Article Text

Abstract
Background Patients with advanced rare cancers have poor prognosis and few treatment options. As immunotherapy is effective across multiple cancer types, we aimed to assess pembrolizumab (programmed cell death 1 (PD-1) inhibitor) in patients with advanced rare cancers.
Methods In this open-label, phase 2 trial, patients with advanced rare cancers whose tumors had progressed on standard therapies, if available, within the previous 6 months were enrolled in nine tumor-specific cohorts and a 10th cohort for other rare histologies. Pembrolizumab 200 mg was administered intravenously every 21 days. The primary endpoint was non-progression rate (NPR) at 27 weeks; secondary endpoints were safety and tolerability, objective response rate (ORR), and clinical benefit rate (CBR).
Results A total of 127 patients treated between August 15, 2016 and July 27, 2018 were included in this analysis. At the time of data cut-off, the NPR at 27 weeks was 28% (95% CI, 19% to 37%). A confirmed objective response (OR) was seen in 15 of 110 (14%) evaluable patients (complete response in one and partial response in 14). CBR, defined as the percentage of patients with an OR or stable disease ≥4 months, was 38% (n=42). Treatment was ongoing in 11 of 15 patients with OR at last follow-up. In the cohort with squamous cell carcinoma (SCC) of the skin, the NPR at 27 weeks was 36%, ORR 31%, and CBR 38%. In patients with adrenocortical carcinoma (ACC), NPR at 27 weeks was 31%, ORR 15%, and CBR 54%. In the patients with carcinoma of unknown primary (CUP), NPR at 27 weeks was 33%, ORR 23%, and CBR 54%. In the paraganglioma–pheochromocytoma cohort, NPR at 27 weeks was 43%, ORR 0%, and CBR 75%. Treatment-related adverse events (TRAEs) occurred in 66 of 127 (52%) patients, and 12 (9%) had grade ≥3 TRAEs. The most common TRAEs were fatigue (n=25) and rash (n=17). There were six deaths, all of which were unrelated to the study drug.
Conclusions The favorable toxicity profile and antitumor activity seen in patients with SCC of skin, ACC, CUP, and paraganglioma–pheochromocytoma supports further evaluation of pembrolizumab in this patient population.
Trial registration number NCT02721732
- oncology
- tumours
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Rare cancers pose many challenges. In the absence of a universal definition,1 the American Cancer Society has defined rare cancers as those with an incidence of fewer than six cases per 100,000 people per year.2 3 Although they are individually uncommon, rare cancers are a major public health problem, as they collectively accounted for 13% of all new cancer diagnoses and 25% of all cancer-related deaths in adults in 2017 in the USA.3 The prognosis for patients with advanced rare cancer is poor; the 5-year survival rate for these cancers is 15%–20% lower than for more common cancers.4 The poor outcomes associated with rare cancers have been attributed to difficulty or delay in diagnosis, limited access to centers with expertise, and limited therapeutic options.4 Despite the significant burden and aggressive nature of these diseases, research initiatives that could lead to the development and approval of new therapies for rare cancers are few. Thus, there is a compelling and urgent need to identify novel therapeutic strategies to improve treatment outcomes in this patient population.
In recent years, the seminal discovery of immune checkpoint inhibitors have revolutionized treatment of cancer.5 6 Pembrolizumab is a humanized IgG4κ monoclonal antibody that targets the programmed cell death (PD-1)/programmed cell death ligand 1 (PD-L1) pathway, which is often hijacked by tumor cells to escape immune surveillance. On the basis of durable objective responses and a favorable safety profile, pembrolizumab has been approved by the US Food and Drug Administration (FDA) for the treatment of several tumor types.7 Accumulating evidence indicates that pembrolizumab produces durable responses in a tumor-agnostic fashion, as it targets the immune system rather than the tumor itself. Recently, the FDA provided its first tissue-agnostic/site-agnostic drug approval—for pembrolizumab in patients with unresectable or metastatic microsatellite instability—high or mismatch repair deficient solid tumors.8 This approval suggests that pembrolizumab may have therapeutic potential in patients with advanced rare cancers. However, the efficacy and the safety profile of the drug in rare cancers are unknown. To address this critical, unmet clinical need and overcome the challenges associated with the rarity of these cancers, we designed an open-label, investigator-initiated, phase 2 basket study of pembrolizumab in patients with advanced rare cancers. As enrolling patients with advanced rare cancers remains a challenge, we chose to publish the findings from the four tumor-specific cohorts (squamous cell carcinoma (SCC) of the skin, adrenocortical carcinoma (ACC), carcinoma of unknown primary (CUP), and paraganglioma–pheochromocytoma) that met the protocol-specified criteria for interim analysis rather than to wait for completion of enrollment in all the 10 cohorts.
Methods
Study design and patients
Patients enrolled in the study from August 15, 2016, to July 27, 2018 (cut-off date) were included in this analysis. Eligible patients were adults 18 years and older, with histologically confirmed advanced rare cancers, whose disease had progressed while on standard therapies (if available) within the previous 6 months. Patients had to have measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) V.1.19 or immune-related RECIST (irRECIST),10 except for cohorts 9 and 10, in which patients could have evaluable disease. Additional inclusion criteria were an Eastern Cooperative Oncology Group performance status of 0 or 111 and adequate organ and bone marrow function. Patients previously treated with an anti-PD-1, anti-programmed cell death ligand (PD-L)1, or anti-PD-L2 agent were excluded from this study. A full list of inclusion and exclusion criteria is in the online supplementary file (eMethods).
