Article Text

Abstract
Background Immune checkpoint inhibitors (ICIs) have expanded treatment options for metastatic renal cell carcinoma (mRCC); however, there are limited predictive biomarkers for response to ICIs in this indication, with programmed death-ligand 1 (PD-L1) status demonstrating little predictive utility in mRCC. While predictive of ICI response in other tumor types, the utility of tumor mutation burden (TMB) in mRCC is unclear. Here, we assess TMB, loss of antigen presentation genes and PD-L1 status correlated with outcomes to ICI treatment in mRCC.
Methods Tumor samples from 34 patients with mRCC treated with ICI therapy at Duke Cancer Institute were retrospectively evaluated using Personal Genome Diagnostics elio tissue complete (RUO version), a tumor genomic profiling assay for somatic variants, TMB, microsatellite status and genomic status of antigen presentation genes. Tumor samples were also analyzed with the Dako 28-8 PD-L1 immunohistochemistry assay. Deidentified clinical information was extracted from the medical record, and tumor response was evaluated based on the Response Evaluation Criteria In Solid Tumors (RECIST) V.1.1 criteria.
Results Patients were stratified by overall response following ICI therapy and designated as progressive disease (PD; n=18) or disease control groups (DC; n=16). TMB scores ranged from 0.36 to 12.24 mutations/Mb (mean 2.83 mutations/Mb) with no significant difference between the PD and DC groups (3.01 vs 2.63 mutations/Mb, respectively; p=0.7682). Interestingly, 33% of PD patients displayed loss of heterozygosity of major histocompatibility complex class I genes (LOH-MHC) vs 6% of DC patients. Nine of 34 samples were PD-L1-positive (4 in the PD group; 5 in the DC group), suggesting no correlation between PD-L1 expression and response to ICI therapy. Notably, the DC group displayed an enrichment of mutations in DNA repair genes (p=0.04), with 68.8% exhibiting at least one mutated homologous recombination repair (HRR)-related gene compared with only 38.9% of the PD group (p=0.03).
Conclusions Overall, neither TMB nor PD-L1 correlated with ICI response and TMB was not significantly associated with PD-L1 expression. The higher incidence of LOH-MHC in PD group suggests that loss of antigen presentation may restrict response to ICIs. Separately, enrichment of HRR gene mutations in the DC group suggests potential utility in predicting ICI response and a potential therapeutic target, warranting future studies.
- genitourinary medicine
- oncology
- molecular immunogene
- immunology
- tumors
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Renal cell carcinoma (RCC) represents approximately 90% of all renal cancers, with the incidence of RCC steadily increasing over the past 65 years1 2 At diagnosis, nearly one-third of patients present with advanced or metastatic RCC (mRCC). There are estimated to be 73,820 new cases of RCC and 14,770 deaths from RCC in 2019. Although patients with metastatic disease have previously exhibited poor 5-year survival rates,3 targeted therapies over the last decade have greatly improved outcomes. Tyrosine kinase inhibitors that inhibit vascular endothelial growth factor receptor and mammalian target of rapamycin have long been the mainstay of therapy for patients with mRCC4 5; however, immune checkpoint inhibitors (ICIs) have improved clinical outcomes with both nivolumab monotherapy and ipilimumab-nivolumab combination treatments. In a registrational trial of patients with treatment-naïve mRCC randomised to ipilimumab-nivolumab versus sunitinib, the combination immunotherapy improved median overall survival (OS; not reached vs 26.6 months, p<0.0001) and objective response rates (42% vs 27%, p<0.0001), with a remarkable complete response (CR) rate of 10% in patients with intermediate-risk and poor-risk disease.6 The combination of ipilimumab-nivolumab has therefore changed first-line treatment of patients with intermediate-risk to high-risk mRCC.7 However, although there is now the possibility of robust responses with ICIs, the majority of patients do not achieve objective responses. There is currently no clinically validated biomarker to predict ICI response in patients with mRCC.
Nivolumab targets the programmed cell death receptor 1 (PD-1) expressed on T cells, disrupting interaction with its ligand PD-L1, allowing T cells to target and kill cancer cells.8 Several Food and Drug Administration-approved immunotherapies use this dynamic to treat multiple tumor types. In mRCC, PD-L1 is found to be aberrantly expressed in primary and metastatic tumors9 and has demonstrated utility as a prognostic biomarker. In the CheckMate 025 study comparing nivolumab versus everolimus treatment, patients with mRCC expressing higher PD-L1 had worse OS rates regardless of treatment arm (median OS 21.8 months for patients with ≥1% PD-L1 expression vs 27.4 months for patients with <1% PD-L1 expression).10 Nivolumab improved median OS when compared with everolimus across all patients, regardless of PD-L1 status.10 Similarly, in the CheckMate 214 study, PD-L1 expression was associated with a greater benefit from ipilimumab-nivolumab, but both PD-L1-negative and PD-L1-positive patients had improved survival from combination immunotherapy.7 Furthermore, each PD-1/PD-L1 targeting drug has its own diagnostic assay for PD-L1, complicating clinical interpretation of PD-L1 status due to lack of standardization across assays.11 Overall, there is more evidence for PD-L1 expression as a prognostic marker rather than a predictive marker for treatment response in patients with mRCC.
