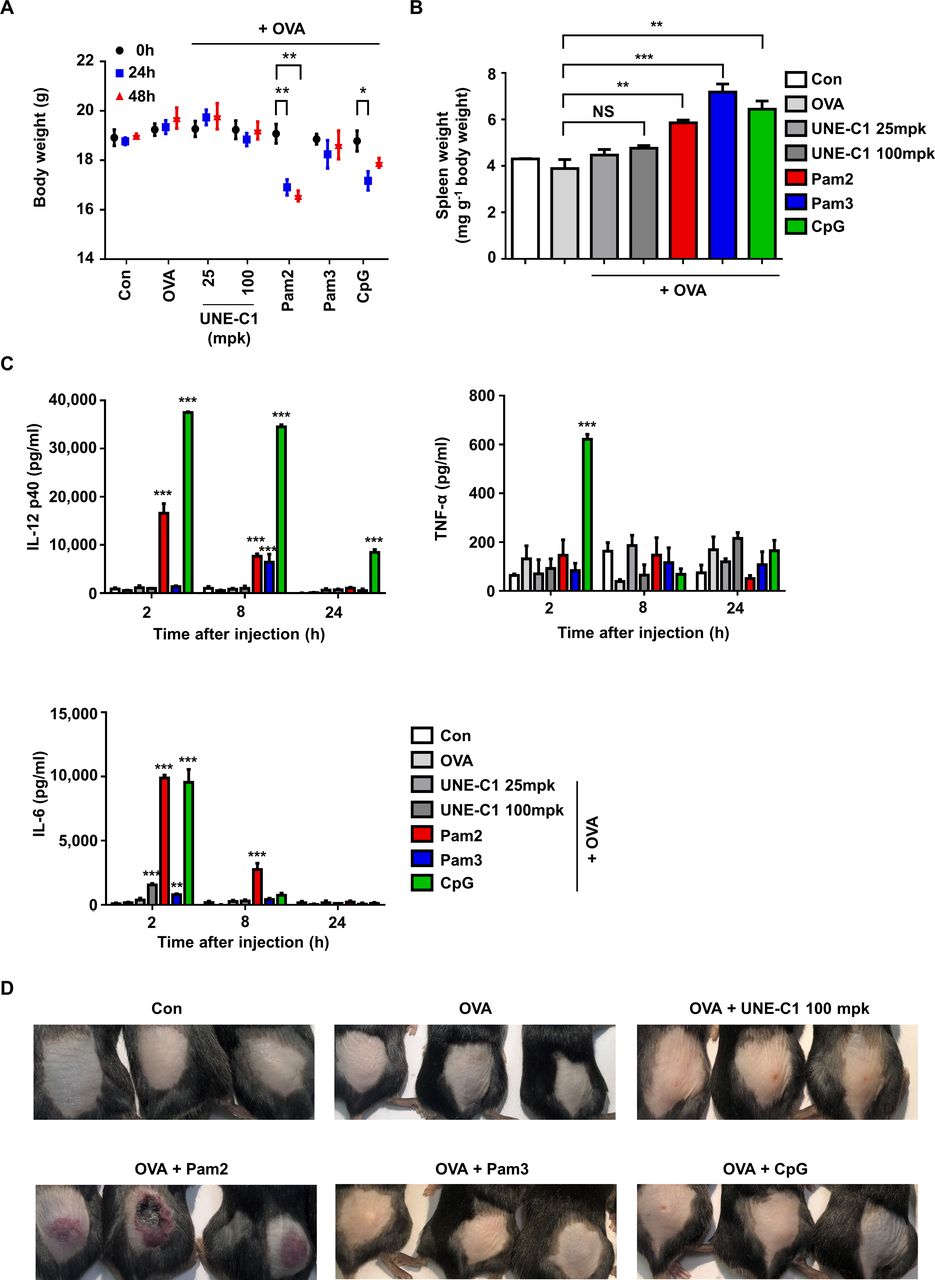
Since known TLR ligands often cause rapid systemic toxicity,30 a novel TLR agonist with safety and potency is needed for cancer vaccine development. To evaluate the potential toxicity of UNE-C1, we injected UNE-C1 into mice at 25 mpk (28.6 nmol) and 100 mpk (114.4 nmol). The TLR2 agonists, Pam2, Pam3 (114.4 nmol) and TLR9 agonists, CpG (28.6 nmol) were also injected as controls. UNE-C1 did not affect body weight at both doses, while the known TLR2 or TLR9 agonists induced body weight loss and/or splenomegaly (figure 7A,B). We also examined systemic toxicity by checking cytokines in the bloodstream after injection. While all of the known TLR agonist-treated groups showed a significant release of inflammatory cytokines into their serum as previously reported,13 31 UNE-C1 did not induce systemic toxicity at both doses (figure 7C). Particularly, CpG showed significant systemic toxicity even at a low dose of injection (28.6 nmol), although it produced antigen-specific Th1 and CTL responses superior or comparable to UNE-C1 (figure 4C–E). Furthermore, Pam2 provoked significant skin irritation, whereas an equal amount of UNE-C1 did not (figure 7). These results suggest that UNE-C1 could work as a novel immunogenic booster with higher safety, compared with known TLR agonists, in vivo.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Low systemic toxicity of UNE-C1 (A–D) C57BL/6 mice (n=3) were injected subcutaneously with OVA plus an excess doses of UNE-C1 or well-established TLR ligands. Two days after the injection, body (A) and spleen weights were measured and presented as a mass of spleen per body weight (B). At the indicated time, blood was collected, and the levels of IL-12p40, TNF-α, and IL-6 were measured by ELISA (C). Photos were taken 2 days after the injection (D). Three mice in each group are shown. Data are representative of three independent experiments. Results are presented as mean±SEM, and statistical significance was analyzed with Student’s t-test (*p<0.05, **p<0.01, ***p<0.001). IL, interleukin; NS, not significant; OVA, ovalbumin; TLR, Toll-like receptor.
Discussion
Here we unveiled the immune-stimulating activity of a unique domain (UNE-C1) inserted into the catalytic region of human CARS1. Considering that many newly acquired domains have been found at the terminal ends and non-catalytic regions of tRNA synthetases,5 6 10 32 it is intriguing that UNE-C1 is located within the catalytic domain of CARS1. Although it is not clear whether UNE-C1 would play any positive role in the catalytic process of CARS1, CARS1 lacking UNE-C1 lost its catalytic activity, suggesting its potential role in substrate binding or enzymatic reactions. To our surprise, the isolated UNE-C1 is expected to form a rigid conformation with extremely high stability based on CD and thermal denaturation analysis (online supplementary figure S1A), implying that its homologs may exist as a distinct entity. So far, it appears to be uniquely present in CARS1, although the functional reason for the integration of UNE-C1 and CARS1 into one polypeptide remains to be answered. We previously reported that the peptide containing the WHEP domain added to the N-terminal end of human WARS1 can prime innate immune responses on bacterial infection via its specific interaction with TLR4–MD27 32 and now report another domain, UNE-C1, as a specific ligand of TLR2. Perhaps, human ARSs have provided a unique platform to which endogenous TLR ligands can be functionally linked to coordinate immune response and protein synthesis during evolution.
Antigen-based cancer vaccines have recently attracted much interest as effective cancer therapy as manifested by many clinical trials.33–35 However, antigens alone often do not reach sufficient therapeutic potency due to poor immunogenicity.36 Therefore, numerous strategies have been explored to improve vaccine efficacy, especially by boosting immune responses with immune-stimulating adjuvants.37 38 Among these immune-activating adjuvants, TLR agonists have been suggested as they can induce immune responses by activating APCs.29 39 40 Particularly, it is well known that DCs, one of the main professional APCs, provide robust activating signals to antigen-specific CD8+ T cells on TLR ligation.26 29 As a consequence, CTLs are generated and infiltrate into the microenvironments of target tumors with specific antigens.35 40 41 Since the sufficient generation of tumor-specific CTL responses determines anticancer efficacy, TLR adjuvants with these properties are intensively investigated in the cancer vaccine field.38 42
Currently, TLR agonists based on microbial components or small chemicals are mainly tested as cancer vaccine adjuvants.29 43 However, little is known about protein-based TLR agonists, although they may have advantages over non-protein TLR ligands for conjugation with vaccine antigens.44 For example, antigens conjugated with proteins can lower the cost of vaccine production using a recombinant system. Further, it is possible to avoid poor immunogenicity and toxicity by modifying protein structures. Moreover, immune-stimulating proteins with specific receptors can also function as carrier proteins, leading efficient antigen delivery to APCs. In this study, we demonstrated that UNE-C1 activates the innate immune system via the TLR2/TLR6 complex (figure 3B–G) and the efficacy and toxicity of UNE-C1 in cancer vaccine models (figures 5–7). Coadministration of UNE-C1 with antigens induced potent adjuvant efficacy by upregulating antigen presentation, antigen-specific CTL priming and cytotoxicity (figure 4). Together, UNE-C1 can be considered a cancer vaccine adjuvant that can be conjugated with antigens for its application.
