Article Text

Abstract
Background Neuroblastoma (NB) is a childhood cancer for which new treatment options are needed. The success of immune checkpoint blockade in the treatment of adult solid tumors has prompted the exploration of immunotherapy in NB; however, clinical evidence indicates that the vast majority of NB patients do not respond to single-agent checkpoint inhibitors. This motivates a need for therapeutic strategies to increase NB tumor immunogenicity. The goal of this study was to evaluate a new immunotherapeutic strategy for NB based on potent activation of the stimulator of interferon genes (STING) pathway.
Methods To promote STING activation in NB cells and tumors, we utilized STING-activating nanoparticles (STING-NPs) that are designed to mediate efficient cytosolic delivery of the endogenous STING ligand, 2’3’-cGAMP. We investigated tumor-intrinsic responses to STING activation in both MYCN-amplified and non-amplified NB cell lines, evaluating effects on STING signaling, apoptosis, and the induction of immunogenic cell death. The effects of intratumoral administration of STING-NPs on CD8+ T cell infiltration, tumor growth, and response to response to PD-L1 checkpoint blockade were evaluated in syngeneic models of MYCN-amplified and non-amplified NB.
Results The efficient cytosolic delivery of 2’3’-cGAMP enabled by STING-NPs triggered tumor-intrinsic STING signaling effects in both MYCN-amplified and non-amplified NB cell lines, resulting in increased expression of interferon-stimulated genes and pro-inflammatory cytokines as well as NB cell death at concentrations 2000-fold to 10000-fold lower than free 2’3’-cGAMP. STING-mediated cell death in NB was associated with release or expression of several danger associated molecular patterns that are hallmarks of immunogenic cell death, which was further validated via cell-based vaccination and tumor challenge studies. Intratumoral administration of STING-NPs enhanced STING activation relative to free 2’3’-cGAMP in NB tumor models, converting poorly immunogenic tumors into tumoricidal and T cell-inflamed microenvironments and resulting in inhibition of tumor growth, increased survival, and induction of immunological memory that protected against tumor re-challenge. In a model of MYCN-amplified NB, STING-NPs generated an abscopal response that inhibited distal tumor growth and improved response to PD-L1 immune checkpoint blockade.
Conclusions We have demonstrated that activation of the STING pathway, here enabled by a nanomedicine approach, stimulates immunogenic cell death and remodels the tumor immune microenvironment to inhibit NB tumor growth and improve responses to immune checkpoint blockade, providing a multifaceted immunotherapeutic approach with potential to enhance immunotherapy outcomes in NB.
- immunology
- tumours
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Neuroblastoma (NB) is the third most common form of childhood cancer, and the most common extracranial solid tumor of childhood, accounting for 15% of all pediatric cancer deaths each year.1 Approximately 50% of these children are diagnosed with an advanced and high-risk form of the disease, and approximately half of patients with stage 4 NB are expected to relapse, with little likelihood of long-term survival.2 Over the past decade, there have been significant advances in treatment for patients with high-risk disease, which typically comprises chemotherapy, surgery, radiotherapy, autologous hematopoietic stem cell transplantation, and anti-GD2 antibody immunotherapy.3 Despite this multimodal treatment regimen, which is often associated with significant treatment-related toxicity and complications,1 the 5 year overall survival for high-risk NB is only ~50%.3 Thus, new treatment options for NB are urgently needed, particularly for children with advanced and high-risk NB.
Immune checkpoint blockade (ICB) antibodies targeting CTLA-4, PD-1, or PD-L1 are revolutionizing the treatment of an expanding diversity of adult solid tumor types (eg, melanoma, lung cancer).4 5 While clinical trials of ICB in NB and other pediatric solid tumor malignancies are ongoing, initial results indicate that response rates are likely to be less than 10% for single-agent ICB.6–8 Such poor responses can be largely attributed to NB tumors being immunologically ‘cold’ as they lack significant T cell infiltration and instead contain high densities of immunosuppressive cell populations.9–12 This is particularly evident in high-risk NB, where amplification of N-Myc, which occurs in 25% to 30% of high-risk NB,3 has been linked to low tumor immunogenicity, poor T-cell infiltration, and lack of responsiveness to ICB.13 This has motivated recent efforts to identify agents and therapeutic strategies that can convert immunosuppressive NB tumors to a more immunogenic and T cell-inflamed (‘hot’) phenotype, which is associated with favorable outcome in NB.14–16
The stimulator of interferon genes (STING) pathway has recently been identified as an important mechanism by which the innate immune system is capable of sensing tumors, in order to initiate a type I interferon (IFN-I)-driven inflammatory program that stimulates dendritic cell (DC) cross-presentation of tumor antigens, ultimately leading to mobilization of tumor-specific CD8+ T cells.17–19 Owing to this critical role in cancer immune surveillance, cyclic dinucleotide (CDN) STING agonists have emerged as a promising new class of therapeutics that activate innate immunity to increase tumor immunogenicity.20–24 While STING agonists have been explored in preclinical models of several adult solid tumors with variable degrees of efficacy and are also being advanced in the clinic,25 the immunologic and therapeutic impact of STING activation in NB has not yet been explored. This is particularly significant in light of recent findings that MYCN amplification in NB is associated with reduced IFN-I pathway activity,13 which likely contributes to impaired endogenous T cell priming and infiltration and, hence, lack of responsiveness to ICB. We therefore postulated that pharmacological activation of STING in NB tumors could enhance antitumor immunity and response to ICB.
While promising immunotherapeutic agents, CDNs—anionic, highly water-soluble molecules—suffer from low bioavailability and poor drug-like properties.26–31 As a result, CDNs do not readily cross the cellular plasma membrane leading to limited access to the cytosol where STING is located.29 30 To address these drug delivery barriers, we have recently described STING-activating nanoparticles (STING-NPs)—endosome-destabilizing polymer vesicles (polymersomes) optimized for intracellular delivery of 2’3’-cGAMP (cyclic guanosine monophosphate-adenosine monophosphate, cyclic(G(2’,5’)pA(3’,5’)p)), the natural and endogenous high affinity ligand of STING.26 At physiologic pH, membrane-destabilizing segments are sequestered in the polymersome bilayer, shielded by a poly(ethylene glycol) (PEG) corona. In response to cellular uptake and endosomal acidification, the polymersomes disassemble to release CDNs and reveal membrane lytic domains that mediate endosomal escape of CDNs to the cytosol. Consequent to this design, STING-NPs are among the most potent platforms for STING activation reported to date, increasing the biological potency of cGAMP by two to three orders of magnitude. Herein we leverage this technology to determine if STING activation in NB cells and within the NB tumor microenvironment (TME) can increase tumor immunogenicity and stimulate antitumor T cell immunity. Our results provide the first evidence that STING agonists can be used as an immunotherapy for NB. Significantly, we find that the efficient cytosolic delivery of cGAMP enabled by STING-NPs potently activates STING signaling in both MYCN-amplified and non-amplified NB cells, resulting not only in increased expression of IFN-I and other interferon-stimulated genes (ISGs) but also immunogenic cell death (ICD) at therapeutically relevant nanomolar concentrations that are 2000-fold to 10000-fold lower than free cGAMP. Importantly, administration of STING-NPs increases antitumor T cell activation and infiltration, resulting in inhibition of tumor growth, prolonged survival, and stimulation of immunological memory that protects from tumor re-challenge. Furthermore, in a MYCN-amplified model of NB, we demonstrate that STING-NPs synergize with PD-L1 ICB in inhibiting tumor growth and increasing survival, providing preclinical validation of a novel combination immunoregimen for treating high-risk NB.
Results
STING expression correlates with T cell infiltration in NB patients
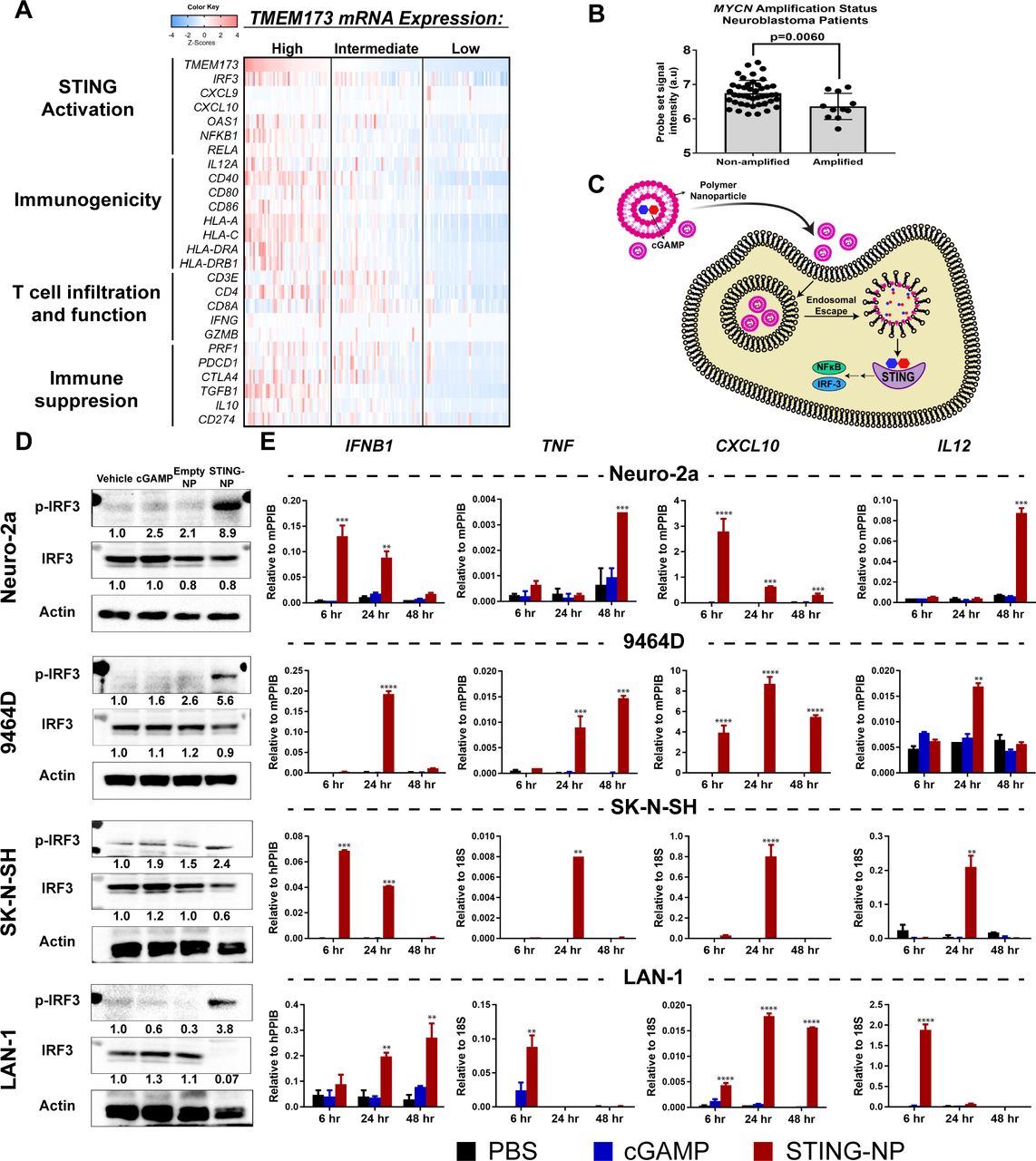
In mouse models of several cancers, activation of cGAS/STING signaling, either endogenous or treatment induced, has been shown to play an important role in stimulating the production of IFN-I and other ISGs that are critical to inducing antitumor T cells and in the response to several immunotherapies, including ICB.18 19 24 While this link between innate and adaptive antitumor immunity has been further supported by transcriptome analyses correlating STING-driven IFN-I gene signatures to T cell infiltration in some human cancers (eg, melanoma),32 such relationships are not observed ubiquitously across all tumor types that have been evaluated33 and, importantly, correlations between STING (TMEM173) expression and gene signatures of IFN-I and/or T cell infiltration have not yet been investigated in NB. We therefore first analyzed gene expression data in The Cancer Genome Atlas (TCGA) TARGET database (141 NB patients; figure 1A and online supplementary figure S1) to determine relationships between expression levels of TMEM173 and markers of STING activation and IFN-I responses (IRF3, CXCL9, CXCL10, OAS-I, NFKB1, RELA), immunogenicity (IL-12A, CD40, CD80, CD86, HLA-A, -C, -DRA, -DBRB1), T cell infiltration and function (CD3E, CD4, CD8A, IFNG, GZMB, PRF1), and immunosuppression (PDCD1, CTLA-4, TGB1, IL10, CD247). Our analysis revealed that intermediate/high expression of TMEM173 were associated with increased levels of several ISGs as well as NF-κB. Accordingly, this tended to correlate with increased markers of immunogenicity, including significant increases in levels of HLA-associated genes and costimulatory molecules. Consistent with increased expression of T cell chemokines (CXCL9 and CXCL10), levels of CD3, CD4, CD8A expression and markers of T cell activation (IFNG, GZMB, PRF1) also tended to be elevated in TMEM173 high/intermediate tumors. Additionally, STING expression, as well as the expression of STING pathway genes and ISGs, was significantly lower in MYCN-amplified NB (figure 1B and online supplementary figure S2), consistent with a previous report describing low T cell infiltration and poorer response to ICB in MYCN-amplified NB.13 While we did not identify STING expression as a prognostic indicator of survival in NB, as is the case for a number of tumor types,33 these analyses suggest that STING expression in NB largely correlates with T cell infiltration and activation, which has previously been correlated with increased survival in stage 4 NB.13 Taken together, these findings offer a potential link between STING activation and T cell responses in NB and serve to motivate the exploration of STING agonists as a strategy to increase tumor immunogenicity, T cell infiltration, and response to ICB in NB.
