Article Text

Abstract
Background Pancreatic cancer (PC) is a common malignancy of the digestive system and is characterized by poor prognosis and early metastasis. Tumor immune escape plays an important role in PC progression. Programmed death 1 (PD1) blockade therapy is a promising treatment for patients with PC, but is yet to achieve significant clinical effects so far. Interferon gamma (IFN-γ) is a soluble dimeric cytokine that is closely associated with tumor immune surveillance and cytotoxicity. IFN-γ suppresses a variety of tumor-derived cytokines in PC, such as CXCL8. In the present study, we investigated the therapeutic efficacy of combined anti-PD1 and IFN-γ treatment in PC.
Methods BxPC-3 and Panc-1 human PC cell lines were used to construct a murine PC model. Blood samples (n=44) and surgical resection specimens (n=36) from human patients with PC were also collected. χ2 test, two-tailed unpaired t-test or Kaplan-Meier survival analysis was used to calculate p values.
Results PD1/PD-L1 signaling was overexpressed in PC tumor-bearing mice. Anti-PD1 prevented tumor growth if initiated early after tumor inoculation; however, delayed anti-PD1 treatment showed limited benefit. Murine PC model had a preferential expansion of CXCR2+CD68+ macrophages, and these cells showed an immunosuppressive nature (M2 polarization). PC tumors overexpressed CXCL8 and tumor-derived CXCL8 deficiency prohibited the trafficking of CXCR2+CD68+ macrophages. IFN-γ suppressed the expression of tumor-derived CXCL8, and combined with IFN-γ treatment, delayed anti-PD1 treatment showed significant antitumor effects. Thus, we conclude that murine CXCR2+CD68+ macrophages traffic to PC tumors by tumor-derived CXCL8 and mediate local immunosuppression, which limits the efficacy of PD1 blockade therapy. IFN-γ suppresses tumor-derived CXCL8 and inhibits the tumor trafficking of CXCR2+CD68+ macrophages by blocking the CXCL8–CXCR2 axis to enhance anti-PD1 efficacy. Human PC also produces high levels of CXCL8. Patients with PC present elevated CXCR2 expression on peripheral and tumor-infiltrating CD68+ macrophages, which are associated with advanced tumor stage and poor prognosis.
Conclusion Our findings suggest that IFN-γ is a translatable, therapeutic option to improve the efficacy of PD1 blockade therapy by preventing trafficking of CXCR2+CD68+ macrophages via blocking the CXCL8–CXCR2 axis.
- interferon-gamma (IFN-γ)
- pancreatic cancer (PC)
- CXCR2
- CXCL8
- macrophages
- programmed death 1 (PD1)
- cancer immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- interferon-gamma (IFN-γ)
- pancreatic cancer (PC)
- CXCR2
- CXCL8
- macrophages
- programmed death 1 (PD1)
- cancer immunotherapy
Background
Pancreatic cancer (PC) is one of the most lethal types of cancer and carries a dire prognosis with a 5-year survival rate of only 3%–6%.1 2 PC is characterized by immune tolerance and immunotherapeutic resistance.3 Programmed death 1 (PD1) signaling of host T cells in the tumor is a major mechanism of tumor immune escape.4–7 Tumor expression of programmed death-ligand 1 (PD-L1) inhibits tumor-reactive T cells in PC animal models, and several clinical studies show a close relationship between tumor expression of PD-L1 and adverse outcomes in patients with PC.8–10 Although accumulating evidence suggests that the PD1/PD-L1 axis plays an important role in PC immune escape, clinical application of anti-PD1 therapy alone shows optimistic antitumor effects in a minority of cases,11 suggesting that other mechanisms also contribute to immune evasion in patients with PC.
Tumor-associated macrophages (TAMs) constitute the most important component of the tumor microenvironment (TME),12 13 which can be polarized into a classically activated (M1) or alternatively activated (M2) status depending on different stimuli.14 15 M1 macrophages are tumoricidal, whereas M2 macrophages facilitate tumor progression.16 17 Expansion of M2 TAMs is another mechanism of tumor immune escape and a number of approaches have been investigated to reprogram the polarization of TAMs to an immunogenic phenotype,18–20 which may hold promise in the field of tumor immune therapy in patients with PC.
Interferon gamma (IFN-γ) is a dimeric soluble cytokine and is involved in tumor immune surveillance and cytotoxicity.21 IFN-γ exerts its immunostimulatory and immunomodulatory effects primarily by activating the Janus kinase 1 (JAK1) and signal transducer and activator of transcription 1 (STAT1) signaling pathways.22 IFN-γ is an important activator of macrophages and a key stimulus for inducing M1 polarization of macrophages.14 23 Our previous study proved that IFN-γ suppresses a variety of tumor-derived cytokines, such as CXCL8, in PC tumors.24 CXCL8 is an important pro-inflammatory chemokine and mediates its signals via extracellular binding to two G protein-coupled receptors, C-X-C chemokine receptor type 1 (CXCR1) and C-X-C chemokine receptor type 2 (CXCR2).25 TAMs widely express CXCR1 and CXCR2,13 26 so the CXCL8–CXCR1/2 axis appears critical for TAMs mobilization and recruitment in the PC TME and plays a crucial role in tumor immune escape.18 27 28
Based on the aforementioned data, we could reasonably hypothesize that IFN-γ may prohibit the tumor trafficking and infiltration of CXCR1/2-expressing TAMs by downregulating tumor-derived CXCL8, thus reducing resistance to PD1 blockade. In the present study, we report that murine PC induces the preferential expansion of CXCR2+CD68+ macrophages, and these immunosuppressive cells traffic to the PC tumor bed by tumor-derived CXCL8 and mediate local immunosuppression to limit the efficacy of PD1 blockade therapy. IFN-γ suppresses the expression of PC tumor-derived CXCL8, thus, inhibiting the trafficking of CXCR2+CD68+ macrophages by blocking the CXCL8–CXCR2 axis. Combined with IFN-γ treatment, PD1 checkpoint blockade showed significant antitumor effect. This study shows that IFN-γ is a rational agent for improving the potency of PD1 checkpoint blockade, which could prevent TAM-mediated immune suppression in cancer therapy.
Methods
Tumor cell lines and mice
Human PC cell lines BxPC-3, Panc-1, Miapaca-2 and healthy pancreatic cell line HPDE6-C7 were obtained from Cancer Institute of Chinese Academy of Sciences (Beijing, China). All cell lines were maintained in an appropriate cell culture medium (see online supplementary additional file 2: table S1), supplemented with 10% fetal bovine serum (HyClone, Logan, USA) and 100 µg/mL each of penicillin and streptomycin (Gino, Hangzhou, China) in 5% CO2 at 37℃. All cell lines were used within 6 months of thawing. The animal experiment abided by national guidelines for the care and use of animals and approved by the ethics committee of Huzhou Central Hospital (HzhcLS2018-0807). C57BL/6 (B6) mice were purchased from Experimental Animal Center of Weitonglihua Co., Ltd. (Beijing, China), and were maintained in laminar airflow cabinets under specific pathogen-free conditions, and provided free access to irradiated pellet food and sterilized water. An ectopic inoculation method was used to establish a tumor xenograft model (six per group). Anti-PD1 blocking antibody (BioXcell Life Sciences RMP1-14, West Lebanon, USA) was used at a dose of 200 mg per mouse and performed intraperitoneally on the indicated days. Recombinant human IFN-γ (Wanbang Biopharma, Shanghai, China) was diluted in 2.5 mL of PBS (4×105 U/bottle) and given intraperitoneally with 25 µL (1×104 U) per mouse on continuous 5 days per week. Tumor tissues were harvested on indicated day after tumor implantation (day 21 after tumor implantation). Tumor volumes and survival time of each mouse was documented in the duration of the experiments. Tumor volumes were calculated using the formula: volume=length×width2/2. The endpoint for tumor growth and survival analysis was day 42 after tumor implantation.
