Article Text

Abstract
Background We previously reported that dendritic cell-based mRNA vaccination plus ipilimumab (TriMixDC-MEL IPI) results in an encouraging rate of tumor responses in patients with pretreated advanced melanoma. Here, we report the TriMixDC-MEL IPI-induced T-cell responses detected in the peripheral blood.
Methods Monocyte-derived dendritic cells electroporated with mRNA encoding CD70, CD40 ligand, and constitutively active TLR4 (TriMix) as well as the tumor-associated antigens tyrosinase, gp100, MAGE-A3, or MAGE-C2 were administered together with IPI for four cycles. For 18/39 patients, an additional vaccine was administered before the first IPI administration. We evaluated tumor-associated antigen specific T-cell responses in previously collected peripheral blood mononuclear cells, available from 15 patients.
Results Vaccine-induced enzyme-linked immunospot assay responses detected after in vitro T-cell stimulation were shown in 12/15 patients. Immune responses detected in patients with a complete or partial response were significantly stronger and broader, and exhibited a higher degree of multifunctionality compared with responses in patients with stable or progressive disease. CD8+ T-cell responses from patients with an ongoing clinical response, either elicited by TriMixDC-MEL IPI or on subsequent pembrolizumab treatment, exhibited the highest degree of multifunctionality.
Conclusions TriMixDC-MEL IPI treatment results in robust CD8+ T-cell responses in a meaningful portion of stage III or IV melanoma patients, and obviously in patients with a clinical response. The levels of polyfunctional and multiantigen T-cell responses measured in patients with a complete response, particularly in patients evidently cured after 5+ years of follow-up, may provide a benchmark for the level of immune stimulation needed to achieve a durable clinical remission.
Trial registration number NCT01302496.
- dendritic cells
- immunotherapy
- melanoma
- T-lymphocytes
- vaccination
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The immune tolerance seen in late stage cancer patients recapitulates the multiple failures that accumulated in the cancer-immunity cycle.1 Reinvigoration of a completely functional cancer immunity cycle with sufficient amplitude is believed to be necessary to eradicate advanced malignancies. Immune checkpoint pathways regulate the immune system and are crucial to maintain self-tolerance. Interference with these pathways has the potential to stimulate antitumor immune responses. The potency of the immune system to reject cancer on blockade of the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or programmed death (ligand) 1 (PD-(L)1) immune checkpoint molecules was first shown for anti-CTLA-4 antibodies and led to the approval of ipilimumab (IPI) for the treatment of advanced melanoma. Next, PD-(L)1 blockers demonstrated a higher level of activity across different solid and hematological malignancies.2 Blockade of other immune checkpoints such as T cell immunoreceptor with Ig and ITIM domains (TIGIT), T-cell immunoglobulin and mucin domain 3 (TIM-3), lymphocyte-activation gene 3 (LAG3) and natural killer group 2 member A (NKG2A) is currently under intensive investigation.3 4 Although impressive clinical responses can be observed on immune checkpoint blockade, these only occur in a fraction of patients. Insufficient immune activation is considered as a main reason for treatment failure.5 Thus, combining immune checkpoint blockade with tumor-associated antigen (TAA) specific T-cell stimulation could increase clinical response rates.
We previously demonstrated that autologous monocyte-derived dendritic cells (DCs) electroporated with mRNA encoding a mixture of three immune modulating molecules (TriMix), that is, a constitutively active TLR-4, CD40 ligand (CD40L) and CD70, have a superior T-cell stimulatory capacity.6 TriMixDCs coelectroporated with mRNA encoding melanoma-associated antigens fused to the human leukocyte antigen (HLA) class II targeting signal DC-lysosomal membrane protein (DC-LAMP) (TriMixDC-MEL) were shown to be safe and immunogenic and to result in durable tumor responses in 4/15 patients with pretreated advanced melanoma on combined intravenous and intradermal administration.7
To evaluate a potential added benefit of combining immune checkpoint blockers with TriMixDCs, 39 patients with pretreated advanced melanoma were enrolled to receive TriMixDC-MEL vaccines using mRNA coding for four TAAs (tyrosinase, gp100, MAGE-A3, and MAGE-C2) in combination with IPI (TriMixDC-MEL IPI) to overcome the immune tolerance. Results concerning primary and secondary objectives of the study showed that 6 months disease control rate was 51% with 8/39 (20.5%) patients obtaining a complete response (CR), 7/39 (17.9%) a partial response (PR), 6/39 (15.4%) a stable disease (SD), and 18/39 (46.2%) having progressive disease (PD). Grade 3 or 4 immune-related adverse event occurred in 36% of the patients. Grade 5 adverse events were not reported.8 We report here the long-term clinical outcome of responding patients after more than 5 years of follow-up and the vaccine-specific immune responses that were evaluated in 15/39 patients.
Methods
Patients
Patients with histologically confirmed, unresectable, American Joint Committee on Cancer stage III or IV melanoma with measurable disease, who experienced treatment failure with at least one prior line of systemic treatment, were eligible. Other key inclusion criteria included the following: age ≥18 years; WHO performance status of 0, 1, or 2; normal hematologic, liver, and renal function tests; and negative serologic tests for HIV, syphilis, hepatitis B, and hepatitis C. Exclusion criteria included prior treatment with an anti-CTLA-4 antibody, history of autoimmune disease, primary uveal melanoma, untreated or symptomatic CNS metastasis, and the need for permanent therapeutic anticoagulation.