Supplemental material

Nanoparticle-enabled cytosolic delivery of 2’3’-cGAMP activates the STING pathway in neuroblastoma cells. (A) Integrated molecular analysis of mRNA expression of genes from the pediatric neuroblastoma TARGET dataset that have been distinguished by functional significance and clustered into evenly split tertiles based on high (upper tertile, n=47), intermediate (median tertile, n=47), and low (bottom tertile, n=47) TMEM173 mRNA expression. (B)TMEM173 mRNA expression in MYCN non-amplified and amplified samples profiled by microarray in the TARGET pediatric neuroblastoma (n=55) datasets. Data were accessed through the cBioPortal.69 Mann-Whitney U test (two-tailed) was used for statistical comparison.(C) Schematic representation of STING-NPs designed to enhance cytosolic delivery of cGAMP via endosomal escape, resulting in potent activation of STING signaling. (D) Neuroblastoma cell lines (Neuro-2a, 9464D, SK-N-SH, and LAN-1) were treated with vehicle (PBS), empty nanoparticles (NP), 200 nM (or 400 nM for 9464D) cGAMP, or STING-NPs for 48 hours; cells were collected for western blot analysis using anti-IRF3 and anti-phospho-IRF3 (p-IRF3) antibodies. Gel loading was normalized for equal actin; representative blots from one of two independent experiments. The relative density of bands is shown under each immunoblot, after normalization to the levels of actin. (E) qRT-PCR gene expression of IFNB1, TNF, CXCL10, and IL12 in neuroblastoma cell lines at 6, 24 and 48 hours after treatment with cGAMP or STING-NPs. (n=2, data shown as mean±SD; data was analyzed by two-way ANOVA followed by Dunnett’s posthoc test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 indicate a statistically significant difference relative to vehicle (PBS). ANOVA, analysis of variance; cGAMP, cyclic guanosine monophosphate–adenosine monophosphate; qRT-PCR,quantitative real-time PCR; STING, stimulator of interferon genes; STING-NPs,STING-activating nanoparticles.
Nanoparticle-enabled cytosolic delivery of cGAMP activates STING signaling in NB cells
Unlike the toll-like receptors (TLRs), which are expressed primarily in hematopoietic cells and may be restricted to specific immune cell subsets,34 STING is constitutively expressed at variable levels in many cancer cell types.33 35 This offers the potential to activate cancer cell-intrinsic mechanisms of innate immunity to increase immune recognition of tumors as well as potentially exploit STING-mediated tumor cell death to stimulate antitumor T cell responses.36–39 Moreover, the more ubiquitous expression profile of STING offers a mechanism for stimulating innate immunity that is independent of specific infiltrating immune cell subsets, which may be heterogeneous across patients.40 We found no mutations or alterations in STING in our analysis of NB tumors (0% of 814 profiled tumors in TCGA), providing a potential opportunity to harness cancer cell-intrinsic STING activation in the vast majority of NB patients. However, in analyzing gene expression data in the Cancer Cell Line Encyclopedia we found that mRNA expression of TMEM173 in human NB cell lines are among the lowest of all tumor types (online supplementary figure S3A); whether or not this correlates with protein-level expression or this trend holds true for murine cancer cell lines remains to be investigated. This motivated the need for a highly efficient strategy of activating STING signaling in NB cells. Based on the capacity of STING-NPs to enhance cytosolic delivery of cGAMP via endosomal escape as well as promote CDN uptake by tumor cells in the TME,26 we hypothesized that STING-NPs could be used to potently activate STING signaling in NB tumor cells (figure 1C). We first evaluated basal STING expression levels in both murine (9464D and Neuro-2a) and human (SK-N-SH and LAN-1) NB cell lines using western blot analysis and confirmed that STING was expressed in both MYCN-amplified (9464D, LAN-1) and non-amplified (Neuro-2a, SK-N-SH) cell lines (online supplementary figure S3B). We next evaluated the ability of STING-NPs to activate STING signaling in NB lines, which were treated with empty nanoparticles (NP), free cGAMP, or STING-NPs for 48 hours. In all NB lines tested, western blot analysis demonstrated that treatment with STING-NPs, but not empty NPs, resulted in significant increases in phosphorylation of interferon regulatory factor 3 (IRF3) (figure 1D). Activation of STING signaling by STING-NPs was further confirmed by quantitative real-time PCR (qRT-PCR) analysis of transcript levels of IFNB1, and other downstream antitumor effectors of STING activation including TNF, CXCL10, and IL12 in NB cell lines (figure 1E). Significantly, free cGAMP and empty NPs (online supplementary figure S4) had no effect on IRF3 phosphorylation or downstream gene expression, demonstrating the importance of nanoparticle-mediated cytosolic delivery of cGAMP on activation of STING signaling in NB cells.
STING activation triggers NB cell apoptosis and immunogenic cell death
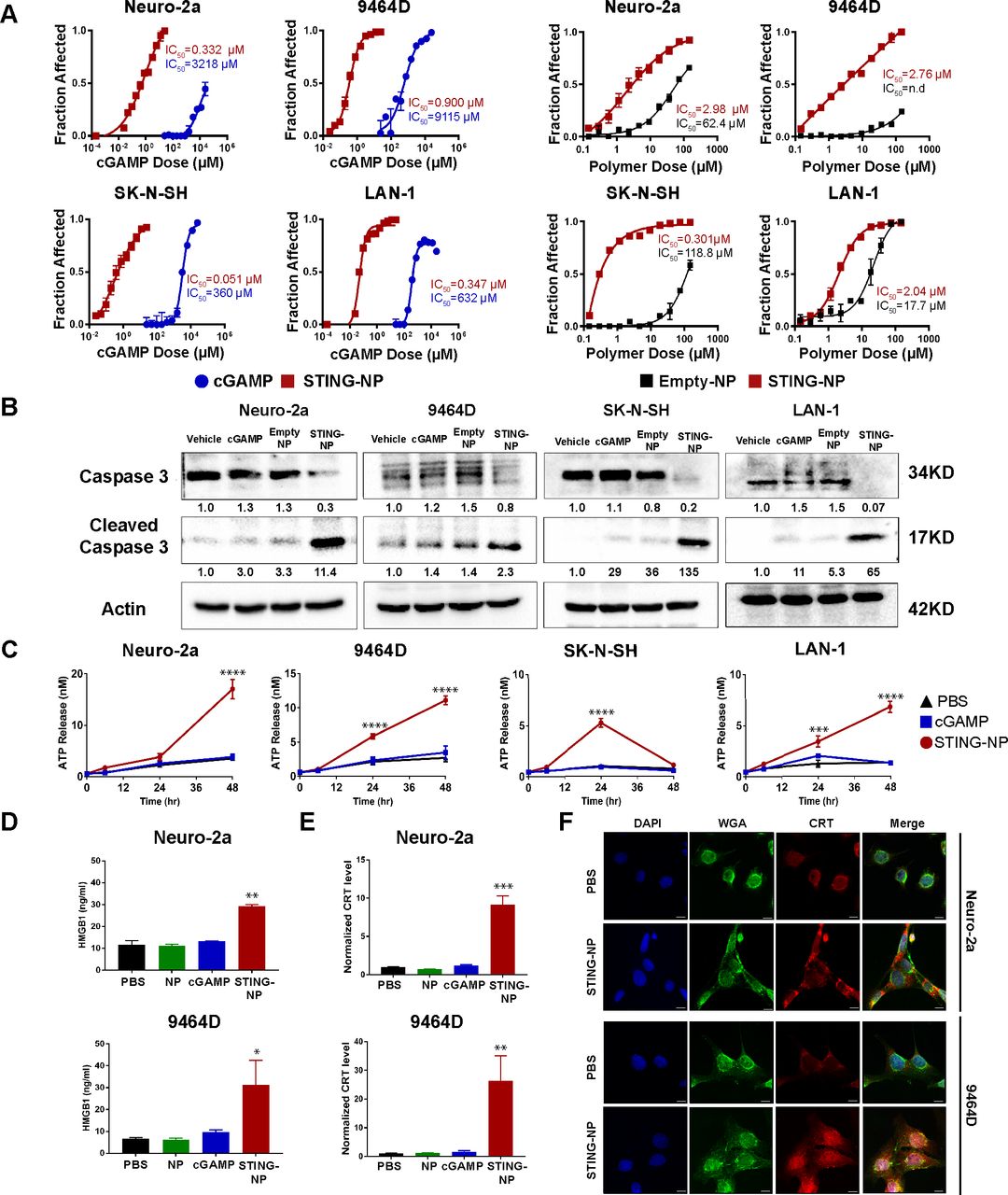
Activation of cGAS/STING signaling has recently been shown to induce cell death in both immune cells as well as some cancer cells,36 37 39 41 but the possibility that STING agonists may exert direct cytotoxic effects on NB cells has not yet been explored. We therefore investigated the effect of STING-NPs and free cGAMP on the viability of both MYCN-amplified and non-amplified NB cell lines. Owing to their capacity for highly efficient cytosolic cGAMP delivery, STING-NPs significantly abrogated NB cell viability, resulting in a reduction of the cGAMP IC50 by 2000-fold to 10000-fold to ~50 to 1000 nM (figure 2A). STING-NPs also significantly reduced NB cell viability to a greater extent than empty NPs in all NB lines, further implicating activation of STING signaling via cytosolic delivery of cGAMP in NB cell death. Furthermore, Annexin V/7-AAD staining revealed that STING-NP treatment induced 25-fold and 18-fold higher levels of apoptosis relative to controls in Neuro-2a and 9464D cells, respectively (online supplementary figure S5). Consistent with this finding, western blot analysis confirmed that STING-NPs triggered NB cell apoptosis as evidenced by increased levels of cleaved-caspase 3 in all cell lines tested (figure 2B). Therefore, in addition to their immunostimulatory capacity, STING-NPs also induce cell death in all of the NB cell lines tested here (9464D, Neuro-2a, SK-N-SH and LAN-1).