Supplemental material
Patients were enrolled in ten cohorts: (1) SCC of the skin, (2) anaplastic thyroid carcinoma, later revised to small cell malignancies of non-pulmonary origin, (3) ACC, (4) medullary renal cell carcinoma, (5) CUP, (6) penile carcinoma, (7) thymic carcinoma, later revised to vascular sarcoma, (8) testicular cancer, later relabeled as germ cell tumor, (9) paraganglioma–pheochromocytoma, and (10) other rare tumor histologies (excluding tumor types included in NCT02054806). As no patients were enrolled in the anaplastic thyroid carcinoma (cohort 2) and thymic carcinoma (cohort 7) cohorts in the first 9 months of the study, the protocol was revised to change these cohorts to include small cell malignancies of non-pulmonary origin and vascular sarcoma, respectively. The testicular cancer cohort (cohort 8) was relabeled as the germ cell tumor cohort to include germ cell tumors arising not only in the testicles but also in other locations.
Procedures
All patients received 200 mg of pembrolizumab intravenously every 3 weeks. Treatment continued until documented disease progression, unacceptable adverse event(s), investigator’s decision to stop treatment, withdrawal of consent, or completion of 24 months of treatment with pembrolizumab. Patients who had radiographic progression but were clinically stable were allowed to continue pembrolizumab until progressive disease (PD) was confirmed by imaging at least 4 weeks after the first PD assessment. Patients who had a confirmed complete response (CR) could discontinue pembrolizumab after receiving at least 27 weeks of treatment. No dose modifications were allowed, but in the event of adverse events dose delays were permitted.
Tumor imaging was performed at baseline and every 9 weeks for the first 6 months of treatment and every 12 weeks thereafter at the discretion of treating physician if patients had had CR, partial response (PR), or stable disease (SD) for more than 27 weeks. irRECIST was applied to direct clinical management and to report the interim results of this study. Response to treatment was also assessed using RECIST V.1.1 to capture the atypical responses observed in patients treated with immunotherapeutic agents. Objective response (OR), defined as irCR or irPR was confirmed by a repeat radiographic assessment at least 4 weeks after the criteria for OR were first met.
The adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events V.4.03.12
Biomarker analysis was performed on fresh tissue sample obtained at baseline or an archival tissue sample. The PD-L1 characterization of baseline tumor samples by immunohistochemistry was performed at a central laboratory on formalin-fixed, paraffin-embedded tissue sections using Merck 22C3 antibody. Based on the percentage and intensity of membrane staining, an H-score ranging from 0 to 300 was assigned. By using recursive partitioning analysis, a score of 42·5 was identified as the optimal cut-off point for PD-L1 H-score. Tumor infiltrating lymphocytes (TILs) within tumor nests were scored on a scale from 0 to 3: 0 for absence of TILs, 1 for a few TILs, 2 for a moderate amount of TILs, and 3 for intense intratumoral lymphocytic infiltration.
Outcomes
The primary endpoint was non-progression rate (NPR) at 27 weeks, defined as the proportion of patients who were alive and progression free at 27 weeks as assessed by irRECIST. The secondary endpoints were safety and tolerability of pembrolizumab; objective response rate (ORR), defined as the percentage of patients with irCR or irPR; clinical benefit rate (CBR), defined as the percentage of patients with irCR, irPR, or irSD ≥4 months; and association of NPR at 27 weeks and baseline PD-L1 status. The exploratory objective was evaluation of the potential of TILs as a predictor of therapy effectiveness. As the study remains open, the final response rates may change with additional follow-up.
Statistical analysis
We used Simon’s optimal two-stage design,13 developed by one of the co-authors (RMS), for each of the nine tumor-specific cohorts. At least three of the first 12 treated patients had to be alive and progression free at 27 weeks before an additional 13 patients were enrolled. The null success rate was set at 20% and the alternative rate at 40% for each cohort. This design has a 10% Type I error rate, 82% power, and 56% probability of stopping after the first stage if the true 27-week progression-free survival rate is 20%. Because the tenth cohort was made up of a heterogeneous mix of rare tumors, the protocol dictated that at least 25 eligible patients would be enrolled. If a positive outcome (alive and progression free at 27 weeks) was observed in a tumor type, that tumor type would be expanded to include an additional 14 patients. With 14 patients, the exact 95% CI for the success rate will have half-width no larger than 0.27.
Clinical efficacy was measured by NPR at 27 weeks. Patients who discontinued the study before 27 weeks for reasons other than disease progression or death were considered non-evaluable for assessment of the primary efficacy endpoint. The best overall response was defined as the best response observed from the start of the treatment until disease progression or discontinuation of treatment for any reason. A waterfall plot was used to illustrate the maximum percentage of change in tumor measurements from baseline per irRECIST. All patients who received at least one dose of pembrolizumab were included in safety analysis. The Pearson χ2 test was used to assess the association between NPR at 27 weeks and baseline PD-L1 expression and TIL scores.
Futility interim analyses were planned for each of the tumor-specific cohorts (cohorts 1–9) after the first 12 evaluable patients enrolled in that cohort had 27-week data available to assess NPR at 27 weeks or when there were three or more responders in a cohort, even when fewer than 12 patients were enrolled.
Results
A total of 128 patients with advanced rare cancers were enrolled between August 15, 2016 and July 27, 2018. One patient enrolled in the ACC cohort was excluded from the study due to change in the diagnosis. Therefore, 127 patients were included in this analysis. The numbers of patients enrolled in each of the ten cohorts at the time of data cut-off and baseline patient characteristics are shown in table 1.