Supplemental material
Antibodies and flow cytometry
Rat anti-mouse CD4 (clone GK1.5), rabbit anti-mouse CD68 (clone 2449C) and rabbit anti-mouse CXCR1 (clone 1122A) antibodies were purchased from R&D Systems Inc. Rat anti-mouse CD8 antibody (clone 53-6.7) was purchased from BD Pharmingen. Mouse anti-human CD68 (clone Y1/82A) and mouse anti-human PD-L1 (clone MIH1) antibodies were purchased from eBioscience Inc. Mouse anti-human CXCL8 (clone E8N1), rat anti-mouse CXCR2 (clone SA045E1) and mouse anti-human CXCR2 (clone 5E8/CXCR2) antibodies were purchased from BioLegend. All antibodies were used according to the manufacturer’s instructions. Intracellular cytokine staining was performed after the whole cells incubated with soluble anti–IL-2 (clone JES6-5H4; BioLegend) or anti–TNF-α (clone MP6-XT22; eBioscience) antibodies and monensin at 37°C and 5% CO2 for 6 hours. Flow cytometry was performed with FACS Fortessa (BD Biosciences), analyzed by FlowJo V.9.0 software. In all flow cytometry assays, background versus positive staining was ascertained using isotype controls and fluorescence minus one (FMO) controls. Boxed populations quantified were identified as positive in relation to isotype and/or FMO controls.
Immunostaining and immunoblotting
Tissue sections from isolated mice tumor xenografts and human PCs were deparaffinized in xylene and rehydrated with ethanol, followed by antigen repair with EDTA buffer (PH 8.0) boiling for 10 min, then samples were incubated with 2% BSA for 30 min and added primary antibody at 4°C overnight. After three times PBS washes, HRP-conjugated secondary antibody (corresponding to species of primary antibody) was added for 30 min. Following three times PBS washes, fluorophore was added for 10 min. The fluorophores used for dual-immunofluorescence assays were FITC for CD68 and CY3 for CXCR1/CXCR2 (figure 1C), while the fluorophores used for three-marker immunofluorescence assays were CY3 for CD68, FITC for CXCR2 and CY5 for CXCL8 (figure 2). After DAPI staining for 10 min, immunofluorescence of tissue sections was evaluated on inverted fluorescent microscope (BX-51, TR32000 Olympus, Japan) and analyzed by Image J 2X software (Rawak Software, Germany). Immunoblotting for detection of CXCL8 expression in cells and tumor tissues was performed as previously described.24 The primary antibodies used for immunoblotting were mouse anti-human CXCL8 (1:1000; BioLegend) and mouse anti-human GAPDH (1:1000, clone 1E6D9; ProteinTech). PE-conjugated goat anti-mouse IgG (1:2000; Santa Cruz) was used as secondary antibody. Reactive bands were detected using enhanced chemiluminescence (ECL; Amersham Bioscience, USA) immunoblotting detection reagent and analyzed by Image Lab V.5.2 software (Bioscience, USA).

Effect of anti-PD1 therapy to murine pancreatic cancer (PC) tumor. Mice were ectopic inoculated with BxPC-3 and Panc-1 PC cells, control immunoglobulin G (No treatment) or anti-PD1 blocking antibody were administered intraperitoneally (200 mg per mouse, two times per week). (A) Survival and tumor volumes were monitored when anti-PD1 treatment starting on day 0 after BxPC-3 PC cells inoculation. (B) PD1 expression on circulating CD4+ and CD8+ T cells in BxPC-3 and Panc-1 tumor-bearing mice on day 14 after tumor inoculation. (C) Surface expression of PD-L1 on BxPC-3 and Panc-1 tumor xenograft cells. (D) Survival and tumor volumes were monitored when anti-PD1 treatment starting on day 7 after BxPC-3 PC cells inoculation. (E and H) Intracellular production of IL-2 and TNF-α on tumor-infiltrating CD8+ T cells from BxPC-3 (E) and Panc-1 (H) tumor-bearing mice on day 21 after delayed anti-PD1 treatment. F and G Survival and tumor volume were monitored when anti-PD1 treatment starting on day 0 (F) or on day 7 (G) after Panc-1 PC cells inoculation. Experiments consist of six mice per group and are representative of at least three separate experiments. The three-dimensional data of mice tumors were measured by somatometry method. Tumor volumes were calculated using the formula: volume=length×width2/2. The endpoint for tumor growth and survival analysis is day 42 after tumor implantation. Two-tailed unpaired t-test or Kaplan-Meier survival analysis was used to calculate p values. NS, not significant.

CXCR1/2+CD68+ macrophages expand in murine PC model and associated with tumor progression. (A) Representative fluorescence-activated cell sorting (FACS) plots and percentages of CXCR1/2+CD68+ macrophages from mice blood on day 14 after BxPC-3 and Panc-1 PC cells inoculation. (B) Relationship between tumor volumes and the percentages of CXCR1/2+CD68+ macrophages from tumor-bearing mice blood. (C) Tumor tissue sections immunofluorescence staining images and percentages of tumor-infiltrating CXCR1/2+CD68+ macrophages from BxPC-3 and Panc-1 tumor xenografts (day 14). Scale bars represent 100 µm. (D) Relative quantification of arginase-1 and iNOS transcripts in CXCR1+CD68+ and CXCR2+CD68+ macrophages sorted from BxPC-3 and Panc-1 tumor xenografts (day 14) versus circulating macrophages of non-tumor-bearing mice. (E) FACS histograms showing surface expression of PD-L1 on gated populations of circulating CXCR1/2+CD68+ macrophages from BxPC-3 and Panc-1 tumor-bearing mice blood (day 14). MFI, mean fluorescent intensity. Experiments consist of five mice per group and data are representative of at least two independent experiments. Two-tailed unpaired t-test or Pearson r correlation analysis was used to calculate p values.
Real-time fluorescence quantitative PCR and ELISA
Total RNA was extracted from cultured cells, sorted CXCR1+/CXCR2+ macrophages and tumor xenografts from mice using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Complementary DNA was synthesized using M-MLV reverse transcriptase (Takara Bio, Dalian, China). Real-time fluorescence quantitative PCR (RTFQ-PCR) was performed with specific primers for detecting murine arginase-1, iNOS, PD-L1, GAPDH and human CXCL8, GAPDH (see online supplementary additional file 2: table S2). The primers were obtained from Wuhan Servicebio Technology Co., Ltd., and the assays were performed according to the manufacturer’s protocol. The PCR reactions were performed in a Bio-Rad Hard Shell 96-well microplate on a Bio-Rad CFX96 Real-Time System C1000 Touch Thermal Cycler using iTaq Universal SYBR Green Supermix (Bio-Rad) and amplified for 40 cycles. CXCL8 levels of serum samples and cell culture supernatants were detected by using the human CXCL8 sandwich ELISA kits (Dakewe Biotech, Shenzhen, China) according to the manufacturer’s instructions. Normal ranges for human CXCL8 were defined by the range of values spanning (means±2 SDs) obtained from the CXCL8 level data of serum from 12 healthy donors.