TriMixDC-MEL production
Immature DCs were generated by culturing monocytes in the presence of 1% autologous plasma, 1000 U/mL of granulocyte-macrophage colony-stimulating factor, and 500 U/mL of IL-4. After leukapheresis, monocytes were enriched by plastic adherence. On day 6, DCs were harvested and coelectroporated with TriMix-mRNA and mRNA encoding MAGE-A3, MAGE-C2, tyrosinase, or gp100 linked to an HLA class II targeting signal (DC-LAMP). After electroporation, the four different TriMixDC-MEL cellular constituents (DCs expressing one of the four TAAs) were mixed at equal ratios and cryopreserved. DCs were thawed 2 to 3 hours before injection. An in-process quality control of the final product was carried out, as reported previously.8
Study design
TriMixDC-MEL IPI study was an open-label, single-arm, single-centre, two-stage phase II clinical trial. The primary objective of this study was to estimate the disease control rate at 6 months according to immune-related response criteria (irRC). Secondary objectives included safety, estimation of the overall response rate (ORR) by irRC, duration of response and estimation of the median progression-free survival (PFS) and median overall survival (OS).8
Treatment schedule
The treatment schedule was previously described8 and is depicted in figure 1.
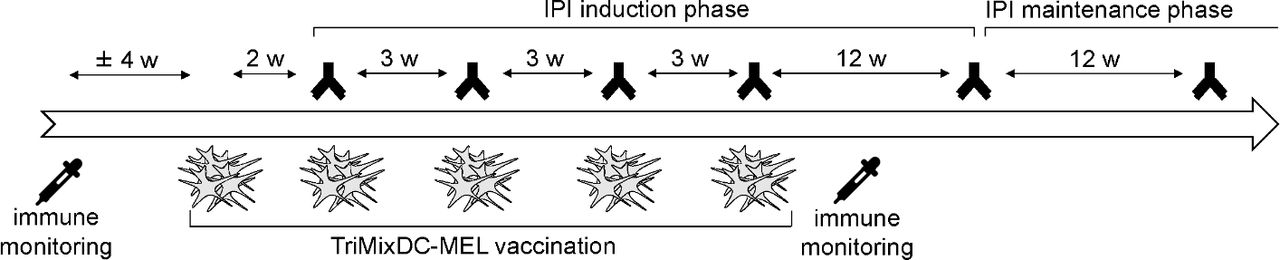
TriMixDC-MEL IPI treatment schedule TriMixDC-MEL vaccines were administered both intradermally (4 million cells) and intravenously (20 million cells) together with IPI every 3 weeks for a total of four administrations. For 18/39 patients, a first vaccine was performed 2 weeks before the first IPI administration. This first administration was omitted for the remaining 21 patients. Patients with SD, PR, or CR at week 24 were eligible to enter an IPI maintenance phase every 12 weeks. IPI was given at 10 mg/kg. CR, complete response; IPI, ipilimumab; PR, partial response; SD, stable disease; TriMixDC-MEL, dendritic cell -based mRNA vaccination plus ipilimumab.
Immune monitoring
Samples
Peripheral blood mononuclear cells (PBMC) were collected by leukapheresis (before treatment, “PreVac”) and by a buffy coat at week 12 (“PostVac”). PBMC were isolated using lymphoprep medium (Fresenius-Kabi) and frozen in freezing medium consisting of 88% autologous plasma, 10% dimethyl sulfoxide (DMSO) (WAK-chemie) and 2% glucose (Glucose Sterop). The freezing container was placed at −80°C and vials were transferred into the vapor phase of liquid nitrogen. On thawing, PBMCs were used as a source of T cells and B cells.
B-cell expansion
PBMC were thawed and resuspended in prewarmed Iscove’s Modified Dulbecco Medium (IMDM, Gibco) with 10% heat-inactivated human AB serum (Innovative Research, lot pretested for performance for B-cell expansions), penicillin-streptomycin, L-glutamine (both from Sigma), and insulin-transferrin-selenium (Thermo Fisher Scientific), recombinant human multimeric CD40L (Human CD40-ligand multimer kit, Miltenyi Biotec), recombinant human IL-4 (Miltenyi Biotec) and recombinant human IL-21 (ImmunoTools) (B-cell medium), supplemented with cyclosporine A (Novartis). On days 6–8, B cells were enriched using CD19 MicroBeads (Miltenyi Biotec) and resuspended in prewarmed B-cell medium. Afterwards, B cells were counted every 72–96 hours and resuspended in prewarmed B-cell medium. When B-cell numbers were sufficient to perform T-cell assays, B cells were frozen in Cryostor CS10 medium (StemCell Technologies) prior to their use as antigen-presenting cells (APCs) in T-cell assays.
B-cell electroporation
B cells were thawed, washed twice with prewarmed in an improved minimal essential medium (Opti-MEM), resuspended in 200 µL Opti-MEM medium containing 20 µg antigen-encoding mRNA or 5 µg enhanced green fluorescent protein (EGFP)-encoding mRNA and transferred to 4 mm electroporation cuvettes (cell projects). Following mRNA constructs were used: peTheRNAv2-EGFP, peTheRNAv2-tyrosinase-DC-LAMP, peTheRNAv2-gp100-DC-LAMP, peTheRNAv2-sig-MAGE-A3-DC-LAMP, peTheRNAv2-sig-MAGE-C2-DC-LAMP, pST1-CEF, and peTheRNAv2-Gag. The CEF mRNA used for B-cell transfection encodes 32 HLA class I-restricted epitopes of Cytomegalovirus (CMV), Epstein–Barr Virus (EBV) and Influenza Virus.9 Per condition, 3–8 million B cells were electroporated using the Gene Pulser Xcell Electroporation system (Bio-Rad). Afterwards, B cells were resuspended in T-cell medium (see below) and rested at 37°C, 5% CO2. The electroporation efficiency was 87.8% on average as measured by EGFP expression using flow cytometry 20–24 hour after electroporation.
PBMC thawing, resting, and T-cell isolation
PBMC were thawed the day before performing the ex vivo enzyme-linked immunospot assay (ELISPOT) and in vitro T-cell stimulation (IVS) (see below). PBMC were resuspended in IMDM supplemented with 5% heat-inactivated human AB serum (lot pretested for performance), penicillin-streptomycin, L-glutamine, and MEM non-essential amino acid solution (all from Sigma) (T-cell medium), supplemented with 25 U/mL recombinant human IL-2 (ImmunoTools) and 10 U/mL DNAse (Roche) and rested at 37°C, 5% CO2 in a humidified incubator. T cells were isolated using the Pan T-cell isolation kit (Miltenyi Biotec).