Characteristics of 127 patients with advanced rare cancers treated with pembrolizumab
The primary endpoint, NPR at 27 weeks assessed by irRECIST in 105 patients with advanced rare cancer, was 28% (29 patients; 95% CI, 19% to 37%). NPR at 27 weeks was not assessed in nine patients who had come off treatment prior to 27 weeks due to withdrawal of consent (n=5), patient/investigator choice (n=2), or toxicity (n=2) and in 13 active patients who had not reached week 27 on treatment by the cut-off date. Of the 127 patients, 110 patients were evaluable for response by irRECIST. Seventeen patients were not evaluable as they had either not reached the time of first restaging (n=10) or had come off treatment prior to first restaging due to withdrawal of consent (n=4), patient/investigator choice (n=2), or toxicity (n=1). Four of the 110 patients did not have measurable disease. Two of them were in cohort 10, which allowed for patients with evaluable disease. The remaining two patients with SCC and CUP who were initially said to have measurable disease were found to have evaluable disease on review. However, they were included in the analysis, as they were followed for response using non-target lesions. Fifteen of the 110 patients had OR (one had irCR and 14 had irPR), representing an ORR of 14% (95% CI, 8% to 21%). An additional 27 (25%) patients had SD ≥4 months, representing a CBR of 38% (n=42; 95% CI, 29% to 48%). All the patients with OR had remained on the study for at least 8 months (range, 8·1 to 23·5 months), with 11 of them (73%) continuing on the study at last follow-up.
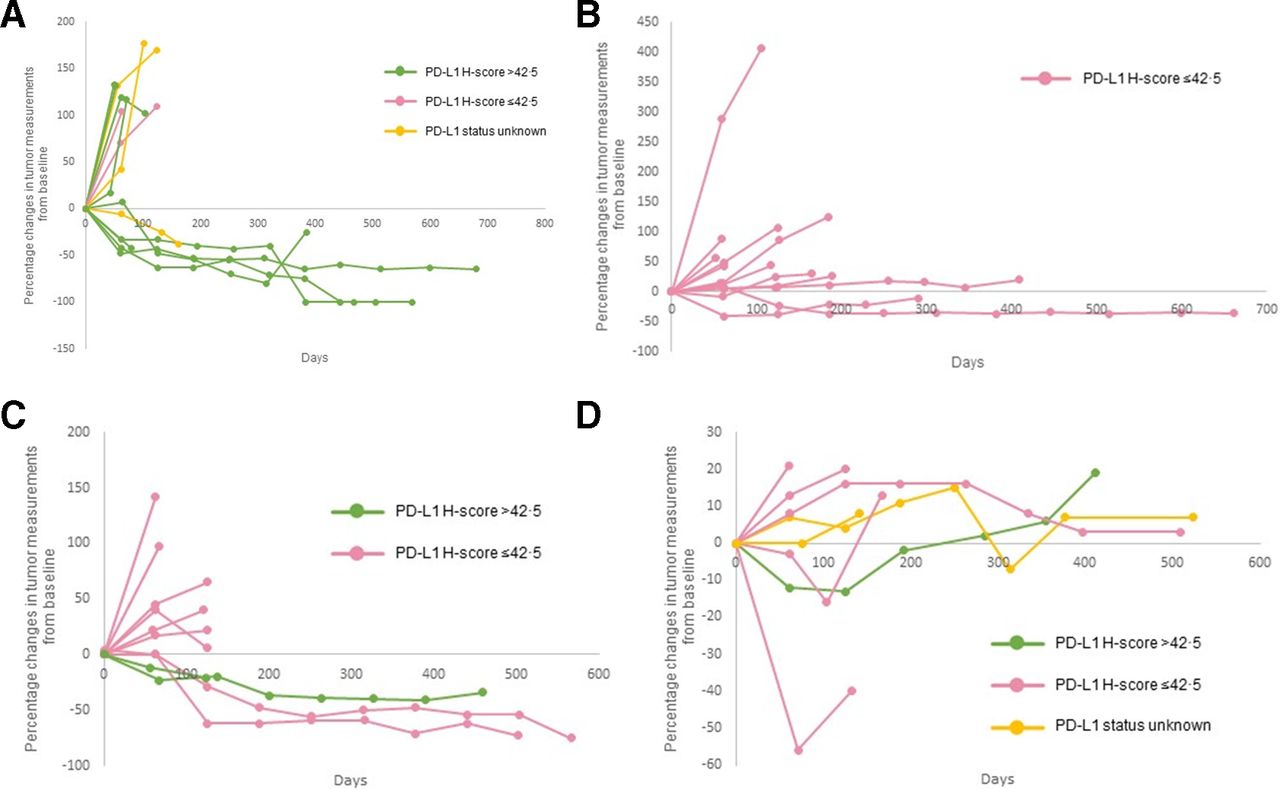
The pre-specified criteria for interim analyses were met in four tumor-specific cohorts: cohort 1 (SCC of the skin), cohort 3 (ACC), cohort 5 (CUP), and cohort 9 (paraganglioma–pheochromocytoma) (table 2). The best overall responses of the evaluable patients are illustrated in figure 1. Percentage changes in tumor measurements from baseline during the course of treatment with pembrolizumab is shown in figure 2. Thirteen additional patients will be enrolled in each of the four cohorts as the criteria for non-futility was met.