Tumor mutation burden (TMB), typically measured as mutations per megabase (mutations/Mb), is an emerging biomarker that measures the number of tumor-specific somatic mutations in the tumor’s exome. It has been demonstrated in some indications that high TMB tumors present more surface neoantigens, thereby inducing an enhanced immune response.12 In metastatic melanoma and non-small cell lung cancer (NSCLC), high TMB scores have served as a stratification biomarker for predicting ICI responses in several studies,13–15 demonstrating potential as a predictive biomarker. Additionally, the utility of TMB as a biomarker of ICI response may be influenced by other biological factors. Restriction or loss of antigen presentation caused by mutations or loss of heterozygosity (LOH) of the beta-2-microglobulin (B2M) and major histocompatibility complex (MHC) class I genes may contribute to tumor evasion of CD8+ T-cell-mediated destruction.16 17
Overall, with the increasing incidence of RCC and high proportion of metastatic disease, along with the expanding use of costly ICIs and potential adverse events, more reliable biomarkers are needed to predict sensitivity and resistance. In this study, we aimed to evaluate genomic alterations and signatures as well as PD-L1 status in a cohort of patients with mRCC treated with standard of care immunotherapy.
Methods
Retrospective analysis was performed on patients with mRCC treated with standard of care ICI therapy, with archival pathology specimens at Duke Cancer Institute. Clinical characteristics and outcomes were extracted from the electronic medical record. Pathological specimens were characterized by tumor histology, primary versus metastatic biopsy site and anatomical location of biopsy site, and tumor samples for PD-L1 expression and genomic analysis were obtained from formalin-fixed paraffin-embedded (FFPE) slides. Clinical record review abstracted patient characteristics, including sex, race, ethnicity, Eastern Cooperative Oncology Group performance score, diagnosis age, time from diagnosis to sample collection, time from diagnosis to last follow-up, time from diagnosis to death if applicable and the International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) risk score. Treatment information collected included ICI treatment type, line of therapy, time from diagnosis to first and last doses of ICI, number of doses of ICI and reason for ICI discontinuation. Best response scan for each patient was determined radiographically by independent evaluation by a radiologist and re-scored per RECIST V.1.1 criteria.
FFPE tissue from 34 patients were evaluated at Personal Genome Diagnostics (PGDx; Baltimore, Maryland, USA). Sample processing from tissue, library preparation, hybrid capture and sequencing were performed at PGDx. Samples were run on the PGDx elio tissue complete RUO tumor profiling next-generation sequencing (NGS) assay, screening for somatic alterations covering 500+ genes for sequence mutations comprising single nucleotide variants (SNVs) and insertion-deletion events (indels), gene amplifications and gene translocations, as well as TMB and microsatellite status. DNA was extracted from FFPE tissue and following shearing, genomic libraries were prepared using end-repair, A-tailing and adapter ligation modules. Genomic libraries were amplified and captured in-solution, targeting the predefined regions of interest across full exonic regions. Captured libraries were sequenced on an Illumina NextSeq platform.
The 500+ gene panel encompasses 2.23 Mbp with an average distinct coverage of ~900 x. Somatic variant identification was performed using PGDx’s proprietary machine learning software, which has previously demonstrated high sensitivity and specificity across multiple variant and tissue types while using algorithms to discriminate germline mutations as well as sequencing artifacts/errors.18
Results from mutational profiling were analyzed using the International Cancer Genome Consortium Data Portal.19 Mutated gene sets were grouped according to overall clinical response and analyzed for gene pathway enrichment using the Reactome pathway curated database. Enriched pathways identified among the mutated genes were confirmed via other publicly available pathway databases (Cytoscape, Classification System, Gene Set Enrichment Analysis).
Tissue specimens were also analyzed for PD-L1 status at Duke using the Dako 28-8 PD-L1 immunohistochemistry (IHC) assay. PD-L1 positivity was defined as >1% tumor cell membrane staining. Two pathologists scored each sample to control for interobserver variability.
Results
Cohort overview
Tissue specimens from 34 patients with mRCC treated with standard of care ICI at Duke Cancer Institute from January 1, 2012 to December 1, 2017 were collected, sequenced and analyzed (table 1). Of the 34 samples, 71% (n=24) were from the primary tumor and 29% (n=10) were from distant metastases. Most patients (n=26, 76%) had intermediate-risk disease based on IMDC risk score, and the majority of patients (n=31, 91%) were treated with nivolumab. Two patients (6%) were treated with nivolumab and ipilimumab and one patient (3%) was treated with pembrolizumab. The median number of ICI doses was 9.5 (1–50 doses), with an average time on therapy of 4.8 months (0–24 months). ICI therapy was primarily used in the second-line (n=14, 44%) or third-line setting (n=14, 44%). Clinical outcomes and disease progression were scored based on RECIST V.1.1 criteria as follows: CR (n=2), partial response (PR; n=5), stable disease (SD; n=9) or progressive disease (PD; n=18). For further analysis of this cohort, CR, PR and SD patients were grouped as disease control (DC; n=16) and compared with PD patients (n=18). For patients who discontinued treatment, the majority (24/26 (92%)) discontinued due to disease progression.
Baseline patient characteristics, treatment selections and clinical outcomes
Overall, 360 somatic mutations were identified in the 34-patient cohort, across 166 unique genes, from the 500+ targeted gene panel (figure 1A,B, online supplementary tables 1 and 2). Mutations that were identified as potential germline variants and mutations with mutant allele frequencies of <5% were filtered from the dataset and excluded in the correlative analyses to clinical outcomes. Of these 360 mutations, the majority were SNVs and indels (n=350, 97.22%). A small number of amplification events were identified (n=10, 2.78%) across seven genes (BRCA1, CCND1, CCND3, CCNE1, ERBB2, FGFR3, FGFR4, ERBB2 (online supplementary table 2)). No translocation events were identified within this cohort.