In vitro migration assay
Peripheral blood CXCR1+/CXCR2+ macrophages (1×104) which electronically sorted from tumor-bearing mice were placed in the top chamber of a Transwell (Millipore, USA). BxPC-3 or Panc-1 cells were plated in the bottom chamber. Blocking antibodies for CXCL8, CXCR1 and CXCR2 (50 µg/well) were added at the beginning of the experiment. The total number of macrophages migrating from the top to the bottom chamber after 12 hours was calculated. The migrated cells counting method was performed as previously described.24
Patient samples
Serum samples (n=44) from patients with PC were obtained at the time of hospitalization in Huzhou Central Hospital from May 2016 to January 2019, following informed consent. In these patients, 36 patients underwent radical resection and the surgical resection specimens (n=36) were collected during the operation. All fresh specimens containing pancreatic tumor or adjacent normal pancreatic tissue were selected for further study. There are 25 men and 19 women in these patients (male:female=1.32:1); the median age was 52 years (range, 37–84 years). Serum samples from healthy donors (n=12) were obtained after informed consent and following approved protocols (ChiCTR1800017665).
Statistical analysis
SPSS V.19.0 was used for clinical data statistical analysis. The χ2 test and Fisher’s exact test were used for quantitative data (table 1). Graphs for experimental data were created using GraphPad Prism software. Data in graphs were expressed as mean±SD. P values were calculated in each respective figure using paired or unpaired t-test for intergroup comparison and log-rank test for survival analysis. P value <0.05 was considered statistically significant.
Correlation of CXCR2 expression on peripheral CD68+ macrophages with clinicopathological parameters in patients with pancreatic cancer
Results
PD1 blockade therapy shows diminished effectiveness in established PC
First, we analyzed the effect of PD1 blockade therapy in PC xenografts using BxPC-3 cells. BxPC-3 tumors grow rapidly after tumor inoculation into the axilla of mice, contributing to 100% mortality by day 27 (figure 3A and online supplementary additional file 1: figure S1a). BxPC-3 tumors constitutively overexpressed PD-L1 (figure 3C and online supplementary additional file 1: figure S2, left panel) and enhanced PD1 expression on circulating CD4+ or CD8+ T cells by day 14 (figure 3B and online supplementary additional file 1: figure S3a), leading us to hypothesize that the PC-induced PD1/PD-L1 signal pathway on T cells contributes to immune escape in BxPC-3 tumors. Subsequently, we administered anti-PD1 blocking monoclonal antibodies (mAbs) to mice on the day of tumor inoculation and the mice showed 100% survival without evidence of tumor development (figure 3A). This indicated a role for PD1/PD-L1 signaling in immune escape by PC. To detect whether anti-PD1 treatment could lead to the regression of established BxPC-3 tumors, anti-PD1 mAbs were administered 7 days after tumor inoculation, at which time the PC tumors were well established. We found the delayed treatment modestly decreased the tumor growth and lead to a minor statistically significant improvement in survival (figure 3D). Although delayed anti-PD1 treatment showed minor therapeutic efficacy on tumor regression, BxPC-3 tumor-bearing mice after delayed anti-PD1 treatment had significantly more interleukin-2–positive (IL-2+) and tumor necrosis factor-α–positive (TNF-α+) CD8+ T cells infiltrating the tumors than untreated control mice (figure 3E and online supplementary additional file 1: figure S4a), suggesting that delayed anti-PD1 treatment could induce immune activation of tumor-bearing mice but it was insufficient to mediate tumor regression.
Supplemental material
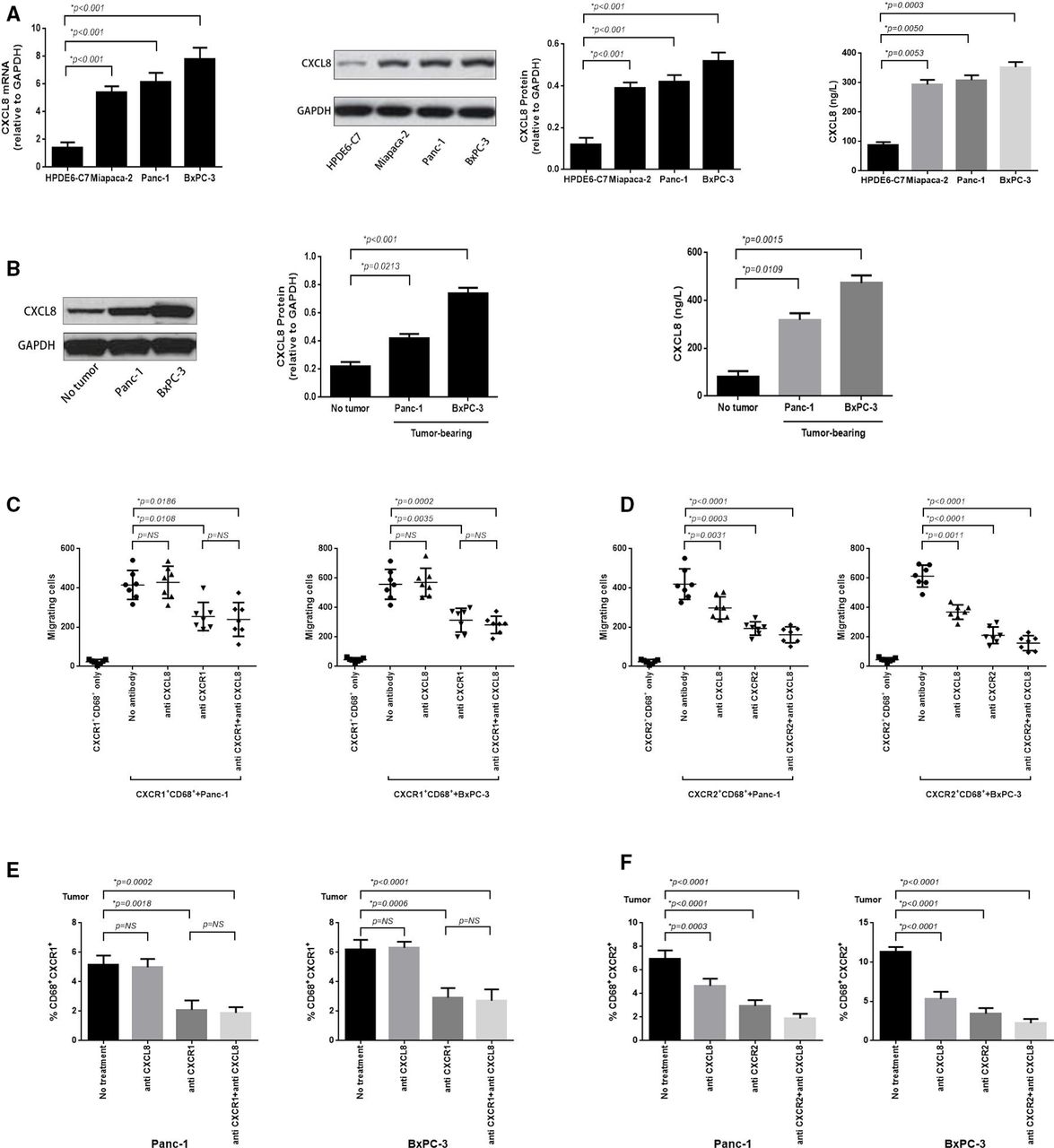
Murine pancreatic cancer (PC) tumor overexpresses CXCL8 and which predominantly induces CXCR2+CD68+ macrophages tumor trafficking. (A) RT-PCR was performed to assess the expression of CXCL8 mRNA in BxPC-3, Panc-1, Miapaca-2 PC cell lines and normal pancreatic cell line HPDE6-C7 (left panel); protein analysis (immunoblotting) for CXCL8 protein expression (middle panel); and ELISA analysis for secretion of CXCL8 in cell culture supernatants (right panel). (B) Immunoblotting was performed to detect the expression of CXCL8 protein in murine BxPC-3 and Panc-1 tumor, or adjacent non-tumor tissue (left panel); ELISA for CXCL8 detection in serum obtained from blood samples taken from BxPC-3 and Panc-1 tumor-bearing mice or nomal mice (right panel). (C) In vitro migration assay was performed to calculate the absolute number of CXCR1+CD68+ macrophages which electronically sorted from blood of BxPC-3 or Panc-1 tumor-bearing mice migrating to corresponding PC cells (12 hours) when non-antibody or blocking antibodies for CXCL8, CXCR1 and CXCL8+CXCR1 (5 mg/mL) were added at the beginning of the experiment. (D) The in vitro migration assay was performed as in (C) and the migrated CXCR2+CD68+ macrophages were enumerated whereas blocking antibodies for CXCL8, CXCR2 and CXCL8+CXCR2 (5 mg/mL) were added. (E) Tumor tissue sections immunofluorescence staining was performed to calculate the tumor-infiltrating CXCR1+CD68+ macrophages from BxPC-3 and Panc-1 tumor when blocking antibodies for CXCL8, CXCR1 and CXCL8+CXCR1 (10 mg/per mouse) were used in tumor-bearing mice for 6 hours. (F) Immunofluorescence staining was performed as in (E) and tumor-infiltrating CXCR2+CD68+ macrophages were enumerated when blocking antibodies for CXCL8, CXCR2 and CXCL8+CXCR2 (10 mg/per mouse) were used. Two-tailed unpaired t-test was used to calculate p values.