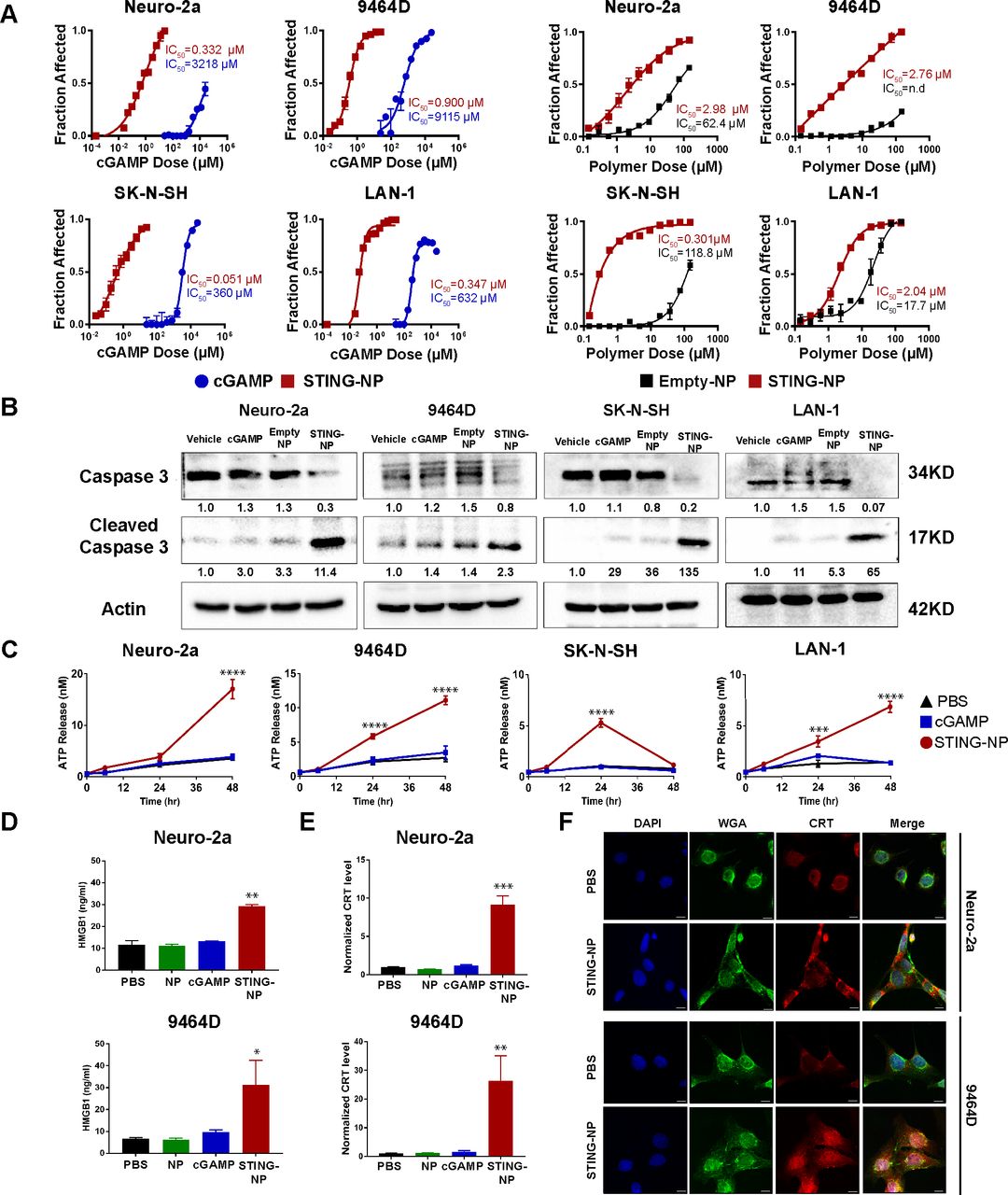
STING activation induces apoptosis associated with hallmarks of immunogenic cell death in neuroblastoma cell lines. (A) Neuroblastoma cells (Neuro-2a, 9464D, SK-N-SH and LAN-1) were treated with cGAMP, STING-NPs, or empty NPs for 72 hours and cell viability was measured. IC50 values were obtained by non-linear regression curve fitting; n=2–4 biologically independent samples. (B) Neuroblastoma cells were treated with vehicle (PBS), empty NP, cGAMP, and STING-NPs for 24 hours followed by western blot analysis of caspase 3 and cleaved caspase 3 in cell lysates. Gel loading was normalized for equal actin; representative blots from one of two independent experiments. The relative density of bands is shown under each immunoblot, after normalization to the levels of actin. (C) ATP release in neuroblastoma cells following treatment with vehicle (PBS), 500 nM cGAMP, or STING-NPs for 6, 24 and 48 hours (n= 3 biologically independent samples; one-way ANOVA with Dunnett’s post hoc test). (D) Concentration of HMGB1 released into culture supernatants by Neuro-2a or 9464D cells 48 hours after treatment (n= 3 biologically independent samples; one-way ANOVA with Dunnett’s post hoc test). (E) Flow cytometric detection of cell surface calreticulin (CRT) on Neuro-2a and 9464D cells in response to PBS, NP, 500 nM cGAMP, or STING-NPs after 48 hours. Data is represented as fold increase in median fluorescent intensity over unstained control (n= 3 biologically independent samples; one-way ANOVA with Dunnett’s post hoc test). (F) Representative fluorescence micrographs demonstrating CRT surface localization in Neuro-2a and 9464D cells following treatment with PBS or STING-NPs (500 nM) for 48 hours. Cell nuclei (blue), surface membrane (green), and CRT (red) were detected by DAPI, Alexa Fluor 488-conjugated Wheat Germ Agglutinin, and Alexa Fluor 647-conjugated anti-CRT antibody staining, respectively. All data shown are mean±SD. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance; ATP, adenosine triphosphate; cGAMP, cyclic guanosine monophosphate–adenosine monophosphate; STING, stimulator of interferon genes; STING-NPs,STING-activating nanoparticles.
Immunogenic cell death (ICD) is an inflammatory form of cell death that is implicated in the generation of antitumor adaptive immunity.42 During ICD, dying cells upregulate expression and/or release of damage-associated molecular patterns (DAMPs) that act through a diversity of mechanisms to increase antigen cross-presentation and provide an inflammatory context for generating antigen-specific T cell responses. ICD may be associated with multiple mechanisms of cell death (eg, pyroptosis, necroptosis) and can be triggered via a myriad of therapeutic agents and interventions (eg, radiation, anthracyclines, viral infection).42 Recent evidence has also demonstrated that ICD can be induced in some cancer cells via activation of the cGAS-STING pathway;36 however, this has not yet been explored in the majority of cancer types, including in NB. Given the potent cytotoxic effects of STING-NPs in multiple NB lines, we next investigated if STING-mediated cell death was associated with several common and widely accepted biochemical correlates of ICD, including adenosine triphosphate (ATP) release, HMGB-1 release, and surface expression of calreticulin (CRT), an ‘eat me’ signal that enhances phagocytosis of dying tumor cells.42 In all NB lines tested (Neuro-2a, 9464D, SK-N-SH, and LAN-1), STING-NPs, but not free cGAMP, triggered release of ATP 24 to 48 hours post-treatment (figure 2C). Similarly, STING-NPs, but not free cGAMP or empty NPs, stimulated the secretion of HMGB1 (figure 2D) and promoted translocation of CRT to the cell surface, which was assessed by both flow cytometry (figure 2E) and immunofluorescence staining (figure 2F).