Radiological response to pembrolizumab treatment. The figure illustrates the best overall response to pembrolizumab treatment using the immune-related Response Evaluation Criteria in Solid Tumors (irRECIST) in four tumor-specific cohorts. The values shown are the maximum percentage change from baseline in the sum of the longest diameters of all target lesions and any new lesions in patients receiving pembrolizumab. Each bar represents a patient. Five of six patients who derived benefit from continuing on the treatment after an initial assessment of PD by RECIST V.1.1 (pseudoprogression) are represented in the figure. The sixth patient who had pseudoprogression was enrolled in 10th cohort (not shown). Patients with clinical progression of disease were arbitrarily assigned 20% increase in tumor burden. The dashed line indicates the 30% reduction in tumor burden that is consistent with an objective response to treatment according to irRECIST. In squamous cell carcinoma of skin cohort, response data is not shown for three of the 19 patients enrolled in this cohort; two patients came off treatment prior to first restaging due to withdrawal of consent and patient choice. The third patient had not reached the 9-week response assessment period. Two patients with 100% reduction in target lesions were considered as irPR as irCR could not be confirmed in one patient at the time of data cut-off and the presence of non-target lesion in the other patient precluded the assignment of irCR. Two patients with unconfirmed irPR were considered as irSD for the analysis as one patient withdrew consent from the study and another patient came off treatment due to toxicity prior to the date of data cut-off. In the adrenocortical cancer cohort, response data is not shown for two of the 15 patients enrolled in this cohort as they came off treatment prior to first restaging due to toxicity and PI choice. In the carcinoma of unknown primary cohort, response data is not shown for nine patients as two patients came off treatment prior to first restaging due to withdrawal of consent and seven patients had not reached the 9-week response assessment period. In the paraganglioma–pheochromocytoma cohort, response data is not shown for one patient who had not reached the 9-week response assessment period. One another patient with 56% decrease in the size of the target lesions was considered to have irPD due to the presence of new non-target lesions in the liver, which was confirmed in the subsequent scan. CR, complete response; ir, immune-related; PD, progressive disease; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease.

{kind=link}
{kind=link}
Kinetics of tumor measurement during treatment with pembrolizumab. Percentage changes in tumor measurements from baseline per irRECIST during the course of treatment with pembrolizumab by PD-L1 expression levels are shown for all the patients represented in figure 1. Panel (A) squamous cell carcinoma of the skin. Panel (B) adrenocortical carcinoma. Panel (C) carcinoma of unknown primary and Panel (D) paraganglioma–pheochromocytoma. Of the patients represented in figure 1, five patients are not represented in figure 2. Three patients had clinical progression of disease (squamous cell carcinoma of the skin (n=1) and carcinoma of unknown primary (n=2)) and two patients had no measurable disease (squamous cell carcinoma of the skin (n=1) and carcinoma of unknown primary (n=2)). irRECIST, immune-related Response Evaluation Criteria in Solid Tumors; PD-L1, programmed cell death-ligand 1.
Response to treatment with pembrolizumab as assessed by irRECIST in four tumor-specific cohorts
In the cohort of patients with SCC of the skin, 19 patients received at least one dose of pembrolizumab. The NPR at 27 weeks was 36% (5 of 14 evaluable patients; 95% CI, 13% to 65%). Sixteen patients were evaluable for response. Three patients were not evaluable for response as two patients came off treatment prior to first restaging due to withdrawal of consent and patient choice. The third patient had not reached the 9-week response assessment period. One patient had irCR and four had irPR, for an ORR of 31% (95% CI, 11% to 59%). In addition, one of two patients with irSD had disease control for ≥4 months, representing a CBR of 38% (95% CI, 15% to 65%). At the time of data cut-off, four of the five patients with OR had an ongoing response, at 15·9, 17·3, 20·1, and 23·4 months since treatment initiation. Despite a 100% decrease in target lesions, two patients were assessed as irPR as irCR could not be confirmed at the time of data cut-off in one patient and presence of non-target lesions precluded the assignment of irCR in the other patient. Two other patients with unconfirmed irPR were assessed to have irSD as one patient withdrew consent from the study and the other patient had come off the study due to toxicity just prior to the date of data cut-off.
In the cohort of patients with ACC, 15 patients received at least one dose of pembrolizumab. The NPR at 27 weeks was 31% (four of 13 evaluable patients; 95% CI, 9% to 61%). Of the 15 patients, two were not evaluable for response as they came off treatment prior to first restaging due to toxicity and principal investigator (PI) choice. Two of the 13 patients evaluable for response had irPR for an ORR of 15% (95% CI, 2% to 45%). In addition, five of six patients with irSD had disease control for ≥4 months, representing a CBR of 54% (95% CI, 25% to 81%). Of the two patients with an OR, one was still continuing treatment at 23·5 months since treatment initiation.
In the cohort of patients with CUP, 22 patients received at least one dose of pembrolizumab. The NPR at 27 weeks was 33% (four of 12 evaluable patients; 95% CI, 10% to 65%). Of the 22 patients, nine were not evaluable for response as two patients came off treatment prior to first restaging due to withdrawal of consent and seven patients had not reached the 9-week response assessment period. Of the 13 patients evaluable for response, three had irPR, for an ORR of 23% (95% CI, 5% to 54%), and all three patients had ongoing responses, at 16·7, 17·6, and 21·3 months. In addition, four patients had irSD ≥4 months, representing a CBR of 54% (95% CI, 25% to 81%).
In the cohort of patients with paraganglioma-pheochromocytoma, nine patients received at least one dose of pembrolizumab. The NPR at 27 weeks was 43% (three of seven evaluable patients; 95% CI, 10% to 82%). At the time of data cut-off, one patient had not reached the 9-week response assessment period. Of the eight patients evaluable for response, six patients had irSD, and all of them had disease control for ≥4 months, representing a CBR of 75% (95% CI, 35% to 97%). Three of these six patients were still continuing treatment at 5·0, 18·5, and 19·8 months after treatment initiation. One patient with 56% decrease in target lesions was considered to have irPD due to the presence of new non-target lesions in the liver, which was confirmed in the subsequent scan.