Moreover, elaborated tumor inhibition was shown when UNE-C1 with antigen was combined with CTLA-4 blockade (figure 6B). Numerous studies have stated the significance of blocking immune suppression in tumor microenvironments for successful cancer immunotherapy. It is known that activated T cells are negatively regulated by checkpoint molecules such as CTLA-4, PD-1, and PD-L1.45–47 In this regard, the development of immune checkpoint inhibitors has achieved remarkable success in treating patients with metastatic melanoma.48 However, response rates are only 10%–20%, possibly due to a lack of CTL generation and infiltration into the tumor.49 On this basis, enhanced tumor inhibition by the combination of UNE-C1 vaccination with CTLA-4 blockade suggests the potential of UNE-C1 as an immunoadjuvant for various checkpoint inhibitors.
Injection of an excess dose of UNE-C1 did not change body or organ mass in acute toxicity tests (figure 7A,B). In addition, UNE-C1 induced low systemic cytokine production even if it exerted comparable or superior efficacy compared with the known TLR2 activators (figure 7C). The low systemic toxicity of UNE-C1 is not clearly understood at this moment. Perhaps, it could be because UNE-C1 is the endogenous TLR2/TLR6 ligand as opposed to other known ligands with exogenous origins. Alternatively, UNE-C1 may not be well spread to the whole body due to its higher molecular weight, compared with other TLR2 ligands or to proteolytic cleavage. Whatever the reason may be, UNE-C1, showing high efficacy and low toxicity, suggests its great potential as an immune booster platform for various cancer vaccines with checkpoint inhibitors.
Conclusions
Here, we discovered a novel immune-activating domain incorporated into the catalytic region of human CARS1. While UNE-C1 is crucial for the protein synthesis activity of CARS1, it could be isolated as a highly stable entity and work as a potent immune activator with high safety. Injection of UNE-C1 with cancer antigen strongly augmented the antitumor activities of immune checkpoint inhibitor in vivo. This work thus shows functional convergence of a unique immune-activating domain to a protein synthesis enzyme during evolution toward higher-order organisms and suggests its potential as a novel immune-activating agent.
Acknowledgments
We thank Eun-Kyeong Jo and Shizuo Akira for providing TLR2–/– mice.
References
Footnotes
SC and SBK contributed equally.
Contributors SC, SBK and SK designed the study; SC and SBK developed the methodology; SC, SBK, YL, ECS, UK, HYK, JHS, PCG, YHK, IY, NYS, DK, I-KK, C-YK, SYJ, and MHK acquired the data; SC, SBK, and SK interpreted the data and wrote the paper. All authors read and approved the final manuscript. SC and SBK contributed equally.
Funding This work was supported by the Global Frontier Project grant (NRF-M3A6A4-2010-0029785) of National Research Foundation funded by the Ministry of Science and ICT of Korea.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Mouse experiments were performed under guidelines by the recommendations in the Guide for the Care and Use of Laboratory Animals of ‘Animal and Plant Quarantine Agency’, and the protocol was approved by the Institutional Animal Care and Use Committee of WOOJUNG BIO CROWISE (Suwon, Korea).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. The RNA sequencing data reported in this paper have been deposited in the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo, accession number GSE138488).