Supplemental material
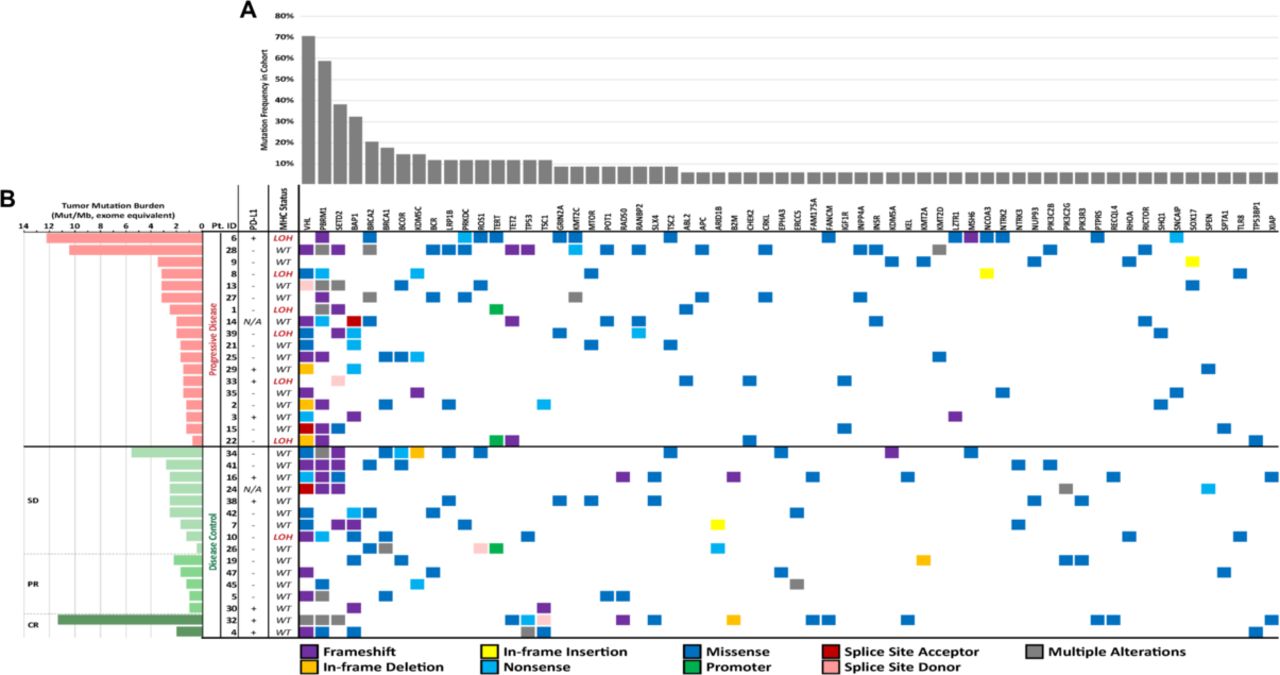
Comprehensive mutational profile of metastatic renal cell carcinoma (mRCC) cohort. (A) Mutational profile determined by Personal Genome Diagnostics (PGDx) elio tissue complete 500+ gene RUO tumor profiling next-generation sequencing assay (currently under development) and programmed death-ligand 1 (PD-L1) status determined by Dako 28-8 PD-L1 immunohistochemistry (IHC) assay. Mutated genes identified in <3 distinct patients in this cohort were excluded from this display. The type of sequence mutation identified is denoted below. Tumor mutation burden, PD-L1 status and major histocompatibility complex (MHC) genomic status was determined and stratified by overall clinical response across the cohort. (B) Patient overall response was categorized into either the progressive disease (PD) group or the disease control (DC) group, with the latter being further subdivided into stable disease (SD), partial response (PR) or complete response (PR) groups. PD-L1 overexpression is denoted with (+) and normal levels of PD-L1 expression is denoted with (−); N/A denotes cases where PD-L1 status was indeterminate or unevaluable. MHC genomic status is categorized as either wild-type (WT) or loss of heterozygosity (LOH).
Correlation of ICI biomarkers to clinical outcomes: TMB, neoantigen presentation capability and PD-L1 status
TMB scores were assessed from somatic mutations (SNVs and indels) identified by the PGDx elio tissue complete targeted NGS panel, calculated as mutations/Mb and standardized to whole exome sequencing.20 This mRCC cohort displayed TMB scores ranging from 0.37 to 12.24 mutations/Mb (figure 1), with a mean and median TMB score of 2.83 and 1.97 mutations/Mb, respectively. TMB scores were then compared between the PD (mean of 3.01 mutations/Mb) and DC groups (mean of 2.63 mutations/Mb); however, no significant difference between the two groups was observed (p=0.77, t-test) (figure 2). LOH of MHC class I genes (LOH-MHC) was also assessed to determine neoantigen presentation functionality and 7 of 34 patient samples (21%) were positive for LOH-MHC. Interestingly, LOH-MHC was present in 33% of patients with PD (6/18) vs 6% of responders (DC, 1/16) (figure 1). One PD patient (Pt. 6) had high TMB and exhibited LOH-MHC, suggesting that while the tumor could produce neoantigens to stimulate an immune response, antigen presentation was likely compromised and no response to ICI was observed. Conversely, one DC patient (Pt. 32) showed high TMB and functional MHC class I genes (intact antigen presentation), with CR to ICIs. Pt. 28 also displayed a relatively high TMB score in this cohort (10.43 mutations/Mb) and had a normal MHC (wtMHC) status, suggesting potential for a favorable response, but was observed to be PD. However, this sample was also low for PD-L1, which may explain the lack of response to ICIs.