Ex vivo ELISPOT
IFN-γ ELISPOT kits with precoated plates were used (Diaclone). ELISPOT plates were washed with PBS (Lonza) and then blocked with T-cell medium containing 10% heat-inactivated human AB serum for 2 hours at 37°C, 5% CO2. Afterwards, the plates were washed with PBS. T cells were cocultured with autologous expanded B cells electroporated with TAA-encoding mRNA at a 3:1 ratio in a total volume of 200 uL per well in T-cell medium. Following negative controls were tested: (1) T-cell medium only, (2) T cells+T-cell medium, and (3) T cells+B cells electroporated with the HIV antigen Gag. Following positive controls were evaluated: (1) T cells+microbeads coated with anti-CD3 and anti-CD28 (Gibco) and (2) T cells+B cells electroporated with mRNA encoding CEF. In addition, reference T cells from a healthy donor with known specificity for viral epitopes were tested on each plate to evaluate overall ELISPOT performance. ELISPOT plates were incubated for 16–20 hours at 37°C, 5% CO2. Afterwards, ELISPOT plates were developed. First, cell suspensions were removed and the plates were washed with PBS containing Tween-20 (Sigma) (ELISPOT washing buffer). Biotinylated detection antibody was diluted in PBS containing bovine serum albumin (BSA) (ELISPOT assay diluent). The plates were incubated for 3 hours at 37°C. Then, ELISPOT plates were washed with ELISPOT washing buffer and streptavidin-alkaline-phosphatase diluted in ELISPOT assay diluent was added to the wells. The plates were incubated for 1 hour at 37°C, after which they were washed with ELISPOT washing buffer and with PBS. 5-Bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) substrate were added to the wells and the plates were incubated protected from light until spot development was visible in positive control wells. Finally, the wells were thoroughly washed with water and were dried for at least 20 hours at 4°C protected from light, after which spots were counted using an Immunospot S6 Ultimate Analyzer with accessory software (CTL Europe GmbH).
In vitro T-cell stimulation
IVS was performed in 48-well microtiter plates. On day 1, purified T cells were stimulated with freshly electroporated autologous B cells at a 3:1 ratio. Plates were incubated in a humidified incubator at 37°C, 5% CO2. On day 2, half of the medium was replaced by fresh T-cell medium containing recombinant human IL-2 (ImmunoTools) and recombinant human IL-15 (PeproTech). On days 6–8, recombinant human IL-2 was added. On days 11–13, T cells were counted and tested in ELISPOT and intracellular cytokine staining (ICS).
ELISPOT after in vitro T-cell stimulation (IVS ELISPOT)
After IVS, T cells were cocultured with freshly electroporated autologous B cells at a 3:1 ratio in an IFN-γ ELISPOT plate. Positive and negative controls, reference sample, and ELISPOT incubation and development were identical to ex vivo ELISPOT (see above).
ICS after in vitro T-cell stimulation (IVS ICS)
After IVS, T cells were cocultured with autologous B cells freshly electroporated with TAA-encoding mRNA at a 3:1 ratio in a 48-well microtiter plate in T-cell medium supplemented with Golgi Plug protein transport inhibitor (Beckton Dickinson). Following reagents or cells were added to the T cells for the negative controls: (1) T-cell medium only and (2) B cells electroporated with the HIV antigen Gag; for the positive controls: (1) microbeads coated with anti-CD3 and anti-CD28 and (2) B cells electroporated with mRNA encoding CEF. In addition, reference T cells were tested on each plate to evaluate overall ICS performance. Plates were incubated during 16–20 hour in a humidified incubator at 37°C, 5% CO2. Next, T cells were harvested, washed with PBS, and stained with the Zombie Yellow Fixable Viability Kit (BioLegend) for 25 min at room temperature. Subsequently, the cells were washed with PBS and incubated for 25 min at 4°C with antibodies for membrane marker staining prediluted in PBS containing BSA and sodium azide (flow cytometry buffer): anti-CD4 PerCP/Cy5.5, anti-CD8 APC-H7 (both from Beckton Dickinson), anti-CD3 PE/Dazzle 594, anti-CD14 BV785, and anti-CD19 BV785 (all from Biolegend). Afterwards, the cells were washed with flow cytometry buffer and Cytofix/Cytoperm buffer (Fixation/Permeabilization solution kit, Beckton Dickinson) was added to each sample followed by an incubation step of 25 min at 4°C. Then, the cells were washed with Perm/Wash buffer (Fixation/Permeabilization solution kit) and incubated with antibodies for intracellular staining (anti-IFN-γ PE, anti-TNF-α FITC, and anti-IL-2 APC, all from Biolegend), prediluted in Perm/Wash buffer, for 25 min at 4°C. Finally, cells were washed with Perm/Wash buffer, resuspended in flow cytometry buffer, and acquired using a Cytoflex flow cytometer (Beckman Coulter), and analyzed using the CytExpert V.2.0 software (Beckman Coulter). Compensation was calculated using VersaComp Antibody Capture Kit following manufacturer’s instructions (Beckman Coulter) or using single stained cells in case of Zombie Yellow. Gating strategy is shown in online supplementary figure 1.
Supplemental material
T-cell receptor (TCR) repertoire sequencing after in vitro T-cell stimulation (IVS–TCRseq)
After IVS, T cells that were not used for IVS ELISPOT or IVS ICS were collected at day ±14 and RNA was extracted. A minimum of 200 ng of RNA was engaged in a reverse transcription with Superscript III reverse transcriptase (Invitrogen). A library of TCRβ (TCR, T-cell receptor) rearrangements was built from the obtained cDNA after an amplification with a set of primers specific for TRBV and TRBC genes. After addition of required adaptors, library was sequenced in an Illumina platform. The library setup was based on a molecular barcoding or “digital sequencing” approach. This one consists to tag each initial TCRβ molecules with a unique genetic barcode (Unique Molecular Identifier, UMI) before library amplification. UMIs allowed to compile reads derived from the same initial molecule and to correct for amplification biases or sequence errors introduced during the sequencing process. In addition, digital TCRseq provide an absolute quantification of molecules sequenced. The TCRβ repertoire was evaluated for T cells stimulated with TAAs tyrosinase, gp100, MAGE-A3 and MAGE-C2 and with HIV antigen Gag as a negative control. Enrichment of TCRβ rearrangements in the culture well stimulated with one of the TAAs compared with the negative control well allowed to identify T cells clonotypes specifically amplified by the TAAs stimulation.