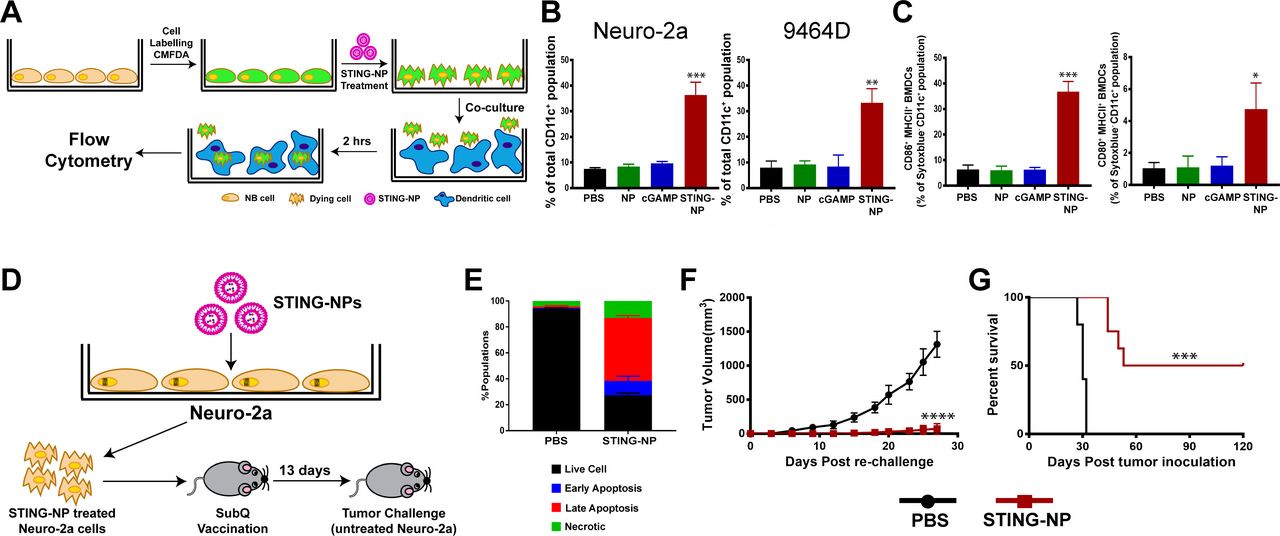
We further evaluated whether activation of STING in NB cells could enhance dendritic cell activation and phagocytosis of tumor cells. 9464D and Neuro-2a cells were fluorescently labeled with CellTracker green CMFDA dye, treated with STING-NPs, empty NPs, free cGAMP, or PBS (vehicle) for 48 hours, and co-cultured with bone marrow-derived dendritic cells (BMDCs) at a 1:1 ratio for 2 hours, followed by flow cytometric analysis of tumor cell uptake by BMDCs (figure 3A). Consistent with increased CRT expression, STING-NP-treated cells were phagocytosed by BMDCs to a significantly greater extent than all other treatment groups (figure 3B). We further evaluated the ability of NB cells undergoing ICD in response to STING-NPs to induce maturation of BMDCs. Consistent with STING-mediated expression of proinflammatory cytokines and DAMPs, co-culture of BMDCs with STING-NP-treated NB cells resulted in upregulation of the costimulatory molecules CD86 and CD80 (figure 3C), markers of BMDC maturation. Together, these results indicate that STING-mediated induction of ICD in NB cells enhances tumor cell phagocytosis and BMDC maturation, therefore demonstrating the potential of STING-NPs to enhance priming of tumor antigen-specific T cell responses.

Activation of STING in neuroblastoma increases tumor cell phagocytosis and generates protective antitumor immunity. (A) Schematic representation of tumor cell phagocytosis assay. (B) Flow cytometric quantification of relative NB cell uptake by BMDCs after co-culture with Neuro-2a or 9464D cells that were treated with PBS, empty NPs, 800 nM cGAMP, or STING-NPs and stained with CMFDA. (C) Percentage of CD11c+MHCII+CD86+ and CD11c+MHCII+ CD80+ BMDCs after 18 hours of co-culture with Neuro-2a cells treated with PBS, NPs, 800 nM cGAMP and STING-NPs; **p<0.01 and ***p<0.001, one-way ANOVA followed by Dunnett’s multiple comparison test. (D) Schematic representation of cell-based tumor vaccination model. A/J mice were vaccinated with Neuro-2a cells pre-treated with STING-NPs at 800 nM cGAMP for 48 hours (n=8 mice/group) or vehicle control (PBS, n=5 mice/group) and 13 days later mice were challenged with 106 live Neuro-2a cells in the opposite flank and tumor growth was measured. (E) Annexin V/7-AAD staining was used to confirm that~70% of NB cells were dying (~15% necrotic,~10% early apoptotic,~45% late apoptotic) prior to vaccination. Tumor growth curves (F) and percent survival (G) of naïve tumor-free mice that were vaccinated with Neuro-2a cells pre-treated with STING-NPs or vehicle control (PBS), and subsequently challenged with Neuro-2a cells on the opposite flank. Tumor volumes were compared on day 27 by Student’s t-test and log-rank (Mantel-Cox) test was used to compare Kaplan-Meier survival curves. ***p<0.001, ****p<0.0001 indicate a statistically significant difference. All data shown is mean±SD. ANOVA, analysis of variance; BMDCs, bone marrow-derived dendritic cells;cGAMP, cyclic guanosine monophosphate–adenosine monophosphate; NB, neuroblastoma; NP, nanoparticles;STING, stimulator of interferon genes; STING-NPs, STING-activating nanoparticles.
While these in vitro data supported the ability of STING-NPs to induce ICD in NB, the gold standard for testing ICD relies on vaccinating immunocompetent, syngeneic mice with cells killed in vitro followed by challenge of vaccinated mice with live cells of the same type43 (figure 3D). We treated Neuro-2a NB cells with STING-NPs for 48 hours to induce ~70% cell death (figure 3E), subsequently inoculated A/J mice with dying NB cells, and challenged mice 13 days later with 1×106 live Neuro-2a cells injected subcutaneously into the opposite flank (figure 3D). Immunization of tumor-free naïve mice with STING-NP-treated Neuro-2a cells provided significant protection from tumor growth (figure 3F), with 50% (4/8) of mice remaining tumor-free for at least 120 days (figure 3G). Collectively, these data validate STING activation as a novel mechanism for inducing ICD in NB, a process that is significantly augmented via nanoparticle-enabled cytosolic delivery of cGAMP.
STING activation reprograms the NB tumor immune microenvironment
While pharmacologic modulation of STING has been explored in murine models of several adult solid tumors,20 23 44–46 with diverse and multifaceted effects on the TME and variable levels of therapeutic efficacy reported, the effect of STING activation on the NB tumor microenvironment has not yet been investigated. Harnessing the capacity of STING-NPs to enhance in vivo cGAMP activity, we first validated the ability of STING-NPs to stimulate STING signaling in the NB TME. In a Neuro-2a model of NB, treatment with STING-NPs resulted in significant increases in expression of IFN-I (IFNB1), proinflammatory cytokines (TNF, IL12), and T cell chemokines (CXCL10), consistent with a STING-driven inflammatory response (figure 4A). Increased STING activation was further validated via western blot analysis that demonstrated increased protein level expression of phospho-IRF3 and phospho-STAT1 in STING-NP treated tumors (figure 4B). Additionally, consistent with in vitro findings demonstrating STING-NP-driven NB cell death, western blot analysis also confirmed increased levels of cleaved-caspase 3 in NB tumors (figure 4B). This was further supported by immunohistochemical analysis, which also demonstrated increased levels of cleaved-caspase 3 and reduced cell proliferation as determined via Ki67 staining of Neuro-2a tumor sections (figure 4C). We next evaluated the effect of STING activation on T cell infiltration. Consistent with the increased expression of CXCL10, immunohistochemical staining demonstrated a significant increase in CD8+ T cell infiltration into Neuro-2a tumors in response to STING-NP treatment (figure 4D). Interestingly, this was also accompanied by a reduction in FoxP3 staining, indicative of decreased levels of immunosuppressive regulatory T cells (Treg). Similar results were observed in a MYCN-amplified 9464D model (online supplementary figure S6) in which we also demonstrated that free cGAMP had an insignificant effect on STING activation, consistent with our previous findings in a melanoma model.26 Collectively, these data demonstrate that STING-NPs convert NB tumors to a more tumoricidal, immunogenic, and T cell inflamed phenotype.
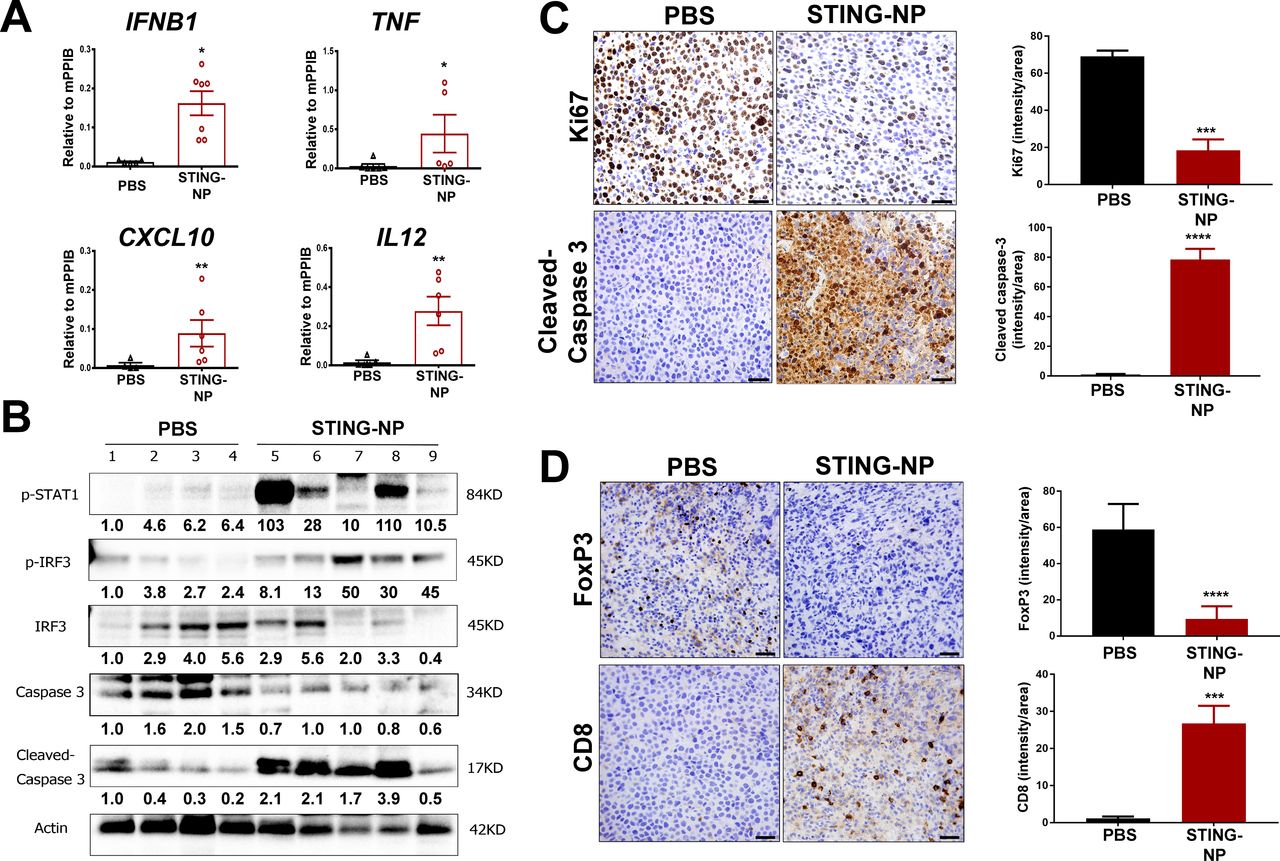
STING activation in neuroblastoma generates an immunogenic, tumoricidal, and T-cell inflamed tumor microenvironment. Mice with 200 mm3 Neuro-2a tumors were administered PBS or STING-NPs (10 µg cGAMP) intratumorally three times, spaced 3 days apart, and tumors were harvested 48 hours following the last treatment. (A) qRT-PCR analysis of IFNB1, TNF, CXCL10, and IL12 gene expression in injected Neuro-2a tumors. n=5 to 7 mice per group represented as mean±SEM, *p<0.05, **p<0.01 indicate a statistically significant difference using a two-tailed Mann-Whitney U test. (B) Neuro-2a tumor lysates (n=4–5) were analyzed using western blot for phospho-STAT1 (p-STAT1), IRF3, and phospho-IRF3 (p-IRF3), caspase 3, and cleaved caspase 3. Gel loading was normalized for equal actin. The density of bands is shown under each immunoblot, after normalization to the levels of actin. (C/D) Immunohistochemical staining of tumor sections for cleaved caspase-3 (apoptosis), Ki-67 (proliferation), CD8, and FoxP3 and corresponding quantification of staining intensity using ImageJ software. Data shown as mean±SD for n=3 Neuro-2a tumors, ***p<0.001, ****p<0.0001 indicate a statistically significant difference using a Student’s t-test. cGAMP, cyclic guanosine monophosphate–adenosine monophosphate; qRT-PCR,quantitative real-time PCR; STING, stimulator of interferon genes; STING-NPs, STING-activating nanoparticles.