Tumor mutation burden does not correlate with clinical response in patients with metastatic renal cell carcinoma (mRCC) treated with immune checkpoint inhibitor (ICI) therapy. Mean tumor mutation burden was 3.01 mutations per megabase DNA in patients with progressive disease (PD), compared with mean tumor mutation burden of 2.63 mutations per megabase DNA for patients in the disease control (DC) group (p =0.76820). ns, not statistically significant.
The mRCC samples were also assessed for PD-L1 status, to evaluate for possible correlation to TMB or treatment outcomes. Within the 34-sample cohort, 9 samples (26%) stained PD-L1-positive, 23 (68%) were PD-L1-negative and 2 (6%) were indeterminate (figure 1). Among the PD group, 4 of 18 (22%) were PD-L1-positive compared with 5 of 16 (31%) of the DC group. PD-L1 status did not correlate with clinical response to immunotherapy (p=0.69, Fisher’s exact test) (figure 3A) nor did it correlate with TMB scores (p=0.77, t-test) (figure 3B). Additionally, all patients were identified as microsatellite stable (MSS; data not shown).
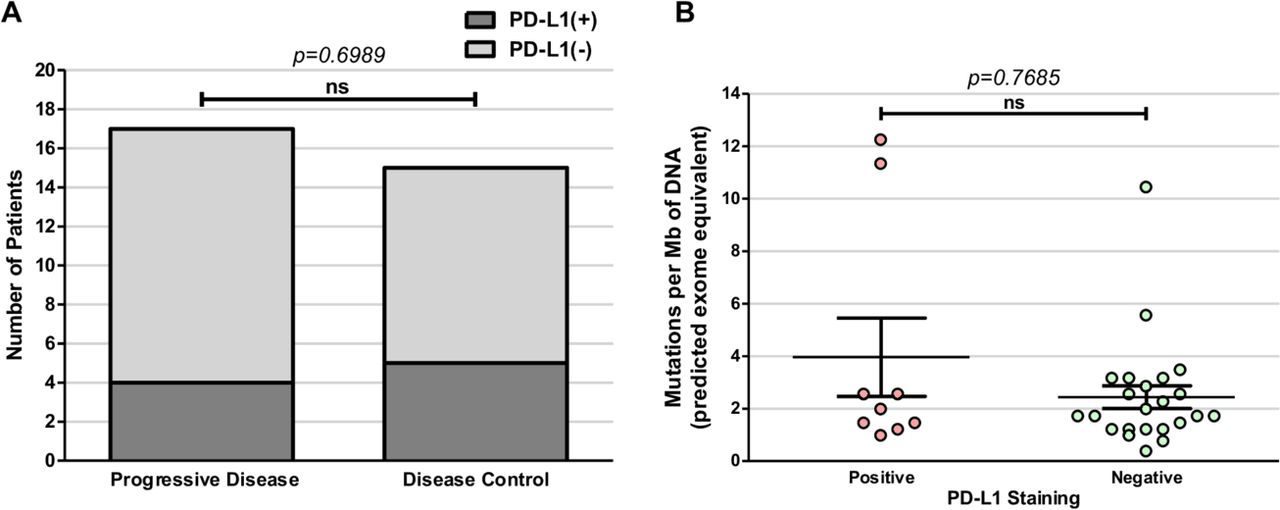
Programmed death-ligand 1 (PD-L1) expression does not correlate with clinical response in patients with metastatic renal cell carcinoma (mRCC) treated with immune checkpoint inhibitors (ICIs). (A) PD-L1 expression does not significantly correlate with clinical outcomes or with (B) tumor mutation burden (p=0.6989). ns, not statistically significant.
Comprehensive genomic profiling in mRCC
The 10 most commonly mutated genes identified in this cohort were VHL, PBRM1, SETD2, BAP1, BRCA2, BRCA1, BCOR, KDM5C, BCR and LRP1B, in descending order (figure 1). Missense mutations comprised 67.71% (n=237) of SNVs and indels. Second most frequent were frameshift mutations, comprising 17.14% (n=60) of SNVs and indel mutations. Nonsense mutations were found at a lower frequency at 7.43% (n=26), and remaining SNV/indel alteration types (in-frame insertion and deletions, splice sites and promoter mutations) were found at frequencies <3%.