Regulatory T-cell (Treg) characterization
PBMCs were thawed, washed with PBS, and stained with the Zombie Yellow Fixable Viability Kit for 25 min at room temperature. Subsequently, the cells were washed with PBS and incubated for 25 min at 4°C with antibodies for membrane marker staining prediluted in flow cytometry buffer: anti-CD3 PE/Dazzle 594, anti-CD4 FITC, anti-CD45RA PE/Cy7, anti-CD27 Brilliant Violet 421, anti-inducible T-cell costimulator (ICOS) Brilliant Violet 421, anti-CD127 Brilliant Violet 510, anti-C-C chemokine receptor type 7 (CCR7) PE/Cy7, anti-HLA-DR PerCP, anti-CD62L PerCP/Cyanine 5.5 (all from Biolegend), and anti-CD25 PE (Miltenyi Biotec). Afterwards, the cells were washed with flow cytometry buffer and fixation/permeabilization reagent (Foxp3/transcription factor buffer set, eBioscience) was added to each sample followed by an incubation step of 25 min at 4°C. Then, the cells were washed with permeabilization buffer (Foxp3/transcription factor buffer set) and incubated with anti-Foxp3 APC (eBioscience), prediluted in permeabilization buffer, for 25 min at 4°C. Finally, cells were washed with permeabilization buffer and resuspended in flow cytometry buffer. Acquisition and compensation was performed as described for ICS.
Data analysis and criteria for response
PFS and OS were estimated by means of Kaplan-Meier statistics using IBM SPSS software V.22.0. Immune responses were analyzed using GraphPad Prism software V.7.03.
Acceptance criteria for the immune assays were as following: (1) viability of PBMC ≥80% on thawing; (2) B-cell electroporation efficiency ≥50%; (3) ELISPOT analyzer/flow cytometer qualified prior to acquisition; (4) ≥15,000 viable CD14− CD19− CD3+ T cells acquired for the ICS; (5) ELISPOT tests performed in ≥2 replicate wells per condition; (6) number of ELISPOT spots measured in T-cell medium only wells ≤10 spots per well; (7) number of ELISPOT spots/million T cells measured on stimulation with anti-CD3 and anti-CD28 coated microbeads ≥1000 or too numerous to count.
Positive vaccine-specific immune reactivity was determined according to a predefined criteria set. For ELISPOT, a sample was considered to show reactivity against a TAA when (1) ≥5 spots were measured in all replicate wells and (2) spot number was ≥spot number measured for the negative control (T cells+B cells electroporated with Gag encoding mRNA) plus a threefold of its SD. For ICS, responses were considered positive when (1) ≥0.23% of CD4+ or CD8+ T cells stained positive for the tested cytokine and (2) percentage of cytokine-positive CD4+ or CD8+ T cells was ≥threefold of the negative control (T cells+B cells electroporated with Gag encoding mRNA).
Patients were considered to have a vaccine response if they showed reactivity to a TAA as described above and (1) when the PreVac sample showed positive reactivity for the TAA: if the T-cell response measured PostVac, after subtraction of its negative control, was ≥ twofold of the T-cell response measured PreVac, after subtraction of its negative control or (2) when the PreVac sample showed no reactivity for the TAA: if the T-cell response measured PostVac, after subtraction of its negative control, was ≥threefold of the values measured PreVac, after subtraction of its negative control.
Concerning TCR sequencing, a clonotype was considered as specifically amplified by the TAAs stimulation if its frequency in the TAA stimulated culture well was ≥200 x higher compare to the frequency in the negative control well stimulated with HIV antigen Gag. Based on previous assessments, it was determined that this threshold was beyond 6 SD from the average clonotypes frequency differences measured by digital TCR sequencing on replicates from a same blood sample.
Results
Long-term clinical outcome of responding patients
After more than 5 years, TriMixDC-MEL IPI treatment resulted in an OS of 28% and a PFS of 18% (figure 2). After a median follow-up of 390 weeks, 11 out of 39 patients are alive of whom seven remain free-from progression and have remained in a complete remission following treatment with TriMIxDC-MEL IPI (range of follow-up for these seven patients is 314 to 405 weeks) (figure 2). Two out of the four surviving patients who progressed following TriMixDC-MEL IPI are currently disease-free and off-therapy, respectively, following treatment with pembrolizumab and dabrafenib plus trametinib followed by a metastasectomy, one additional patients is currently disease-free and continues on pembrolizumab treatment. A fourth patient has progressed on multiple lines of additional therapy (including BRAF-inhibitor/MEK-inhibitor and anti-PD-1 therapy).

Durable clinical responses on TriMixDC-MEL IPI treatment Kaplan-Meier estimates of PFS and OS. IPI, ipilimumab; OS, overall survival; PFS, progression-free survival; TriMixDC-MEL, dendritic cell -based mRNA vaccination plus ipilimumab.
TriMix-DC-MEL IPI treatment results in strong vaccine-specific T-cell responses
Immune monitoring was performed for 15 patients who had sufficient PreVac and PostVac PBMC available (four with CR, four with PR, two with SD, and five with PD). Twelve out of 15 patients (80%) were considered as vaccine responders with IVS ELISPOT. T-cell responses were detected for all of the antigens; in total, 31 vaccine specific T-cell responses were measured of which 7 (22.6%) were detectable in PreVac samples (figure 3A), but much weaker compared with the PostVac responses. A mean fold-increase of 8.5 (range: 1.4–17.5) of spots was measured in PostVac versus PreVac samples (figure 3B). Ten out of 15 patients (66.7%) showed IVS ELISPOT responses directed against at least two antigens. Remarkably, three of these patients showed responses against all four vaccine antigens.