Responses were also seen in other cohorts. irPR was seen in penile carcinoma cohort (n=1) and other rare histologies (n=4; cancer of the orbit, pituitary carcinoma, alveolar soft part sarcoma, and epithelioid neoplasm of the dermis). In addition, irSD ≥4 months was seen in the cohorts for vascular sarcoma (n=2), germ cell tumor (n=3), and other rare histologies (n=6; patients with granulosa cell tumor of the ovary (n=2), mesothelioma in the testicle, intracranial meningioma, sclerosing epithelioid fibrosarcoma, and neuroblastoma (n=1 each)).
Six of 34 patients who continued treatment after an initial assessment of PD by RECIST V.1.1 derived benefit. Two of them with irPR as their best overall response (75% decrease and 73% decrease) continue to receive treatment at week 91 and 75 since treatment initiation. The other four patients had 41% decrease, 38% decrease, 26% decrease, and 16% decrease as best overall response before discontinuing treatment at week 42, 25, 32, and 25 respectively.
Of the 127 patients who received at least one dose of pembrolizumab, 119 (94%) experienced at least one treatment-emergent adverse event (online supplementary table 1). A total of 146 treatment-related adverse events (TRAEs) of any grade occurred in 66 (52%) patients (table 3). Sixteen TRAEs grade ≥3 occurred in 12 (9%) patients. The most common TRAEs (occurred in ≥10% of the patients) were fatigue (n=25), maculopapular rash (n=17), and hypothyroidism (n=14). The most common TRAEs of grade ≥3 were anemia (n=4), pneumonitis (n=3), and increased alanine aminotransferase level (n=2). Six deaths (5%) were reported during the study, five due to PD and one due to acute kidney injury secondary to sepsis; none were treatment-related. Treatment-related irAEs of any grade occurred in 24 (19%) patients; treatment-related irAEs were grade ≥3 in eight (6%) patients (online supplementary table 2). The most common irAEs were hypothyroidism (n=14), pneumonitis (n=4), and hyperthyroidism (n=4). The only irAE grade ≥3 that occurred in more than one patient was pneumonitis.
Treatment-related adverse events in 127 patients treated with pembrolizumab
PD-L1 membrane expression and presence of TILs were assessed in tumor samples from 105 patients (online supplementary table 3). Of these 105 patients, 91 had data on primary endpoint. Seventy-four of the 91 (81%) patients had a PD-L1 H-score ≤42·5, while 17 had a score >42·5. A higher proportion of the patients with a PD-L1 H-score >42·5 were alive and progression free at 27 weeks (8/17 (47%), compared with 15/74 (20%) for H-score ≤42·5; p=0·02). TIL score was not associated with the primary endpoint (p=0·99). Three of the four (75%) patients with a TIL score of 0, eight of the 42 (19%) with a TIL score of 1, five of the 22 (23%) with a TIL score of 2, and seven of the 23 (30%) with a TIL score of 3 were alive and progression free at 27 weeks.
Discussion
The objective of this histology-independent, phase 2 study was to identify promising signals of antitumor activity of pembrolizumab across different rare cancers. As delayed response is characteristically seen with immunotherapeutic agents, landmark survival estimates have been widely used for reporting treatment outcomes with immunotherapy as conventional endpoints may not be ideal.14 15 In our study as imaging was performed at baseline and at 9-week intervals, the primary endpoint NPR at 27 weeks allowed us to observe the tumor for three imaging cycles, which would give us a fair estimate of the drug activity that can be followed up in later studies. Antitumor activity was observed in cohorts of patients with SCC of the skin, ACC, CUP, and paraganglioma-pheochromocytoma.
SCC of the skin accounts for 20% of non-melanoma skin cancers.16 While surgical excision is curative in localized SCC of the skin, no effective treatment was available for locally advanced or metastatic disease,17 until cemiplimab, a PD-1 inhibitor, was recently FDA-approved for this indication.18 Though ORR of 47% was reported in patients with metastatic SCC of the skin treated with cemiplimab, 68% of patients either had received no prior systemic therapy or only one line of prior systemic therapy with curative intent. Interim results from two other phase 2 studies of pembrolizumab in patients with SCC of the skin have been reported. An ORR of 42% at 15 weeks has been reported with pembrolizumab as first line of therapy in 17 evaluable patients with unresectable SCC of skin.19 In another study, an ORR of 40% has been reported in 10 patients with metastatic SCC of skin, majority of whom have not received prior systemic therapy.20 Thus, findings from our study (ORR of 31% and CBR of 38%) is noteworthy given that all of our patients had refractory disease and 88% of them had received ≥1 prior systemic therapy. Studies estimate that SCC of the skin causes a similar number of deaths to melanoma.21 Given that the incidence of SCC of the skin has risen dramatically, by up to 200%, in the past three decades in the USA,16 22 the mortality rate is likely to rise as well, indicating that an effective therapeutic option is needed for this underserved population. Therefore, the above findings indicate that pembrolizumab may be a potential therapeutic option for patients with SCC of the skin, both in treatment naïve and refractory setting.
ACC is an orphan endocrine malignancy characterized by a poor prognosis and limited response to chemotherapy.23 Most patients with ACC experience rapid disease progression, and the median overall survival (OS) remains dismal at 9 months for patients with stage IV disease.24 Mitotane, the only FDA-approved agent for ACC, is often used in combination with etoposide, doxorubicin, and cisplatin.25 However, treatment with mitotane and chemotherapy is associated with considerable toxicity that frequently makes it difficult for patients to continue therapy. Data about immunotherapy in ACC are limited, despite reported PD-L1 expression in 11% of 28 surgically resected primary ACC tumors.26 Recently, 6% ORR was reported in patients treated with avelumab, but almost half of that study’s subjects had concomitant therapy with mitotane, which clouds the interpretation of the efficacy of avelumab.27 In this context, the ORR of 15% and CBR of 54% in our study using single-agent pembrolizumab are clinically meaningful. Given that all therapies for metastatic ACC are considered palliative at best,23 the NPR of 31% in our ACC cohort is encouraging.