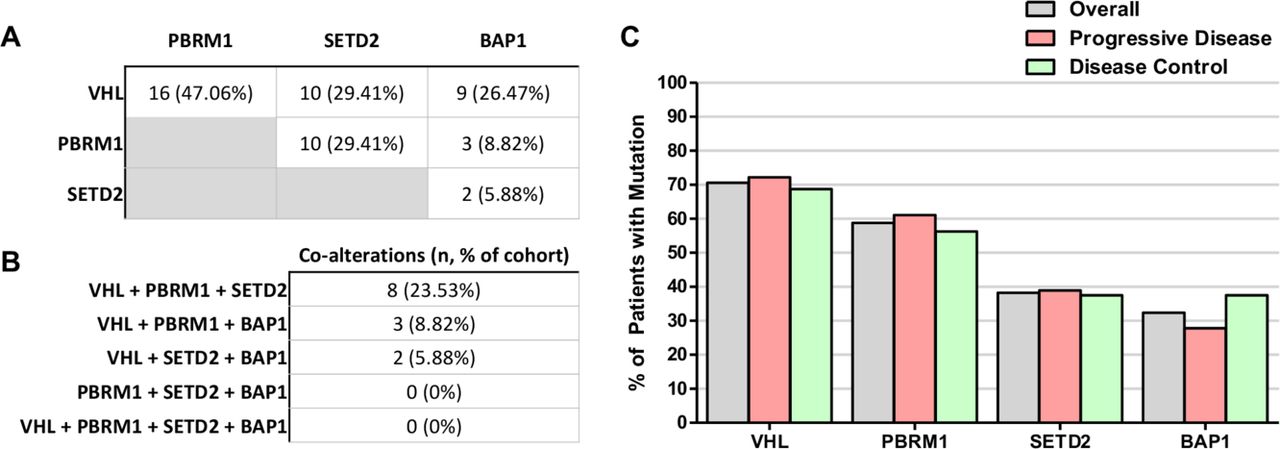
The cohort exhibited a high frequency of co-alterations among VHL, PBRM1, SETD2 and BAP1 (figure 4A), although none of these single gene mutations was found to be significantly different between the PD and DC groups. Sixteen of the 34 patients (47%) had concurrent mutations in VHL and PBRM1 of which 8 (23.53%) had additional mutations in SETD2 (figure 4B). Three patients (8.82%) had concurrent mutations in VHL, PBRM1 and BAP1. No patient had concurrent mutations across all four genes. Despite the high frequency of co-alterations among these genes in this cohort, none significantly correlated with clinical outcomes (figure 4C, online supplementary figure 1). Notably, there appeared to be some mutual exclusivity between SETD2 and BAP1 mutation (figure 4A). Although 32.40% (n=11) had BAP1 mutations and 38.23% (n=13) had SETD2 mutations, only two patients had mutations in both genes (5.88%). Similarly, although PBRM1 mutations were identified in 58.83% (n=20) of the cohort, only 8.82% (n=3) were also mutated in BAP1. No patients were found to have co-occurring mutations in PBRM1, SETD2 and BAP1.
Supplemental material

Frequency of alterations of commonly mutated genes in metastatic renal cell carcinoma (mRCC). (A) VHL, PBRM1, SETD2 and BAP1 are among the most frequently mutated gene mutations in this mRCC cohort and exhibited a high degree of concomitant mutations. (B) A small subset of patients were found to harbor three concurrently altered genes. (C) Mutation frequency of frequently altered mRCC genes did not correlate clinical response.
Finally, comparative mutational analysis between PD and DC groups showed clear exclusivity of several genes in each group (online supplementary figure 1). Using the gene set enrichment analysis methods available through the
ICGC Collaborative Data Portal, the mutational profiles from the 34-patient cohort was curated to identify differences in key genomic signatures and signaling pathways. Most notable between the PD and DC groups was the enrichment of DNA damage response and repair gene mutations in the DC group where several key pathways were flagged as significantly altered compared with the PD group (online supplementary tables 3 and 4).
DNA damage response and repair pathway gene mutations
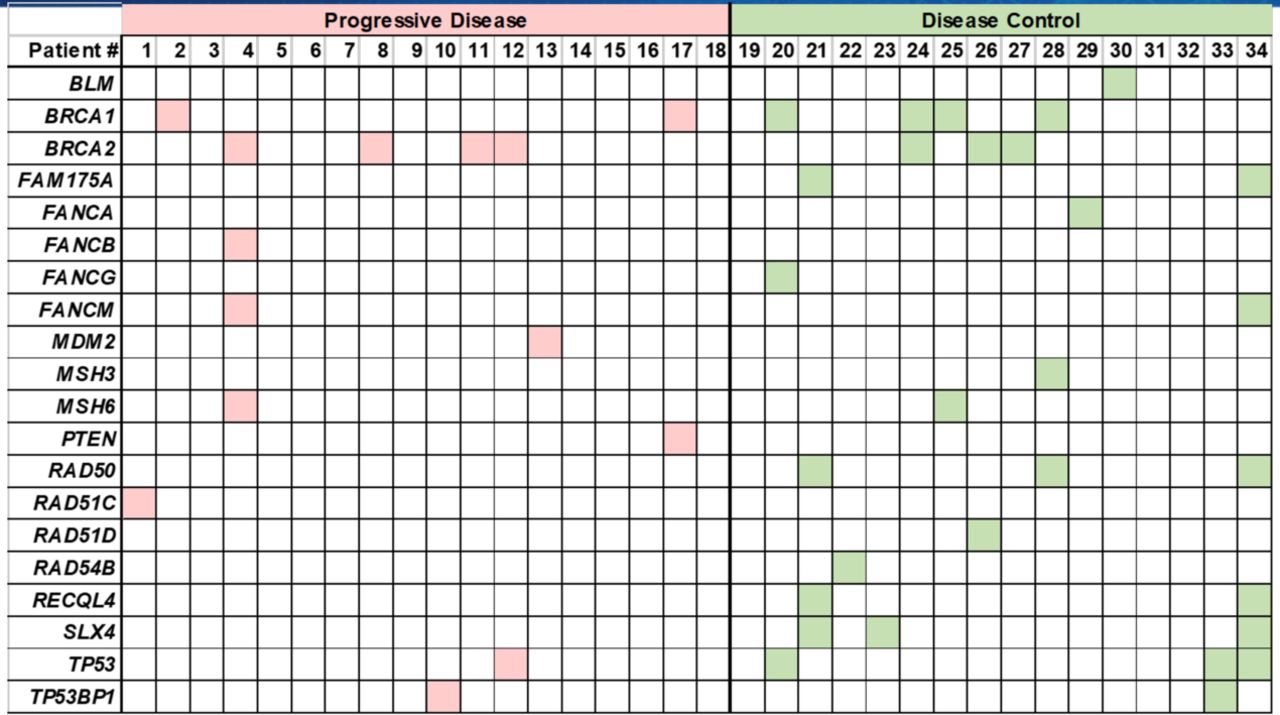
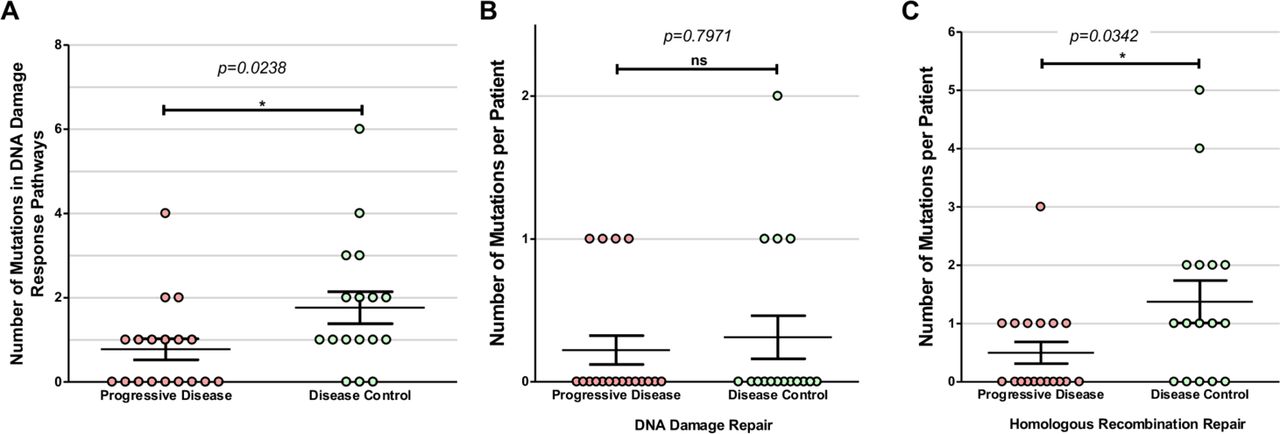
A large number of mutations in genes associated with DNA damage response and repair functions were identified in this cohort (online supplementary figure 1, online supplementary tables 3 and 4), including BLM, BRCA1/2, FAM175A, FANCA, FANCB, FANCG, FANCM, MDM2, MSH3, MSH6, PTEN, RAD50, RAD51C, RAD51D, RAD54B, RECQL4, SLX4, TP53 and TP53BP1 (figure 1 and figure 5). Although these genes constitute several functional pathways, they were grouped together for correlative analysis. Gene mutations in the DNA damage response pathways and processes identified in patients with mRCC were grouped by overall response to ICIs and were found to be significantly associated with the DC group (figure 6A; p=0.02, t-test). Thirteen of the 16 patients in the DC group (81.3%) had at least one mutation in a DNA damage response-related gene, compared with only 50% (9 out of 18) of patients in the PD group.