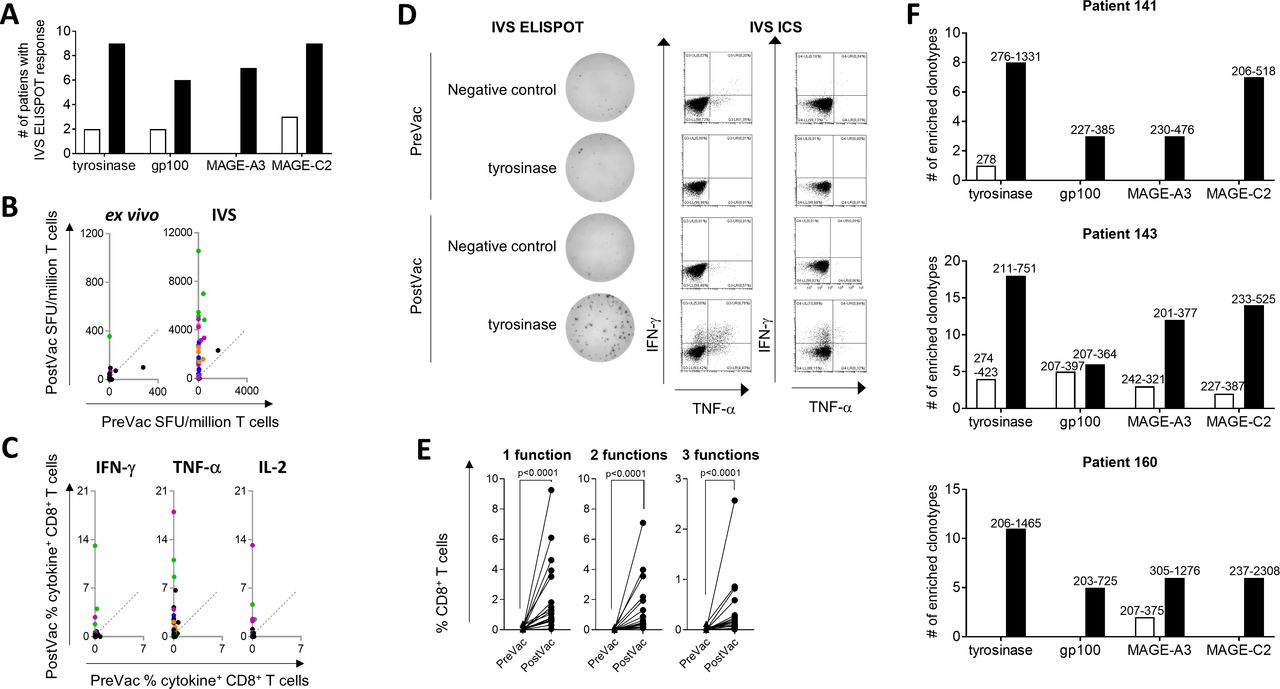
TriMixDC-MEL IPI treatment results in strong vaccine-specific T-cell responses. (A) Bars show the numbers of patients for whom an IVS ELISPOT response was detected PreVac (white bars) or PostVac (black bars). (B and C) TriMixDC-MEL IPI treatment mediated increase of TAA-specific IVS ELISPOT (B) and IVS ICS (C) response height. Each dot represents the PreVac and PostVac response measured for one patient for one TAA. T-cell responses that were considered as vaccine-specific immune responses are indicated in color (green: tyrosinase; orange: gp100; blue: MAGE-A3; purple: MAGE-C2). The line indicates an equal PreVac and PostVac response. (D) Example of a tyrosinase-specific response as measured with IVS ELISPOT and IVS ICS. (E) Detection of multifunctional CD8+ T-cell responses on TriMixDC-MEL IPI treatment. Each dot represents an IVS ICS response that was considered to be vaccine-specific. The percentage of CD8+ T cells producing either one, two, or three cytokines is shown on the graphs (triangles: PreVac, dots: PostVac). A paired Wilcoxon test was performed to determine statistical significance. (F) Breadth of vaccine responses elicited by TriMixDC-MEL IPI in patients showing an immune response against all vaccine TAA. The bar graphs show the number of enriched clonotypes detected per antigen at the PreVac (white bars) and PostVac (black bars) time point. Clonotypes were considered to be enriched if their frequency in a TAA-stimulated well was >200 fold of their frequency in a Gag (negative control)-stimulated well. The range (min–max) of the enrichment as compared with the Gag-stimulated well is shown above the bars. ICS, intracellular cytokine staining; IVS, in vitro T-cell stimulation; IPI, ipilimumab; ELISPOT, enzyme-linked immunospot assay; SFU, spot forming units; TAA, tumor-associated antigen; TNF, tumor necrosis factor; TriMixDC-MEL, dendritic cell -based mRNA vaccination plus ipilimumab.
The majority of the vaccine responses shown with ELISPOT were confirmed with IVS ICS (figure 3, B-C). IVS ICS revealed that the T-cell responses were mainly mediated by CD8+ T cells (figure 3, C and online supplementary figure 2): 21 of the 35 TAA-specific immune responses that were detected PreVac or PostVac with IVS ELISPOT could be confirmed with IVS ICS; 19 were only CD8+ T-cell mediated, one was only CD4+ T-cell mediated and one was both CD8+ and CD4+ T-cell mediated. The CD8+ T-cell vaccine responses were frequently characterized by a multifunctional profile (figure 3, D and E; online supplementary figure 3): next to IFN-γ, vaccine-specific T cells were also producing TNF-α and IL-2. Overall, the number of IFN-γ spots correlated with the percentage of IFN-γ+ CD3+ T cells measured on IVS by, respectively, ELISPOT and ICS. However, ELISPOT presented to be the more sensitive assay as 16/56 (28.6%) IVS ELISPOT responses were not detected with IVS ICS. As shown in online supplementary figure 4, weaker ELISPOT responses were mostly below the ICS detection limit.