STING activation inhibits NB tumor growth and stimulates antitumor immunity
Based on the capacity of STING-NPs to increase NB immunogenicity, promote ICD, and stimulate antitumor innate immunity in the NB TME, we next evaluated therapeutic efficacy in both non-amplified (Neuro-2a) and MYCN-amplified (9464D) models of NB. Here we used an intratumoral (i.t.) administration route being explored clinically for STING agonists and other innate immune activators as an in situ vaccination strategy for eliciting antitumor T cell responses to a broad swath of tumor antigens.47–49 Mice growing subcutaneous NB tumors were treated via i.t. administration of STING-NPs, cGAMP, or vehicle (PBS) three times, spaced 3 days apart (figure 5). In both Neuro-2a (figure 5A–D) and 9464D (figure 5H–K) NB tumor models, STING-NPs inhibited tumor growth, while free cGAMP had no efficacy relative to vehicle control. In Neuro-2a tumors, a 100% complete response rate with no overt evidence of tumor growth 35 days after the final STING-NP injection was observed (figure 5B–D), whereas 50% of mice appeared tumor-free through 80 days in the 9464D tumor model (figure 5I–K). Minimal weight loss was observed in response to i.t. administration of STING-NPs in NB models (online supplementary figure S7). Based on the high complete response rate in the Neuro-2a model, we next re-challenged mice on the contralateral flank to evaluate the ability of in situ vaccination with STING-NPs to protect against recurrence. We found that 75% of mice were completely protected from tumor re-challenge without any evidence of tumor growth through at least day 120, indicative of a T-cell memory response (figure 5E–G). While such a memory response is strongly suggestive of the generation of tumor antigen-specific T cells, the magnitude and antigenic specificity of the response remains to be confirmed. Additionally, the breadth of a T cell response elicited by in situ vaccination with STING agonists has not yet been evaluated in NB or other tumor models. Nonetheless, these studies provide the first demonstration that potent activation of STING in the NB TME can stimulate antitumor immunity that inhibits tumor growth with potential to combat disease recurrence.

STING-NPs inhibit tumor growth in murine models of neuroblastoma. (A) Schematic summary of treatment and re-challenge for Neuro-2a tumor model. Mice were inoculated with 1×106 Neuro-2a cells and were treated intratumorally with 10 µg STING-NPs, free cGAMP, or PBS three times, spaced 3 days apart. Mice demonstrating complete responses were re-challenged on the contralateral flank on day 50 with 1×106 Neuro-2a cells. (B) Individual tumor growth curves of Neuro-2a tumor-bearing mice. (C) Average tumor size of Neuro-2a tumor-bearing mice treated with PBS, cGAMP, or STING-NP (n=6–7 mice/group, tumor volumes of treated groups were compared with vehicle on day 23 by one-way ANOVA followed by Dunnett’s post hoc test ****p<0.0001). (D) Kaplan-Meier survival curves of mice treated with indicated formulation using 1500 mm3 tumor volume as endpoint criteria (n=6-7 mice/group, two-tailed Mantel-Cox test, ****p<0.0001). Mice exhibiting complete responses to STING-NP treatment were re-challenged with Neuro-2a cells on the contralateral flank and tumor growth was compared with treatment-naïve mice inoculated with Neuro-2a cells. (E) Spider plots of tumor naïve and STING-NP re-challenged mice. (F) Average tumor size of re-challenged mice compared with tumor naïve mice (n=7 biologically independent samples, tumor volume was compared on day 23 by Student’s t-test ***p<0.001). (G) Kaplan-Meier survival curves following Neuro-2a challenge for treatment naïve mice vs mice rendered tumor-free (ie, complete responders) by STING-NP treatment of the initial tumor (n=7 mice/group; two-tailed Mantel-Cox test, ***p<0.001). (H) Schematic summary of treatment and re-challenge for 9464D tumors. Mice were inoculated with 1×106 9464D cells and treated as indicated. (I) Individual tumor growth curves of 9464D tumor bearing mice. (J) Average tumor size of 9464D tumor bearing mice treated with PBS, cGAMP or STING-NP (n=9-10 mice/group, tumor volumes of treated groups were compared with vehicle on day 46 by one-way ANOVA followed by Dunnett’s post hoc test ****p<0.0001).(K) Kaplan-Meier survival curves of mice treated with indicated formulation using 1500 mm3 tumor volume as endpoint criteria (n=9-10 mice/group, two-tailed Mantel-Cox test, ****p<0.0001). ANOVA,analysis of variance; cGAMP, cyclic guanosine monophosphate–adenosine monophosphate; STING, stimulator of interferon genes; STING-NPs, STING-activating nanoparticles.
STING-NPs enhance response to PD-L1 blockade in a model of MYCN-amplified NB
While PD-L1 expression in NB is typically low and infrequent,7 8 particularly in MYCN-amplified NB,13 NB cells can upregulate PD-L1 expression in response to IFN-γ produced by tumor infiltrating T cells.50 Indeed, we observed increased levels of PD-L1 via qRT-PCR and western analysis in subcutaneous 9464D tumors following STING-NP treatment (online supplementary figure S5B and D). Additionally, STING activation can directly increase expression of PD-L1 on NB cells (online supplementary figure S5E). This is also consistent with increased PD-L1 (CD274) expression in patients with intermediate/high STING expression (figure 1A and online supplementary figure S1). We reasoned that expression of PD-L1, induced either in direct response to STING activation or subsequently via IFN-γ production by tumor infiltrating T cells, may restrain the efficacy of STING-NPs, particularly in the 9464D model where a lower complete response rate was observed. We therefore investigated the effect of systemic (intraperitoneal) administration of anti-PD-L1 antibodies (αPD-L1) on responses to i.t. STING-NP treatment. To evaluate this, we used a synchronous tumor model in which two 9464D tumors were concurrently established subcutaneously on opposite flanks and STING-NPs were i.t. administered into only one tumor, ie, the treated tumor (figure 6A). This is a strategy being explored in many clinical trials of innate immune agonists as a strategy to induce a systemic immune response capable of inhibiting the growth of a distal (untreated) tumor (ie, abscopal effect).47 The growth of both the treated and untreated tumors was suppressed by administration of STING-NPs, resulting in an increased median survival time (54 days) (figure 6B,C). Consistent with the immune ‘cold’ phenotype of most NB tumors, αPD-L1 monotherapy had no effect on tumor growth relative to vehicle control. However, combining PD-L1 blockade with STING-NPs further reduced growth, an effect that was particularly evident in the untreated (ie, abscopal) tumor, leading to a median survival time of 68 days, a significant increase relative to both vehicle (median survival time of 44 days) and STING-NP only treatment groups. Consistent with these findings, we found that combining STING-NPs with αPD-L1 resulted in significant increases in CD8+ T cell infiltration (figure 6D) and reduced FoxP3 staining in both the treated and untreated tumor (figure 6E), whereas PD-L1 blockade alone had no effect on CD8 staining and only a modest effect on FoxP3 levels. Collectively, these findings offer the first demonstration that STING activation can increase response rates to ICB in MYCN-amplified NB, and provide preclinical evidence supporting a novel combination immunotherapy for treating high-risk NB.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
STING activation sensitizes MYCN-amplified neuroblastoma to PD-L1 immune checkpoint blockade. (A) Schematic summary of treatment for 9464D tumors. Mice were inoculated with 1×106 9464D cells in both flanks, and when tumors became palpable (day 14) the right side was treated with STING-NP (10 µg cGAMP) intratumorally and/or αPD-L1 antibodies (100 µg) intraperitoneally. (B) Average tumor size and spider plots for treated and untreated tumors. (n=8–10 mice/group; tumor volumes of treated groups were compared with vehicle on day 37 by one-way ANOVA followed by Dunnett’s post hoc test ***p<0.001,****p<0.0001. (C) Kaplan-Meier survival curves of mice treated with indicated formulation using a total tumor volume >1500 mm3 as endpoint criteria (n=8-10 mice/group; two-tailed Mantel-Cox test, *p<0.05, ***p<0.001). (D/E) Immunohistochemical staining and quantification of CD8 and FoxP3 staining on treated and untreated tumors 48 hours following the last STING-NP administration; data shown as mean±SD and treated tumors were compared with vehicle control by one-way ANOVA followed by Dunnett’s post hoc test *p<0.05, ***p<0.001, ****p<0.0001 indicate a statistically significant difference. Scale bars: 25 µm. ANOVA,analysis of variance; cGAMP, cyclic guanosine monophosphate–adenosine monophosphate; STING, stimulator of interferon genes; STING-NPs,STING-activating nanoparticles.
Discussion
While low-risk and intermediate-risk NB can often be cured using a combination of conventional therapies such as high-dose chemotherapy, surgery, and radiotherapy, although with considerable toxicities, treatment of high-risk NB remains a challenge owing to the metastatic, heterogeneous, and aggressive nature of the disease.51 Despite their success in a growing number of adult solid tumors, immune checkpoint blockade antibodies targeting CTLA-4, PD-1, and PD-L1 have not yet significantly impacted clinical outcomes in NB.6 Similar to other ‘cold’ tumors, this can be largely ascribed to several intertwined factors including a relatively low mutation load, a highly immunosuppressive TME, and poor infiltration of tumor antigen-specific CD8+ T cells.11 52 53 Therefore, the development of new and more effective immunotherapy strategies for high-risk NB has become a high priority and has prompted recent exploration into alternative immunotherapeutic modalities and combinations. Here we describe the functional and mechanistic roles of STING-NPs—a new class of nanoparticle designed for efficient cytosolic delivery of cyclic dinucleotide STING agonists—in increasing tumor cell immunogenicity, reprogramming the NB TME, and stimulating anti-tumor immunity that inhibits tumor growth and improves survival in murine models of NB.