CUP accounts for 2% of all solid cancers and is associated with poor prognosis. The 2-year survival rate has been reported to be less than 20% with empiric platinum-based combination regimens.28 In the absence of standard of care or FDA-approved agents to treat CUP, there is a critical and unmet need to develop novel therapeutic strategies to treat CUP. Recently, 22·5% of 389 patients with CUP were reported to have PD-L1–positive tumor cells, 58·7% PD-1–positive TILs, and 11·8% a high total mutation load.29 Despite the presence of these promising indicators of response, with the exception of a few case reports,30–32 the role of checkpoint inhibitors has not been investigated in patients with CUP. As such, the results from our study showing early signs of clinical activity resulting in an NPR of 33% at 27 weeks, ORR of 23%, and CBR of 54% are encouraging and must be explored further.
Metastatic pheochromocytomas and paragangliomas are rare neuroendocrine tumors associated with poor prognosis.33 High-specific-activity meta-iodine benzyl guanidine (MIBG) is the only FDA-approved systemic therapy for the treatment of these patients, and it is only effective for the roughly half of patients with MIBG-avid tumors.34 Treatment with the chemotherapeutic agents cyclophosphamide, vincristine, and dacarbazine is at best palliative and is associated with a clinical benefit in approximately 37% of patients.35 Several antiangiogenic agents, such as sunitinib, pazopanib, axitinib, and cabozantinib, are under investigation. Given that PD-L1/PD-L2 were expressed in 50% of the tumors surgically removed,36 NPR of 43% and CBR of 75% in our study, provides a rationale for the development of pembrolizumab for these tumors.
Evaluation of the efficacy of immunotherapeutic agents remains a challenge, as the transient increase in the size of the tumor and/or the development of new lesions due to the inflammatory response (also termed pseudoprogression) may be misdiagnosed as PD by conventional RECIST V.1.1, the only imaging assessment criteria accepted by the FDA for drug approval.37 In our study, adaptations made to RECIST V.1.1 allowed 34 patients to continue treatment beyond the first assessment of PD by RECIST V.1.1, six (18%) of whom derived benefit. These patients would have been removed prematurely from the study by RECIST V.1.1. Nevertheless, for patients with true PD, such a wait to confirm PD delays switching to more effective therapy and exposes these patients to an unnecessary risk of toxicity. Therefore, it is critical to develop a biomarker to distinguish in real time between patients with atypical responses and those with true PD.
Currently, there are no validated markers of response to treatment, and the predictive potential of PD-L1 as a biomarker is much debated. Different cut-off values such as ≥1%, ≥5%, ≥10% and≥50% have been used in different studies as summarized by Diggs and Hsueh38 to stratify patients by PD-L1 expression. In the absence of a standardized cut-off value for assessing PD-L1-positivity, we had used recursive partitioning analysis to identify the cut-off value for PD-L1 H-score. Therefore, our finding that a higher proportion of patients with a PD-L1 H-score ≥42·5 were alive and progression free at 27 weeks should be considered preliminary. Nevertheless, given that pembrolizumab has been approved for first-line treatment of advanced non-small cell lung cancer in patients with high PD-L1 expression according to an FDA‐approved test in the absence of EGFR or ALK molecular alterations,39 PD-L1 expression likely reflects an immune-active tumor milieu that should be investigated further. Contrary to the conventional belief that the presence of TILs is a favorable prognostic indicator of response to treatment with checkpoint inhibitors, in our study TIL score was not associated with NPR at 27 weeks. Though unexplained, similar findings have been reported in a phase 2 biomarker study in patients with unresectable melanoma treated with ipilimumab40 and in patients with melanoma, non-small cell lung cancer, renal cell carcinoma, colorectal carcinoma, or castration-resistant prostate cancer treated with nivolumab.41 One possible explanation could be the presence of other immunosuppressive factors such as indoleamine 2,3-dioxygenase (IDO) and arginase in the tumor microenvironment. Thus, additional investigations are needed to identify immune correlates of response to treatment with pembrolizumab.
The safety profile of pembrolizumab in patients with advanced rare cancer is consistent with that previously reported for other common cancers, such as melanoma and non-small cell lung cancer.
We acknowledge that our study has a few limitations. First, the small sample size of the tumor-specific cohorts precluded us from making inferences applicable to all rare cancers. Second, the site from which the tissue was procured (primary vs metastatic) for biomarker assessment may have influenced the immune marker levels. Third, the differences in antitumor activity between different cancers is likely a reflection of differences in the tumor microenvironment. Nonetheless, because these are rare tumors, the period until the final analysis may be prolonged. Considering the poor prognosis associated with the lack of evidence-based treatment options for many of these tumor types, we chose to report the results from the interim analysis to inform the scientific community. In conclusion, the current study provides early indications of antitumor activity of pembrolizumab in four tumor-specific cohorts of patients with SCC of the skin, ACC, CUP, and metastatic pheochromocytoma–paraganglioma. This finding is significant given that these patients had experienced disease progression while on another treatment within the previous 6 months or had no standard-of-care therapies available. Findings from our study support further investigation to confirm the clinical activity of pembrolizumab in advanced rare cancers and to identify immune signatures predictive of response to treatment.