Samples from the three patients showing an IVS ELISPOT PostVac response against all four TAAs were subjected to TCR sequencing after IVS. The results showed a high abundance of multiple TAA-specific T-cell clones in all three patients (figure 3F). The most immunogenic TAA of the vaccine antigens used in this study was tyrosinase with a high number of patients responding to the antigen (tyrosinase: nine patients; gp100: five patients; MAGE-A3: seven patients, and MAGE-C2: nine patients showing vaccine specific IVS ELISPOT response) and with strongest PostVac immune responses for tyrosinase although no significant differences were found with the other TAA (figure 3, B and C and online supplementary figure 5). The TriMixDC-MEL IPI treatment regimen did not affect the immune responses measured for the viral recall antigens encoded by the CEF mRNA (online supplementary figure 6), confirming that the detected immune responses were highly TAA-specific.
Next to the assays performed on in vitro stimulated T cells, ex vivo ELISPOT assays were performed. As expected, the number of responses detected with this assay was much lower compared with the IVS ELISPOT results; 2/14 patients showed a vaccine-specific ex vivo ELISPOT response; only 3/38 (7.9%) of the PreVac or PostVac tumor antigen responses measured with IVS ELISPOT were also detectable with ex vivo ELISPOT (one tyrosinase, one gp100, and one MAGE-C2 response) (figure 3B).
TriMix-DC-MEL IPI treatment results in higher numbers of peripheral blood CD62Lhigh Tregs
As an increase in the frequency of Tregs in the peripheral blood was previously shown on TriMixDC-MEL IPI treatment,8 we enumerated and characterized Tregs in 12 patients based on a CD3+ CD4+ CD127low CD25high Foxp3high expression profile. We could confirm that in the PostVac samples, the Treg frequency was significantly higher compared with the PreVac sample (figure 4A). Furthermore, by analyzing the phenotype of the Tregs, we found significantly higher percentages of Tregs expressing CD62L in the PostVac compared with the PreVac samples while the treatment did not affect CD45RA, HLA-DR, CD27, ICOS, or CCR7 expression by Tregs (figure 4B).

TriMix-DC-MEL IPI treatment results in higher numbers of peripheral blood CD62Lhigh Tregs. (A) Percentage of Tregs within CD4+ T-cell population of peripheral blood. Each data point represents the Treg frequency in one patient. Data were analyzed by a Wilcoxon matched-pairs signed rank test. (B) Left: percentage of CD45RA+, HLA-DR+, CD27+, CCR7+, CD62L+, and ICOS+ cells within Treg population of peripheral blood. Each data point shows the PreVac (triangles) or PostVac (dots) result. Results were analyzed with a two-way ANOVA with Sidak’s multiple comparison test. Right: example of CD62L expression profile of Tregs from PreVac (black histogram) and PostVac (red histogram) samples from patient 141. The filled grey histogram shows the negative control not stained with anti-CD62L. ANOVA, analysis of variance; IPI, ipilimumab; TriMixDC-MEL, dendritic cell -based mRNA vaccination plus ipilimumab.
TriMixDC-MEL IPI-mediated immune responses correlate with clinical responses
Patients with CR or PR showed significantly higher numbers of T cells producing cytokines in a TAA-specific fashion compared with patients with SD or PD (figure 5, A and C). In addition, all patients with CR or PR demonstrated an IVS ELISPOT vaccine response for at least two out of the four vaccine TAA (figure 5B). The quality of the vaccine-specific T-cell responses measured in patients with CR or PR also differed from the responses measured in patients with SD or PD as they showed a higher degree of multifunctionality with higher percentages of T cells producing ≥2 cytokines. Patients who are currently in complete remission exhibited TAA-specific CD8+ T-cell responses with a particularly high degree of multifunctionality (figure 5, C–F), which significantly correlates with a prolonged OS (figure 5, E and F). There was a trend towards a higher degree of multifunctionality exhibited by CD8+ T cells from patients with the longest PFS, but this trend was not significant (online supplementary figure 7). The height of the CEF-specific immune responses nor their degree of multifunctionality showed significant differences depending on clinical response (online supplementary figure 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation between immune responses and clinical responses towards TriMixDC-MEL IPI treatment. (A) Each symbol represents an IVS ELISPOT response measured PreVac (triangles) or PostVac (dots) for one patient for one TAA. The bars indicate the mean. A two-way ANOVA with Sidak’s multiple comparisons test was performed to determine statistical significance. (B) The graph indicates the number of TAA for which a vaccine-specific immune response was detected by IVS ELISPOT. Each dot represents one patient. (C) Multifunctional profile of vaccine-specific IVS ICS responses. Each bar shows the percentage of CD8+ T cells producing either one, two or three of the tested cytokines in a TAA-specific fashion on vaccination. (D) The graph shows the percentage of the total CD8+ T-cell IVS ICS response characterized by a multifunctional profile (≥2 cytokines). Each dot represents one vaccine-specific IVS ICS response. The bars indicate the median. A Mann-Whitney test was performed to determine statistical significance. (E and F) The percentages of the total CD8+ T-cell IVS ICS response characterized by a multifunctional profile (E: at least two cytokines, F: three cytokines). Each dot represents one vaccine-specific IVS ICS response. A linear regression was performed to analyze the data. Panels A–F: Red symbols indicate immune responses of patients that showed a CR to the TriMixDC-MEL IPI treatment which is still ongoing after >314 weeks. Blue symbols indicate immune responses of patients having experienced PD after, respectively, 6.77 and 20.45 months but that afterwards obtained CR to pembrolizumab treatment, which is currently still ongoing. Orange symbols indicate patients with a mixed response (complete regression of some of their metastases while others progressed). ANOVA, analysis of variance; CR, complete response; ICS, intracellular cytokine staining; IPI, ipilimumab; IVS, in vitro T-cell stimulation; ELISPOT, enzyme-linked immunospot assay; OS, overall survival; PD, progressive disease; PR, partial response; SFU, spot forming units; SD, stable disease; TAA, tumor-associated antigen; TriMixDC-MEL, dendritic cell -based mRNA vaccination plus ipilimumab.