The important role of STING in cancer immune surveillance has recently motivated the development of cGAMP and other structurally-related CDNs as therapeutics that activate innate immunity to enhance antitumor T cell responses, including in models of non-immunogenic tumors that are resistant to ICB.24 44 46 However, similar to other nucleic acid therapeutics, CDNs suffer from poor intracellular bioavailability and suboptimal pharmacokinetic properties that restricts their activity and clinical utility.29 30 To address these challenges, we recently developed STING-NPs to enhance the cytosolic delivery of CDNs via an active endosomal escape mechanism,26 a drug carrier design that results in significant enhancement of immunostimulatory potency. Additionally, nanoparticle-based STING agonists offer a number of potential advantages over their small molecule counterparts, including enhanced delivery to tumor infiltrating macrophages and dendritic cells,26 31 which have been implicated as critical responders to STING agonists,22 26 31 as well as an enhanced capacity to exploit lymphatic drainage to enrich CDN delivery and STING activation in sentinel lymph nodes,26 28 a primary site of T cell priming that may also be immunosuppressed.54 Therefore, we leveraged this technology to achieve potent stimulation of STING signaling in NB tumors, a poorly immunogenic tumor that is largely resistant to checkpoint blockade and in which STING agonists have not yet been evaluated.
While several molecularly-defined pattern recognition receptor agonists are being advanced in the clinic (eg, CpG ODN) and have been also been explored in NB,55 56 a potential advantage of targeting STING, which is expressed in a majority of cancer cells, is the capacity to activate innate immune signaling not only in infiltrating immune cells, but also in the tumor cell compartment. While the importance of tumor-intrinsic STING activation in response to STING agonists remains a topic of debate and may depend on tumor type,57 direct activation of STING in cancer cells may nonetheless obviate requirements for infiltration of distinct immune cell populations (eg, plasmacytoid DCs responding to TLR-9 agonists58). Indeed, despite low expression levels of STING relative to other cancer types (online supplementary figure S2A), the efficient cytosolic delivery of cGAMP enabled by STING-NPs potently activated STING signaling in all NB cell lines evaluated, resulting in expression of type I interferons, antitumor cytokines, and T cell chemokines. Recent work has also demonstrated that activation of the cGAS/STING pathway can induce apoptosis in some cancer cell lines, whereas other cell types are largely resistance to STING-mediated cell death.57 As this has not been investigated in NB, we examined the cytotoxic effects of STING-NPs in both mouse and human cell lines representing both MYCN-amplified and non-amplified NB. We found that STING-NPs triggered NB cell apoptosis at concentrations several thousand-fold lower than free cGAMP, which demonstrated in vitro toxicity only at high micromolar or millimolar concentrations. Notably, the use of STING-NPs resulted in IC50 values for cGAMP that are comparable to those of chemotherapeutics commonly employed in treatment of NB (eg, doxorubicin, etoposide).59 Accordingly, intratumoral administration of STING-NPs, but not free cGAMP, stimulated apoptosis and reduced cell proliferation in NB tumors at therapeutically relevant doses of cGAMP. The susceptibility of NB cells to STING-NP-mediated apoptosis relative to other cancer types or to stromal and immune cells in the TME remains to be elucidated.
Significantly, we also found that STING-mediated cell death of NB cells was associated with several hallmarks of ICD, including the release of ATP and HMGB1 as well as surface expression of CRT, an important mediator of tumor cell phagocytosis. Importantly, we validated STING-mediated ICD using STING-NP-treated Neuro-2a cells as a cell-based vaccine that conferred a high degree of protection against tumor challenge. Notably, this was achieved using only a single vaccine dose and without the addition of an exogenous adjuvant, highlighting the potential to use STING-NPs as a platform for augmenting immune responses to cell-based vaccines. Collectively, these findings demonstrate that potent induction of STING signaling in NB can trigger ICD, which further contributes to a growing body of evidence supporting the use of ICD-inducers as a strategy to stimulate antitumor immunity in NB.60 It should also be noted that ICD is not a specific cell death pathway nor implies any particular mechanism(s) of cell death; therefore, while our studies demonstrate that induction of STING signaling in NB cells can induce ICD, the cell death pathways underlying this response remain to be determined.
Correlations between increased T cell infiltration and improved clinical outcomes in NB has motivated significant preclinical exploration into strategies to abrogate immunosuppression in the NB TME and/or engage immunostimulatory mechanisms to enhance T cell infiltration and function.13–15 Consistent with our analysis demonstrating correlation between STING expression and increased markers of T cell infiltration in human NB (figure 1A), we found that administration of STING-NPs increased CD8+ T cell infiltration, likely a result of increased expression of T cell chemokines, immunosupportive cytokines (eg, IL-12, TNF-α), and reduced infiltration of Tregs. Accordingly, STING-NPs exhibited a strong antitumor response in both the Neuro-2a and 9464D models with no evidence of tumor growth in 100% and 50% of mice, respectively. This difference in response rate between the two models may reflect the relative frequency and immunogenicity of tumor antigens, differential levels of MHC-I expression, inherent variability between immune systems of different mouse strains, and/or a more immunosuppressive TME that is associated with MYCN-amplified NB. Strikingly, the majority (75%) of complete responders in the Neuro-2a model were protected from tumor re-challenge, a strong indication that in situ vaccination with STING-NPs can induce an endogenous T cell memory response in NB.
While PD-L1 expression has been reported as a negative prognostic indicator in NB8 and clinical trials of PD-1/PD-L1 blockade in NB have yet to be completed, the relatively infrequent expression of PD-L1 in NB tumors7 8 and the poor response to ICB in mouse models of NB already suggest that single agent PD-1/L1 ICB is unlikely to provide a major impact to high-risk NB patients. Based on the ability of STING-NPs to increase both T cell infiltration and PD-L1 expression in NB, we hypothesized that improved response rates to PD-L1 ICB could be achieved. We investigated this in mice bearing synchronous subcutaneous contralateral MYCN-amplified 9464D tumors, administering STING-NPs intratumorally into only one of the tumors in order to induce a systemic immune response capable of inhibiting distal (untreated) tumor growth (ie, abscopal effect), a strategy that is being widely explored clinically for innate immune agonists.47 As a single agent, STING-NPs reduced tumor growth of both the treated and untreated tumor, an effect that was augmented through systemic administration of αPD-L1, which, as anticipated, had no effect on tumor growth alone. While we, and others, have observed similar effects in models of adult cancers,26 44 61 this represents the first evidence that STING agonists can be leveraged to improve response to ICB in NB, and also offers among the first accounts of stimulating a tumor-intrinsic mechanism of innate immunity to improve response to ICB in a MYCN-amplified model of NB.
In these studies, we used an intratumoral administration route that is being explored clinically for a diversity of immunostimulatory agents across a range of solid tumor types to stimulate systemic antitumor immunity (ie, in situ vaccination).47 While there can be significant clinical and economic challenges associated with intratumoral therapy for less accessible tumor sites, in principle the majority of solid tumors can be directly injected using image-guided techniques.62 NB most commonly arises in and around the adrenal glands and frequently metastases to sites that are accessible via image-guided injection, such as the skin, lymph nodes, and liver.3 Additionally, intratumoral administration may be applied in a post-surgical setting as a strategy to treat un-resectable or partially-resectable tumors, and recent advancements in injectable depot and tumor-tethering technologies can improve drug retention and control release profiles to minimize the number of injections that may be required.63 Nonetheless, we also have previously demonstrated that STING-NPs can be administered systemically (intravenously) with minimal evidence of toxicity,26 offering a potential strategy for increasing the immunogenicity of metastatic sites without the need for local injection. This possibility merits further investigation in metastatic and/or orthotopic models of NB, as there may be important differences in the immunological profile between primary and metastatic tumors, between orthotopic and subcutaneous tumor models, and between tumor sites that can influence response to therapy. This was not explored in this work, which establishes an initial preclinical foundation for future exploration of STING agonists to enhance immunotherapy outcomes in NB in these additional contexts. It should also be noted that treatment was initiated when mice had relatively small tumors (<100 mm3), which are typically easier to treat with in situ vaccination approaches than large and more established tumors.64 For example, in an NXS2 model of NB, Sondel and colleagues demonstrated that only mice with smaller tumors (<34 mm3) were responsive to intratumoral injection of an IL-2 immunocytokine.65 The therapeutic efficacy of STING-NPs, as well as their capacity to induce local NB cell death and promote T cell infiltration, in mice with larger tumors remains to be investigated. Furthermore, while intratumoral administration of STING-NPs inhibited the growth of an untreated, distal tumor, an abscopal effect that was further augmented via the addition of αPD-L1 ICB, none of the mice in this study exhibited complete responses, due primarily to growth of the untreated tumor after cessation of therapy. Therefore, additional research is still required to determine how to further improve and maximize responses to STING-NPs, particularly for generating an abscopal response and/or in the setting of a high tumor burden. This may include further optimization of STING-NP dose and/or administration schedule as well as incorporation of other agents (eg, chemotherapy, other checkpoint inhibitors, cytokines) or treatment modalities (eg, radiation) into the regimen.
In addition to their direct therapeutic effects and capacity to enhance response to αPD-L1 ICB in NB, STING-NPs may also have utility as an adjunctive therapy for improving responses to approved and emerging treatments for NB. Notably, based on their capacity to increase T cell infiltration into NB tumors, STING-NPs may offer a technology platform for improving outcomes of T cell-based immunotherapies such as cancer vaccines, adoptive T cell transfer, or chimeric antigen receptor (CAR)-T cell therapy that are restrained by poor T cell infiltration and function.66 For example, CAR-T cells targeting GD2 are being explored in the clinic for treatment of NB.67 Additionally, like PD-L1, our TCGA analysis revealed several other immunosuppressive factors (eg, CTLA-4, TGF-β, IL-10) that are correlated with high STING expression in human NB (figure 1A) and may therefore also be promising targets in the design of combination regimens leveraging STING agonists. Similarly, STING-NPs may synergize with approved treatments for NB (eg, chemotherapy, radiation therapy, anti-GD2 therapy) by generating an immunosupportive context for antigen cross-presentation and T cell priming of locally liberated tumor antigen.
In summary, we have established pharmacological activation of STING signaling as a multifaceted therapeutic for NB, a childhood cancer for which new treatments are urgently needed. Using a nanotechnology designed to enable the efficient cytosolic delivery of cGAMP, we have demonstrated that potent activation of STING can induce apoptosis in NB cells, resulting in immunogenic cell death that can be leveraged to generate antitumor immunity. Additionally, STING activation converts highly immunosuppressive NB tumors to immunogenic and tumoricidal TMEs, ultimately leading to increased T cell infiltration, induction of antitumor immunity, and improved response to αPD-L1 ICB in a MYCN-amplified model of NB. As the elusive search for safe and effective therapies for high-risk NB continues and the development and clinical evaluation of STING agonists accelerates, our findings support the future exploration of CDNs, and delivery technologies that enhance their efficacy and utility, as versatile and multimodal immunotherapeutics for improving clinical outcomes for children with NB.