Acknowledgments
Sunita Patterson and Amy Ninetto, Department of Scientific Publications at MD Anderson, provided editorial assistance. We thank the patients and their families and caregivers for participating in the study.
References
Footnotes
Twitter @AnaingMD
Deceased Deceased: July 26, 2019 after approval of the final draft of the manuscript
Contributors AN contributed to study design, conduct of the study, enrollment of patients, data and biospecimen collection, data analysis, data interpretation, and writing of the manuscript; FM-B, JH contributed to study design, data interpretation and writing; BS contributed to literature search, data collection, data analysis, data interpretation and writing the manuscript; DDK, SAP-P, AMT, EEID, SP, FJ contributed to acquisition and interpretation of data; JRA, TAY contributed to acquisition, and interpretation of data and provided critical feedback for intellectual content of manuscript; RRC collected and reviewed the radiographic data, performed the quantitative imaging analysis, interpreted the data; CJ contributed to study design, data collection, data analysis, data interpretation and writing the manuscript; KPR contributed to patient accrual, and data interpretation; RF contributed to data analysis, data interpretation and writing the manuscript; S-MT contributed to protocol design and patient selection; MC contributed to patient accrual, data analysis, data interpretation, and writing; LW contributed to biostatistics data analysis; SHS contributed to patient selection and data collection; CT contributed to data collection; CB reviewed the manuscript and provided feedback; MF contributed to recruitment of patients and writing of manuscript; NT contributed to data collection and data interpretation; VR contributed to patient accrual, and data interpretation; SK contributed to data collection and tabulation; JMP helped the investigators to assess patients for protocol eligibility, consent and enroll patients on the study, supervised the acquisition of biospecimens from patients and provided documentation in the electronic medical record per protocol and institutional requirements; AA (Abonofal) contributed to data collection, tabulation and writing; JG contributed to clinical research team daily operation to ensure patient safety and quality of research data; AA (Alshawa) contributed to data collection, data interpretation, and writing; LMM contributed to protocol development and regulatory affairs; MX contributed to data collection and data analysis; SA collected and reviewed the radiographic data and performed the quantitative imaging analysis; VS contributed to study design, provision of study materials, patient enrollment, data analysis, and data interpretation; DSH contributed to data collection, data analysis and data interpretation; SF contributed to data acquisition, data analysis, data interpretation and writing; RMS contributed to statistical analysis; KRH contributed to study design, data analysis and data interpretation; GRV contributed to patient enrollment; MAH contributed to patient accrual, data collection, data analysis, data interpretation, and writing. All authors critically reviewed and approved the final manuscript.
Funding Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. provided the study drug and funded the study. Support was also provided by the National Institutes of Health/National Cancer Institute under award number P30CA016672 (used the Biostatistics Resource Group) and The University of Texas MD Anderson Cancer Center through the Molecular Evaluation and/or Biopsy Related Support Program (used for performing biopsy in select patient cohorts).
Competing interests AN reports research support and non-financial support from Merck Sharpe grants from NCI, research support from EMD Serono, MedImmune, Healios Onc. Nutrition, Atterocor, Amplimmune, Armo BioSciences, Karyopharm Therapeutics, Incyte, Novartis, Regeneron, Merck, Bristol Myers Squibb, Pfizer, CytomX Therapeutics, Neon Therapeutics, Calithera BioSciences, TopAlliance BioSciences, Eli Lilly, Kymab, and PsiOxus, non-financial support for travel and accommodation from Armo BioSciences, and has served as an advisory board member for Novartis and CytomX Therapeutics outside the submitted work; FM-B reports grants from Novartis/Aduro, Calithera, Bayer, Jounce, CytoMx, eFFECTOR, PUMA Biotechnology, Curis, Millennium, GlaxoSmithkline, Daiichi Sankyo, Abbvie, Guardant Health, Takeda, and Aileron, personal fees for advisory from Inflection Biosciences, Darwin Health and Spectrum, personal fees for consulting from GRAIL, Clearlight Diagnostics, Dialectica, Samsung Bioepis, Aduro, Xencor, Jackson Laboratory, personal fees from OrigiMed, Kolon Life Science and Parexel International, personal fees for consulting/travel related from Pieris, Sumitomo Dainippon, and OrigMed, personal fees for advisory/travel related from Mersana, grants and personal fees for travel related from Taiho, grants and personal fees for Consulting/travel related from Genentech, Debio, and Pfizer, grants and personal fees for consulting from Zymeworks, grants and personal fees for advisory from Seattle Genetics, grants from AstraZeneca outside the submitted work; JH reports grant from Immune Deficiency Foundation, outside the submitted work; JRA reports personal fees from Novartis, Eli Lilly, Orion Pharmaceuticals, Servier Pharma, Peptomyc, and Merck Sharpe, on the advisory board for Novartis, Eli Lilly, Orion Pharmaceuticals, Servier Pharma, Peptomyc, Merck Sharpe & Dome, Kelun Pharma/Klus Pharma, Pfizer, Roche Pharma, and Elipses Pharma, research funding from Bayer, Novartis, Spectrum