We also detected the antitumor effects of PD-1 blockade in mice inoculated with Panc-1, another PC cell line which overexpresses PD-L1 in tumor xenografts (figure 3C and online supplementary additional file 1: figure S2, right panel) and demonstrated upregulated PD1 expression on peripheral T cells (figure 3B and online supplementary additional file 1: figure S3b). The therapeutic efficacy of anti-PD1 treatment was similar to that observed in mice with BxPC-3 tumors when PD-1 blockade therapy was initiated on the day of tumor inoculation (figure 3F and online supplementary additional file 1: figure S5a). The delayed treatment initiated on day 7 had no significant impact on tumor growth and survival rate (figure 3G and online supplementary additional file 1: figure S5b); however, immune activation was observed (figure 3H and online supplementary additional file 1: figure S4b).
Expansion of immunosuppressive CXCR1/2+CD68+ macrophages contributes to PC progression
To explore the underlying mechanisms by which PC may limit the efficacy of immune-based therapy, we analyzed TAMs in the tumors and peripheral blood of tumor-bearing mice. We found that CXCR1/2+CD68+ macrophages were increased in the peripheral blood of mice as early as day 7 after tumor inoculation (figure 1A). The proportion of CXCR1/2+CD68+ macrophages in peripheral blood increased following the growth of BxPC-3 and Panc-1 tumors (figure 1). By day 21, CXCR1/2+CD68+ macrophages represented a sizeable fraction of the monocytes in BxPC-3 and Panc-1 tumors compared with healthy tissue (figure 1C). The expansion of CXCR1/2+CD68+ macrophages in Panc-1 tumors was less significant than in BxPC-3 tumors, and the expansion of CXCR2+CD68+ macrophages greater than CXCR1+CD68+ macrophages in PC.
To investigate whether the CXCR1/2+CD68+ macrophages expanded in PC tumor-bearing mice were suppressive cells, we sorted CXCR1/2+CD68+ cells from tumor-bearing mice and evaluated their ability to produce arginase-1 and inducible nitric oxide synthase (iNOS), mediators of macrophages immune suppression. We found that higher levels of arginase-1 were observed in tumor-infiltrating CXCR1/2+CD68+ macrophages which from BxPC-3 and Panc-1 tumor-bearing mice, whereas the levels of iNOS were decreased (figure 1D), suggested M2 polarization. Both circulating and tumor-infiltrating CXCR1/2+CD68+ macrophages from BxPC-3 and Panc-1 tumor-bearing mice expressed higher levels of PD-L1 compared with macrophages from healthy mice (figure 1E and online supplementary additional file 1: figure S6). We also detected the fraction of tumor-infiltrating T-cell subsets, such as Th1 (CD4+IL-2+, CD4+TNF-α+), Th2 (CD4+IL-4+, CD4+IL-10+), Treg (CD4+FoxP3+), CD8+IL-2+ and CD8+TNF-α+ cells. We observed a significant reduction of Th1, CD8+IL-2+ or CD8+TNF-α+ cells, whereas we observed an increase of Th2 or Treg cells in the tumor bed (see online supplementary additional file 1: figure S7).
CXCL8 overexpressed in PC cell lines and murine PC mediates tumor trafficking of CXCR2+CD68+ macrophages
We next investigated the mechanisms by which TAMs traffic to the PC tumor bed to mediate their immunosuppressive effect. We detected whether PCs produce and secrete ligands for CXCR1/2 and observed that BxPC-3, Panc-1 and Miapaca-2 PC cell lines overexpressed CXCL8 mRNA and protein, and the culture supernatant taken from these cell lines contained high concentrations of CXCL8 protein compared with a healthy pancreatic cell line HPDE6-C7 (figure 4A). In tumor-bearing mice, BxPC-3 and Panc-1 tumors overexpressed CXCL8 protein and high concentrations of CXCL8 protein was also detected in the serum (figure 4B).

Human pancreatic cancer (PC) tumors secrete CXCL8. Surface expression of CXCR2 on peripheral and tumor-infiltrating CD68+ macrophages are associated with tumor progression and negatively correlate with survival in patients with PC. (A) CXCL8 were measured by ELISA in serum samples collected from healthy donors (n=12) and patients with PC (n=44) (left panel); flow cytometry for detection of CXCR2+CD68+ macrophages percentage in peripheral blood of healthy donors (n=12) and patients with PC (n=44) (middle panel); tissue section immunofluorescence staining cells counting was performed in at least four random high-power fields (original magnification, ×400) to calculate the percentage of infiltrated CXCR2+CD68+ macrophages in PC tumor tissues or adjacent non-tumor tissues (n=36) (right panel). CXCR2+CD68+ macrophages percentage was calculated as follows: %=(CXCR2+CD68+ macrophages/CD68+ macrophages)×100. (B) Patients with PC have no significant survival differences between serum CXCL8 high (n=19) and low (n=25) groups (left panel); whereas patients with PC with higher expression of CXCR2 on circulating (high: n=18, low: n=26; middle panel) and tumor-infiltrating (high: n=13, low: n=23; right panel) macrophages have diminished overall survival. High and low levels were defined using the median level of serum CXCL8, CXCR2 expression on peripheral or tumor-infiltrating CD68+ macrophages as the cut-off value. Two-tailed unpaired t-test or Kaplan-Meier survival analysis with log-rank test was used to calculate p values. NS, not significant.
To confirm whether CXCL8 induced a chemokine gradient used for CXCR1/2+CD68+ macrophages trafficking to the tumor bed, we used an in vitro migration assay where BxPC-3 and Panc-1 cells were plated in the bottom chamber, and sorted CXCR1/2+CD68+ cells isolated from the blood of BxPC-3 and Panc-1 tumor-bearing mice were added to the top chamber of a 96-well Transwell plate. Subsequently, blocking antibodies toward CXCL8, CXCR1/2 and CXCL8+CXCR1/2 were added. After 12 hours, the migration of CXCR1/2+CD68+ cells was enhanced in the presence of BxPC-3 and Panc-1 cells (p<0.0001 and p<0.0001, respectively) (figure 4C,D). Using CXCL8 or CXCR2 blocking antibody significantly inhibited the migration of CXCR2+CD68+ cells in the Panc-1 (p=0.0031 and p=0.0003, respectively) and BxPC-3 groups (p=0.0011 and p<0.0001, respectively) when used alone, and their combined effects were additive (p<0.0001 in Panc-1 group and p<0.0001 in BxPC-3 group, respectively) (figure 4D). Furthermore, the blocking antibody for CXCR1 significantly prohibited the migration of CXCR1+CD68+ cells in the Panc-1 and BxPC-3 groups (p=0.0108 and p=0.0035, respectively), and CXCL8 blockade shown no inhibitory effects, similar to untreated controls (p=0.6437 in Panc-1 group and p=0.8180 in BxPC-3 group, respectively). The blockade of CXCR1 and CXCL8 simultaneously was equivalent to anti-CXCR1 used alone (p=0.3846 in Panc-1 group and p=0.3819 in BxPC-3 group, respectively) (figure 4C), demonstrating that CXCL8, as a ligand of CXCR1/2, mediates a chemokine gradient effect for CXCR2+CD68+, but not CXCR1+CD68+ macrophages trafficking to the tumor bed. The same results mirrored in the in vivo experiments (figure 4E,F and online supplementary additional file 1: figures S8 and S9).