Methods
Analysis of published data sets
A comparison of TMEM173 mRNA expression z-scores among all cancer tumor types profiled by microarray in the Cancer Cell Line Encyclopedia (n=877) was performed.68 Integrated molecular analysis of mRNA expression of genes from the Pediatric Neuroblastoma TARGET RNA-seq data set was performed and clustered evenly based on tertiles of high (upper tertile, n=47), intermediate (median tertile, n=47), and low (bottom tertile, n=47) TMEM173 mRNA expression. Data were accessed through the cBioPortal.69 Statistical analysis was carried out using GraphPad Prism 6 software and Mann-Whitney U tests (two-tailed) were used for statistical comparison. P <0.05 was considered statistically significant.
Cell culture and reagents
Human (LAN-1 and SK-N-SH) NB cell lines were obtained from Dr. Dai Chung (Vanderbilt University). Neuro-2a cells were obtained from American Type Culture Collection (ATCC) (Manassas, Virginia). 9464D cells were derived from spontaneous TH-MYCN tumors70 and obtained from Dr. Yonghzi Cui (National Cancer Institute). Cells were maintained in Dulbecco’s modified Eagle’s medium (GIBCO) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, California) and 1% penicillin/streptomycin (GIBCO). Antibodies against STING (13 647S), p-IRF3 (4997S), IRF3 (4302S), Cleaved-Caspase 3 (9664T), and p-STAT1 (9167S) were purchased from Cell Signaling Technology (Beverly, Massachusetts). Antibodies for β-Actin (A5411) and Caspase (SC 56053) were purchased from Sigma Aldrich (St. Louis, Missouri) and Santa Cruz Biotechnology, respectively. Anti-PD-L1 antibody (BE0101) was purchased from BioXcell (West Lebanon, New Hampshire).
Synthesis of STING-activating nanoparticles (STING-NPs)
STING-NPs were formulated as previously described.26 Briefly, poly((ethylene glycol)-block-((2-diethylaminoethyl methacrylate)-co-(butyl methacrylate)-co-(pyridyl disulfide ethyl methacrylate))) polymers were synthesized via reversible addition fragmentation transfer (RAFT)polymerization by dissolving in dioxane a 2 kDa poly(ethylene glycol)-(4-cyanopentanoic acid) macro chain transfer agent, 2,2’-Azobis(4-methoxy-2,4-dimethylvaleronitrile, 2-(Diethylamino)ethyl methacrylate, butyl methacrylate, and pyridyl disulfide ethyl methacrylate, at a molar ratio of 1:0.2:23:15:3, respectively. The reaction vessel was sealed with a septum, purged with N2, and reacted at 30°C for 24 hours before 3x precipitation in pentane and vacuum drying overnight. To formulate the NPs, the synthesized polymer was dissolved in the minimum amount of ethanol. To this solution an equivalent volume, relative to the amount of ethanol used, of 50 mg/mL concentrated 2’,3’-cyclic GMP-AMP (cGAMP) solution in deionized water was added, mixed, and allowed to equilibrate at 37°C. cGAMP was synthesized as described previously.26 The resultant gel was gradually diluted in DI water with mixing and sonication to disperse the colloidally stable nanoparticles. Finally, 0.5 molar equivalents of dithiothreitol, relative to the total number of pyridyl disulfide moieties in the formulation, were added to crosslink the solution. Centrifugal filtration, using a 3 kDa cut-off Amicon filter was used to remove any unencapsulated drug and byproducts of the crosslinking reaction to yield STING-NPs. cGAMP quantification was performed using a Waters e2695 HPLC equipped with a Waters 2298 PDA detector at 50°C using a SeQuantR ZIC-cHILIC column (100 Å, particle size, 3 µm; 150×2.1 mm inside diameter; Millipore) with a SeQuantR ZIC-cHILIC PEEK coated guard column (20×2.1 mm inside diameter; Millipore). A two-solvent mobile phase consisted of mobile phase A (15 mM ammonium acetate, 0.1% glacial acetic acid and 10% methanol) and mobile phase B (15 mM ammonium acetate in 90% acetonitrile, 5% methanol, 5% water) with a run time of 25 min at a flow rate of 0.3 mL/minute and an injection volume of 20 µL. Gradient profile was as follows: time, 0 min: 30% A:70% B; time, 2 min: 70% A:30% B; time, 8 min: 70% A:30% B; time, 20 min: 30% A:70% B. Absorbance of cGAMP was measured at 260 nm. The mean encapsulation efficiency from 10 unique batches of STING-NPs was determined to be 49.8%±10.9%. Particle diameter and polydispersity was characterized using dynamic light scattering (Malvern ZS), and the mean intensity-average particle diameter for 10 independently prepared batches of STING-NPs was determined to be 127.1±52.4 nm.
Cell viability
Cell viability was measured using CellTiter-Glo Luminescent Cell Viability Assay (Promega). Briefly, cells were seeded at 5000 cells/well in a 96-well plate and treated with free cGAMP or STING-NPs for 72 hours. CellTiter-Glo reagent was added, the plate was incubated for 15 mins, and luminescence was measured using a BIO-TEK Synergy HI plate reader. The IC50values were extrapolated from non-linear regression (curve fit) of the cytotoxicity curves using Prism 7.0 (GraphPad software). The data is presented as mean±SD from three independent experiments.
Flow cytometric analysis of apoptosis
Cell apoptosis was detected by FlowCellect Annexin Red Kit (MilliporeSigma, Billerica, Massachusetts) according to the manufacturer’s instructions. Briefly, cells were seeded at 2×105 cells/well in a 12-well plate and treated with empty particle (NP), free cGAMP, or STING-NPs for 48 hours. Cells were washed with 1X Assay Buffer HSC followed by staining with Annexin Red for 15 mins at 37°C. The treated cells were then washed with 1X Assay Buffer HSC, and stained with 7-AAD for 5 mins at RT in the dark, and run on a BD-FACS flow cytometer. Data from three independent experiments/replicates were analyzed using FlowJo software.
Western blotting
NB cells were scraped on ice, centrifuged, and pellets were re-suspended in RIPA lysis buffer (Santa Cruz). Frozen tumor tissues (~30 mg) were homogenized in RIPA lysis buffer using TissueLyser II (Qiagen). Protein concentration was determined using BCA (Thermo Scientific, Waltham, Massachusetts). Equal amounts of protein were subjected to SDS/PAGE and transferred onto nitrocellulose membranes using the semi-dry transfer protocol (Bio-Rad Laboratories, Hercules, California). After transfer, membranes were probed with each respective primary antibody (against p-STAT1, p-IRF3, IRF3, Cleaved-Caspase 3, Caspase 3, PD-L1 and β-actin) overnight at 4°C. Following incubation, the membranes were probed with HRP-conjugated secondary antibodies (Promega). Protein bands were visualized using the commercial Immobile Western Chemiluminescent HRP Substrate Kit (Millipore Sigma, Billerica, Massachusetts). Images of immunoblots were obtained using the ChemiDoc XRS+system (Bio-Rad). Gel loading was normalized for equal actin and all shown are representative from at least two independent experiments. The relative density of each band was quantified using ImageJ and indicated under each immunoblot after normalization to density of the actin band in the corresponding lane.
Quantitative real-time PCR (qRT-PCR)
NB cells were treated with vehicle (PBS), empty nanoparticles, cGAMP (200 nM) or STING-NPs (200 nM) for 48 hours. Neuro 2a or 9464D tumors were grown to 200 mm3 and treated with vehicle or STING-NPs (10 µg) every 3 days for three injections, tumors were harvested 48 hours after final injection, and homogenized using TissueLyser II (Qiagen). Total RNA from cells or tumor homogenates were isolated using the RNeasy Mini kit (Qiagen, Germantown, Maryland). Total RNA (1 µg) was reverse transcribed by an iScript cDNA synthesis kit (Bio-Rad). Real-time PCR was conducted with the SYBR Green PCR kit (Bio-Rad) or TaqMan kits (Thermo Fisher Scientific) using a Bio-Rad CFX Connect Real-time System, with the threshold cycle number determined by Bio-Rad CFX manager software V.3.0. Reactions were performed in triplicate and the threshold cycle numbers were averaged. The results of the genes were normalized to housekeeping genes Ppib, 18 S, or Hmbs. The SYBR Green gene expression primers were purchased from Integrated DNA Technologies (IDT) (Coralville, Iowa): mouse Ifnb1 (Mm.PT.58.30132453.g), mouse Cxcl10 (Mm.PT.58.4357827), mouse Il12b (Mm.PT.58.12409997), and mouse Ppib (Mm.PT.58.29807961). Human Ifnb1 (Hs.PT.58.39481063), and human Ppib (Hs.PT.58.40291667). The TaqMan gene expression primers were purchased from Thermo Fisher Scientific (Waltham, Massachusetts): mouse Tnf (Mm00443258_m1); mouse Ifnb1 (Mm00439552_s1); mouse Cxcl10 (Mm00445235_m1); mouse cd274 (Mm03048248_m1); mouse Il12b (Mm00434174_m1) and mouse Hmbs (Mm01143545_m1). Human Ifnb1 (Hs01077958_s1); human Cxcl10 (Hs00171042_m1); human Il12 (Hs00168405_m1); human Tnf (Hs00174128_m1); 18 S (4 310 893E, applied biosystems).
ATP detection assay
Extracellular ATP was measured using the ATP Determination Kit (Molecular Probes; A22066), a luciferin-based bioluminescent assay as per manufacturer’s instructions. Supernatant from treated cells or ATP standard were mixed with reaction buffer and luminescence was measured using the SYNERGY H1 microplate reader (BioTek). ATP concentrations were normalized to background from untreated samples. Results are the mean±SD from three experiments.
Flow cytometric analysis of calreticulin
Neuro-2a or 9464D cells were seeded in 12-well plates and treated with vehicle, NP, cGAMP, or STING-NPs at the indicated concentrations for 24 hours. To assess calreticulin (CRT) expression by flow cytometry, cells were trypsinized, washed in cold PBS, and stained with an Alexa Fluor 647-conjugated anti-CRT antibody (Ab196159, Abcam) for 1 hour at room temperature. Cells were then washed three times and re-suspended in ice cold PBS with 3% BSA and 0.5% Sodium Azide, 50 µl/mL propodium iodide and analyzed on a LSRII flow cytometer (BD Biosciences). Data is expressed as fold-increase in mean fluorescence intensity compared with vehicle control. The experiment was repeated three times. Representative flow cytometry dots plots, histograms, and gating strategies for determining relative CRT levels are shown in online supplementary figure S8.