Mutations in DNA damage repair pathway genes identified in patients with metastatic renal cell carcinoma (mRCC) grouped by best response to immune checkpoint inhibitors (ICIs). The DNA repair genes identified were: BLM, MDM2, PTEN, TP53, TP53BP1, BRCA2, FAM175A, FANCA, FANCB, FANCM, MSH3, MSH6, RAD50, RAD51C, RAD51D, RAD54B, RECQL4 and SLX4. In the disease control (DC) group, 81.3% of patients were found to have at least one mutation in a DNA damage response pathway gene compared with only 50% of patients in the progressive disease (PD) group.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Number of DNA response-related gene mutations in progressive disease (PD) vs disease control (DC) groups. (A) The PD group were found to have significantly more mutations in DNA damage response pathways gene than the DC group (p=0.0238, t-test). Further delineating the DNA damage response gene mutations found that those categorized as (B) DNA damage repair (DDR) was not found to be significantly different between the PD and DC groups (p=0.7971), ns, not statistically significant. However, there was a significant difference in the number of mutations in (C) homologous recombination repair (HRR) genes between the PD and DC groups (p=0.0342).
In order to further examine the correlation to clinical outcomes, these mutated genes were divided into two functional pathways: DNA damage repair (DDR) and homologous recombination repair (HRR) genes (online supplementary table 5). DDR genes include BLM, MDM2, PTEN, TP53 and TP53BP1. HRR genes include BRCA2, FAM175A, FANCA, FANCB, FANCM, RAD50, RAD51C, RAD54B, RECQL4 and SLX4. There was no significant difference in the overall number of DDR gene mutations between the DC and PD groups (p=0.80, t-test) (figure 6B). Interestingly, within HRR-related genes, the DC group was found to have significantly higher number of mutations/patient compared with the PD group (figure 6C; p=0.34, t-test). Eleven of the 16 patients (69%) in the DC group had at least one HRR-related gene mutated compared with only 38.9% (n=7) in the PD group.
Discussion
TMB, antigen presentation capability and PD-L1 status
Recent data surrounding combination therapy with VEGF inhibitors and immunotherapy in mRCC have shown that PD-L1 may demonstrate a signal for improved objective response rates.21 However, the prognostic utility of PD-L1 expression in patients with mRCC treated with ICIs remains equivocal. In this study, PD-L1 expression did not correlate with high TMB nor with clinical response in patients with mRCC treated with ICI therapy. This agrees with results comparing nivolumab monotherapy with everolimus, which showed no significant difference in OS based on PD-L1 status.10 PD-L1 faces several challenges as a biomarker, including lack of standardization, tumor heterogeneity of PD-L1 expression, temporal evolution of PD-L1 status and variation in PD-L1 expression.9 Ultimately, PD-L1 has not demonstrated sufficient and reliable value as a predictive biomarker for mRCC.