Discussion
Between May 2011 and November 2013, 39 patients with pretreated advanced melanoma were enrolled in the TriMixDC-MEL IPI study. The vaccination with TAA mRNA electroporated DCs plus IPI resulted in an encouraging 6-month ORR of 38%.8 Long-term follow-up after more than 5 years following the initiation of TriMixDC-MEL IPI indicates that 7/39 patients, who all obtained a CR, are still disease-free, that is, apparently cured. Retrospectively, analyzing the correlation between immune cell samples from 15 patients and clinical outcome, we show that TriMixDC-MEL IPI elicited strong TAA-specific immune responses. In addition, the immune and clinical responses correlated very well. All patients that were cured had multiantigen T-cell responses with mostly high numbers of multifunctional CD8+ T cells specific to the TAAs included in the vaccine.
The immunogenicity of DC vaccines has been established in most clinical trials; a meta-analysis of DC vaccination for prostate cancer and renal cancer has shown that DC vaccines have induced tumor-specific immune responses in, respectively, 77% and 61% of the vaccinated patients.10 However, the clinical efficacy of DC vaccines remains mostly unsubstantiated. We now show that treatment of advanced melanoma patients with TriMixDC-MEL IPI elicits objective long-term clinical responses, with an OS of 28% after 5 years of follow-up which is substantially higher to survival rates observed on IPI treatment alone.11 12 Apart from the TriMixDC-MEL IPI study, the number of trials investigating combinations of checkpoint blockade agents with DC vaccination remains very limited. A first phase I trial was performed by Ribas et al, who investigated vaccination with immature DCs pulsed with a MART-1 peptide administered to patients with advanced melanoma with a dose escalation of the CTLA-4 blocker tremelimumab. On treatment, 4/16 (25%) patients had an objective tumor response of which two PRs and two CRs. In this study, ex vivo ELISPOT and HLA tetramer assay results did not differ between patients with or without a tumor response.13 A smaller phase I study investigated vaccination with neoantigen-loaded DCs, matured through culture with CD40L-expressing cells, IFN-γ, poly I:C and R848, preceded by IPI treatment in three stage III melanoma patients. This treatment promoted a diverse antigen-specific TCR repertoire,14 which is in line with our results. A recent study investigated the combination of adoptively transferred NY-ESO-1 transgenic T cells administered with a DC vaccine and IPI showing no apparent clinical benefit of the addition of IPI.15 When the TriMixDC-MEL IPI study was initiated, IPI was the only approved immune checkpoint inhibitor available for malignant melanoma. In the meantime, approved monoclonal antibodies against PD-1 have demonstrated their superiority over IPI in terms of efficacy and safety in patients with metastatic melanoma.16 17 Therefore, more studies are now focusing on the combination of DC vaccines with PD-1 blocking antibodies, as reviewed in.18 To our knowledge, none of these studies have published results yet. Interestingly, a recent publication showing that expanded T-cell clones detected on anti-PD-1 treatment are derived from clones that were absent in the tumor before the treatment, further supporting the combination of PD-1 blockade and active vaccination.19
Next to the CD8+ T-cell responses, we also monitored TAA-specific CD4+ effector T-cell responses. We detected weak CD4+ T-cell responses in only two patients, although the TAA-encoding mRNA used for the vaccine preparation encodes the HLA class II targeting sequence DC-LAMP. TriMixDC-MEL vaccination has previously shown its capability to stimulate robust TAA-specific CD4+ T-cell responses detectable in delayed type hypersensitivity (DTH) biopsies.20 21 One reason for the lack of robust TAA-specific CD4+ T-cell responses measured in the current study could be that these migrated to the tumor site at the time of the PostVac blood sampling since CTLA-4 blockade could stimulate melanoma-specific T-cell motility.22 Unfortunately, no patient samples were available at time points earlier after vaccination nor DTH biopsies to further elucidate kinetics of TAA-specific CD4+ T cells. In addition to our studies on TAA-specific CD4+ effector T cells, we investigated the frequency and phenotype of Tregs in 12 patients showing significantly higher frequencies of Tregs after treatment, which is in accordance with previously published results.8 Furthermore, PostVac Tregs were characterized by a higher expression of CD62L, which is associated with a high Treg proliferative and suppressive potential.23 Overall, the increased number of Tregs and the higher number of CD62Lhigh Tregs in the peripheral blood on TriMixDC-MEL IPI treatment could be caused by the increased CD8+ T-cell mediated IL-2 production measured after vaccination. It has indeed been shown that IL-2 is particularly required to maintain CD62Lhigh Tregs.24 However, as it has previously been shown in mice that the number of splenic Tregs increases on CTLA-4 blockade while their frequency in the tumor microenvironment decreases, it is possible that the increased number of Tregs in the peripheral blood on TriMixDC-MEL IPI treatment does not reflect their number in the tumor. Although the significant increase of functional CD8+ T-cell responses at the PostVac compared with the PreVac timepoint argues against strong Treg-mediated immune suppression, we cannot exclude that the induction of TAA-specific T cells might have been dampened by Tregs. Unfortunately, we were not able to investigate the suppressive function of the Tregs, nor their antigen specificity in this study. More detailed studies with more samples could provide insight as to whether the efficacy of DC-based vaccination and/or anti-CTLA-4-based treatment regimens could be enhanced through combination with therapeutic that abrogates Treg-mediated immune suppression, as previously suggested.25
The initial report on the TriMixDC-MEL IPI study included an ex vivo peptide-HLA multimer-based assay to measure TAA-specific immune responses towards the vaccine.8 This assay showed vaccine-specific immune responses only in 1/10 tested patients. Similarly, only ex vivo immune monitoring assays were performed in the study by Ribas et al and immune responses were comparable in the group of clinical responders versus non-responders.13 In our current study, we also detect only low numbers of ex vivo ELISPOT responses as opposed to the strong responses we detected with ELISPOT and ICS after a 10-day IVS. The discrepancy between the results of the ex vivo and IVS assays is probably both reflecting a difference in sensitivity and these assays are also measuring different types of T-cell responses. The strong responses detected after IVS suggest that the treatment regimen mainly stimulated central memory rather than effector T cells,26 27 which may result in superior, durable antitumor immunity.26 28 29 The fact that the results of the IVS assays show a strong correlation with the clinical results demonstrates their value. These observations underline the importance of detailed immune monitoring studies, preferably based on more than one type of assay.