Immunofluorescence staining of CRT
Immunofluorescence was performed to visualize CRT surface localization. Neuro-2a and 9464D cells (1×104 cells) were seeded in 4-well chamber slides (Lab-Tek). NB cells were treated with vehicle or STING-NPs for 24 hours, fixed with 4% paraformaldehyde solution for 20 min, incubated with PBS (0.5% Triton-X) for 15 min, and blocked using 10% normal goat serum blocking solution (Thermo Fisher Scientific, Waltham, Massachusetts) for 1 hour at room temperature. Cells were stained with an Alexa Fluor 647-conjugated anti-CRT antibody (ab196159, 1/200, Abcam) overnight at 4°C, washed three times, followed by staining with 5 µg/mL Alexa Fluor 488-conjugated wheat germ agglutinin for 30 mins to visualize the cell surface membrane. Slides were washed three times and mounted with DAPI nuclear dye and visualised under an Olympus FV-1000 confocal microscope. High magnification images were obtained under the 40× objective lens.
HMGB1 ELISA
Neuro-2a or 9464D cells were seeded in 12-well plates overnight and incubated with vehicle, NP, cGAMP, or STING-NPs at the indicated concentrations for 48 hours. Supernatants were collected for high mobility group box 1 (HMGB-1) detection by ELISA (Tecan trading AG, Switzerland), according to the manufacturer’s instructions.
Generation of mouse BMDCs
BMDCs were obtained from 7-week-old A/J or C57BL/6 mice. Briefly, mouse femurs and tibias were flushed with cold PBS through a 70 μm-wide cell strainer, centrifuged for 5 min at 450 x g, and re-suspended in RPMI 1640 medium (supplemented with 10% HI FBS, 2 mM L-glutamine, 0.4 mM sodium pyruvate, 50 µM 2-mercapthoethanol, and 20 ng/mL mGM-CSF). Cells amounting to 9×106 were seeded in 100×20 mm non-treated cell culture plates in 10 mL of conditioned medium and incubated at 37°C with 5% CO2. Fresh pre-warmed conditioned medium was added on day 4 and 7. On day 8, the percentage of CD11c+ cells was at least 90% as measured by flow cytometry and used for further experimentation.
Tumor cell phagocytosis assay
Neuro-2a and 9464D cells were labeled with 5 mM CellTracker Green CMFDA (Molecular Probes, C7025) for 30 min and then treated with vehicle, NP, cGAMP or STING-NPs for 48 hours. The cells were collected, washed, and co-cultured with BMDCs at a ratio 1:1 for 2 hours.71 The co-cultured cells were harvested, immunostained with PE-anti-CD11c (BD Pharmingen, 550261), dead cells were excluded by SYTOX Blue staining, and, phagocytic efficiency was determined by flow cytometry (BD FACS Verse) and analysis performed using FlowJo (v.10.0.8) software. The phagocytic BMDCs are calculated as the percentage of double-positive CMFDA+CD11c+ cells of all CD11c+ cells (the total BMDC population). Representative flow cytometry dots plots, histograms, and gating strategies for determining tumor cell phagocytosis by DCs are shown in online supplementary figure S9.
Analysis of BMDC maturation
To induce cell death, Neuro-2a cells were treated with vehicle, NP, cGAMP, or STING-NPs for 48 hours and apoptosis measured by flow cytometric analysis using FlowCellect Annexin Red Kit. The dying Neuro-2a cells were collected, washed in RPMI culture medium, and co-cultured with 50,000 BMDCs in a ratios of 10:1. In parallel, control BMDCs were left untreated. Co-culture was performed for 18 hours at 37°C in 6-well plates with 2.5 mL BMDCs culture medium. After co-culture, all cells were collected, centrifuged (600xg, 5 min, 4°C), and washed once in PBS with 1% BSA. Maturation of BMDCs was analyzed by immunostaining using FITC-anti-CD11c, APC/Cy7-anti-MHC class II, PE-anti-CD86, and APC-anti-CD80 (Biolegend). Dead cells were excluded from the flow cytometry analysis by staining with SYTOX Blue (Molecular Probes, S11348). Mature BMDCs cells were identified as a percent of CD11c+MHCII+ CD86+and CD11c+MHCII+ CD80+. All samples were acquired on the BD FACS Verse flow cytometer and analyzed with FlowJo V.10.0.8 software. Representative flow cytometry dots plots, histograms, and gating strategies for determining the percentage of cells that were CD11c+MHCII+CD86+ and CD11c+MHCII+CD80+ are shown in online supplementary figure S10.
Ethics statement
Studies involving the use of animals were completed under an Animal Care Protocol (M1800129) approved by Vanderbilt University Animal Care and Use Committee. The health assessment of animals was completed using a standard operating procedure also approved by the Vanderbilt University Animal Care and Use Committee.
Cell-based cancer vaccination
Neuro-2a cells were treated with STING-NPs at 800 nM for 48 hours and cell death was detected by Annexin V/7AAD apoptosis assay (see above). The percentage of apoptotic cells was at least 70% to 80% as measured by flow cytometric analysis. Dying cells amounting to 1×106 were collected, re-suspended in cold PBS, and injected subcutaneously into A/J mice on the right flank. On day 14 after vaccination, the mice were challenged subcutaneously in the opposite flank with 5×105 untreated live Neuro-2a cells. Tumor growth at the challenge site was evaluated using electronic caliper measurements for up to 6 weeks after challenge. Mice were sacrificed when the tumor volume exceeded 1500 mm3.
Tumor immunohistochemistry
Tumors were resected from mice, fixed in 10% formalin, embedded in paraffin wax, and sectioned (5 µm) onto microscope slides. Sections were de-waxed in xylene, rehydrated, boiled in citric acid buffer (10 mM, pH 6) for antigen retrieval, and incubated with 3% hydrogen peroxide to quench endogenous peroxidase. After blocking with 10% goat serum, samples were incubated overnight at 4°C with anti-CD8 (eBioscience Inc, San Diego, California, 1:100 dilution), anti-cleaved caspase 3, anti-Ki-67 (Cell Signaling Technology, Danvers, Massachusetts, 1:200) and anti-Foxp3 (Abcam, Cambridge, UK, 1:100) primary antibodies. Following this step, samples were incubated with biotinylated secondary antibodies, incubated with the diaminobenzidine kit, and counterstained with hematoxylin. After mounting, sections were imaged using brightfield microscopy. Representative images for each section were taken at 20x magnification and used for quantification, which was evaluated in FIJI (Image-J-based open-source software).72 Briefly, color deconvolution was used to extract positively stained areas. Integrated density values were calculated from the resulting binary images.
Cellular and molecular analysis of tumors
A/J mice were inoculated with 1×106 Neuro-2a cells and when the tumor volume reached 200 mm3, mice were treated intratumorally every 3 days for three total injections and sacrificed 48 hours after final treatment. Tumors were collected for immunohistochemical staining (see above), qRT-PCR (see above) and Western blot (see above) analyses.
Tumor growth
In single tumor models, Neuro-2a (5×105) cells or 9464D (1×105) cells were suspended in 100 µl of PBS and subcutaneously injected into right flank regions of 6- to 7-week-old female A/J mice or C57BL/6 mice (The Jackson Laboratory, Bar Harbor, Maine), respectively. Tumor volume was measured every 3 days via caliper measurements, and tumor volumes were calculated using Vtumor=L × W2×0.5, in which Vtumor is tumor volume, L is tumor length and W is tumor width. At an average tumor volume of 68 mm3 (Neuro-2a) or 24 mm3 (9464D), mice were administered 100 µL vehicle (PBS), 2’3’-cGAMP (10 µg), or STING-NPs (normalized to 10 µg cGAMP dose) intratumorally every 3 days for three doses. Following treatment, mice were weighed and tumor sizes were measured every 3 days until reaching a tumor burden endpoint (>1500 mm3). To demonstrate generation of immune memory, surviving mice were re-challenged by injection 0.5×106 cells in the contralateral flank. For synchronous, contralateral tumor models, 9464D (1×105) cells were implanted subcutaneously on left and right flanks of C57BL/6 female mice. Treatment began when tumors reached 4 to 6 mm in largest diameter. STING-NPs (10 µg) were injected intratumorally into the tumor grown on the right flank every 3 days for three injections. Mice were treated with anti-PD-L1 (100 µg, BioXcell, West Lebanon, New Hampshire) intraperitoneally every 3 days for five total injections beginning on same day as STING-NPs. Mice were sacrificed when tumor volume reached >1500 mm3.
Acknowledgments
We gratefully acknowledge Dr. Dai Chung for providing SK-N-SH and LAN-1 cells, Dr. Yonghzi Cui for providing 9464D cells, and Dr. Scott Guelcher assistance with microscopy. We thank the Vanderbilt Translational Pathology Shared Resource (supported in part by the NCI/NIH Cancer Center Support Grant 5P30 CA684850-19), the Vanderbilt Cell Imaging Shared Resource (CISR) for use of Olympus FV-1000 confocal microscope, and the VUMC Flow Cytometry Shared Resource, supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404), for the usage of flow cytometers. 2’3’-cGAMP was provided by the Vanderbilt Institute of Chemical Biology Chemical Synthesis Core. MW acknowledges a postdoctoral fellowship from the Canadian Institutes of Health Research (CIHR).
References
Footnotes
Twitter @WilsonLabVU
Contributors LWB conceived of and designed experiments, analyzed and interpreted the majority of the data, drafted the manuscript, and provided technical support. MW synthesized materials and assisted in acquisition of data, data analysis, and drafting the manuscript. DS developed the STING-NP technology and synthesized materials for these studies. JLJ and JMB performed genomic and transcriptomic analyses. BCH and MR conducted immunohistochemical staining and assisted in image analysis. KMG assisted with in vivo evaluation of STING-NPs in NB models. PC synthesized and characterized 2’3-cGAMP. JTW conceived of the study concept and design, oversaw research activities, and drafted and critically revised the manuscript for intellectual content.
Funding This research was supported by grants from Alex’s Lemonade Stand Foundation ‘A’ Award (SID924 to JTW) and Pediatric Oncology Student Training (POST) Award cosponsored by Love Your Mellon (KMG), an American Cancer Society Institutional Research Grant (IRG-58-009-56 to JTW), the National Science Foundation (CBET-1554623 to JTW), the National Institutes of Health (R00CA201304 to MR), the Canadian Institutes of Health Research (MW), and also supported by a Stand Up To Cancer Innovative Research Grant (SU2C-AACR-IRG 20-17 to JTW). Stand Up To Cancer (SU2C) is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
Competing interests DS and JTW are coinventors of a US Patent Application 15/615753 on 'Reversibly Crosslinked Endosomolytic Polymer Vesicles for Cytosolic Drug Delivery'. JMB receives research support from Genentech/Roche, Bristol Myers Squibb, and Incyte Corporation, has received consulting/expert witness fees from Novartis, and is an inventor on provisional patents regarding immunotherapy targets and biomarkers in cancer.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data available from authors upon request.