CXCL8 levels and CXCR2 expression on peripheral and tumor-infiltrating CD68+ macrophages are elevated in patients with PC, which are associated with clinicopathological and prognostic significance
Our study revealed a critical role for CXCR2+CD68+ macrophages in murine PC progression and CXCL8 is a key chemokine in recruiting CXCR2+CD68+ macrophages to tumors. We next explored whether human PCs use the same axis. We have demonstrated that human PC cell lines secrete high concentrations of CXCL8 protein into the culture supernatants previously; thus, we measured the levels of CXCL8 in serum from patients with PC (n=44) and healthy donors (n=12). We found that patients with PC had higher levels of CXCL8 compared with healthy donors (patients with PC: 146.0±16.11; healthy donor: 44.42±3.747, p=0.0019) (figure 5A, left panel). We divided patients with PC into high/low (19/25) CXCL8 level groups according to the median value of CXCL8 levels of the total patients with PC, with a follow-up of 24.3 months, the survival rate of the two groups had no significant differences (HR 1.248; 95% CI 0.7017 to 2.414; p=0.4391) (figure 5B, left panel).

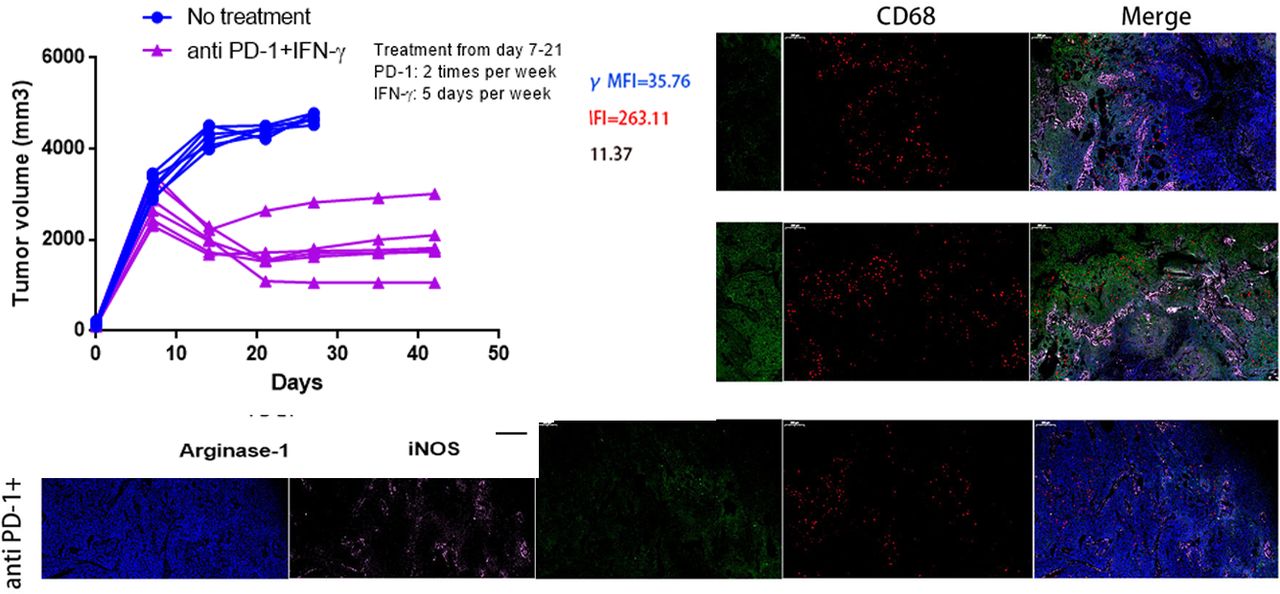
PD1 checkpoint blockade is more effective in tumor-bearing mice when combinatorial using IFN-γ. (A) RT-PCR was performed to assess the expression of CXCL8 mRNA in murine BxPC-3 tumors when IFN-γ (1×104 U per mouse, 5 days per week) treated for 7, 14, 21 days (left panel); immunoblotting assay for detection of CXCL8 protein expression after IFN-γ treatment (middle panel); CXCL8 in serum samples collected from BxPC-3 tumor-bearing mice with IFN-γ treatment were measured by ELISA (right panel). (B) BxPC-3 tumor volumes were monitored when anti-PD1 (200 mg per mouse, two times per week) (left panel), IFN-γ (1×104 U per mouse, 5 days per week) (middle panel) or anti-PD1+IFN-γ (right panel) treatment starting on day 7 after tumor inoculation (n=6 per group). The endpoint for tumor growth analysis is day 42 after tumor inoculation. (C) Kaplan-Meier survival curves showed the overall survival of no treatment, anti-PD1, IFN-γ or anti-PD1+IFN-γ treatment groups and evaluated using log-rank statistics (n=6 per group). The endpoint for mice survival analysis is day 42 after tumor inoculation. (D) The percentages of CXCR2+CD68+ macrophages in BxPC-3 tumor-bearing mice peripheral blood from four experimental groups as in (C) were measured by flow cytometry (day 14). NS, not significant. (E) Tumor tissue immunofluorescence tri-markers staining images of CXCL8 and tumor-infiltrating CXCR2+CD68+ macrophages from BxPC-3 tumor xenografts after anti-PD1, anti-PD1+IFN-γ treatment or no treatment (day 21) (left panel). Scale bars represent 200 µm. IOD sums of CXCL8 (right top panel) and the percentages of tumor-infiltrating CXCR2+CD68+ macrophages (right bottom panel) from three groups were calculated using Image-Pro Plus V.6.0 software (day 21). IOD, index of distribution. (F) RT-PCR was performed to evaluate levels of arginase-1 and iNOS produced by sorted tumor-infiltrating CXCR2+CD68+ macrophages from cohorts of mice as in (E) on day 21. NS, not significant. (G) FACS histograms showing surface expression of PD-L1 on gated populations of circulating CXCR2+CD68+ macrophages from cohorts of mice as in (E) on day 21. (H) RT-PCR was performed to detect the mRNA expression of PD-L1 in sorted tumor-infiltrating CXCR2+CD68+ macrophages from cohorts of mice as in (E) on day 21. MFI, mean fluorescent intensity. Two-tailed unpaired t-test or Kaplan-Meier survival analysis with log-rank test was used to calculate p values.