TMB is a surrogate marker of immune response and high TMB is thought to be associated with increased number of neoantigens and be predictive for ICI response,13 but its utility predicting response has not been as thoroughly elucidated in mRCC as compared with metastatic melanoma and NSCLC.14 15 In this study, we found that TMB did not correlate with clinical response in patients with mRCC treated with ICI. These results are consistent with prior observations and is likely due to the lower TMB typically observed in mRCC.22 Clear cell RCC (ccRCC) tumors have, on average, only 1.1 mutations/Mb, consistent with our data.23 Additionally, TMB was not found to correlate with PD-L1 status concurring with a recently published study analyzing 9987 samples across multiple indications, including RCC, finding no relationship between PD-L1 expression and median TMB (r2=0.065, p>0.1).24 What is striking about mRCC, however, is that a high rate of insertion/deletions (indels) has been associated with increased neoantigen load and may be predictive of response to immunotherapy in mRCC.25 Additionally, the RCC tumor microenvironment has been shown to have high immune cytolytic activity, immune infiltration score and T-cell infiltration score compared with other tumor types.26 27 Overall, TMB alone may not correlate with indels, immune-cell infiltration or immune recognition in mRCC.
The utility of TMB as a biomarker of ICI response may be influenced by other factors, one being compromised antigen presentation capabilities at the tumor cell surface due to mutation or LOH of B2M and LOH-MHC. This has been shown to be a means of evading CD8+ T-cell destruction.16 17 Therefore, high TMB tumors may be dependent on their antigen presentation capabilities to respond to ICIs. In this cohort, two patients had high TMB, exhibiting vastly different responses to ICIs, where the PD patient had LOH-MHC (Pt. 6; figure 1), while the responder with wtMHC exhibited CR (Pt. 32). An additional patient with high TMB in this cohort was found to have wtMHC but did not respond to ICI treatment (Pt. 28). This, however, may be due to the patient being PD-L1-negative, suggesting low likelihood of benefit from ICI therapy. Within the cohort, most patients with LOH-MHC were PD. Overall, these results suggest that the wide range of responses to ICIs observed in mRCC may be due to an increased complexity of this indication and highlights the need for more comprehensive approaches, which includes assessing neoantigen presentation capability, rather than relying on single biomarkers like PD-L1 or TMB alone.
Lastly, although microsatellite instability (MSI-H) is a promising marker for predicting response to pembrolizumab, mRCC is rarely found to be MSI-H.28 Our data are consistent with the literature as all patients were MSS.
Genetic profiling
There is speculation that the presence of frequently mutated genes in mRCC may serve as potential biomarkers for immunotherapy response. In this cohort, the most mutated genes identified included VHL, PBRM1, SETD2 and BAP1, consistent with existing literature.29 Notably, PBRM1, SETD2 and BAP1 are located on chromosome 3p21 in close proximity to VHL at chromosome 3p25.30 However, none of these gene mutations was found to be significantly different between responders and non-responders.
VHL, the most commonly mutated gene in RCC, regulates the hypoxia response pathway.31 Given its association with RCC tumorigenesis, VHL mutational status has been investigated as a potential prognostic biomarker. One study showed improved outcomes in patients with stage III RCC with VHL alterations, but not stage IV, suggesting that VHL status may contribute to determining metastatic potential.32 However, other studies did not support these findings.33
PBRM1 has also been highlighted as a potential biomarker in RCC. PBRM1 codes for a subunit of the PBAF complex that suppresses the hypoxia transcriptional signature in VHL-loss RCC. Loss of function alterations in PBRM1 occur in 41% of ccRCC tumors.34 However, there are discrepancies in the literature regarding the predictive value of PBRM1 mutations. In one study, biallelic PBRM1 loss demonstrated improved OS (p=0.0074) and progression-free survival (p=0.029) compared with those without PBRM1 loss.35 In the Checkmate 025 trial, PBRM1 loss was associated with clinical benefit to nivolumab, although the presence of PBRM1 loss alone was not sufficient for responses.10 However, in a larger cohort of patients with mRCC, PBRM1 loss was not associated with improved OS (HR 1.37; 95% CI 0.79 to 2.4, p=0.265).36 Similarly, our results found that PBRM1 mutation did not predict immunotherapy responses.
SETD2 and BAP1 code for epigenetic tumor suppressors and mutation has been associated with worse cancer-specific survival thus playing a role in disease progression.30 However, neither have been demonstrated to be predictive biomarkers. Furthermore, despite finding a significant number of co-alterations, these did not correlate with clinical outcomes.