Pharmaceuticals, Tocagen, Symphogen, BioAtla, Pfizer, GenMab, CytomX, KELUN-BIOTECH, Takeda-Millenium, GlaxoSmithkline, Ipsen, from null, outside the submitted work. SAP-P reports grants from AbbVie, Inc., Aminex Therapeutics, BioMarin Pharmaceutical, Inc., Boehringer Ingelheim, Bristol Myers Squibb, Cerulean Pharma, Inc., Chugai Pharmaceutical Co., Ltd, Curis, Inc., Five Prime Therapeutics, Flex Bio, Inc., Genmab A/S, GlaxoSmithkline, Helix BioPharma Corp., Incyte Corp., Jacobio Pharmaceuticals Co., Ltd, Medimmune, LLC, Medivation, Inc., Merck Sharpe & Dome Corp., NewLink Genetics Corporation/Blue Link Pharmaceuticals, Novartis Pharmaceuticals, Pieris Pharmaceuticals, Inc., Pfizer, Principia Biopharma, Inc., Puma Biotechnology, Inc., Seattle Genetics, Taiho Oncology, Tesaro, Inc., Transthera Bio, and XuanZhu Biopharma, outside the submitted work; RF reports personal fees for serving on advisory board from Ayala and Regeron-Sanofi, personal fees for consultation from Cellestia, and other from Merck, outside the submitted work; MC reports personal fees for consulting from Pfizer Inc., Genentech, Inc., and Apricity Health LLC, personal fees for serving as scientific/advisory committee member from EMD Serono, Inc., and Genentech, Inc., outside the submitted work; SHS reports personal fees from Angiodynamics, non-financial support from Neuwave Medical, Medtronic, and Merit Medical, outside the submitted work; CT reports salary support from Merck, during the conduct of the study; salary support from Merck, and for contract work to perform correlatives from Armo Bioscience, outside the submitted work; MF reports personal fees and non-financial support for speaking engagements and research funding from Stryker, personal fees for serving on advisory board from Biom’Up, Genetech, and Ipsen, outside the submitted work; VS reports clinical trial research funding from Novartis, Bayer, GlaxoSmithkline, Nanocarrier, Vegenics, Celgene, Northwest Biotherapeutics, Berghealth, Incyte, Fujifilm, Pharmamar, D3, Pfizer, Multivir, Amgen, Abbvie, Alfa-sigma, Agensys, Boston Biomedical, Idera Pharma, Inhibrx, Exelixis, Blueprint medicines, Loxo oncology, Takeda and Roche/ Genentech, National Comprehensive Cancer Network, NCI-CTEP and UT MD Anderson Cancer Center, outside the submitted work; DSH reports research/grant funding from Abbvie, Adaptimmune, Amgen, Astra-Zeneca, BMS, Daiichi-Sankyo, Eisai, Fate Therapeutics, Genmab, Ignyta, Kite, Kyowa, Lilly, Medimmune, Merck, Merrimack, Mirati, MIRNA, Molecular Templates, Mologen, NCI-CTEP, Novartis, Pfizer; personal fees from Axiom, Baxter, GLG, Group H, Guidepoint Global, Jannsen, Medscape, Numab, Trieza Therapeutics; research/grant funding and personal fees from Bayer, Genentech, Infinity, LOXO, Seattle Genetics, Takeda; and other from Molecular Match, OncoResponse, Presagia Inc, during the conduct of the study; SP reports personal fees and other for financial relationship/speakers bureau consultant from Tyme, Inc., and 4-D Pharma, outside the submitted work; TAY reports personal fees and other for research support, consulting, speakers bureau from AstraZeneca and Pfizer, personal fees and other for research support, consulting from Bayer, Seattle Genetics, and Vertex Pharmaceuticals, personal fees and other for research support, speakers bureau from Tesaro, personal fees for consultant, speakers bureau from Merck, research support from Jounce, Eli Lilly and Kyowa, personal fees for consultant services from Aduro, Almac, Atrin, Bristol-Meyers Squibb, Calithera, Clovis, Cybrexa, EMD Serono, Ignyta, Jansen, and Roche, outside the submitted work; AMT reports grants from NIH/NCI, during the conduct of the study; grants from EMD Serono, Boston Biomedical, Inc., Verastem Oncology, Karus Therapeutics, Ltd., Immatics Biotechnologies, CPRIT, Tvardi Therapeutics, OBI Pharma, Parker Institute, Tempus, Foundation Medicine, and Placon Therapeutics, for consulting/advisory role from Genentech, Roche Europe, and Covance, outside the submitted work; FJ reports grants from Novartis, Genentech, BioMed Valley Discoveries, Plexxikon, Piqur, Symphogen, Bayer, and Fujifilm Corporation and Upsher-Smith Laboratories, research funding & SAB from Deciphera, SAB from IFM Therapeutics, Synlogic, Gaurdant Health, services as paid consultant & ownership interests in Trovagene, and paid consultant in Immunomet, outside the submitted work; SF reports clinical trial research support from Polaris Pharmaceuticals, Inc., Takeda., Lilly, Astra Zeneca, Endocyte, Novartis NIH/NCI, Aprea Therapeutics, Aneropharma Science, OncoMed Pharmaceuticals, Huya Bioscience International, Parexel International, LLC, Medivir AB, New Pharma, Inc, BioAtla LLC, MacroGenics, BeiGene, IMV, Inc, and Tolero Pharmaceuticals, outside the submitted work; RMS reports fees for consulting services from Amgen, Bristol-Myers Squibb, Jansen, Abbvie, Pfizer, Innocrin Therapeutics, Tessa Therapeutics during the conduct of the study; MAH reports grants from Exelixis Inc, grants and personal fees from Eisai Inc, and HRA Pharma, outside the submitted work. BS, DDK, RRC, CJ, KPR, S-MT, LW, CB, NT, VR, SK, JMP, AA (Abonofal), JG, AA (Alshawa), LMM, MX, SA, EEID, KRH, and GRV declare no competing interests.
Patient consent for publication Not required.
Ethics approval The protocol was approved by the FDA and the Institutional Review Board at The University of Texas MD Anderson Cancer Center. The study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. All the study participants provided written informed consent before enrollment.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request and approval from study sponsor according to available guidelines at time of request.