We next detected CXCR2 expression on peripheral and tumor-infiltrating CD68+ macrophages and found that patients with PC have a higher percentage of CXCR2+CD68+ cells in the peripheral blood compared with healthy donors (p<0.0001) (figure 5A, middle panel). Furthermore, CXCR2+CD68+ macrophages represent a higher fraction in tumor tissues compared with adjacent normal pancreatic tissue (p<0.0001) (figure 5A, right panel). We divided patients with PC into high/low CXCR2+CD68+ expression groups in peripheral blood (18/26) and tumor (13/23) according to the median value respectively, and all groups were compared with clinicopathological parameters (age, sex, tumor size, tumor site, histological grade, T classification, N classification, M classification, clinical stage) by χ2 test. We found that CXCR2 expression on peripheral CD68+ macrophages was closely correlated with T classification (χ2=6.631, p=0.010), N classification (χ2=4.400, p=0.036) and clinical stage (χ2=5.107, p=0.024). There was no significant correlation between CXCR2 expression and age, sex, tumor size, tumor site, histological grade and M classification (table 1). The same results were observed for CXCR2 expression on tumor-infiltrating CD68+ macrophages (see online supplementary additional file 2: table S3). As CXCL8 levels seemed to have no relationship with the overall survival of patients with PC, we next explored whether increased CXCR2 expression on peripheral and tumor-infiltrating CD68+ macrophages correlated with diminished overall survival in patients with PC. With a follow-up of 24.3 months, the median survival of patients with high (n=18) versus low (n=26) CXCR2 expression on peripheral CD68+ macrophages was 17.0 months versus 24.0 months (HR 3.358; 95% CI 3.138 to 14.99; p<0.0001) (figure 5B, middle panel). Furthermore, high (n=13) versus low (n=23) CXCR2 expression on tumor-infiltrating CD68+ macrophages showed a median survival of 18.0 months versus 24.0 months (HR 2.416; 95% CI 1.588 to 8.434; p=0.0053) (figure 5B, right panel). These results demonstrated that CXCL8 is an essential chemokine for the recruitment of CXCR2+CD68+ macrophages in murine PC tumors as well as in human patients with PC. Furthermore, the expansion of CXCR2+CD68+ macrophages contributes to human PC progression and poor prognosis.
IFN-γ suppresses the secretion of CXCL8 in PC tumors, preventing CXCR2+CD68+ macrophage trafficking to the tumor microenvironment and enhancing PD1 blockade efficacy
CXCL8 and CXCR2+CD68+ macrophages are capable of promoting PC progression and limiting antitumor T-cell responses and our previous studies showed that IFN-γ suppresses CXCL8 in BxPC-3 and Panc-1 cell lines24 (see online supplementary additional file 1: figure S10); thus, we sought to investigate whether anti-PD1 therapy combined with using IFN-γ was more effective at reducing tumor progression. We focused on the BxPC-3 model because PD1 blockade therapy is highly efficacious and has a significant expansion of CXCR2+CD68+ macrophages. We tested the expression and secretion of CXCL8 in IFN-γ treated tumor-bearing mice and found that the expression of CXCL8 mRNA and protein decreased on days 7, 14 and 21 after IFN-γ treatment. These results mirrored the concentrations of CXCL8 protein in the serum of tumor-bearing mice (figure 2A). Mice were inoculated with BxPC-3 cells on day 0, and treatment begun on day 7, when the tumors were well established. Anti-PD1 or IFN-γ treatment alone was ineffective at preventing tumor growth (figure 2B, left and middle panel). In contrast, mice treated with anti-PD1+IFN-γ experienced significant reductions in tumor growth (figure 2B, right panel) (see online supplementary additional file 1: figure S11). Kaplan-Meier survival analysis in BxPC-3 tumor-bearing mice showed a modest but significant difference in median survival between no treatment and the anti-PD1 groups (median survival: 24.0 days vs 30.0 days; HR 3.704; 95% CI 1.539 to 24.05; p=0.0145), while mice receiving IFN-γ treatment did not have an increase in the median survival (median survival: 24.0 days vs 26.0 days; HR 1.885; 95% CI 0.6659 to 7.179; p=0.2303). Conversely, mice treated with anti-PD1+IFN-γ experienced a significant increase in median survival (median survival: 24.0 days vs 39.0 days; HR 8.103; 95% CI 8.199 to 249.2; p<0.0001) (figure 2C).
We next explored whether the efficacy of combinatorial therapy was related to the downregulation of CXCL8 and reduction of CXCR2+CD68+ macrophages in tumor-bearing mice. We found that the percentage of circulating CXCR2+CD68+ macrophages in mice were no different between control, anti-PD1 or IFN-γ used alone and the combined treatment groups (figure 2D). The expression of CXCL8 was modestly increased in BxPC-3 tumors after anti-PD1 treatment but was significantly decreased following anti-PD1+IFN-γ treatment; however, tumor-infiltrating CXCR2+CD68+ macrophages represented a minor increased fraction in tumor tissue with PD1 blockade therapy but were largely diminished after anti-PD1 +IFN-γ treatment (figure 2E).
The CXCR2+CD68+ macrophages expanded in tumor-bearing mice were suppressive cells; thus, we further investigated whether these cells would change the immunosuppressive nature after anti-PD1+IFN-γ treatment. To this end, we sorted CXCR2+CD68+ macrophages from tumor-bearing mice and evaluated their capacity to produce arginase-1 and iNOS. We observed that increased, but not significant, levels of arginase-1 were detected in tumor-infiltrating CXCR2+CD68+ macrophages after anti-PD1 treatment, which significantly decreased following anti-PD1+IFN-γ treatment. The levels of iNOS in tumor-infiltrating CXCR2+CD68+ macrophages were no different between no treatment and anti-PD1 treated mice; however, iNOS was significantly increased in these cells from anti-PD1+IFN-γ treated tumor-bearing mice (figure 2F), suggesting these cells showed M1 polarization. We also observed higher levels of PD-L1 expression in circulating and tumor-infiltrating CXCR2+CD68+ macrophages from tumor-bearing mice after anti-PD1 treatment, whereas the expression of PD-L1 was significantly decreased in these cells following anti-PD1+IFN-γ treatment (figure 2G,H). We also observed that the fraction of tumor-infiltrating Th1 (CD4+IL-2+, CD4+TNF-α+), CD8+IL-2+ and CD8+TNF-α+ cells minor increased after anti-PD1 or IFN-γ treatment alone while significantly increased following anti-PD1+IFN-γ treatment; whereas Th2 (CD4+IL-10+) and Treg (CD4+FoxP3+) cells minor decreased after anti-PD1 or IFN-γ treatment alone while significantly reduced following anti-PD1+IFN-γ treatment in tumor bed (see online supplementary additional file 1: figures S12 and S13). Together, these results suggest that IFN-γ improves the efficacy of anti-PD1 therapy. IFN-γ could suppress the expression of tumor-derived CXCL8 to inhibit tumor-induced trafficking of CXCR2+CD68+ macrophages to the tumor bed. In addition, the peripheral and tumor-infiltrating CXCR2+CD68+ macrophages alter the immunosuppressive properties, while the T-cell subsets turn to a state of immune activation after anti-PD1 combined with IFN-γ treatment.