This study correlates a multiparameter immunological assessment with a highly robust endpoint of durable complete remission of metastatic melanoma. Correlations with transient tumor responses and even more progression-free and OS can be more easily biased. The durable complete remissions leave no doubt about the causal relation with the experimental therapy. The study is however limited mainly by its relatively small sample size. A total of 39 patients was included in the TriMixDC-MEL IPI study; 8/39 (20.5%) obtained CR, 7/39 (17.9%) obtained PR, 6/39 (15.4%) obtained SD, and 18/39 patients (46.2%) showed PD.8 From 15 patients, we could perform the immune analyses: 4/15 (26.7%) had CR, 4/15 (26.7%) had PR; 2/15 (13.3%) had SD, and 5/15 (33.3) had PD. Despite the somewhat higher proportion of patients with CR of PR and lower proportion with SD or PD among the patients analyzed in the current study, overall this is a relatively accurate representation of the whole study population. Another weakness is that we have no data available to show the impact of the TriMixDC-MEL vaccine on the one hand and IPI on the other hand on the TAA-specific T-cell responses. The strength of the immune responses provoked by the TriMixDC-MEL IPI treatment and their correlation with clinical outcome is, as far as we are aware, superior compared with results from other TAA-targeting vaccines: 8/8 patients with PR/CR show a response directed against at least two of the vaccine TAA, and several patients with objective clinical responses exhibit highly multifunctional T cells. The impressive increase in the magnitude of the CD8+ T-cell responses on TriMixDC-MEL IPI treatment is most probably caused by the TriMixDC-MEL vaccine and supported by the IPI treatment as (1) it was previously shown in several studies that anti-CTLA-4 treatment alone does not alter the magnitude of TAA-specific CD8+ T-cell responses in patients with advanced melanoma;30 31 (2) anti-CTLA-4 dosing had no effect on expansion of TAA-specific T cells in the study performed by Ribas et al; 13 and (3) the magnitude of CEF-specific T-cell responses remained unaltered after treatment, which is in line with previous observations.30 However, some studies suggested that CTLA-4 blockade could enhance NY-ESO-1-specific T-cell responses, mainly in patients obtaining a durable clinical response or SD32 33 and it has been shown that IPI treatment alone is capable to broaden TAA-specific CD8+ T-cell responses.30 Thus, it would be interesting to also monitor T cells with specificity for TAAs not included in the vaccine formulation in future similar studies.
Since the initiation of the TriMixDC-MEL IPI study, the cancer immunotherapy field has evolved at a fast rate; as mentioned above, next to IPI, blockers of the PD-1/PD-L1 pathway have been approved. As CTLA-4 and PD-1 pathways are working on, respectively, the priming and the effector phase of the immune response, which allows a synergy between blockers of these pathways, combinations of anti-CTLA-4 and anti-PD-1 were tested and were shown to be successful in melanoma with OS rates in the range of 50%–60% after 5 years of follow-up in previously untreated advanced melanoma.12 However, this combination treatment is associated with severe adverse events in up to 60% of the patients.34 Thus, a combination of TriMixDC-MEL with a blocker of the PD-1/PD-L1 pathway would be very attractive to evaluate given the favorable safety profile of DC-based vaccines and since blockade of the PD-1 pathway was shown to be safer and more potent compared with CTLA-4 blockade.
Conclusion
TriMixDC-MEL IPI results in highly functional TAA-specific CD8+ T-cell responses, mainly in patients obtaining durable clinical responses. These results show the power of TAA-specific vaccination in combination with immune checkpoint blockade to break cancer immune tolerance and to give long-term clinical response.
Acknowledgments
The authors would like to thank Peggy van den Zegel, Kenneth Verhaegen, Sandy Haussy, and Manuel Constant for excellent technical assistance, Dorien Autaerts and Carlo Heirman for preparing the mRNA, Bertil Lindmark and Marina Cools for critically reviewing the manuscript.
References
Footnotes
Presented at Parts of this work were presented at the CIMT Annual Meeting 2019.
Contributors Conception and design: BDK, SC, BN, and KT. Development of methodology: BDK, SC, JCa, IB, and JCo. Acquisition of data (acquired and managed patients, provided facilities, etc): BDK, SC, JCa, IB, JCo, SW, BN, and KT. Analysis and interpretation of data (eg, statistical analysis, biostatistics, computational analysis): BDK, SC, JCa, IB, SW, and BN. Writing, review and/or revision of the manuscript: BDK, SC, JCa, IB, JCo, SW, BN, and KT. Administrative, technical, or material support (ie, reporting or organizing data, construct databases): BDK, SC, JCa, IB, SW, and BN. Study supervision: BDK, BN, and KT.
Funding This study was supported by a TBM grant awarded by Agentschap voor Innovatie door Wetenschap en Technologie (IWT), Flanders, Belgium, to Bart Neyns and Kris Thielemans.
Competing interests The use of dendritic cells electroporated with tumor antigen mRNA and TriMix is topic of a patent (W2009/034172) on which KT is filed as inventor. This patent has been licensed to eTheRNA immunotherapies. BDK and SC are respectively current and former employees of eTheRNA immunotherapies. BN has received financial compensation from Bristol-Myers Squibb for public speaking and participation in advisory board meetings.
Patient consent for publication All patients provided signed informed consent.
Ethics approval This trial was approved by the institutional ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.