DNA damage response mutations
Tumors with high levels of clonal neoantigens have been shown to have improved response to ICIs and loss leading to ICI resistance.37 There is limited data, however, indicating that DNA damage response mutations alone correlate with improved response to ICIs in RCC. In a large cohort, 17% had mutated DNA damage repair genes and had significantly longer OS in the ICI cohort (HR 0.29, log rank p=0.04), but not in those receiving a tyrosine kinase inhibitor (HR 0.74, log rank p=0.44).38
Although a small cohort, the data presented here suggest an association between mutations in DNA damage response genes, in particular HRR genes, and response to ICI therapy in mRCC. Specific DNA damage mutations may yield different immunological consequences on the tumor microenvironment and affect ICI response, although these are not yet understood. Double-stranded breaks in DNA are typically repaired via homologous recombination and dysregulated HRR pathways may lead to genomic instability and neoantigen generation. These mutated surface proteins have been shown to activate inflammatory cytokines, generating further oxidative stress and DNA damage.39 Another theory involves DDR mutations and their direct involvement in immunity. The STING pathway is thought to be the primary innate immunity pathway for detecting tumors, driving T-cell priming against tumor-specific antigens.40 DDR mutations impair the STING pathway, potentially impeding host T-cell recognition of tumor cells. ICIs, which inhibit the tumor cell’s immune evasion capabilities, can enable host T cells to better recognize tumor cells for destruction.40 Therefore, tumors with a higher number of DNA damage response mutations may respond well to ICIs (online supplementary figure 2). Due to the small size of the cohort, it is unclear if this association is subject to a gene-dose effect, such that a greater number of DNA damage response mutations correlates to increasing response to ICIs. Although DC patients were found to have a significantly greater number of mutations in HRR genes (figure 6C), a greater variation of responses within the DC group is necessary to further elucidate a potential gene-dose effect.
Supplemental material
Overall, single biomarkers such as PD-L1 and TMB status did not correlate with ICI outcomes. However, the correlation of LOH-MHC with ICI resistance and correlation of HRR gene mutations with ICI outcomes in this cohort may be hypothesis generating for future studies. While this study did reveal multiple interesting associations, there are several limitations and remaining questions that require further inquiry. First, this analysis was based on archival tissue specimens and performed retrospectively; therefore, observations should be confirmed in a prospectively collected patient cohort.
A correlation between DNA damage response gene mutations and increased TMB was observed, aligning with previous reports in several indications that defects in HRR/DDR pathways may lead to increased TMB.41–44 It should be noted, however, that in this study, this association may in part be due to the fact that the mutations in these genes contribute to the TMB calculation. Therefore, it is challenging to specifically attribute increased TMB to HRR/DDR mutations or discern a causal relationship from these data alone. Given the biological implications of mutations in DNA damage response genes leading to neoantigen generation, patients who are concomitant with LOH-MHC might indicate a subset of non-responders to ICI treatment. Although this correlation was not observed in this study, only seven patient samples were identified as LOH-MHC and therefore this study is not sufficiently powered for this analysis, warranting future studies.
Despite TMB scores being relatively low in mRCC,45 46 patients do benefit from ICI therapy. Although TMB is merely a surrogate measure for neoantigen burden, the actual estimation remains difficult due to imperfect bioinformatics approaches, highly polymorphic MHC genes and an overall lack of understanding of neoantigen immunogenicity.47 48 Direct assessment of neoantigenic burden is beyond the scope of this study; however, given the clinical implications, further exploration would be beneficial.
Conclusions
This study associated clinical outcomes of patients with mRCC treated with ICI with tissue analyzed for several biomarkers including genetic correlates, TMB, MHC loss and PD-L1 expression. TMB alone, PD-L1 status alone and combination TMB with PD-L1 status did not correlate with ICI outcomes, which is consistent with prior observations. Interestingly, non-responders showed an increased incidence of LOH-MHC, warranting future investigations to determine if antigen presentation may serve as a predictor of ICI response in patients with mRCC. Finally, ICI responders had more frequent mutations in DNA damage response genes than non-responders, particularly within HRR genes. Ultimately, further research on the tumor microenvironment and engagement of RCC tumors with the immune system is necessary to better understand response and predict for ICI treatment outcomes.
References
Footnotes
Twitter @MattLabs831
Contributors MKL, JZ, TZ abstracted clinical data. RG interpreted imaging using RECIST. SMC prepared pathology slides. JJ, EFK, GC, JRW analyzed samples and performed statistical analysis. MKL, EFK, JJ, KG wrote the manuscript with support from TZ, DG, JKS. All authors read and approved the final manuscript. TZ conceived the study and was in charge of overall direction and planning.
Funding Personal Genome Diagnostics Inc. Duke Cancer Institute.
Competing interests TZ: research funding from Acerta, Novartis, Merrimack, AbbVie, Merck, Regeneron, Mirati Therapeutics, Janssen, AstraZeneca, Pfizer, OmniSeq and PGDx. Speakers bureau for Genentech Roche, Exelixis, Sanofi-Aventis and Genomic Health. Advisory board/consultant for Genentech/Roche, Merck, Exelixis, Sanofi-Aventis, Janssen, AstraZeneca, Pfizer, Amgen, BMS, Pharmacyclics, Bayer, Foundation Medicine and Seattle Genetics. Employee/stockholder of Capio Biosciences (spouse) JZ: consulting: Bayer, NGM Biopharmaceuticals.Travel assistance: UroTodayRG: consulting: Bayer, Philips, Bard. Speakers bureau: Bayer. DG: research funding: Acerta, Astellas, Bayer, BMS, Calithera. Exelixis, Janssen, Novartis, Pfizer, Sanofi, Seattle Genetics. Speakers bureau: Bayer, Exelixis. Advisory board/consultant: Astellas, AztraZeneca. Bayer, BMS, Exelixis, Flatiron. Janssen, Merck, Michael J Hennessey Associates, Modra, Myovant Sciences, Nektar, Physician Education Resource LLC, Pfizer, Sanofi, Vizuri Health, NCI. Editor: American Association for Cancer Research. Millennium Medical Publishing, Clinical Advances in Hematology and Oncology. Independent contractor: Axess Oncology, Honorarium: Bayer, EMD Serono, Exilixis, Ipsen, Michael J Hennessey Associates, Pfizer, Sanofi, UroGPO, UroToday. The remaining authors have no competing interests.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.