Discussion
PD1 receptor is an important immune checkpoint molecule, which controls inflammation and self-reactivity in physiological conditions.4–6 However, cancers often use the PD1/PD-L1 signaling axis to induce the apoptosis of tumor-reactive cytotoxic T cells to mediate tumor immune escape.7 10 29 T-cell checkpoint-based immunotherapy has widely been used in the treatment of patients with melanoma, non-small cell lung cancer and renal cell carcinoma with objective antitumor responses observed in 6%–28% of these patients.30 31 Although PD1 immune checkpoint blockade has shown promise in cancer therapy and induced durable responses in several tumor types, its efficacy is limited for patients with PC.11 In the present study, the murine model of PC presented here also illustrates the limitations of PD1 checkpoint blockade as a single-agent treatment. Tumor development was completely inhibited when anti-PD1 treatment began at the time of tumor inoculation but had only minor effects on established tumors. These results showed that factors beyond PD1 signaling contribute to tumor immune evasion in PC tumors. PC is characterized by a dense, fibrotic stroma that can trap tumor-infiltrating cytotoxic T cells in peritumoral tissues and thus can prevent them from reaching the vicinity of tumor cells. Moreover, the immunosuppressive TME within the stroma also dampens the activity of tumor-infiltrating T lymphocytes.3 32 A minor percentage of patients with PC benefited from PD1 checkpoint inhibitors used alone, suggesting that other mechanisms also contribute to immune evasion. Combinatorial strategies may be a rational option to overcome the resistance of cancers to PD1 immune checkpoint blockade by enhancing cytotoxic T-cell tumor infiltration, rendering T cells resistant to PD1/PD-L1-mediated anergy or simultaneously blocking other mechanisms of tumor immune escape.29 33 34 Several clinical trials demonstrated that combination regimens appear to be more potent than single agents, with objective response rates of 53% in metastatic melanoma, 30.8% in gastric or esophagogastric junction cancer and 50% in advanced hepatocellular carcinoma.35–37 Despite the progress of combinatorial treatment with PD1 blockade in melanoma or non-melanoma solid cancers, PC has proven refractory to PD1 checkpoint-based immunotherapy even when combined with chemotherapy (capecitabine, gemcitabine or nab-paclitaxel) or combined with other immunotherapy (tremelimumab: antibody targeting CTLA-4; acalabrutinib: bruton tyrosine kinase inhibitor; mogamulizumab: antibody targeting CC chemokine receptor 4; cabiralizumab: antibody targeting CSF-1 receptor).11 These pieces of clinical evidence encourage us to pay more attention to combinations of checkpoint inhibitors with other modalities that could potentially change the tumor-generated TME or target other mechanisms of immune escape.
TAMs constitute an important component of the TME.12 13 M2 polarized TAMs are considered immunosuppressive cells and can induce a favorable environment for cancer by inhibiting the immune system. Expansion of M2 polarized TAMs is a major mechanism used by cancers to escape immune surveillance, by upregulating the expression of checkpoint molecules, prohibiting tumor-reactive cytotoxic T-cell activation, or producing more cytokines and growth factors that directly sustain tumor cells.15–17 19 In PC tumor-bearing mice, we observed significant increases in the frequency of CXCR1/2+CD68+ macrophages in peripheral blood and tumors. CXCR1+CD68+ macrophages accounted for up to 8.3% (BxPC-3) and 6.1% (Panc-1) of the total cells in the blood, 6.1% (BxPC-3) and 5.5% (Panc-1) of the total cells in the tumor, whereas CXCR2+CD68+ macrophages increased to 9.8% and 6.7% in the blood of BxPC-3 and Panc-1 tumor-bearing mice and to 10.6% and 6.4% in the tumors of these mice, respectively. The fraction of circulating CXCR1/2+CD68+ macrophages positively correlates with tumor volume. These findings illustrated that tumor-induced expansion of CXCR1/2+CD68+ macrophages is associated with PC progression and CXCR2+CD68+ macrophages were preferentially expanded in two types of PC cell-based tumors. Moreover, the tumor-infiltrating CXCR1/2+CD68+ macrophages in our model produced higher levels of arginase-1 and decreased levels of iNOS. Arginase-1 is an amino acid–degrading enzyme which has influence on immunogenic tolerance and contributes to the functional failure of T cells.38 Conversely, iNOS significantly reversed T cells suppression,39 and this result suggested that PC tumor-infiltrating CXCR1/2+CD68+ macrophages are M2 polarized TAMs and mediate immunosuppressive effects on T cells. In humans with PC tumors, similar patterns were found, demonstrating that the expansion of CXCR2+CD68+ macrophages also contributes to human PC progression and poor prognosis.
In humans, CXCL8 is one of the most important CXC chemokine ligands and its signal is mediated through both CXCR1 and CXCR2.25 CXCL8 expression is significantly upregulated in human colon, head and neck, pancreatic and esophageal cancers compared with healthy tissues based on previously published literature.25 27 In the present study, we also confirmed this result in a murine model of PC. As CXCL8 expression is significantly different between mice and humans (mice lack a CXCL8 homolog), so the CXCL8 detected in tumor-bearing mice was totally derived from the human PC cells. CXCR1/2-expressing TAMs are migratory hematopoietic cells and have an immunosuppressive nature (M2 polarization). They have been shown to expand in murine and human PC tumors; thus, these cells may be recruited to the tumor bed by tumor-derived CXCL8 through the CXCL8–CXCR1/2 axis to facilitate PC immune escape. In our in vitro migration assay experiment shown in figure 4C,D, we present a significant reduction in the number of CXCR2+CD68+ macrophages that migrate toward a tumor attractant when we use anti-CXCL8 and/or anti-CXCR2 blockade, whereas the migration of CXCR1+CD68+ macrophages is not completely inhibited when CXCL8 is absent, suggesting that CXCL8 predominantly mediated the tumor trafficking of CXCR2-expressing TAMs. This result also been proved by our in vivo experiments. In a review of previous published articles, Ijichi showed that CXCL8 overexpressing human PC cells mixed with pancreatic fibroblasts increased tumor growth in a mouse model. In addition, CXCR2 blockade treatment in the stromal cells delayed tumor progression in a mixed cells xenograft model, demonstrating the interaction between tumor-derived CXCL8 and stromal CXCR2 promotes tumor progression, invasion and metastasis.28 Trellakis et al indicated that deletion of CXCR2 in a CXCL8 upregulated colitis-associated tumorigenesis mouse model reduced the number of granulocytic myeloid-derived suppressor cells (GrMDSCs), which mediated a marked effect on T-cell function.40 Chen et al showed expression of CXCR1 positively correlates with expansion of cancer stem cells (CSCs) in PC, exogenous CXCL8 promoted PC cells proliferation, invasion ability, tumorsphere formation and CSCs population in vitro, and CXCR1 blockade treatment reversed these effects.27 Finally, one study indicated that CXCR1 predominantly engaged mouse CXCL6, another CXC chemokine ligand, which binds to both CXCR1 and CXCR2.41 CXCR1 and CXCR2 likely both play a role in modulating tumor stromal cell trafficking to the tumor bed via CXCL8 or other CXC chemokine ligands depending on the model system studied. In our model, PC tumor-derived CXCL8 mainly mediated CXCR2+CD68+ macrophage trafficking to the tumor bed.
IFN-γ is an important activator of innate and adaptive immunity and possesses antiviral, immunoregulatory, and anti-tumor properties. It modulates transcription in up to 30 genes via an IFN-γ signaling pathway to mediate a variety of physiological and cellular responses.22 The in vitro studies on the direct killing effect of IFN-γ on cancer cells are extensive and the results indicate that IFN-γ has anti-proliferative activity leading to growth inhibition or cell death by apoptosis or autophagy.24 42 However, the evidence to support the contribution of IFN-γ to significant tumor regression is sparse in in vivo studies and clinical trials.43 Based on its immunoregulatory characteristics, it is theorized that IFN-γ induces the activation of the immune system in multiple ways to promote anti-tumor immune responses, including enhancing natural killer (NK) cell activity, antigen presentation and lysosome activity of macrophages, inducing M1 polarization and iNOS production and the production of IgG2a and IgG3 from activated plasma B cells, increasing the expression of MHC-I molecules in tumor cells, and increasing the expression of MHC-II molecules on antigen-presenting cells.44 On the other hand, there are opposing effects of IFN-γ that attenuate immune function based on findings that IFN-γ promotes T-cell exhaustion through upregulating the coregulatory molecules B7-H1 (PD-L1) and B7-DC (PD-L2), inducing T-cell apoptosis via the PD1/PD-1 checkpoint signaling pathway.45 Benci et al recently showed that blocking tumor IFN-γ signaling decreased interferon-stimulated genes (ISGs) in cancer cells, while in immune cells, ISGs were increased by enhancing IFN-γ production in exhausted T cells (T-EXs). In non-immunogenic tumors, T-EXs use IFN-γ to drive the maturation of innate immune cells. Thus, IFN-γ signaling in cancer cells and immune cells has opposing effects, establishing a regulatory relationship that limits both adaptive and innate immune killing, and the perturbation of this relationship is associated with the immune checkpoint blockade response, which is independent of the tumor mutational burden. These results presented a possible mechanism in which the opposing effects of IFN-γ were integrated and impacted PD1 checkpoint-based immunotherapy.46 Although the pleiotropic activities of IFN-γ mostly involve its immunostimulatory and immunomodulatory effects that enhance antitumor immune responses, accumulating evidence indicates that IFN-γ may regulate the expression and secretion of tumor-derived cytokines and growth factors, thereby contributing to the alteration of the tumor microenvironment.47 48
In our previous study, we proved that IFN-γ suppressed the expression and secretion of CXCL8 in PC cell lines and inhibited the proliferation and migration of these cells.24 In the present study, we confirmed that IFN-γ treatment also reduced tumor-derived CXCL8 expression in PC xenograft tumors and its secretion in mouse peripheral blood. As the CXCL8–CXCR1/2 signaling axis is essential for the pathogenesis of cancer, and the interaction between CXCL8 secreted by cancer cells and CXCR1/2 in the tumor microenvironment is critical for cancer progression and metastasis,25 28 we next investigated the biologic ramifications of IFN-γ combined with anti-PD1 treatment on murine PC tumors. Remarkably, combining PD1 checkpoint blockade with IFN-γ treatment showed significantly enhanced antitumor effects and prolonged the survival time of the mice compared with anti-PD1 or IFN-γ treatment used alone, despite systemic accumulation of CXCR2+CD68+ macrophages in the peripheral blood of the mice. CXCL8 was overexpressed and CXCR2+CD68+ macrophages accumulated in the mouse tumor bed when anti-PD1 monotherapy was administered, while combinatorial treatment significantly reduced the expression of CXCL8 and diminished CXCR2+CD68+ macrophage tumor infiltration. Interestingly, the retained tumor-infiltrating CXCR2+CD68+ macrophages converted and produced less arginase-1 and more iNOS and then showed an M1 polarization. These results indicated that when PD1 blockade monotherapy promotes a cytotoxic T cell–mediated tumoricidal effect, the expression of tumor-derived CXCL8 increases, thus activating the CXCL8–CXCR2 axis, enhancing CXCR2-expressed M2 TAM tumor trafficking and infiltration and inducing immunosuppressive effects on T cells as a rebound mechanism. However, IFN-γ reverses this opposing effect by blocking the CXCL8–CXCR2 axis by downregulating the expression of CXCL8 and inhibiting CXCR2-expressing M2 TAM tumor trafficking and infiltration, thus promoting the antitumor effects of PD1 blockade in murine PC tumors. As IFN-γ has increasingly been regarded as a key stimulus that induces the M1 polarization of TAMs, IFN-γ may act directly on tumor-infiltrating macrophages and then convert these cells to the M1 subtype. This may be another mechanism by which IFN-γ enhances the efficacy of PD1 blockade therapy.
The clinical relevance of these findings is provided by studies in human patients with PC. We demonstrated that CXCL8 levels are elevated in peripheral blood of patients with PC. Meanwhile, patients with PC presented elevated CXCR2 expression on peripheral and tumor-infiltrating CD68+ macrophages, which are associated with clinicopathological and prognostic significance. These results suggest that the tumor-induced CXCL8 upregulation and CXCR2+CD68+ macrophage expansion observed in tumor-bearing mice is also active in patients with PC and the expansion of CXCR2+CD68+ macrophages also contributes to human PC progression and poor prognosis. Thus, our results showed a close correlation between our in vivo study and clinical observation.
The potential for clinical application of these findings is notable because there are currently no clinically relevant evidence to show that anti-PD1 immunotherapy is effective in treating PC.11 Conversely, IFN-γ is also not approved for the treatment of any cancer, although studies showed improved survival when IFN-γ was administered to patients with bladder carcinoma, malignant melanoma and advanced ovarian carcinoma.43 In the present study, we reported that IFN-γ suppresses the expression and secretion of tumor-derived CXCL8, which inhibits tumor-infiltrating of CXCR2+CD68+ macrophages by blocking the CXCL8–CXCR2 axis. This ultimately enhances the anti-PD1 efficacy in a murine PC model. Conversely, human PC also produces high levels of CXCL8. Patients with PC had elevated CXCR2 expression on peripheral and tumor-infiltrating CD68+ macrophages, which are associated with advanced tumor stage and poor prognosis. Our findings indicate that IFN-γ is a translatable, therapeutic option to improve the efficacy of PD1 blockade therapy in patients with PC by preventing the tumor trafficking of CXCR2+CD68+ macrophages blocking the CXCL8–CXCR2 axis.
Limitations of this work include the absent information regarding whether IFN-γ also influences the differentiation of tumor-infiltrating T-cell subsets (such as Th1, Th2, Th17 and Treg cells) and whether the variable CXCR2+CD68+ macrophage infiltration has associations with the tumor-infiltrating T-cell differentiation. Nonetheless, the findings presented here strongly suggest that clinical studies on IFN-γ in combination with PD1 checkpoint blockade are beneficial to human patients with PC.
Conclusion
We report that IFN-γ suppresses the expression of tumor-derived CXCL8 and inhibits tumor-induced CXCR2+CD68+ macrophage tumor trafficking by blocking the CXCL8–CXCR2 axis, thereby abolishing the local immunosuppressive effect of these cells. Combined with IFN-γ treatment, PD1 checkpoint blockade showed significant antitumor effects in an established murine PC model (figure 6). Human PC also produces high levels of CXCL8 and patients with PC present elevated CXCR2 expression on peripheral and tumor-infiltrating CD68+ macrophages, which are associated with advanced tumor stage and poor prognosis. These findings suggest that IFN-γ is an important agent to improve the potency of anti-PD1 therapy in patients with PC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Graphical abstract.
Acknowledgments
We thank Dr Ping Xie at the Department of Hepatobiliary Surgery, Huzhou Hospital, School of Medicine, Zhejiang University (Huzhou Central Hospital) for his assistance of providing blood and surgical samples of the patients with PC. We also thank Dr Hongfei Liu at the Key Laboratory of Biomedicine and Health, School of Medicine, Hangzhou Normal University for his assistance of immunofluorescence experiments.
References
Footnotes
MZ, LH and GD contributed equally.
Contributors MZ, GD, LC and QY developed the original hypothesis and designed the study. LH, HH, GC, TS, XX performed the experiments. XS, NL and QW performed the data analysis and provided statistical interpretation. MZ and LH wrote the manuscript. All authors read and approved the final manuscript.
Funding This work was supported by the Natural Science Foundation of Zhejiang Province (grant no. LGF18H160004 to MZ); the National Natural Science Foundation of China (grant no. 81772548 to LC); the Public Welfare Technology Application Research Project of Zhejiang Province (grant no. 2017C33189 to QY); the Funds of Huzhou Municipal Science and Technology Bureau (grant no. 2018GY11 to MZ).
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval This study was approved by the ethics committee of Huzhou Hospital, School of Medicine, Zhejiang University (Huzhou Central Hospital) (HzhcLS2018-0807), China. Patients from Huzhou Central Hospital were obtained in our preclinical research with informed consent (ChiCTR1800017665).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. The datasets used and analyzed during the present study are available from the corresponding author on reasonable request.